
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation of biologically important peptides and proteins: inhibition or acceleration depending on protein and metal ion concentrations
Benjamin Gabriel Poulson†
a,
Kacper Szczepski†a,
Joanna Izabela Lachowiczb,
Lukasz Jaremko*a,
Abdul-Hamid Emwas*c and
Mariusz Jaremko *a
*a
aDivision of Biological and Environmental Sciences and Engineering (BESE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: mariusz.jaremko@kaust.edu.sa; lukasz.jaremko@kaust.edu.sa
bDepartment of Medical Sciences and Public Health, University of Cagliari, Cittadella Universitaria, 09042 Monserrato, Italy
cCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: abdelhamid.emwas@kaust.edu.sa
First published on 24th December 2019
Abstract
The process of aggregation of proteins and peptides is dependent on the concentration of proteins, and the rate of aggregation can be altered by the presence of metal ions, but this dependence is not always a straightforward relationship. In general, aggregation does not occur under normal physiological conditions, yet it can be induced in the presence of certain metal ions. However, the extent of the influence of metal ion interactions on protein aggregation has not yet been fully comprehended. A consensus has thus been difficult to reach because the acceleration/inhibition of the aggregation of proteins in the presence of metal ions depends on several factors such as pH and the concentration of the aggregated proteins involved as well as metal concentration level of metal ions. Metal ions, like Cu2+, Zn2+, Pb2+ etc. may either accelerate or inhibit aggregation simply because the experimental conditions affect the behavior of biomolecules. It is clear that understanding the relationship between metal ion concentration and protein aggregation will prove useful for future scientific applications. This review focuses on the dependence of the aggregation of selected important biomolecules (peptides and proteins) on metal ion concentrations. We review proteins that are prone to aggregation, the result of which can cause serious neurodegenerative disorders. Furthering our understanding of the relationship between metal ion concentration and protein aggregation will prove useful for future scientific applications, such as finding therapies for neurodegenerative diseases.
1. Introduction
The rate of aggregation of proteins depends strongly on the concentration of the aggregating proteins, but this relationship is not always straightforward.1 This dependence is also true for the most common protein in human blood, albumin (HSA, at concentrations of ca. 0.63 mM), which is a universal carrier of various substances in the blood of organisms, including metal ions in their complex forms.2 HSA aggregation, which normally does not occur under physiological conditions, is induced by the presence of metal ions such as Co2+, Cr3+ and Ni2+ (with a metal ion ratio up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 at pH = 7.3), with Cr3+ promoting the strongest aggregation rate.3 Metal ions like Cu2+ participate in pathological transformations that lead to aggregation, such as prion (PrPC) proteins for example, which bind to tandem octapeptide repeats,4–6 leading to numerous severe neurological pathologies.7–10 Recently, some authors have postulated that on the molecular level, the N-terminal domain of PrPC may act as a toxic effector whose activity is normally auto-inhibited by metal ion-assisted intramolecular association with the C-terminal domain.6 Therefore, it should be pointed out that at the higher concentrations of Cu2+ ion, the individual tandem repeats are able to coordinate with different geometries up to a total of four Cu2+ ions, mainly by imidazole rings of histidine, together with the amide nitrogen of these residues,6,11 as well as most likely by tryptophan side-chains,4 preventing the PrP molecule from misfolding into the pathological PrPC form4,11,12 with weaker micromolar affinity,6 suggesting that the influence of the Cu2+ ions on the transformation of the prion protein into its pathological forms depends on the concentration of their free accessible form in solution.6 On top of that, it is still not known, if PrP binds Cu2+ ions within the positive or negative cooperativity effects.11
:8 at pH = 7.3), with Cr3+ promoting the strongest aggregation rate.3 Metal ions like Cu2+ participate in pathological transformations that lead to aggregation, such as prion (PrPC) proteins for example, which bind to tandem octapeptide repeats,4–6 leading to numerous severe neurological pathologies.7–10 Recently, some authors have postulated that on the molecular level, the N-terminal domain of PrPC may act as a toxic effector whose activity is normally auto-inhibited by metal ion-assisted intramolecular association with the C-terminal domain.6 Therefore, it should be pointed out that at the higher concentrations of Cu2+ ion, the individual tandem repeats are able to coordinate with different geometries up to a total of four Cu2+ ions, mainly by imidazole rings of histidine, together with the amide nitrogen of these residues,6,11 as well as most likely by tryptophan side-chains,4 preventing the PrP molecule from misfolding into the pathological PrPC form4,11,12 with weaker micromolar affinity,6 suggesting that the influence of the Cu2+ ions on the transformation of the prion protein into its pathological forms depends on the concentration of their free accessible form in solution.6 On top of that, it is still not known, if PrP binds Cu2+ ions within the positive or negative cooperativity effects.11
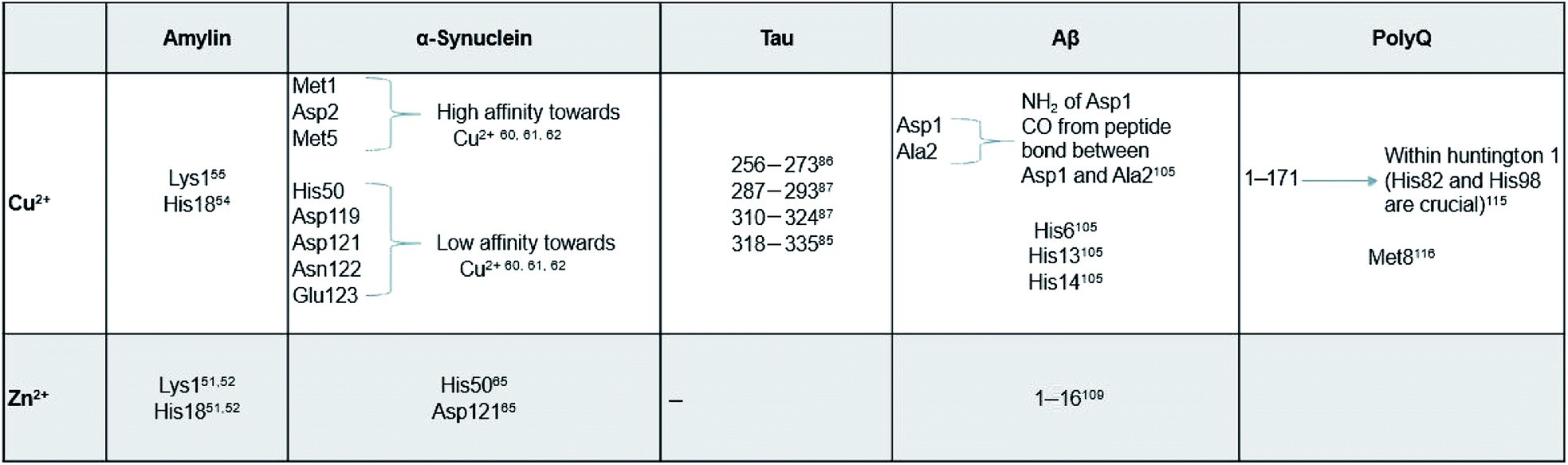
Here, we review a number of biomolecules whose aggregation rates are dependent on their concentration and metal ion coordination properties. The biomolecules reviewed are the following: islet amyloid polypeptide (IAPP), which contributes to glycemic control and has implications for Type II diabetes,13,14 Aβ peptide and Tau protein, which are the main components of amyloid deposits found within the neuronal cells of patients with Alzheimer's disease (AD).15,16 α-Synuclein, which is strongly associated with Parkinson's disease (PD).17 Table 1 lists select metal ions and their binding sites to the proteins discussed in this review. Schemes 1 and 2 give a visual representation of these binding sites.
 |
 | ||
| Scheme 1 Graphical representation of residual binding sites of Cu2+ and their respective proteins. (Bolded text represents the PDB IDs of the proteins). | ||
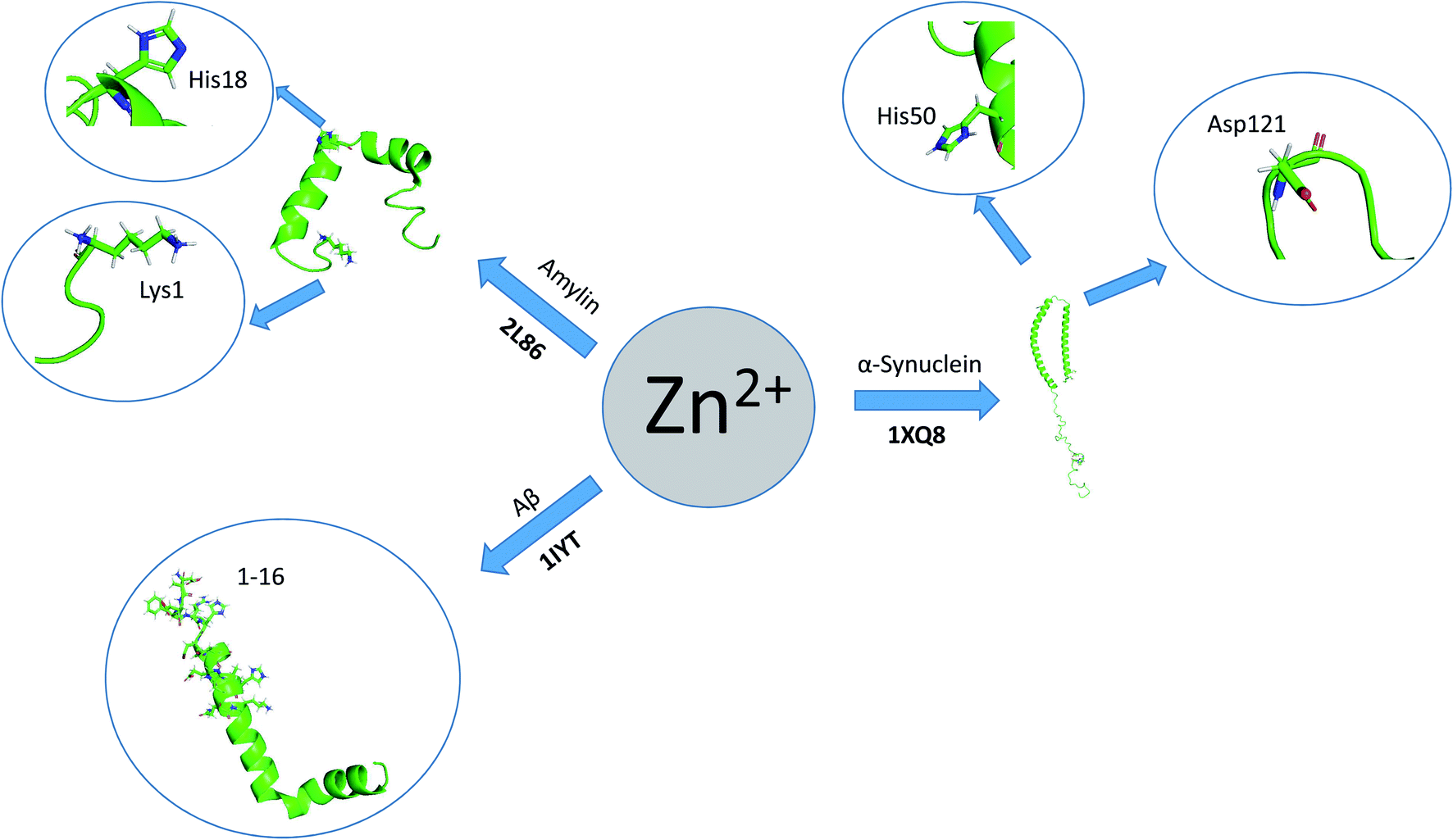 | ||
| Scheme 2 Graphical representation of residual binding sites of Zn2+ and their respective proteins. (Bolded text represents the PDB IDs of the proteins). | ||
2. General conditions of peptide aggregation
There are more than 20 amyloid diseases‡ characterized by the deposition of amyloid fibrils and plaques in central nervous system (CNS) and in some peripheral tissues.18 Moreover, there are other misfolding/conformational pathologies (e.g. cystic fibrosis, Marfan syndrome, amyotrophic lateral sclerosis), featured by the presence of “wrongly” folded proteins (with respect to non-pathological conditions).19 Also, in some cancer cells, certain proteins have “incorrect” structure. Surprisingly, amyloid fibrils and plaques are more toxic at the early stages of polymerization rather than the final product.18At the beginning the protein aggregates are soluble, but gradually become insoluble when they exceed solubility limits. Protein–protein interactions in the aggregates can be electrostatic and/or hydrophobic and can lead to minor conformational changes. Lowering the surface charge of protein can increase aggregation. Most aggregation processes are nucleation-dependent.20
The primary amino acid sequence of proteins is an inherent feature of aggregation processes.21 In many aggregation processes, the initial reaction is the formation or exchange of intermolecular disulfide bond.22 Cysteines located on the protein surface are more easily involved in the aggregation than cysteine residues in the inert part. The disulphide bond aggregation of human serum albumin was studied by Wetzel et al., (1980) who showed that unfolding of the pocket containing the free –SH group of cysteine-34 prevent the formation of disulphide bridges and leads to stable aggregates and irreversible structural alterations.23
Amyloids share common structure (high β-sheet content)24 and the aggregation process occurs in the extracellular space of the CNS (e.g. Alzheimer's and Creutzfeldt–Jakob diseases), and some peripheral tissues and organs (e.g. liver, heart and spleen-systemic amyloidosis and type II diabetes).25,26 Primary or secondary amyloidosis, can also be found in skeletal tissue and joints (e.g. haemodialysis-related amyloidosis) and in some organs (e.g. heart and kidney). Surprisingly, the plaques' formation is less frequent in peripheral nervous system.
