
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of crystalline viologen-based porous ionic polymers with hydrogen-bonded water for efficient catalytic CO2 fixation under ambient conditions†
Yadong Zhang,
Ke Zhang,
Lei Wu,
Ke Liu,
Rui Huang,
Zhouyang Long,
Minman Tong * and
Guojian Chen*
* and
Guojian Chen*
School of Chemistry and Materials Science, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou 221116, China. E-mail: gjchen@jsnu.edu.cn; tongmm@jsnu.edu.cn
First published on 22nd January 2020
Abstract
In this work, we report a series of crystalline viologen-based porous ionic polymers (denoted VIP-X, X = Cl or Br), that have in situ formed dicationic viologens paired with halogen anions and intrinsic hydrogen-bonded water molecules, towards metal-free heterogeneous catalytic conversion of carbon dioxide (CO2) under mild conditions. The targeted VIP-X materials were facilely constructed via the Menshutkin reaction of 4,4′-bipyridine with 4,4′-bis(bromomethyl)biphenyl (BCBMP) or 4,4′-bis(chloromethyl)biphenyl (BBMBP) monomers. Their crystalline and porous structures, morphological features and chemical structures and compositions were fully characterized by various advanced techniques. The optimal catalyst VIP-Br afforded a high yield of 99% in the synthesis of cyclic carbonate by CO2 cycloaddition with epichlorohydrin under atmospheric pressure (1 bar) and a low temperature (40 °C), while other various epoxides could be also converted into cyclic carbonates under mild conditions. Moreover, the catalyst VIP-Br could be separated easily and reused with good stability. The remarkable catalytic performance could be attributed to the synergistic effect of the enriched Br− anions and available hydrogen bond donors –OH groups coming from H-bonded water molecules.
Introduction
Currently, the chemical fixation and utilization of CO2 has attracted increasing attention, because CO2 is an environmentally unfavourable greenhouse gas as well as an abundantly available C1 resource.1 Among various strategies for handling the above issue, the synthesis of high value-added cyclic carbonates by CO2 cycloaddition with epoxides is one of the most efficient and atom-economic routes to achieve catalytic CO2 fixation.2–5 To date, various homogeneous and heterogeneous catalytic systems have been developed for CO2 conversion.3–5 In general, homogeneous catalysts can exhibit high catalytic activities under mild conditions but with complicated processes of separation, purification and recycling, which would limit their practical application.3,4 In contrast, heterogeneous catalysts can solve these problems, but often the reactions are performed under high temperatures and/or CO2 pressures to obtain the desired catalytic activities.5 Meanwhile, there are also some problems in preparing efficient heterogeneous catalysts with available active sites via an easily-achieved synthetic method. Therefore, it is necessary to develop a facile method to construct the desired heterogeneous catalysts toward CO2 fixation under mild conditions.Porous ionic polymers (PIPs) are a class of rising functional polymeric materials, which have aroused much attention owing to their intriguing characters including abundant ionic sites, controllable porous structures, chemical functionalities, and ion exchange properties.6,7 In particular, ionic moieties of halogen-based PIPs have strong dipole quadrupole interactions with CO2, which endow intrinsic CO2-philicity for PIPs, while the specific halogen anions can be used as active nucleophiles for catalytic conversion of CO2.8,9 Further, a lot of ionic liquids (ILs) or IL-like structures in the solid polymers have intrinsic hydrophilicity, which can chemically absorb water molecules nearby ionic moieties via hydrogen bond interactions between water and halogen anions (such as Cl− and Br−).10–13 It was reported that a suitable amount of water can be regarded as hydrogen bond donors (HBD) co-catalysts for enhancing the coupling of CO2/epoxide by forming H-bonds with the oxygen atom of epoxides.4,14 Therefore, we intend to explore the targeted synthesis of task-specific porous ionic polymers with halogen anions and H-bonded water molecules for enhancing the CO2 activation and conversion.
Recently, different types of ionic polymers that containing diverse ionic moieties such as imidazolium,15–18 pyridinium,19–21 viologen,22–25 quaternary phosphonium,26–28 and others,6,7,29–31 have been reported. Amongst, viologen-based ionic polymers have been regarded to be one type of promising functionalized polymers towards diverse applications, due to their unique properties of adjustable redox states and viologen ionic active sites.22,23 In terms of synthesis, a series of viologen-based ionic polymers have been prepared using various strategies including C–C coupling reactions (e.g., Heck reaction,32 and Sonogashira coupling reaction33,34), ionothermal trimerization,35 imine condensation,36,37 Zincke reaction,38–41 and Menshutkin reaction.11,42–48 Through coupling reactions and ionothermal reactions can afford viologen-based porous ionic polymers with high surface areas, these processes are complicated and environmental-unfriendly that require noble-metal catalysts or additives or high reaction temperatures.32–37 Very recently, Zincke reaction has been used as a catalyst-free method to prepare viologen-based porous ionic networks with moderate surface areas.38–41 However, this strategy is highly dependent on using high-cost multi-aniline-based monomers and viologen Zincke salts. Besides, this reaction is not an atomic economic reaction, which inevitably releases the toxic by-product 2,4-dinitroaniline.38,39 Thus, we think the Menshutkin reaction (i.e., nucleophilic substitution) can afford a convenient, catalyst-free, high-efficient and atomic economic route to achieve viologen ionic polymers. Nevertheless, direct synthesis of viologen-based ionic polymers with crystalline and porous structures is still a challenging task. In aspects of heterogeneous catalysis in the CO2 conversion, only a few viologen-based ionic polymers prepared by various reactions have been employed in the catalytic CO2 fixation, but which often performed under harsh conditions at high temperatures or high CO2 pressures.34,35,39 Therefore, it is very desirable to develop a low-cost and facile method to prepare the targeted viologen-based porous ionic polymers for the CO2 conversion at a low temperature and an atmospheric pressure.
Herein, we report a facile and direct synthesis of crystalline viologen-based porous ionic polymers (denoted VIP-X, X = Cl or Br) simultaneous with enriched halogen anions and H-bonded water molecules. As depicted in Scheme 1, the targeted VIP-X were fabricated by the Menshutkin reaction of 4,4′-bipyridine (4,4′-BPy) with commercially-available linkers BCMBP or BBMBP. It is noted that some viologen-based ionic polymers synthesized by the Menshutkin reaction usually have amorphous and non-porous structures with low surface areas.44,46–48 The present VIP-Cl not only has an obvious crystalline ordered structure and but also behaves a flower-like sheet morphology with considerable surface area up to 56 m2 g−1. Besides, the specific contents of stable H-bonded water molecules were found in the obtained VIP-X. Though chemically absorbed water is often observed in ILs and IL-derived solid materials,10–13 few attentions have paid on the valuable role of these intrinsic H-bonded water molecules for enhancing HBD-based catalysis. In this work, the optimal VIP-Br exhibited remarkable heterogeneous catalytic activities in the CO2 cycloaddition with various epoxides under mild conditions without adding any co-catalysts and solvents, which attributed to the synergistic catalysis of the abundant Br− anions and HBD co-catalyst H2O.
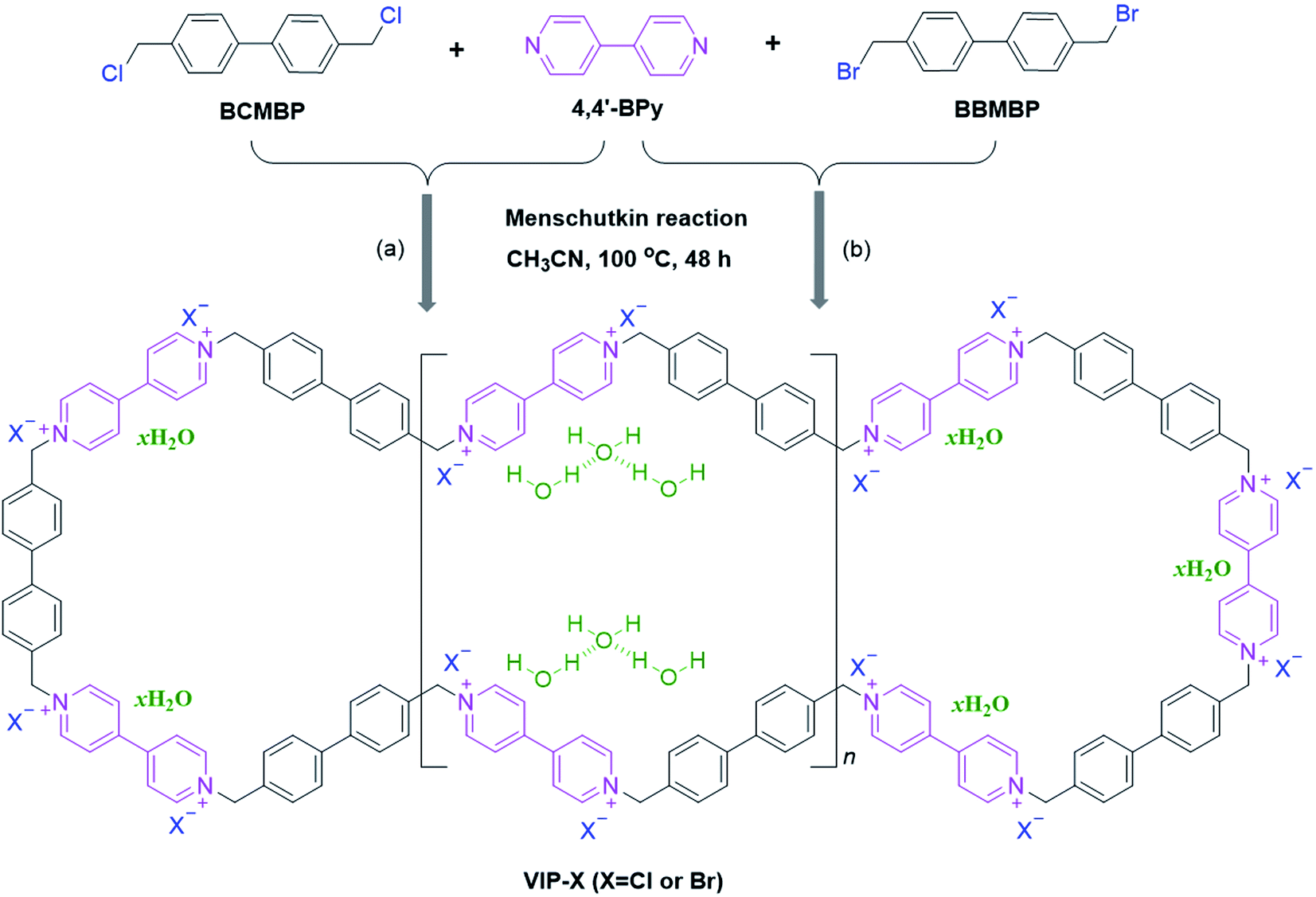 | ||
| Scheme 1 Synthetic route to crystalline viologen-based porous ionic polymers VIP-X (X = Cl or Br) with intrinsic H-bonded water molecules via the Menschutkin reaction of 4,4′-bipyridine with (a) BCMBP and (b) BBMBP under solvothermal conditions (i.e., CH3CN, 100 °C, 48 h). | ||
Experimental
Materials
Raw materials 4,4′-bipyridine, 4,4′-bis(chloromethyl)biphenyl, 4,4′-bis(bromomethyl)biphenyl and the required substrates and solvents are commercially available and used without further purification.Methods
13C cross-polarization/magic angle spinning (CP/MAS) NMR spectra was measured using a Bruker AVANCE III 600 spectrometer. Liquid-state 1H spectra of the product cyclic carbonates were performed on a Bruker DPX 500 spectrometer. Fourier transform infrared spectroscopy (FTIR) was performed on a Bruker Vertex 80 V instrument. Chemical compositions and states of the samples were determined by the X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250Xi). Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMX-10/12 spectrometer at the X-band at room temperature. CHN elemental analysis was conducted on an elemental analyzer Vario EL cube. Thermogravimetry analysis (TGA) was measured with a TA Q50 instrument at a heating rate of 10 °C min−1 under a N2 atmosphere. Scanning electron microscope (SEM) images were performed on the HITACHI S-4800 field emission scanning electron microscope. Transmission electron microscopy (TEM) images were performed on the JEOL JEM-2100F 200 kV field-emission transmission electron microscopes. Powder X-ray Diffraction (PXRD) patterns were collected on a Bruker D8 Advance powder diffractometer. N2 sorption experiments were measured using a Quantachrome autosorb iQ2 analyzer at 77 K, and the surface area of sample was calculated using the Brunauer–Emmett–Teller (BET) method and the pore size distribution was determined was determined by the Barrett–Joyner–Halenda (BJH) model. All the samples were degassed at 150 °C for 6 h in a high vacuum before analysis.Synthesis of viologen-based ionic polymers
Viologen-based ionic polymers were synthesized by one-step Menshutkin reaction. Typically, 4,4′-BPy (1 mmol) and BCMBP (1 mmol) were dissolved in acetonitrile (CH3CN, 20 mL). The static reaction was taken place in a 25 mL Teflon-lined autoclave in a constant temperature oven at 100 °C for 48 h. After reaction, the solid product was collected by filtration and washed with CH3CN, water and ethanol for several times. By drying under vacuum at 80 °C for 12 h. The resultant ionic polymer was named as VIP-Cl (yellow powder solid, yield of 86%). Similarly, VIP-Br was prepared in a similar procedure by the replacement of BCMBP with BBMBP (yellow powder solid, yield of 88%). Besides, a series of VIP-Cl and VIP-Br solid materials were prepared in various solvents including 1,4-dioxane, N-methyl pyrrolidone (NMP) and N,N-dimethylformamide (DMF).Cycloaddition of CO2 with epoxides
Using epichlorohydrin (ECH) as a typical substrate, the catalytic CO2 fixation reaction was carried out under mild conditions. In a typical run, ECH (2 mmol) and the catalyst VIP-Br (0.05 g) were placed in a Schlenk tube connected with a CO2 balloon (1 bar). After then, the mixture was stirred for desired time at the target temperature. After reaction, ethyl acetate (3 mL) was added to the reaction mixture and stirred for 0.5 h, the solid catalyst was separated by centrifugation. The obtained filtrate was analysed by gas chromatograph (GC) to afford the yield and selectivity of the product. For other substrates, the crude products were obtained by concentrating under reduced pressure and then were directly analysed by 1H NMR spectroscopy to determine the yields of cyclic carbonates.For the catalyst recycling experiments, the reaction was performed under the same reaction conditions each time using the recovered catalyst from the previous run. The reusability of the catalyst was tested in five-run cycling experiments. The solid catalyst was collected by centrifuged, washed with ethanol, dried in vacuum and then charged into the next run.