Up to know it is not well established, whether protein aggregation is the cause or consequence of the pathologies. Moreover, early amyloid plaques are similar structurally to pores made of bacterial toxins and pore-forming eukaryotic proteins, which suggests the functional significance of such plaque constructions.18
Aggregation occurs when the normal protein folding machinery does not work correctly. Such black out can be caused by specific mutations, which enhanced protein synthesis or reduced their clearance. Molecular chaperones that process the protein degradation prevents pathologies in normally functioning organisms. Different degenerative diseases have been associated with deterioration of the ubiquitin-proteasome pathway (Alzheimer's disease, Fronto-temporal dementia, Parkinson's disease, dementia with Lewy body, amyotrophic lateral sclerosis, poly-Q extension disorders, Huntington's disease, spinocerebellar ataxias, spinobulbar muscular atrophy).27 It was also shown that 30–33% macromolecular crowding, which can be a result of ageing28 or of progression through the cell cycle,29 can lead to higher molecular binding affinities.30 Amyloid diseases are manifest most frequently late in lifespan, when aging leads to DNA methylation. It could be deduced that DNA changes lead to up-regulation of the expression of some proteins, which in turn accumulate and aggregate inside cells.18
More often protein aggregation is a result of wrong interactions with metal ions, local changes in environmental conditions (e.g. pH, temperature, ionic strength) (Scheme 3) or chemical modification (oxidation, proteolysis). There are five main environmental conditions that influence the aggregation process, and they are directly (temperature and pH) or indirectly (pH and concentration) correlated. It was shown in the experimental studies that even small variation of environmental factors can significantly change the final results. Jha et al., (2014) demonstrated that the amylin fibrillization is directly related to the pH, which is physiologically important.31
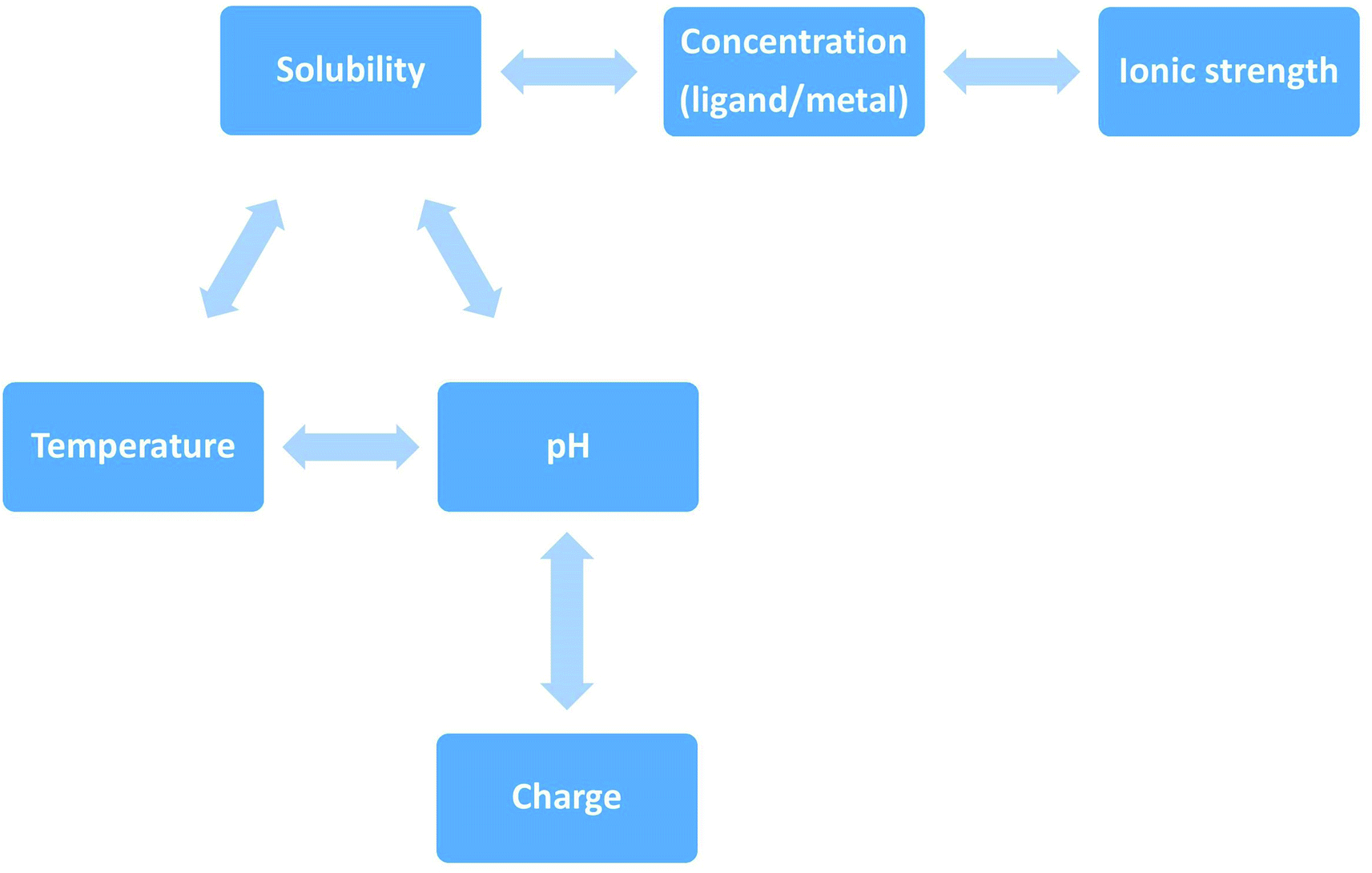 | ||
| Scheme 3 Direct and indirect correlation of environmental factors that influence peptides' aggregation. The solubility of a given solute in a given solvent typically depends on temperature. Depending on the nature of the solute the solubility may increase or decrease with temperature. For most solids and liquids, their solubility increases with temperature. Ionic compounds have limited water solubility, and the amount of soluble products is defined by the solubility product (Ksp). This value depends on the type of salt, temperature, and the common ion effect. Ksp depends directly on ions activity, which is related to the activity coefficient and ion concentration. The pH–solubility profile of a weak acid or base is shown to be a function of its pKsp, and pKa, and uncharged species solubility and was widely described by Streng et al. (1984).34 | ||
Also the final structure of plaques depends on the environmental conditions.32 The pH determines the type and the density of surface charge and the degree of protein structural disruption. Moreover, pH affects intramolecular folding and protein–protein interactions.20 Protein concentration is another important factor in aggregation process, while enhancing protein association or lead to the protein precipitation when it exceeds solubility limit. It is noteworthy that the sequence of the peptide affects its propensity to form or not amyloid structures under specific conditions: aggregation through unfolding intermediates and unfolded states (e.g. protein translocation through the membranes); or aggregation through protein self-association.20 Partially unfolded peptides exhibit hydrophobic sequences and have higher elasticity with respect to the folded state, thus have enhanced susceptibility to aggregation process.33
Bearing in mind arguments described above, it is necessary to conduct the in vitro experiments in the conditions similar as much as possible to that in the physiological conditions.
3. Islet amyloid poly-peptide (hIAPP), amylin
Islet amyloid polypeptide (IAPP) is a specific protein hormone consisting of 37 amino acids (3.9 kDa) in its native form, with the C-terminus amidated, and with a disulfide bridge between Cys-2 and Cys-7. IAPP is secreted from β-cells of the pancreas into the blood along with insulin. Amylin is a primary hormone that regulates and maintains blood glucose levels in the body, and its effects are complementary to insulin.35,36 Human IAPP (hIAPP) plays an active role in glycemic regulation by slowing gastric emptying and promoting satiety, thereby preventing postprandial spikes in blood glucose levels. However, it cannot be used as a drug for the treatment of diabetes because of its tendency to mis-fold and subsequently aggregate, resulting in the formation of cytotoxic fibrils,13,37 which are strongly associated with β-cell degeneration in Type 2 Diabetes Mellitus (T2DM).38 The rate of hIAPP aggregation depends on many factors that we discuss below.It has been reported that His18 acts as an electrostatic switch that inhibits fibrillization (aggregation) in its charged state and is heavily pH-dependent.31 Modulations are observed even in the narrow physiological range of pH of 7.35–7.45.39 This relationship was clearly demonstrated by the usage of ThT dyes for monitoring hIAPP aggregation at different pH, related to the activity of H3O+ ions in solution that is directly related to their concentration in solution.