Results and discussion
Synthesis and characterization of VIP-X
As depicted in Scheme 1, viologen-based ionic polymers VIP-X (X = Cl, Br) were facilely synthesized via the Menshutkin reaction of 4,4′-BPy with BCMBP or BBMBP under solvothermal conditions. For synthesis of VIP-Cl, various solvents including CH3CN, 1,4-dioxane, NMP and DMF were investigated to afford a series of VIP-Cl (solvent) solid materials. These VIP-Cl series are insoluble in water and common organic solvents such as ethanol, ethyl acetate, CH3CN, tetrahydrofuran (THF), DMF and dimethyl sulfoxide (DMSO), which were different from previously reported water or solvent-soluble viologen-based ionic salts,49 oligomers,50 and linear polymers or networks.37,51,52Remarkably, compared with crystalline structures of 4,4′-BPy and BCMBP (Fig. S1A†), XRD patterns (Fig. 1) of the formed VIP-Cl materials lose most of original sharp peaks of monomers, but still display three obvious Bragg diffraction peaks around 10.2°, 21.3° and 25.9°, and also appear broad peaks between 23–25°, indicating VIP-Cl materials are partially-crystalline ordered polymers along with certain amorphous structures. The order crystallization is very rare in the reported viologen-based ionic polymers.38,45 In detail, the sharp peak at 10.2° with a d-spacing value of 0.86 nm corresponds to molecular dimension of viologen moieties, which is in accordance with the molecular structure of 4,4′-BPy monomer (Fig. S2†). The raised broad peaks at around 2θ = 20.3° and 25.9° with corresponding d-spacings of 0.43 and 0.35 nm, implying that viologen and biphenyl moieties are periodically arranged with certain spatial distances.11,53 Unfortunately, our attempts to provide the most probably structure for VIP-Cl materials have failed, because of the lack of enough available cell parameters obtained from XRD patterns, which are similar to previously reported ionic covalent organic polymers.45 These situations mainly due to the reason that VIP-Cl materials are partially-crystalline linear polymers but not long-range order crystalline frameworks, which are different from typical crystalline framework materials such as metal–organic frameworks and covalent organic frameworks with definite topological structures and cell parameters.24,25,29
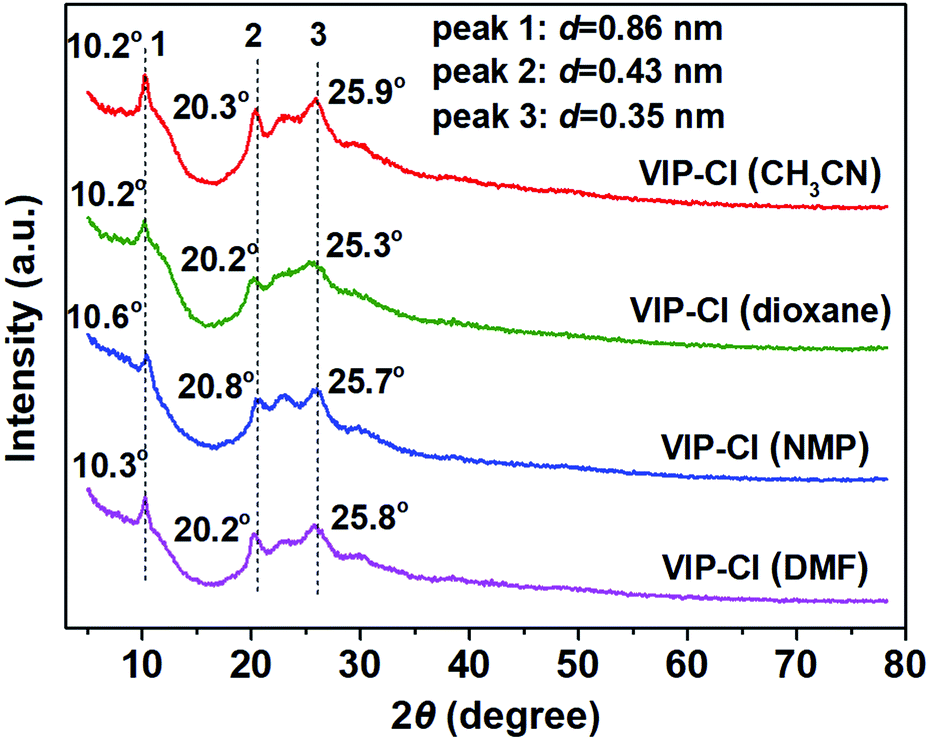 | ||
| Fig. 1 XRD patterns of a series of VIP-Cl materials that were synthesized in different solvents. | ||
Though the VIP-Cl samples obtained in different solvents have similar crystalline structures reflected in the XRD patterns, their porous characters are obviously different, which were determined by N2 adsorption/desorption measurements (Fig. 2 and S3†). For the samples of VIP-Cl (dioxane), VIP-Cl (NMP) and VIP-Cl (DMF), they exhibit type II isotherms (Fig. S3A†) with low N2 uptakes at 77 K, only giving low BET surface areas of 5 m2 g−1, 2 m2 g−1 and 1.5 m2 g−1, respectively. Surprisingly, the sample VIP-Cl (CH3CN) has a larger surface area of 56 m2 g−1 and a total pore volume of 0.33 cm3 g−1. Further, VIP-Cl (CH3CN) exhibit type IV isotherms (Fig. 2A) with an increasing uptake at the higher relative pressure (0.80 < P/P0 < 0.99) and an obvious H1 type hysteresis loop, indicative of a typical mesoporous polymer. The BJH pore size distribution (Fig. 2B) of VIP-Cl (CH3CN) confirm its mesoporous structure with two types of mesopores centered at 3.9 and 24.3 nm. Under the similar synthetic conditions in the above solvents, a series of VIP-Br solid materials were also obtained by replacing BCMBP with BBMBP. The sample VIP-Br prepared in CH3CN has a moderate surface area of 38 m2 g−1 and pore volume of 0.29 cm3 g−1 (Fig. 2), which is higher than those of other VIP-Br samples (Fig. S3B†) but is a bit lower than that of VIP-Cl (CH3CN). These results owe to the larger Br− size than the one of Cl−, which would occupy larger space nearby viologen ionic sites within the polymer. Also, the pore size distribution (Fig. 2B) of VIP-Br displays abundant smaller mesopores ranging from 3.9 to 13.4 nm by pairing with the larger Br− anions. It is noted that VIP-Br series also have partially-crystalline structures with some observable peaks at 10.5°, 20.3°, 22.6° and 26.0° in the XRD patterns (Fig. S1C†), however, they display much lower degree of crystallinity than VIP-Cl series, indicating more amorphous form exists in the VIP-Br series. As a result, the samples VIP-Cl (CH3CN) and VIP-Br (CH3CN) that synthesized in CH3CN have higher surface areas than samples prepared in other solvents, which would appear with the simplified names VIP-Cl and VIP-Br latter. To our knowledge, the prepared crystalline VIP-Cl possesses a relative high surface area of 56 m2 g−1, superior to most of viologen-based polymers with low surface areas that prepared by the Menshutkin reaction,11,45–48 comparable to viologen-based covalent organic networks that synthesized by the Zincke reaction,38–41 but is still inferior to those viologen-based porous polymers that fabricated using various metal catalysts and additives32–35 (see Table S1† for the detailed comparisons).
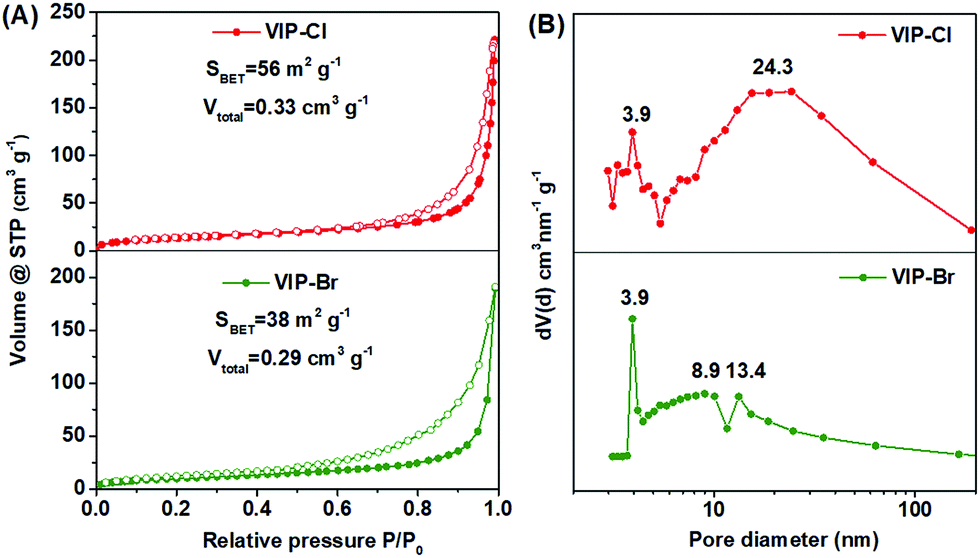 | ||
| Fig. 2 (A) N2 adsorption–desorption isotherms and (B) BJH pore size distributions of VIP-Cl and VIP-Br synthesized in CH3CN. | ||
The morphology of VIP-X (X = Cl or Br) was observed by the scanning electron microscopy (SEM) images. As shown in Fig. 3A–D, the sample VIP-Cl exhibits a hierarchical, flower-like ordered morphology that assembled by the interconnected nanosheets, while VIP-Br also has a flower-like shape consisted of cross-linked aggregates. TEM images exhibit that VIP-Cl and VIP-Br samples are composed of crystalline structures (the dark sections in Fig. 3E and G) and amorphous forms (the light sections in Fig. 3E and G), which are consistent with their partially-crystalline structures reflected by the XRD patterns. From the HRTEM images (Fig. 3F and H), the obvious lattice fringes with a spacing of 0.43 nm for VIP-Cl and VIP-Br were observed, which correspond to the layer d-spacing of 0.43 nm from the Bragg diffraction peaks at 20.3° in the XRD patterns. These ordered spatial arrays should be attributed to aromatic π–π stacking interaction between the periodically repeated viologen and biphenyl units.11,53
 | ||
| Fig. 3 SEM images of (A and B) VIP-Cl and (C and D) VIP-Br, and TEM images of (E and F) VIP-Cl and HRTEM images (G and H) VIP-Br. | ||
The chemical structures and compositions of VIP-X were determined by solid-state 13C NMR, FTIR, XPS and elemental analysis. In the 13C NMR spectra (Fig. 4A) of VIP-Cl and VIP-Br, the obvious multi-peaks in the region of 144.1–126.1 ppm are attributable to aromatic carbon signals in the phenyl and bipyridinium rings of viologen cationic polymers.43,48 The carbon signals located at 61.0 ppm for VIP-Cl and 59.3 ppm for VIP-Br are assigned to methylene carbon atoms (–CH2–) linking the bipyridinium and benzene rings.43,48 The above results of 13C NMR confirm that viologen moieties and biphenyl-methylene units were successfully introduced into the polymers. In addition, FTIR spectra (Fig. 2B and C) of VIP-X were further performed to confirm the chemical structures of the targeted ionic polymers. The presence of viologen ionic moieties were demonstrated by the characteristic peaks at 1634 or 1639 and 1206 cm−1 that assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+ and C–N bonds in viologen units.32,39,48 The successful incorporation of biphenyl-methylene groups was confirmed by the biphenyl aromatic C–H stretching vibrations at 3116, 3034 and 3027 cm−1, methylene C–H stretching vibrations at 2854 cm−1, the skeletal vibrations of the aromatic ring appeared at 1500–1443 cm−1, and obvious out-of-plane C–H bending vibration bands of adjacent hydrogens on aromatic ring at 789 cm−1.54 No obvious wagging absorption peaks of –CH2Cl (1273 cm−1) and –CH2Br (1228 cm−1) groups,42,48,54 stretching absorption peaks for C–Cl (676 cm−1) or