hIAPP is closely related with cytotoxicity, which heavily depends on its concentration as well as on how the “synthetic” peptide sample is prepared. The highest observed cytotoxic potentials of hIAPP is at concentrations of 25 μM for full length hIAPP, and 40 μM for the 8–37 hIAPP fragment.40 The range of reported cytotoxicity for hIAPP, expressed as a percentage of dead cells, is believed to be from 15 to 80% for exposure to 5–25 μM of hIAPP for a duration of 24–48 h.41
In recent years, the importance of the role of the metal ions Cu2+, Zn2+, Al3+ and Fe2+/Fe3+ in the aggregation of hIAPP has been identified (see Fig. 1).42,43 In addition, their ability to modulate the proteolytic activity of hIAPP-degrading enzymes has been extensively studied.14,44–46 It was reported that, Zn2+ plays an important role in glycemic regulation, which is reflected in their high concentrations in the interior of dense granule cores ranging from 10 to 20 mM, confirming their physiological importance.47–50 The effect of concentrations of Zn2+ on hIAPP aggregation has been studied in detail. Several studies have shown that varying concentrations of Zn2+ have different effects on hIAPP aggregation and the different stages of the aggregation process. At high concentrations (10 mM) and in the early stages of aggregation (40 min), Zn2+ promote the formation of large Zn2+–amylin aggregates. In general, it has been reported that Zn2+ ion binds to amylin at the imidazole ring of His18 and the amine group of Lys1.51,52 At low Zn2+ concentrations (100 μM) and in the early stages of aggregation (40 min), Zn2+ induces the formation of even larger Zn2+–amylin aggregates than those formed at high concentrations of Zn2+. During the final stages of aggregation (when the amylin fibrils are formed), fiber formation is inhibited at low concentrations of Zn2+ and accelerated at higher concentrations.14,53 These findings have been supplemented by a study on the effect of Al3+, Fe3+, Zn2+ and Cu2+ at near physiological concentrations (10 μM, i.e., in stoichiometric excess) on amylin at 0.4 and 2 μM (see Fig. 2).42 Cu2+ efficiently inhibited amylin aggregation at certain concentrations. Other studies report that Cu2+ binds to amylin at the imidazole ring of His18 (ref. 54) and to the three preceding amides at the N-terminal side of His18 (ref. 52) and at Lys1.55 An opposite effect was observed for Al3+ and Zn2+ at the same concentration levels. Fe3+ appeared to have very little influence on amylin aggregation for the metal ion and peptide concentration ranges that were tested. Further tests in the same study using sub-stoichiometric concentrations of the metal ions confirmed the inhibitive properties of Cu2+, to a lesser extent for Zn2+, and no influence of Al3+ on hIAPP aggregation.43 A recent study applied several experimental techniques such as ThT fluorescence and Atomic Force Microscopy (AFM) to examine different characteristic changes of hIAPP, and Dynamic Light Scattering (DLS) analysis was used to determine the particular effects of Au3+ complexes on the aggregation of hIAPP.56 Electrospray Ionization-Mass Spectrometry (ESI-MS) and the intrinsic fluorescence method were employed to investigate the binding properties between the Au complexes and hIAPP. He et al. (2015) used NMR spectroscopy to discover that complexes 2-[Au(Ph2bpy)Cl2]Cl (Ph2bpy = 4,4′-diphenyl-2,2′-bipyridyl) and 3-[Au(phen)Cl2]Cl (phen = 1,10-phenanthroline) strongly inhibited the aggregation of hIAPP, compared to complex 1-[Au(bipy)Cl2][PF6] (bipy = 2,2′-bipyridine), which promoted the formation of amylin oligomers/protofibrils at high concentrations (∼30 μM).56 However, at low concentrations (∼5 μM), it inhibited amylin oligomer formation, verifying the concentration dependence of the inhibition process.
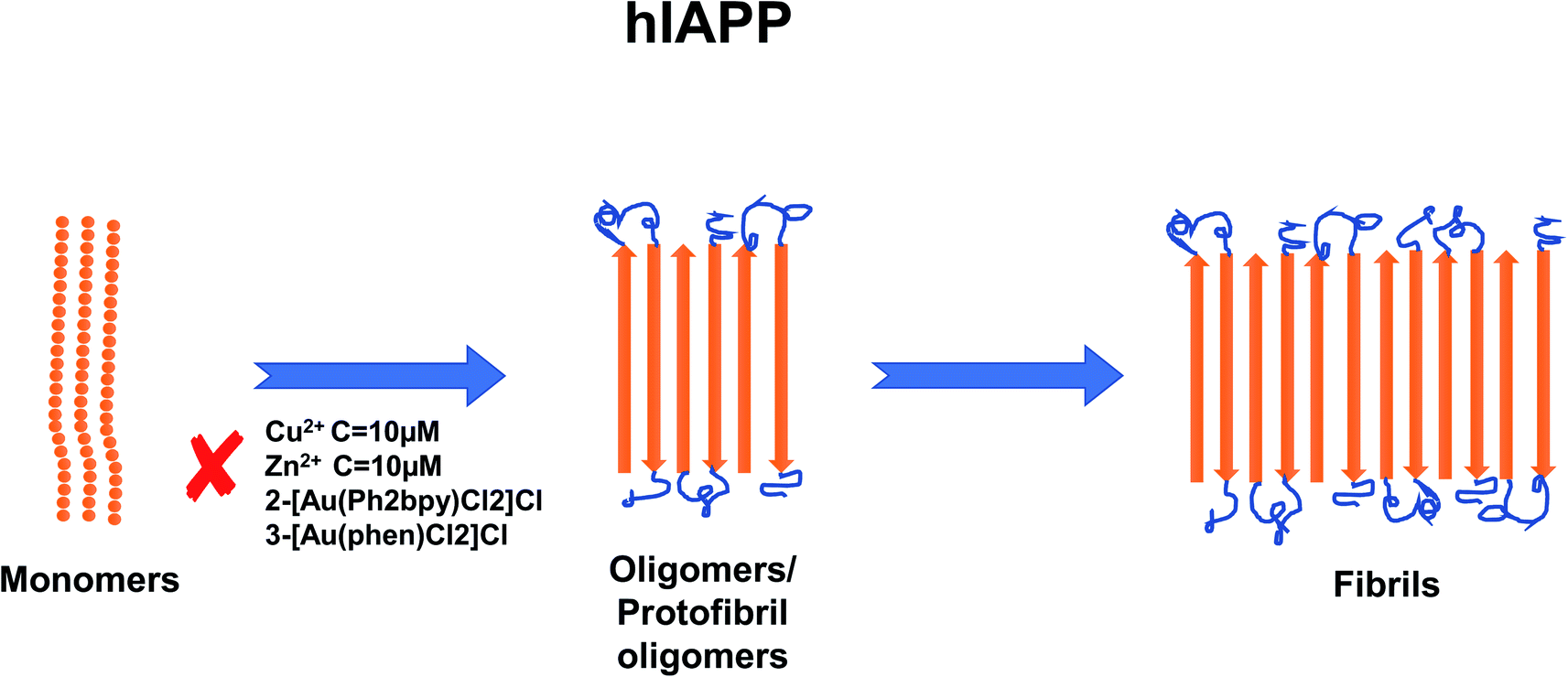 | ||
| Fig. 1 Schematic representation of metal ion concentration-dependent inhibition or acceleration of hIAPP aggregation. Zn2+ (10 μM), Au3+ and Cu2+ (10 μM) inhibit the formation of aggregates. The figure has been copied and adapted with permission from Alghrably et al., (2019).14 | ||
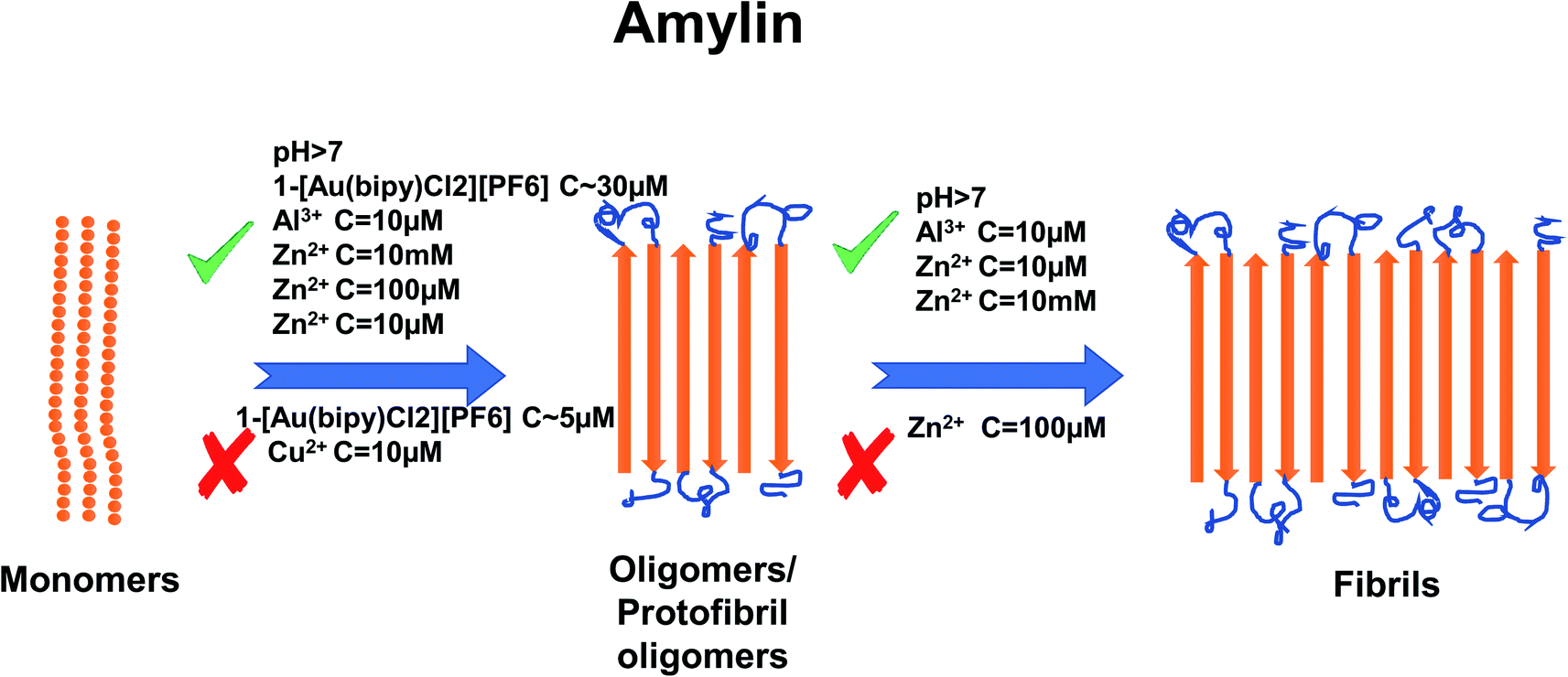 | ||
| Fig. 2 Schematic representation of metal ion concentration-dependent inhibition or acceleration of amylin aggregation. Zn2+ (10 μM, 10 mM), Al3+ (10 μM) and Au3+ (∼30 μM) promote the formation of aggregates, while Cu2+ (10 μM) and Au3+ (∼5 μM) inhibit aggregate formation. Zn2+ (100 μM) promotes the formation of oligomers but inhibits the formation of fibrils. The figure has been copied and adapted with permission from Alghrably et al., (2019).14 | ||
Limited reported results in the scientific literature highlight an urgent need for a systematic and accurate study on the dependence of hIAPP aggregation on peptide and metal ions concentrations, with a particular emphasis on physiological conditions and concentration ranges.