C–Br bonds (596 cm−1) within the monomers BCMBP and BBMBP (Fig. 4B and C) were found in the formed polymers, indicating the formation of closed-loop ionic polymers with high degrees of polymerization, without residual monomers or –CH2Cl/–CH2Br end groups. Moreover, the broad peaks at 3362 and 3358 cm−1 are assigned to H-bonded water associated with halogen anions in viologen-based ionic polymers, which are different from those of free –OH groups in unbounded water.10 The elemental compositions of VIPs were also analysed by elemental analysis (EA), as listed in Table S2.† The EA results of VIP-X are well consistent with the theoretical values for the targeted closed-loop ionic polymers with repeated viologen units and H-bonded water molecules. Based on the results of EA and TGA (Fig. S4†), the contents of water are approximately 14.6 wt% for the VIP-Cl and 8.5 wt% for the VIP-Br. These desired water molecules are originated from the chemical adsorption of moisture from air under ambient conditions that enhanced by the hydrophilic ionic nature of VIP-X,11,12 which are beneficial to accelerate CO2 conversion by HBD catalysis.4,14
N+ and C–N bonds in viologen units.32,39,48 The successful incorporation of biphenyl-methylene groups was confirmed by the biphenyl aromatic C–H stretching vibrations at 3116, 3034 and 3027 cm−1, methylene C–H stretching vibrations at 2854 cm−1, the skeletal vibrations of the aromatic ring appeared at 1500–1443 cm−1, and obvious out-of-plane C–H bending vibration bands of adjacent hydrogens on aromatic ring at 789 cm−1.54 No obvious wagging absorption peaks of –CH2Cl (1273 cm−1) and –CH2Br (1228 cm−1) groups,42,48,54 stretching absorption peaks for C–Cl (676 cm−1) or C–Br bonds (596 cm−1) within the monomers BCMBP and BBMBP (Fig. 4B and C) were found in the formed polymers, indicating the formation of closed-loop ionic polymers with high degrees of polymerization, without residual monomers or –CH2Cl/–CH2Br end groups. Moreover, the broad peaks at 3362 and 3358 cm−1 are assigned to H-bonded water associated with halogen anions in viologen-based ionic polymers, which are different from those of free –OH groups in unbounded water.10 The elemental compositions of VIPs were also analysed by elemental analysis (EA), as listed in Table S2.† The EA results of VIP-X are well consistent with the theoretical values for the targeted closed-loop ionic polymers with repeated viologen units and H-bonded water molecules. Based on the results of EA and TGA (Fig. S4†), the contents of water are approximately 14.6 wt% for the VIP-Cl and 8.5 wt% for the VIP-Br. These desired water molecules are originated from the chemical adsorption of moisture from air under ambient conditions that enhanced by the hydrophilic ionic nature of VIP-X,11,12 which are beneficial to accelerate CO2 conversion by HBD catalysis.4,14