4. α-Synuclein
α-Synuclein is a protein that consists of 140 amino acids and is present in large quantities in the brain.57 α-Synuclein is located within three domains: N-terminal lipid-binding α-helix, amyloid-binding central domain (NAC), and C-terminal acidic tail.58 In the human body, α-synuclein functions as a molecular chaperone for forming SNARE complexes (SNARE is a group of proteins that catalyzes the fusion of membranes in vesicle transport) in synapses, enables the release of neurotransmitters and regulates levels of glucose and the biosynthesis of dopamine.58 α-Synuclein has been identified as the main component of Lewy bodies – aggregates of protein characteristic to Parkinson's disease and other synucleinopathy diseases.59,60 The formation of aggregates of α-synuclein depends on factors such as pH, post translational modifications (PTM), polyamines and concentration of α-synuclein.61Buell et al. (2014) found that the multiplication rate of α-synuclein is suppressed under neutral pH and inert conditions.62 However, changing the pH to mildly acidic (4.8–5.6 pH), i.e., non-physiological pH, strongly affects the multiplication process, with the biggest impact at pH 5.2. Compared to the fibril elongation constant by monomer addition (2 × 103 M−1 s−1 for PBS buffer), an acidic environment increases the fibril elongation rate constant by one order of magnitude and the rate of production of new fibrils (by secondary nucleation) increases by four orders of magnitude.62 Additionally, it was demonstrated that in physiological salt concentrations (150 mM NaCl), α-synuclein tends to form aggregates that can subsequently form gels.62
Another factor favoring the aggregation process is the initial concentration of α-synuclein.63 Uversky et al., (2001) measured the change of ThT fluorescence intensity for various concentrations of α-synuclein: 21 μM, 70 μM, 105 μM and 190 μM. They found that the fluorescence intensity increased with higher concentrations of proteins, which demonstrates an increase in the α-synuclein aggregation rate in the form of fibrillation. Nonetheless, the concentration of 21 μM of α-synuclein was enough to start the fibrillation process.63
Metal ions such as Cu2+, Zn2+, Al3+, Fe3+, Ca2+ and Mg2+ have also been shown to affect aggregation rates.64 For copper, it has been shown that the addition of 40 μM of Cu2+ accelerates the aggregation rate by promoting the nucleation process of α-synuclein.65 In addition, Cu-induced fibrils have been shown to have the same morphology as those formed in the absence of Cu2+.65 There are two regions where Cu2+ binds to α-synuclein. One of them is located at N-terminal site with residues Met1, Asp2, Met5 that have high affinity to copper and residue His50 with low affinity. The other region is at C-terminal part with residues Asp119, Asp121, Asn122, Glu123 and binds copper ions with low affinity.65–67 For His50, the ability to bind Cu2+ is greatly affected by pH. It was shown that lowering the pH to the acidic values cease the ability of His50 to bind copper.68 Additionally the acetylation on N-terminal region of α-synuclein abolished its ability to bind Cu2+ at residue Met1, leaving His50 ability intact in this region.66,69 However, a recent paper67 shows that copper does not bind to His50 in α-synuclein fibrils. Instead, during the fibrillation process Cu2+ has the ability to bind to other residues in N-terminal and C-terminal sites and can “bounce” between them. Zn2+ at concentrations of 100 μM has been proven as an effective promoter of α-synuclein aggregation and specifically α-synuclein fibrillation in vitro.70 It has been proven that Zn2+ binds to residues His 50 with much lower affinity that in case of Cu2+ and Asp121 with similar affinity compared to Cu2+.71 Data shows that the addition of Al3+ to a high concentration of α-synuclein induces the formation of oligomers. Addition of 2.5 mM of AlCl3 shortened the time of fibril formation ∼3-fold and increased the rate of fibril formation ∼1.5 fold63 and these fibrils form structure similar in look to twisted ribbons. On the other hand, Fe3+ (5 μM) has been proven to promote α-synuclein aggregation but only when added in the presence of intermediate concentrations of ethanol (∼5%).72 In the same paper, it was also shown that Al3+ (5 μM) promotes aggregation in 20% ethanol but has a lesser effect on aggregation than Fe3+. Like for most divalent metals, binding site for Fe3+ is postulated to be in C-terminal region, possibly residue Asp121.73 A study from Nath et al. (2011) demonstrated that aggregation is also very dependent on Ca2+ concentrations, whereby higher concentrations of Ca2+ (from 100 μM to 750 μM) resulted in fewer monomers remaining in the sample because of the formation of aggregates.74 However, the concentration of Ca2+ required to induce α-synuclein aggregation in free solution is far higher than that required in order to induce aggregation at a hydrophobic glass surface.74 For a binding site of Ca2+, study shows that Ca2+ binds to the C-terminal domain (126–140) however, currently there is no information which particular residue is involved in the binding process.74 There is currently a lack of information about the effect of Pb on aggregation in vitro, although it has been demonstrated that Zn2+, Al3+ and Pb2+ enable methionine-oxidized α-synuclein,75 to form aggregates at the same rate as the non-oxidized α-synuclein.76 The interaction effects of Mg2+ ions with α-synuclein aggregation have not been investigated we well as Zn and Cu ions hence further investigations are necessary. One study showed that Mg2+ at 500 μM has the ability to inhibit the aggregation process of α-synuclein (23 μM), even under the iron-induced aggregation (50 μM of Fe3+ on 8 μM of α-synuclein).77 On the other hand, Hoyer et al. (2002) have shown that 10 mM of Mg2+ (at pH 7.0) helps to form aggregates composed of densely packed short fibrillary elements.78 For a summary of the effects of metal ion concentration on α-synuclein, refer to Fig. 3.
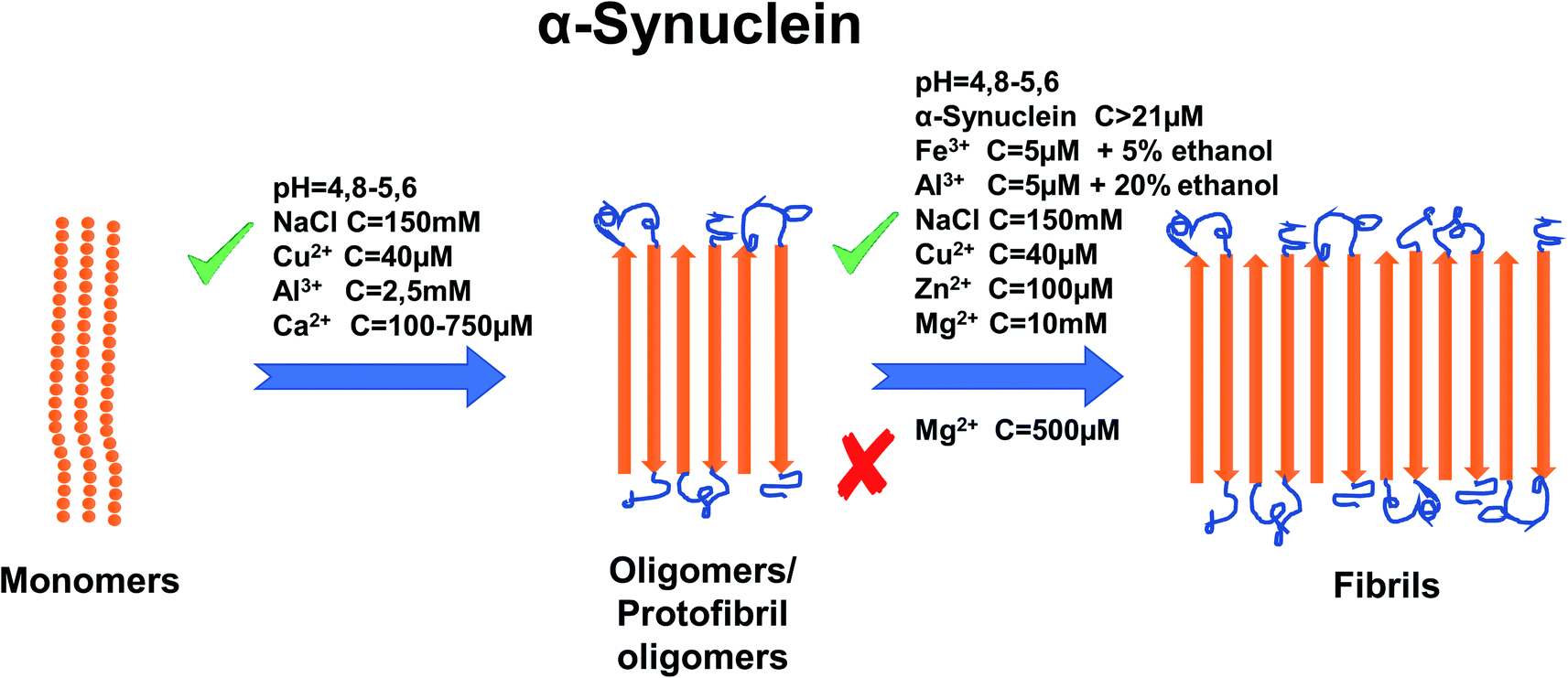 | ||
| Fig. 3 Schematic representation of metal ion concentration-dependent inhibition or acceleration of α-synuclein aggregation. Concentration of α-synuclein (more than 21 μM), Zn2+ (100 μM), Al3+ (2.5 mM, 5 μM in 20% ethanol), Cu2+ (40 μM), NaCl (150 mM), pH (4.8–5.6), Ca2+ (100–750 μM) and Fe3+ (5 μM in 5% ethanol) promote forming aggregates, whereas Mg2+, depending on concentration, inhibits aggregation (500 μM) or promotes (10 mM) formation of fibrils. The figure has been copied and adapted with permission from Alghrably et al. (2019).14 | ||
In conclusion, the evidence shows that metal ions can inhibit or accelerate the aggregation of α-synuclein. Nevertheless, much work remains to be done in order to gather and analyze information on these effects. Our brief literature review indicates a fundamental need for further systematic research on concentration-dependent aggregation of proteins and the influence of metal ions on the aggregation process.
5. Tau protein
The aggregation of Tau protein (TP) in neuronal cells is characteristic of Alzheimer's disease (AD).79 Although there is a clear correlation between the aggregation of TP and the progress of AD,80 the relationship between them still remains elusive, and several scientists are seeking methods to accurately model the exact relationship between them.81,82TP is primarily responsible for stabilizing microtubules in neuronal cells. One of the mechanisms in which TP regulates the stability of these microtubules is via phosphorylation,83,84 though the exact association between TP and microtubules is not completely clear.84,85 Out of the 441 amino acids in Tau's peptide sequence (htau 40 human isomorph), 85 of them are phosphorylation sites. These phosphorylation sites are regulated both by kinase and phosphatase enzymes. A typical TP will have approximately 30 of its 85 phosphorylation sites phosphorylated.86 An abnormal TP will normally contain three times as much phosphate as a normal TP, at which point the TP is “hyperphosphorylated”. In its hyperphosphorylated state, TP cannot properly stabilize microtubules in neuronal cells, and aggregation of TP begins.79
Several studies have reported the effects of metal ion concentrations on TP aggregation, although many have reported contradictory results.87 For example, the mechanism of action of different metal ions are not consistent.88 The consensus, however, is that the higher the concentration of metal ions present in the brain, the more protein aggregation occurs, supporting the progress of AD. Below we discuss the impact of Cu2+, Zn2+ and Li+, as each shows acceleration or inhibition of TP.