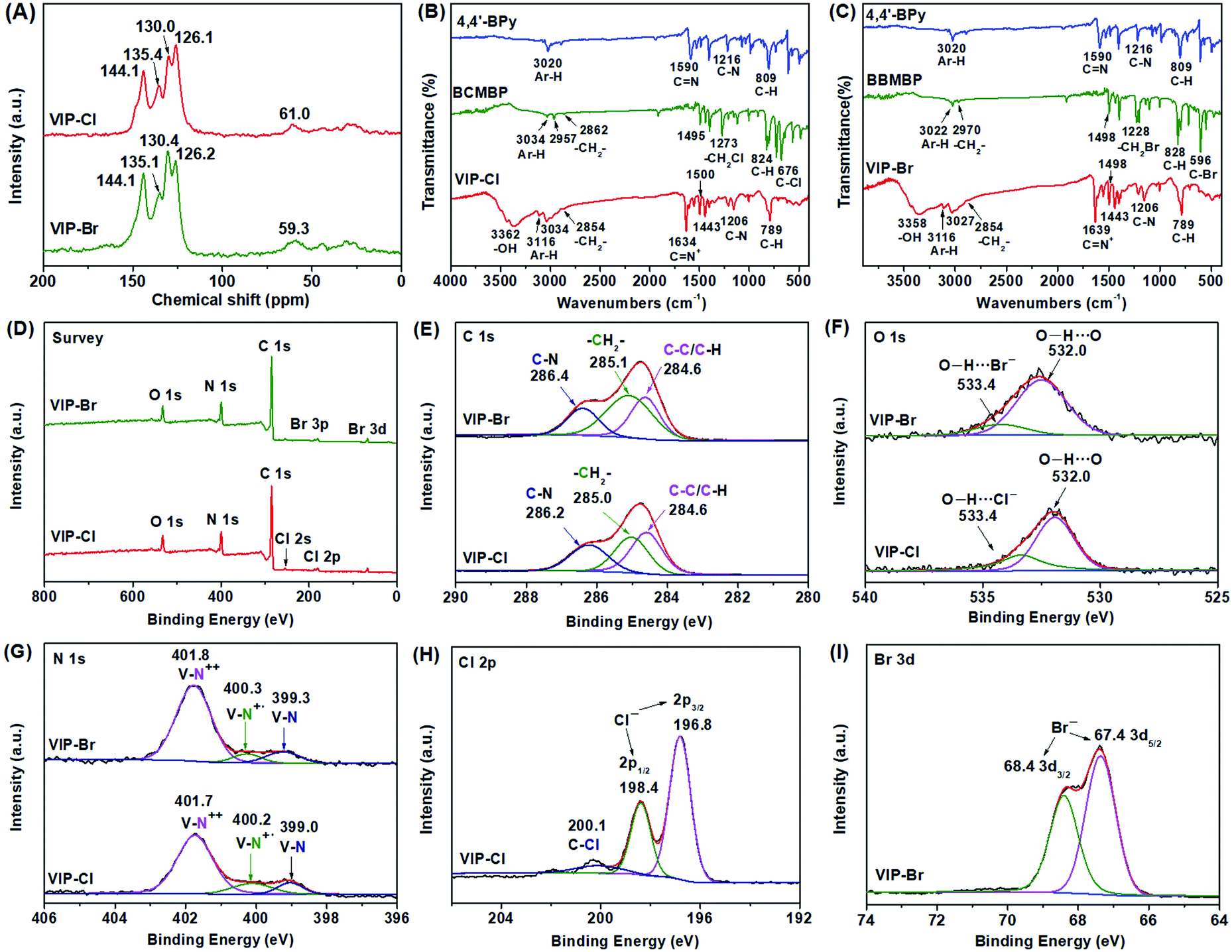 | ||
| Fig. 4 Characterizations for chemical structures of VIP-Cl and VIP-Br: (A) solid-state 13C CP/MAS NMR, (B) and (C) FTIR spectra, (D) XPS spectra survey, (E) C 1s, (F) O 1s, (G) N 1s, (H) Cl 2p and (I) Br 3d. | ||
The chemical compositions of VIP-X were further determined by the XPS spectra. The full survey XPS spectra (Fig. 4D) displayed C, N, Cl or Br elements for VIP-Cl and VIP-Br. As shown in Fig. 4E, the C 1s XPS spectra are divided into three distinct peaks at 286.2/286.4, 285.0/285.1 and 284.6 eV, which are assigned to the carbon atoms of CN bonds in the viologen groups, methylene (–CH2–) and aromatic rings (C–C/C–H) in the VIP-X.32 The obvious O 1s peaks (Fig. 4F) appeared at 532.0 and 533.4 eV that assigned to O–H⋯O and O–H⋯[X−] (X = Cl or Br), further confirming the presence of H-bonded water in the VIP-X.55,56 The N 1s spectra for VIP-X (Fig. 4G) could be fitted to three distinct peaks. For VIP-Cl and VIP-Br, the main peaks appeared at 401.7/401.8 eV with very high fitting area ratios (76.1% and 81.4%) are originated from N atoms of dicationic viologen moieties (V–N++),32,35,40 demonstrating that VIP-X have high contents of viologen dicationic sites. Besides, very small peaks appeared at 400.2/400.3 eV with small fitting area ratios can be assigned to some reduced radical cationic viologen species (V–N+˙) that formed during the reaction.32,35,40 However, these small contents of radical species can't be detected by the solid-state EPR spectroscopy (Fig. S5†). Meanwhile, the small peaks at 399.3/399.0 eV may be assigned to N atoms of fully reduced neutral viologens or unreacted bipyridines.22 In the XPS Cl 2p spectrum (Fig. 4H) of VIP-Cl, the peaks appeared at 196.8 eV (2p3/2) and 198.4 eV (2p1/2) are assigned to free Cl− anion.35 Besides, the peak for C–Cl bond at 200.1 eV was very weak, indicating the monomer BCMBP was almost totally consumed. For the VIP-Br, the Br 3d spectrum (Fig. 4I) displays two strong peaks at 68.4 eV (3d3/2) and 67.4 eV (3d5/2), while no signal for C–Br can be observed,17,32,56 suggesting that all the Br atoms are in the state of Br− anions within the VIP-Br. These results are consistent with FTIR spectra. In other words, these formed VIP-X are highly polymerized linear polymers, not viologen-based oligomers or ionic salts, which possess high contents of ionic moieties paired with halogen anions. Further, the energy-dispersive X-ray absorption spectroscopy (EDS) elemental mapping images (Fig. S6†) describe homogeneous dispersions of the targeted C, N, Cl or Br elements within the VIP-X materials. As a result, the above full characterizations demonstrated that two viologen-based ionic polymers were successfully prepared with high degrees of polymerization and high density of halogen anions and available H-bonded water via one-step Menshutkin reaction.