The scientific literature shows that Cu2+ accelerates the aggregation of TP either by activation of GSK3β kinase89 or activation of CDK5.90 Voss et al. (2014) reported acceleration of TP aggregation with concentrations of 400 μM of Cu2+,89 whereas Crouch et al. (2009) reported acceleration of TP aggregation under concentrations of Cu2+ of 25 μM.90 These numbers seem reasonable, as Cu2+ typically has a concentration of about 10 μM at neuronal synapses, and at this concentration, TP aggregation does not normally occur.91
The literature regarding the precise binding site of Cu2+ is ambiguous, as authors report different binding sites. For example, one paper claims the binding site for Tau protein to be residues 318–335. By binding at this region of the TP, Cu2+ induces fibrillization via formation of alpha helices.92 However, Zhou et al. (2017) claim that Cu2+ simply modulates the aggregation of TP by binding it at residues 256–273 of the htau 441 isoform, and associates it with His-268.93 Still, Soragni et al. (2008) claim that Cu2+ has a minor impact on TP aggregation in vitro, and only binds to TP with micromolar affinity (approximately 0.5 μM).94 The same paper also reports that two sections of TP, amino acids 287–293 and amino acids 310–324, are primarily involved in copper binding.94 A factor that could explain the seemingly contradictory claims is the fact that Cu2+ binding to TP depends both on the stoichiometry of Cu2+ in relation to TP, and the pH of the surrounding environment.92
Zn2+ has also been shown to accelerate the aggregation of TP.95 Huang et al. (2014) claim Zn2+ acts independently of TP phosphorylation.96 This could be possible because Hong et al. (1997) reported Zn2+ inhibits the GSK3 enzyme.97 Most studies have reported acceleration of aggregation at concentrations around 300 μM of Zn2+ (ref. 88) but concentrations as low as 10 μM (ref. 98) and as high as 500 μM are reported.99 Huang et al. (2014) studied the effects of Zn2+ on TP in the presence and absence of Zn2+ and the results suggest that Zn2+ clearly causes aggregation of TP in vitro, and even the fact that removing Zn2+ seems to remove the toxicity of TP.96
The Zn2+ binding site has not been clearly elucidated, though some have proposed that a cysteine residue is involved. It was demonstrated that Zn2+ associates with TP by coordinating with the cysteine residue of the three repeat TP constructs.100 Furthermore, Zn2+ accelerates the fibrillization of human TP by creating a “bridge” between Cys-291 and Cys-322.101
Li+ presents an intriguing case as several studies have reported that it inhibits TP aggregation.88 Fu et al. (2010) reported TP phosphorylation of GSK-3β enzyme at a concentration of 100 mg mL−1 Li+,102 and as mentioned earlier, TP phosphorylation is a key step to TP aggregation.80 Su et al. (2004) reported a reduction in TP phosphorylation at concentrations between 300–600 mg kg−1.103 Though Li+ has not been as extensively studied as Cu2+ or Zn2+, one study has suggested that Li+ reduces Tau phosphorylation by inhibition of glycogen synthase kinase-3.97 For a summary of the effects of metal ion concentration on TP, refer to Fig. 4.
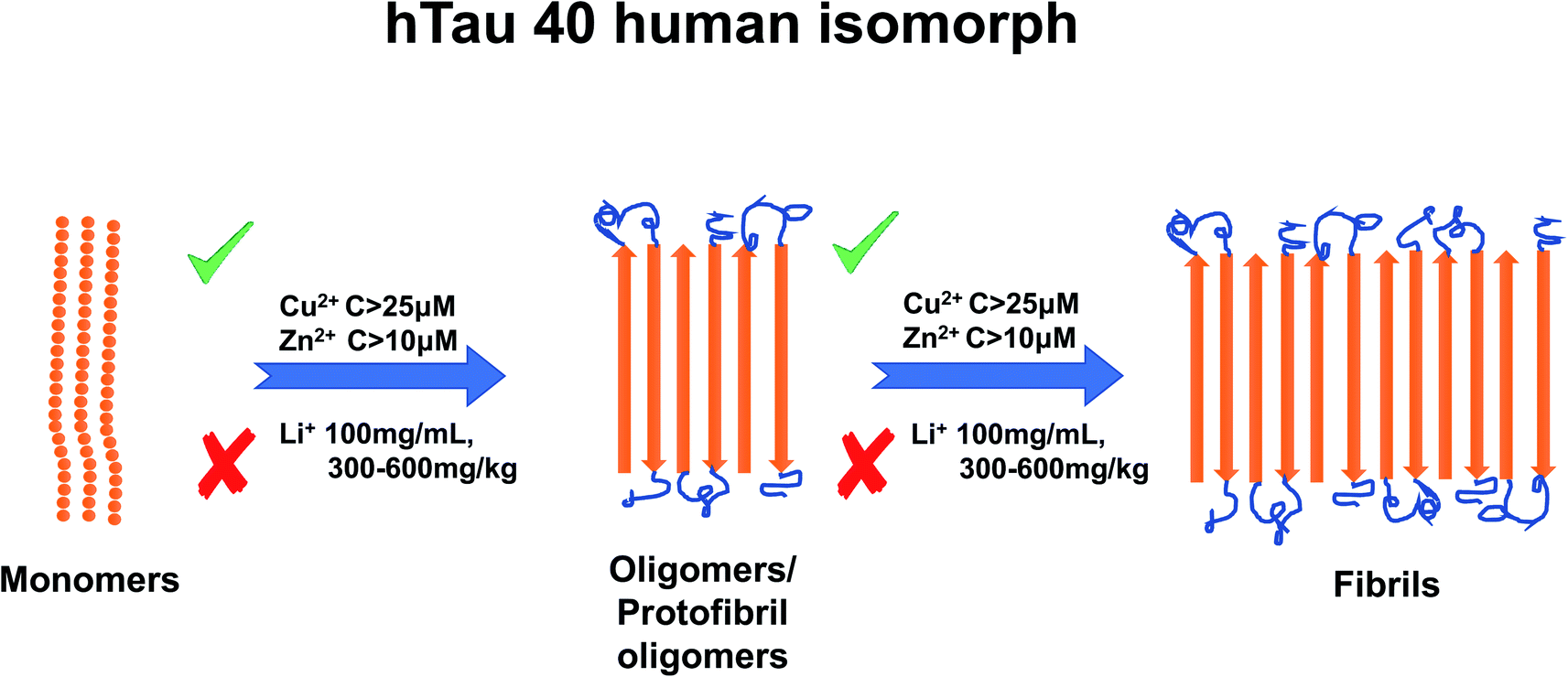 | ||
| Fig. 4 Schematic representation of metal ion concentration-dependent inhibition or acceleration of Tau protein. Zn2+ (concentration higher than 10 μM) and Cu2+ (concentration higher than 25 μM) promote aggregate formation, whereas Li+ (100 mg mL−1, 300–600 mg kg−1) inhibits aggregation formation. The figure has been copied and adapted with permission from Alghrably et al. (2019).14 | ||
6. Amyloid-beta peptide
Like TP, Amyloid-Beta (Aβ) is also characteristic of AD. Unlike TP, Aβ has a much shorter peptide sequence; the two most common isoforms contain a total of 40 or 42 peptides only.104 Nevertheless, its aggregation properties are still of great importance for understanding and finding viable treatments for AD. The Aβ cascade hypothesis proposes that the deposition of Aβ is the precursor to all major stages of AD.105Ha et al., (2007) have shown that Aβ-40 and Aβ-42 must undergo a conformational change before aggregation of this protein can start.106 Novo et al. (2018) studied the effects of Aβ-42 concentrations on Aβ-42 aggregation.107 The relationship is not linear, but rather sigmoidal in nature. They discovered that aggregation of Aβ-42 does not occur until Aβ-42 has reached a critical aggregation concentration of 90 nM. Even at this critical aggregation concentration, only a small percentage (approximately 10%) of Aβ-42 proteins will aggregate, and most Aβ-42 proteins will not aggregate until the concentration of Aβ-42 proteins is considerably higher than 90 nM.107
The effect of metal ion concentrations on Aβ has also been studied extensively,88 and generally must be considered on a case by case basis since the type of ion and its relative amount (stoichiometry) to Aβ can have enormous implications.108 At concentrations of 100 μM, Cu2+ and Zn2+ cause amorphous aggregation of Aβ-42. The presence of Cu2+, Zn2+ and Fe3+ at concentrations of 100 μM increases the volume of aggregated Aβ-42 by a significant percentage.106 The effects of Hg2+ and Pb2+ at concentrations of 0.25 μM, 2.5 μM, 25 μM, and 250 μM were studied, whereby the amount of Aβ-42 also increased.109 Although much less research has been carried out on the impact of Al3+, it has been shown to accelerate the aggregation of Aβ-40.110
In recent years, several efforts have been undertaken to determine how metal ions bind to Aβ, though this is a challenging task because Aβ can change its shape depending on the electronic and structural properties of the binding metal ion.111 Nevertheless, sites for Cu2+ binding to Aβ have been proposed. The most commonly proposed site for Cu2+ binding includes the imidazole ring of a histidine residues at position 6, 13 and 14, the N-terminal amine group, and an adjacent CO functional group from the Asp1–Ala2 peptide bond.112 Many articles argue that the imidazole ring of the histidine residue is required for Cu2+ binding to Aβ, and that the Cu2+ binding mechanism is distinct from the other binding mechanisms of Zn2+, Fe3+, and Al3+.113,114 Cu2+ is also proposed to control Aβ-42 aggregation at submolar concentrations by forming dityrosine linkages between Aβ-42 monomers.115
The binding mechanism of Zn2+ on Aβ also deserves some recognition. Zn2+ binds in the same hydrophilic region (Asp1–Lys16) as Cu2+ (ref. 116) although, perhaps paradoxically, Zn2+ increases the total amount of exposed hydrophobic parts on Aβ, whereas Cu2+ decreases it. Perhaps even more striking is the fact that Zn2+ diminishes the lag time that Aβ experiences upon aggregation, even at small concentrations (5 μM), while Cu2+ at similar concentrations increases the lag time to above 60 hours.113 Several have proposed Zn2+ adopts a tetrahedral coordination, where like its Cu2+ counterpart, associates with histidine residues on Aβ.116
Al3+ presents an interesting case, as in AD patients its concentration is about 1.6 times higher than that of normal people.117 It was reported that toxic amyloid chambers form when Al3+ and Aβ oligomers aggregate in sync with each other.117 This finding may lead future researchers to discover the true binding site of Al3+ to Aβ. Like Cu2+ and Zn2+, it has a distinct, measurable effect on Aβ aggregation113 and therefore, is likely to have its own unique mechanism of binding of Aβ. For a summary of the effects of metal ion concentration on Aβ, refer to Fig. 5.
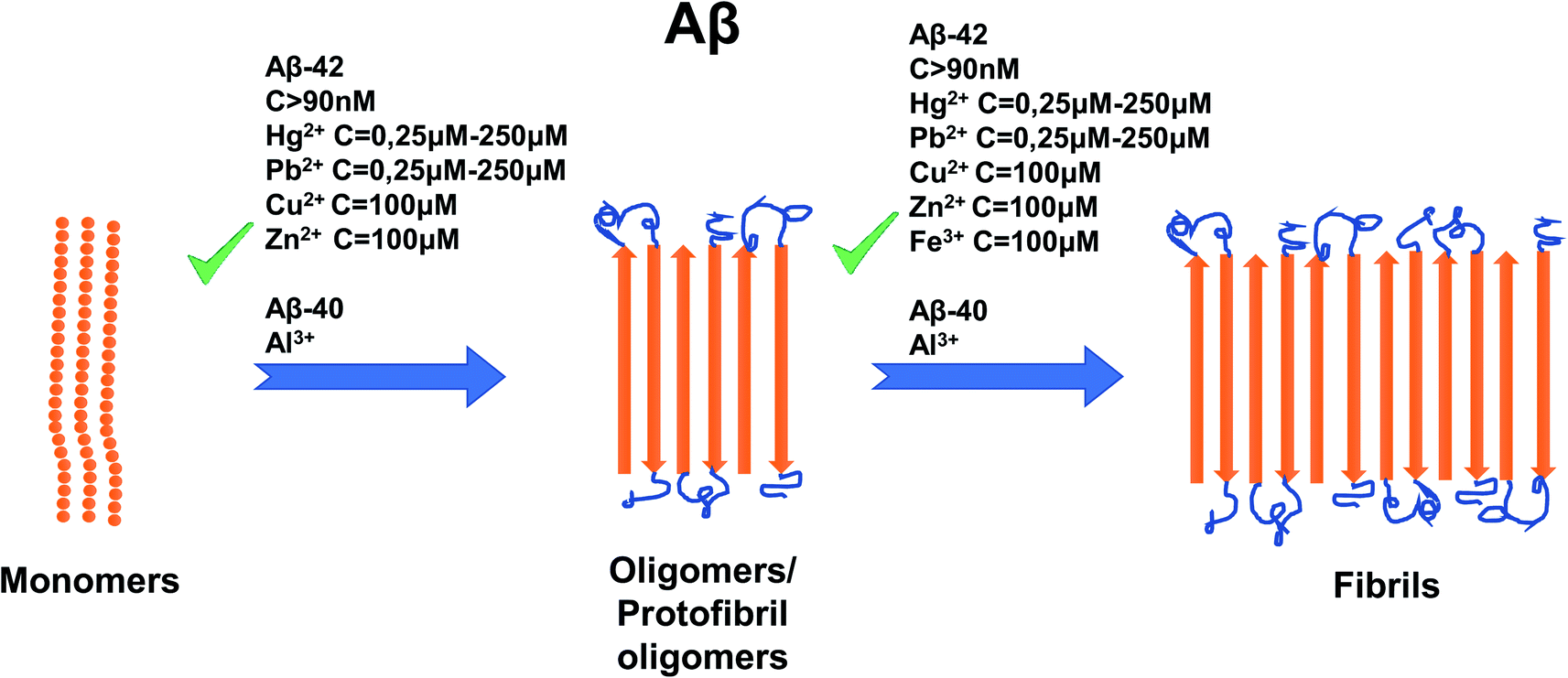 | ||
| Fig. 5 Schematic representation of metal ion concentration-dependent inhibition or acceleration of Aβ. Concentrations of Aβ-42 (more than 90 nM), Hg2+ (0, 25–250 μM), Pb2+ (0, 25–250 μM), Zn2+ (100 mM), Cu2+ (100 μM) and Fe3+ (100 μM) promote the formation of aggregates, whereas for Aβ-42 only, Al3+ favors formation of aggregates. The figure has been copied and adapted with permission from Alghrably et al. (2019).14 | ||
Other metal ions such as Mn2+, Mg2+ and Cd2+ and their effects on the aggregation of Aβ have also been examined. As for the case of TP, some metal ions cause acceleration of aggregation of Aβ-40 or Aβ-42, and others cause inhibition.88 Understanding the precise relationship between concentrations of metal ions and aggregation of Aβ-40 or Aβ-42 will provide interesting research opportunities for the scientific community, as well as helping to find a viable treatment for AD patients.
7. Polyglutamine: Huntington's disease
Polyglutamine (PolyQ) is more complicated than some of the previously presented proteins in this review. PolyQ is associated with at least nine separate diseases, the most prominent one being Huntington's disease (HD).118 Since the most studied disease related to PolyQ is HD, the remainder of this section will focus on the effect of metal ions related to HD, and its protein, huntingtin 1.It is known that PolyQ only becomes toxic in HD only after extending beyond a pathological length119 and that its length is crucial to its aggregation properties.120 PolyQ's aggregation process is distinct from that of other proteins discussed in this review,120 but unlike the other proteins discussed here, its aggregation process is less well understood. As for the factors inducing aggregation of PolyQ in HD, there is a scarcity of information. Currently, it was confirmed that the length of glutamine repeats affects the aggregation process. Yushchenko et al. (2018) demonstrated that repeats of 11 glutamines are not sufficient to cause PolyQ aggregation, however longer sequences of 38 and 56 tend to stimulate aggregation, with 56 repeats having higher aggregation kinetics than 38.121 In terms of metal ions, it was found that copper binds to the first 171 residues on the N-terminal region of huntingtin 1, which contains PolyQ repeats and promotes aggregation of huntingtin 1.122 His82 and His98 were identified as crucial for copper binding. However, there is a lack of information as to whether the length of glutamine residues affects the binding of copper.122 Xiao et al. (2013) also reports a histidine residues being involved in binding, and also suggest that Cu2+ bind to the residue Met8.123 The same authors report that HD arises from a combinatory toxicity of PolyQ and Cu2+, that is, Cu2+ is actually required to cause HD.123 Interestingly enough, zebrafish that lack the huntingtin protein exhibit sizeable defects in iron utilization and development, meaning that huntingtin (PolyQ) may play a role in iron pathways.124
Kar. et al. (2011) propose that aggregation proceeds via a nucleus centered approach, although several other aggregation mechanisms have been proposed.118,120 A beta sheet is likely involved125 and PolyQ only aggregates after reaching a critical aggregation concentration of 3 μM.120 Even so, these results are suggestive at best, and clearly indicate the need for additional studies specifically on HD, its protein huntingtin 1 and the PolyQ repeats it contains.
8. Conclusion and future outlook
There is significant ongoing effort to understand the relationship between metal ions and their effect on protein aggregation. Protein aggregation and misfolding are recurrent in many neurodegenerative diseases (i.e. Parkinson's, Alzheimer's, etc.).126 The relationship between the metal ions and protein aggregation is difficult to describe precisely because even a slight change of the external environment (pH, metal ion/protein concentration, etc.) can disrupt the fragile equilibrium state of the functional protein.126 The disorderliness of Tau and α-synuclein, for example, is context specific,127 including in the presence of metal ions.Some studies have sought to create experiments that might explain more clearly how some proteins aggregate (specifically, TP and α-synuclein) aggregate,128–132 and one paper even claims to have invented a simple and reproducible method for monitoring the aggregation of α-synuclein aggregation133 in a plate-reader based assay. The protocol utilizes Thioflavin T (ThT) fluorescence to measure the kinetics of the aggregation of α-synuclein.133 Protocols such as this could be developed to explain the seemingly obscure relationship between protein aggregation and metal ion concentration. Understanding how protein aggregation works has led some scientists to develop anti-aggregation drugs against TP and α-synuclein.134–136 More systematic experiments designed to clarify this relationship are vital, as they may provide the groundwork to produce better therapeutics. Therefore, further research with more rigorous and detailed studies are necessary to definitively uncover the relationship between metal ions and their effects on the aggregation of proteins, with a particular emphasis on their concentrations and relative ratios.
This detailed knowledge about the link between protein and metal ion concentration and the amount of aggregation would give us a necessary level of understanding of the biochemical processes behind the complex, multi-step aggregation process that would allow us to design better inhibitors (ultimately more efficient and commercially available drugs) of the aggregates formation at the early soluble state. It may result in efficient targeting of the early state of the aggregation process in which smaller and soluble aggregates are formed as a result of the association of β-sheet motifs to each other.31,137
It is an obvious fact that the surrounding environment of the protein must be also considered in these future studies, and not just the proteins in isolation with metal ions. Amylin aggregation, for example, is strongly pH dependent with its two protonable sites at His18 and at the N-terminus.138 α-synuclein fibrils can form under several different solution conditions, but only a handful of these conditions lead to rapid multiplication of α-synuclein fibrils. Clearly, the solution conditions determine the relative importance.62 Designing compounds to successfully inhibit amylin aggregation will require a good amount of strategy because inhibition of amylin aggregation process may not automatically delete its cytotoxicity to islet β-cells.139
Based on the currently available scientific literature, we may speculate about possible aggregation mechanisms of proteins. It was suggested that α-synuclein may aggregate more quickly via oligomer–oligomer interactions than via monomer–monomer interactions.140 Another study corroborates this idea by suggesting that seeding of monomers of α-synuclein is not sufficient to cause α-synuclein aggregation, but rather, exhibits prion-like spreading.141 It is reasonable to speculate that other proteins (TP, amylin, α-synuclein, etc.) may aggregate via oligomer-induced cellular stress, rather than through the precise coordination of the monomers of these proteins.
Metal ions concentrations are only one of several factors that strongly influence the increase or decrease in protein aggregation. Given the current gaps in knowledge relating to this specific factor, and given the potential knowledge that understanding the effect of metal ion concentrations on protein aggregation can provide researchers and scientists regarding the subject of protein aggregation, there is a clear need for further investigation of this topic for the advancement of future therapeutics of protein aggregation related diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank King Abdullah University of Science and Technology (KAUST) for financial support.References
- M. Kodaka, Biophys. Chem., 2004, 109, 325–332 CrossRef CAS PubMed
.
- S. Al-Harthi, J. I. Lachowicz, M. E. Nowakowski, M. Jaremko and Ł. Jaremko, J. Inorg. Biochem., 2019, 198, 110716 CrossRef CAS PubMed
- Y. S. Hedberg, I. Dobryden, H. Chaudhary, Z. Wei, P. M. Claesson and C. Lendel, Colloids Surf., B, 2019, 173, 751–758 CrossRef CAS PubMed
- G. Salzano, G. Giachin and G. Legname, Cells, 2019, 8, 770 CrossRef PubMed
- C. Migliorini, A. Sinicropi, H. Kozlowski, M. Luczkowski and D. Valensin, J. Biol. Inorg Chem., 2014, 19, 635–645 CrossRef CAS PubMed
- A. J. McDonald, D. R. Leon, K. A. Markham, B. Wu, C. F. Heckendorf, K. Schilling, H. D. Showalter, P. C. Andrews, M. E. McComb, M. J. Pushie, C. E. Costello, G. L. Millhauser and D. A. Harris, Structure, 2019, 27, 907–922.e5 CrossRef CAS PubMed
- P. Saá, D. A. Harris and L. Cervenakova, Expert Rev. Mol. Med., 2016, 18, e5 CrossRef PubMed
- G. Ilc, G. Giachin, M. Jaremko, Ł. Jaremko, F. Benetti, J. Plavec, I. Zhukov and G. Legname, PLoS One, 2010, 5, e11715 CrossRef PubMed
- D. Sarnataro, A. Pepe and C. Zurzolo, in Progress in Molecular Biology and Translational Science, ed. G. Legname and S. Vanni, Academic Press, 2017, vol. 150, pp. 57–82 Search PubMed
- G. G. Kovacs, J. Clin. Pathol., 2019, 72, 725–735 CrossRef PubMed
- A.-H. M. Emwas, Z. A. Al-Talla, X. Guo, S. Al-Ghamdi and H. T. Al-Masri, Magn. Reson. Chem., 2013, 51, 255–268 CrossRef CAS PubMed
- C. A. Blindauer, A. H. Emwas, A. Holý, H. Dvořáková, E. Sletten and H. Sigel, Chem.–Eur. J., 1997, 3, 1526–1536 CrossRef CAS
- A. P. Kumar, S. Lee and S. Lukman, Curr. Drug Targets, 2019, 20, 1680–1694 CrossRef CAS PubMed
- M. Alghrably, I. Czaban, Ł. Jaremko and M. Jaremko, J. Inorg. Biochem., 2019, 191, 69–76 CrossRef CAS PubMed
- A. Bernabeu-Zornoza, R. Coronel, C. Palmer, M. Monteagudo, A. Zambrano and I. Liste, Neural Regener. Res., 2019, 14, 2035–2042 CrossRef PubMed
- P. Scheltens, K. Blennow, M. M. B. Breteler, B. de Strooper, G. B. Frisoni, S. Salloway and W. M. V. der Flier, Lancet, 2016, 388, 505–517 CrossRef CAS
- K. Tsukita, H. Sakamaki-Tsukita, K. Tanaka, T. Suenaga and R. Takahashi, Mov. Disord., 2019, 34, 1452–1463 CrossRef CAS PubMed
- M. Stefani and C. M. Dobson, J Mol Med, 2003, 81, 678–699 CrossRef CAS PubMed
- P. J. Thomas, B.-H. Qu and P. L. Pedersen, Trends Biochem. Sci., 1995, 20, 456–459 CrossRef CAS PubMed
- W. Wang, S. Nema and D. Teagarden, Int. J. Pharm., 2010, 390, 89–99 CrossRef CAS PubMed
- M. Vijayan, Prog. Biophys. Mol. Biol., 1988, 52, 71–99 CrossRef CAS PubMed
- V. Cabra, E. Vázquez-Contreras, A. Moreno and R. Arreguin-Espinosa, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1028–1036 CrossRef CAS PubMed
- R. Wetzel, M. Becker, J. Behlke, H. Billwitz, S. Böhm, B. Ebert, H. Hamann, J. Krumbiegel and G. Lassmann, Eur. J. Biochem., 1980, 104, 469–478 CrossRef CAS PubMed
- J. L. Jimenez, J. I. Guijarro, E. Orlova, J. Zurdo, C. M. Dobson, M. Sunde and H. R. Saibil, EMBO J., 1999, 18, 815–821 CrossRef CAS PubMed
- J. W. Kelly, Curr. Opin. Struct. Biol., 1998, 8, 101–106 CrossRef CAS PubMed
- C. M. Dobson, Philos. Trans. R. Soc. London, Ser. B, 2001, 356, 133–145 CrossRef CAS PubMed
- M. Y. Sherman and A. L. Goldberg, Neuron, 2001, 29, 15–32 CrossRef CAS PubMed
- I. Z. Nagy, K. Nagy and G. Lustyik, Exp. Brain Res., 1982,(Suppl 5), 118–122 CAS
- I. J. Conlon, G. A. Dunn, A. W. Mudge and M. C. Raff, Nat. Cell Biol., 2001, 3, 918–921 CrossRef CAS PubMed
- R. J. Ellis, Curr. Opin. Struct. Biol., 2001, 11, 114–119 CrossRef CAS PubMed
- S. Jha, J. M. Snell, S. R. Sheftic, S. M. Patil, S. B. Daniels, F. W. Kolling and A. T. Alexandrescu, Biochemistry, 2014, 53, 300–310 CrossRef CAS PubMed
- J. T. Giurleo, X. He and D. S. Talaga, J. Mol. Biol., 2008, 381, 1332–1348 CrossRef CAS PubMed
- L. Zhang, D. Lu and Z. Liu, Biophys. Chem., 2008, 133, 71–80 CrossRef CAS PubMed
- W. H. Streng, S. K. Hsi, P. E. Helms and H. G. H. Tan, J. Pharm. Sci., 1984, 73, 1679–1684 CrossRef CAS PubMed
- M. Fineman, C. Weyer, D. G. Maggs, S. Strobel and O. G. Kolterman, Horm. Metab. Res., 2002, 34, 504–508 CrossRef CAS PubMed
- C. Weyer, D. G. Maggs, A. A. Young and O. G. Kolterman, Curr. Pharm. Des., 2001, 7, 1353–1373 CrossRef CAS PubMed
- S. Asthana, B. Mallick, A. T. Alexandrescu and S. Jha, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1765–1782 CrossRef CAS PubMed
- F. U. Hartl, Annu. Rev. Biochem., 2017, 86, 21–26 CrossRef CAS PubMed
- J. R. Casey, S. Grinstein and J. Orlowski, Nat. Rev. Mol. Cell Biol., 2010, 11, 50–61 CrossRef CAS PubMed
- B. Konarkowska, J. F. Aitken, J. Kistler, S. Zhang and G. J. Cooper, FEBS J., 2006, 273, 3614–3624 CrossRef CAS PubMed
- M. Magzoub and A. D. Miranker, FASEB J., 2012, 26, 1228–1238 CrossRef CAS PubMed
- B. Ward, K. Walker and C. Exley, J. Inorg. Biochem., 2008, 102, 371–375 CrossRef CAS PubMed
- M. Mold, C. Bunrat, P. Goswami, A. Roberts, C. Roberts, N. Taylor, H. Taylor, L. Wu, P. E. Fraser and C. Exley, J. Diabetes Res. Clin. Metab., 2015, 4, 4 CrossRef
- S. Mukherjee and S. G. Dey, Inorg. Chem., 2013, 52, 5226–5235 CrossRef CAS PubMed
- M. Seal, S. Mukherjee and S. G. Dey, Metallomics, 2016, 8, 1266–1272 RSC
- C. G. Taylor, BioMetals, 2005, 18, 305–312 CrossRef CAS PubMed
- V. Wineman-Fisher and Y. Miller, Phys. Chem. Chem. Phys., 2016, 18, 21590–21599 RSC
- B. Formby, F. Schmid-Formby and G. M. Grodsky, Diabetes, 1984, 33, 229–234 CrossRef CAS PubMed
- M. C. Foster, R. D. Leapman, M. X. Li and I. Atwater, Biophys. J., 1993, 64, 525–532 CrossRef CAS PubMed
- H. W. Davidson, J. M. Wenzlau and R. M. O'Brien, Trends Endocrinol. Metab., 2014, 25, 415–424 CrossRef CAS PubMed
- D. Łoboda and M. Rowińska-Żyrek, J. Inorg. Biochem., 2017, 174, 150–155 CrossRef PubMed
- M. Rowińska-Żyrek, Dalton Trans., 2016, 45, 8099–8106 RSC
- J. R. Brender, K. Hartman, R. P. R. Nanga, N. Popovych, R. de la Salud Bea, S. Vivekanandan, E. N. G. Marsh and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 8973–8983 CrossRef CAS PubMed
- A. Magrì, A. Pietropaolo, G. Tabbì, D. La Mendola and E. Rizzarelli, Chem.–Eur. J., 2017, 23, 17898–17902 CrossRef PubMed
- M. Alghrably, D. Dudek, A.-H. Emwas, Ł. Jaremko, M. Jaremko and M. Rowińska-Żyrek, Copper(II) and amylin analogues - a complicated relationship, Inorg. Chem., 2019 Search PubMed
- L. He, D. Zhu, C. Zhao, X. Jia, X. Wang and W. Du, J. Inorg. Biochem., 2015, 152, 114–122 CrossRef CAS PubMed
- K. Uéda, H. Fukushima, E. Masliah, Y. Xia, A. Iwai, M. Yoshimoto, D. A. Otero, J. Kondo, Y. Ihara and T. Saitoh, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11282–11286 CrossRef PubMed
- F. N. Emamzadeh, J. Res. Med. Sci., 2016, 21, 29 CrossRef PubMed
- M. G. Spillantini, M. L. Schmidt, V. M.-Y. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed
- M. G. Spillantini, R. A. Crowther, R. Jakes, M. Hasegawa and M. Goedert, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6469–6473 CrossRef CAS PubMed
- D. Ghosh, S. Mehra, S. Sahay, P. K. Singh and S. K. Maji, Int. J. Biol. Macromol., 2017, 100, 37–54 CrossRef CAS PubMed
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. Knowles, S. Linse and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7671–7676 CrossRef CAS PubMed
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 44284–44296 CrossRef CAS PubMed
- L. Breydo, J. W. Wu and V. N. Uversky, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 261–285 CrossRef CAS PubMed
- R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4294–4299 CrossRef CAS PubMed
- D. Valensin, S. Dell'Acqua, H. Kozlowski and L. Casella, J. Inorg. Biochem., 2016, 163, 292–300 CrossRef CAS PubMed
- D. N. Bloch, P. Kolkowska, I. Tessari, M. C. Baratto, A. Sinicropi, L. Bubacco, S. Mangani, C. Pozzi, D. Valensin and Y. Miller, Inorg. Chem., 2019, 58, 10920–10927 CrossRef CAS PubMed
- R. De Ricco, D. Valensin, S. Dell'Acqua, L. Casella, P. Dorlet, P. Faller and C. Hureau, Inorg. Chem., 2015, 54, 4744–4751 CrossRef CAS PubMed
- G. M. Moriarty, C. A. Minetti, D. P. Remeta and J. Baum, Biochemistry, 2014, 53, 2815–2817 CrossRef CAS PubMed
- T. D. Kim, S. R. Paik, C.-H. Yang and J. Kim, Protein Sci., 2000, 9, 2489–2496 CrossRef CAS PubMed
- A. A. Valiente-Gabioud, V. Torres-Monserrat, L. Molina-Rubino, A. Binolfi, C. Griesinger and C. O. Fernández, J. Inorg. Biochem., 2012, 117, 334–341 CrossRef CAS PubMed
- M. Kostka, T. Högen, K. M. Danzer, J. Levin, M. Habeck, A. Wirth, R. Wagner, C. G. Glabe, S. Finger and U. Heinzelmann, J. Biol. Chem., 2008, 10992–11003 CrossRef CAS PubMed
- A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernández, J. Am. Chem. Soc., 2006, 128, 9893–9901 CrossRef CAS PubMed
- S. Nath, J. Goodwin, Y. Engelborghs and D. L. Pountney, Mol. Cell. Neurosci., 2011, 46, 516–526 CrossRef CAS PubMed
- V. N. Uversky, G. Yamin, P. O. Souillac, J. Goers, C. B. Glaser and A. L. Fink, FEBS Lett., 2002, 517, 239–244 CrossRef CAS PubMed
- G. Yamin, C. B. Glaser, V. N. Uversky and A. L. Fink, J. Biol. Chem., 2003, 278, 27630–27635 CrossRef CAS PubMed
- N. Golts, H. Snyder, M. Frasier, C. Theisler, P. Choi and B. Wolozin, J. Biol. Chem., 2002, 277, 16116–16123 CrossRef CAS PubMed
- W. Hoyer, T. Antony, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Mol. Biol., 2002, 322, 383–393 CrossRef CAS PubMed
- G. Lippens, A. Sillen, I. Landrieu, L. Amniai, N. Sibille, P. Barbier, A. Leroy, X. Hanoulle and J.-M. Wieruszeski, Prion, 2007, 1, 21–25 CrossRef PubMed
- F. P. Chong, K. Y. Ng, R. Y. Koh and S. M. Chye, Cell. Mol. Neurobiol., 2018, 38, 965–980 CrossRef CAS PubMed
- A. Fardanesh, S. Zibaie, B. Shariati, F. Attar, F. Rouhollah, K. Akhtari, K. Shahpasand, A. A. Saboury and M. Falahati, Int. J. Nanomed., 2019, 14, 901 CrossRef CAS PubMed
- M. Krestova, J. Ricny and A. Bartos, J. Neuroimmunol., 2018, 322, 1–8 CrossRef CAS PubMed
- W. Noble, D. P. Hanger, C. C. Miller and S. Lovestone, Front. Neurol., 2013, 4, 83 Search PubMed
- H. Kadavath, M. Jaremko, Ł. Jaremko, J. Biernat, E. Mandelkow and M. Zweckstetter, Angew. Chem., Int. Ed., 2015, 54, 10347–10351 CrossRef CAS PubMed
- H. Kadavath, Y. Cabrales Fontela, M. Jaremko, Ł. Jaremko, K. Overkamp, J. Biernat, E. Mandelkow and M. Zweckstetter, Angew. Chem., Int. Ed., 2018, 57, 3246–3250 CrossRef CAS PubMed
- T. Kimura, G. Sharma, K. Ishiguro and S. Hisanaga, Front. Neurosci., 2018, 12, 44 CrossRef PubMed
- L. Breydo and V. N. Uversky, Metallomics, 2011, 3, 1163–1180 RSC
- A. C. Kim, S. Lim and Y. K. Kim, Int. J. Mol. Sci., 2018, 19, 128 CrossRef PubMed
- K. Voss, C. Harris, M. Ralle, M. Duffy, C. Murchison and J. F. Quinn, Transl. Neurodegener., 2014, 3, 24 CrossRef PubMed
- P. J. Crouch, L. W. Hung, P. A. Adlard, M. Cortes, V. Lal, G. Filiz, K. A. Perez, M. Nurjono, A. Caragounis and T. Du, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 381–386 CrossRef CAS PubMed
- Y. H. Hung, A. I. Bush and R. A. Cherny, J. Biol. Inorg Chem., 2010, 15, 61–76 CrossRef CAS PubMed
- Q. Ma, Y. Li, J. Du, H. Liu, K. Kanazawa, T. Nemoto, H. Nakanishi and Y. Zhao, Peptides, 2006, 27, 841–849 CrossRef CAS PubMed
- L.-X. Zhou, J.-T. Du, Z.-Y. Zeng, W.-H. Wu, Y.-F. Zhao, K. Kanazawa, Y. Ishizuka, T. Nemoto, H. Nakanishi and Y.-M. Li, Peptides, 2007, 28, 2229–2234 CrossRef CAS PubMed
- A. Soragni, B. Zambelli, M. D. Mukrasch, J. Biernat, S. Jeganathan, C. Griesinger, S. Ciurli, E. Mandelkow and M. Zweckstetter, Biochemistry, 2008, 47, 10841–10851 CrossRef CAS PubMed
- X. Li, X. Du and J. Ni, Int. J. Mol. Sci., 2019, 20, 487 CrossRef CAS PubMed
- Y. Huang, Z. Wu, Y. Cao, M. Lang, B. Lu and B. Zhou, Cell Rep., 2014, 8, 831–842 CrossRef CAS PubMed
- M. Hong, D. C. R. Chen, P. S. Klein and V. M.-Y. Lee, J. Biol. Chem., 1997, 272, 25326–25332 CrossRef CAS PubMed
- Y. Xiong, D.-J. Luo, X.-L. Wang, M. Qiu, Y. Yang, X. Yan, J.-Z. Wang, Q.-F. Ye and R. Liu, Neurosci. Bull., 2015, 31, 331–337 CrossRef CAS PubMed
- K. J. Kwon, E. J. Lee, K. S. Cho, D.-H. Cho, C. Y. Shin and S.-H. Han, Food Funct., 2015, 6, 2058–2067 RSC
- A. C. Jiji, A. Arshad, S. R. Dhanya, P. S. Shabana, C. K. Mehjubin and V. Vijayan, Chem.–Eur. J., 2017, 23, 16976–16979 CrossRef CAS PubMed
- Z.-Y. Mo, Y.-Z. Zhu, H.-L. Zhu, J.-B. Fan, J. Chen and Y. Liang, J. Biol. Chem., 2009, 284, 34648–34657 CrossRef CAS PubMed
- Z.-Q. Fu, Y. Yang, J. Song, Q. Jiang, Z.-C. Liu, Q. Wang, L.-Q. Zhu, J.-Z. Wang and Q. Tian, J. Alzheimer's Dis., 2010, 21, 1107–1117 CAS
- Y. Su, J. Ryder, B. Li, X. Wu, N. Fox, P. Solenberg, K. Brune, S. Paul, Y. Zhou and F. Liu, Biochemistry, 2004, 43, 6899–6908 CrossRef CAS PubMed
- T. Hartmann, S. C. Bieger, B. Brühl, P. J. Tienari, N. Ida, D. Allsop, G. W. Roberts, C. L. Masters, C. G. Dotti and K. Unsicker, Nat. Med., 1997, 3, 1016 CrossRef CAS PubMed
- C. Reitz, Int. J. Alzheimer's Dis., 2012, 2012, 369808 Search PubMed
- C. Ha, J. Ryu and C. B. Park, Biochemistry, 2007, 46, 6118–6125 CrossRef CAS PubMed
- M. Novo, S. Freire and W. Al-Soufi, Sci. Rep., 2018, 8, 1783 CrossRef PubMed
- D. Dharmadana, N. P. Reynolds, C. E. Conn and C. Valéry, Interface Focus, 2017, 7, 20160160 CrossRef PubMed
- D. Meleleo, G. Notarachille, V. Mangini and F. Arnesano, Eur. Biophys. J., 2019, 48, 173–187 CrossRef CAS PubMed
- C. Exley, in Alzheimer's Disease: Cellular and Molecular Aspects of Amyloid β, ed. J. R. Harris and F. Fahrenholz, Springer, US, Boston, MA, 2005, pp. 225–234 Search PubMed
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed
- C. Hureau and P. Dorlet, Coord. Chem. Rev., 2012, 256, 2175–2187 CrossRef CAS
- W.-T. Chen, Y.-H. Liao, H.-M. Yu, I. H. Cheng and Y.-R. Chen, J. Biol. Chem., 2011, 286, 9646–9656 CrossRef CAS PubMed
- G. De Gregorio, F. Biasotto, A. Hecel, M. Luczkowski, H. Kozlowski and D. Valensin, J. Inorg. Biochem., 2019, 195, 31–38 CrossRef CAS PubMed
- D. P. Smith, G. D. Ciccotosto, D. J. Tew, M. T. Fodero-Tavoletti, T. Johanssen, C. L. Masters, K. J. Barnham and R. Cappai, Biochemistry, 2007, 46, 2881–2891 CrossRef CAS PubMed
- M. Rana and A. K. Sharma, Metallomics, 2019, 11, 64–84 RSC
- Y. Kuroda, Journal of Neuroinfectious Diseases, 2017, 8(2), 241 Search PubMed
- A. Michalik and C. Van Broeckhoven, Hum. Mol. Genet., 2003, 12, 173–186 CrossRef PubMed
- E. Scherzinger, R. Lurz, M. Turmaine, L. Mangiarini, B. Hollenbach, R. Hasenbank, G. P. Bates, S. W. Davies, H. Lehrach and E. E. Wanker, Cell, 1997, 90, 549–558 CrossRef CAS PubMed
- K. Kar, M. Jayaraman, B. Sahoo, R. Kodali and R. Wetzel, Nat. Struct. Mol. Biol., 2011, 18, 328 CrossRef CAS PubMed
- T. Yushchenko, E. Deuerling and K. Hauser, Biophys. J., 2018, 114, 1847–1857 CrossRef CAS PubMed
- J. H. Fox, J. A. Kama, G. Lieberman, R. Chopra, K. Dorsey, V. Chopra, I. Volitakis, R. A. Cherny, A. I. Bush and S. Hersch, PLoS One, 2007, 2, e334 CrossRef PubMed
- G. Xiao, Q. Fan, X. Wang and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14995–15000 CrossRef CAS PubMed
- A. L. Lumsden, T. L. Henshall, S. Dayan, M. T. Lardelli and R. I. Richards, Hum. Mol. Genet., 2007, 16, 1905–1920 CrossRef CAS PubMed
- M. Kim, Prion, 2013, 7, 221–228 CrossRef CAS PubMed
- J. T. Marinko, H. Huang, W. D. Penn, J. A. Capra, J. P. Schlebach and C. R. Sanders, Chem. Rev., 2019, 119, 5537–5606 CrossRef CAS PubMed
- F. Yeboah, T.-E. Kim, A. Bill and U. Dettmer, Neurobiol. Dis., 2019, 132, 104543 CrossRef CAS PubMed
- S. L. Shammas, G. A. Garcia, S. Kumar, M. Kjaergaard, M. H. Horrocks, N. Shivji, E. Mandelkow, T. P. J. Knowles, E. Mandelkow and D. Klenerman, Nat. Commun., 2015, 6, 1–10 Search PubMed
- S. Wegmann, B. Eftekharzadeh, K. Tepper, K. M. Zoltowska, R. E. Bennett, S. Dujardin, P. R. Laskowski, D. MacKenzie, T. Kamath, C. Commins, C. Vanderburg, A. D. Roe, Z. Fan, A. M. Molliex, A. Hernandez-Vega, D. Muller, A. A. Hyman, E. Mandelkow, J. P. Taylor and B. T. Hyman, EMBO J., 2018, 37, e98049 CrossRef PubMed
- G. G. Moreira, J. S. Cristóvão, V. M. Torres, A. P. Carapeto, M. S. Rodrigues, I. Landrieu, C. Cordeiro and C. M. Gomes, Int. J. Mol. Sci., 2019, 20, 5979 CrossRef PubMed
- K. Afitska, A. Fucikova, V. V. Shvadchak and D. A. Yushchenko, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 701–709 CrossRef CAS PubMed
- G. Perrino, C. Wilson, M. Santorelli and D. di Bernardo, Cell Rep., 2019, 27, 916–927.e5 CrossRef CAS PubMed
- M. M. Wördehoff and W. Hoyer, Bio-Protoc., 2018, 8, e2941 Search PubMed
- M. Perni, P. Flagmeier, R. Limbocker, R. Cascella, F. A. Aprile, C. Galvagnion, G. T. Heller, G. Meisl, S. W. Chen, J. R. Kumita, P. K. Challa, J. B. Kirkegaard, S. I. A. Cohen, B. Mannini, D. Barbut, E. A. A. Nollen, C. Cecchi, N. Cremades, T. P. J. Knowles, F. Chiti, M. Zasloff, M. Vendruscolo and C. M. Dobson, ACS Chem. Biol., 2018, 13, 2308–2319 CrossRef CAS PubMed
- M. Kurnik, C. Sahin, C. B. Andersen, N. Lorenzen, L. Giehm, H. Mohammad-Beigi, C. M. Jessen, J. S. Pedersen, G. Christiansen, S. V. Petersen, R. Staal, G. Krishnamurthy, K. Pitts, P. H. Reinhart, F. A. A. Mulder, S. Mente, W. D. Hirst and D. E. Otzen, Cell Chem. Biol., 2018, 25, 1389–1402.e9 CrossRef CAS PubMed
- K. Murakami and K. Irie, Molecules, 2019, 24, 2125 CrossRef CAS PubMed
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. Ø. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS PubMed
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed
- Y. Kiriyama and H. Nochi, Cells, 2018, 7, 95 CrossRef PubMed
- X. Li, C. Dong, M. Hoffmann, C. R. Garen, L. M. Cortez, N. O. Petersen and M. T. Woodside, Sci. Rep., 2019, 9, 1–12 CrossRef PubMed
- M. Iljina, G. A. Garcia, M. H. Horrocks, L. Tosatto, M. L. Choi, K. A. Ganzinger, A. Y. Abramov, S. Gandhi, N. W. Wood, N. Cremades, C. M. Dobson, T. P. J. Knowles and D. Klenerman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1206–E1215 CrossRef CAS PubMed
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Alzheimer's disease; spongiform encephalopathies; Parkinson's disease; primary systemic amyloidosis; secondary systemic amyloidosis; Fronto-temporal dementias; senile systemic amyloidosis; familial amyloid polyneuropathy; hereditary cerebral amyloid angiopathy; haemodialysis-related amyloidosis; familial amyloid polyneuropathy; Finnish hereditary systemic amyloidosis; Type II diabetes; medullary carcinoma of the thyroid; atrial amyloidosis; hereditary non-neuropathic systemic amyloidosis; injection-localised amyloidosis; hereditary renal amyloidosis; amyotrophic lateral sclerosis; Huntington's disease; spinal and bulbar muscular atrophy; spinocerebellar ataxias; spinocerebellar ataxia. |
| This journal is © The Royal Society of Chemistry 2020 |