Catalytic performance in CO2 conversion
Owing to the enriched halogen ionic sites and desired bonded water as well as the insolubility in the reaction system, the obtained VIP-X were investigated as heterogeneous catalysts in the cycloaddition of CO2 with epoxides without adding any co-catalysts or solvents. To explore the suitable conditions for the synthesis of value-added cyclic carbonates, epichlorohydrin (ECH) was chosen to the model epoxide for this reaction (Table 1). First, the reaction was directly carried out at a relative low temperature of 60 °C using a CO2 balloon (1 bar), the catalyst VIP-Cl gives a moderate yield of 75% using 48 h but affords a desired yield of 98% using a longer time of 72 h (Table 1, entries 1 and 2), while the control catalysts VIP-Cl (dioxane), VIP-Cl (NMP) and VIP-Cl (DMF) with low surface areas give a bit lower yields of 95–96%, due to the differences of their surface areas. Another control catalyst VIP-Br achieves a high yield of 99% with an ideal selectivity only using 48 h (Table 1, entry 3). Obviously, the catalyst VIP-Br exhibits a better catalytic activity with a high TOF value of 0.453 h−1 than those of VIP-Cl (TOF values: 0.303 and 0.264 h−1) at 60 °C. Stimulated by the good catalytic behaves, the catalysts VIP-Br and VIP-Cl were further performed at near room temperature (40 °C) and room temperature (25 °C). Remarkably, the catalyst VIP-Br achieves the almost full conversion (99%) of ECH only spending 72 h and gives a high TOF of 0.302 h−1 (Table 1, entry 4). In comparison, the catalyst VIP-Cl gives a moderate yield of 60% using 72 h (Table 1, entry 5) and can offer a considerable yield of 90% and a TOF value of 0.146 h−1 by spending a longer time of 120 h (Table 1, entry 6). When the reactions were carried out at 25 °C, VIP-Br can give a moderate yield of 61% but VIP-Cl only shows a low yield of 33% (Table 1, entries 7 and 8). As a result, the catalyst VIP-Br can provide higher catalytic activities that reflected in yields and TOF values than those of VIP-Cl under the same conditions, which can be attributed to the higher nucleophilicity and better leaving ability of the Br− anion to Cl− anion.39,57 By full evaluation of the catalytic activities in TOF values, VIP-Br is considered as the ideal heterogeneous catalyst that can achieve the high yield (99%) and the highest TOF value (0.453 h−1) under the optimal condition (i.e., 1 bar CO2, 60 °C, 48 h). These satisfactory catalytic results are superior to many reported metal-free ionic polymers (IPs)15,16,27,28,34,35 and even surpass to some IPs with electrophiles (metal sites)58–61 or task-specific HBD groups (such as hydroxyl,28,57,62 silanols,17,32,39 carboxylic acids,63 and ureas18) (see Table S3† for details).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | t (h) | Yb (%) | Sb (%) | TOFc (h−1) |
| a Reaction conditions: ECH (2 mmol), CO2 balloon (1 bar), the catalyst VIP-X (0.05 g), temperature (T = 25–60 °C), time (t = 48–120 h).b Yield (Y) and selectivity (S) of the cyclic carbonate were determined by GC and 1H NMR.c Turnover frequency (TOF) = [mmol(product)]/[mmol(V ionic content in the catalyst) × reaction time (h)]. Viologen (V) ionic contents were calculated from EA results (see ESI Table S2). | ||||||
| 1 | VIP-Cl | 60 | 48 | 75 | 99 | 0.303 |
| 2 | VIP-Cl | 60 | 72 | 98 | 99 | 0.264 |
| 3 | VIP-Br | 60 | 48 | 99 | 99 | 0.453 |
| 4 | VIP-Br | 40 | 72 | 99 | 99 | 0.302 |
| 5 | VIP-Cl | 40 | 72 | 60 | 99 | 0.162 |
| 6 | VIP-Cl | 40 | 120 | 90 | 99 | 0.146 |
| 7 | VIP-Br | 25 | 120 | 61 | 99 | 0.112 |
| 8 | VIP-Cl | 25 | 120 | 33 | 99 | 0.053 |
Besides, it is worthy to note that the HBD cocatalyst H-bonded water molecules in VIP-Br play a vital role in activating and enhancing the CO2/ECH coupling by forming H-bonds with the oxygen atom of ECH.4,14,64,65 Fortunately, the high selectivity (99%) of the product cyclic carbonate were still achieved together with high yields catalyzed by the inherent water-contained VIP-Br, indicating that no byproduct diol was formed via a possible hydrolysis process. In this work, no additional water was considered to enhance the conversion of the epoxide, because the selectivity for the product would decrease due to the ring opening of ECH to 3-chloro-1,2-propanediol by hot water.32,66,67 The suitable amount of inbuilt H-bonded water in the catalyst not only can enhance catalytic activities by synergistic catalysis with nucleophilic Br− anions but also has no effect on the high selective synthesis of cyclic carbonates.
The structural stability and reusability of heterogenous catalysts are two key factors that can determine their prospects in practical applications. As shown in Fig. 5, a five-cycle experiments were conducted to assess the catalytic reusability of VIP-Br in the conversion of CO2 with ECH at 60 °C for 48 h. No obvious decrease in both the yield and selectivity were observed, owing to its well-preserved chemical structure and composition, and morphology of the recovered catalyst, which were confirmed by its similar results of elemental analysis (C%, 53.1; H%, 4.8; N%, 5.2), FTIR (Fig. S7†) and SEM image (Fig. S8†) of the reused VIP-Br. These above results demonstrate the excellent stability and reusability of the catalyst VIP-Br.
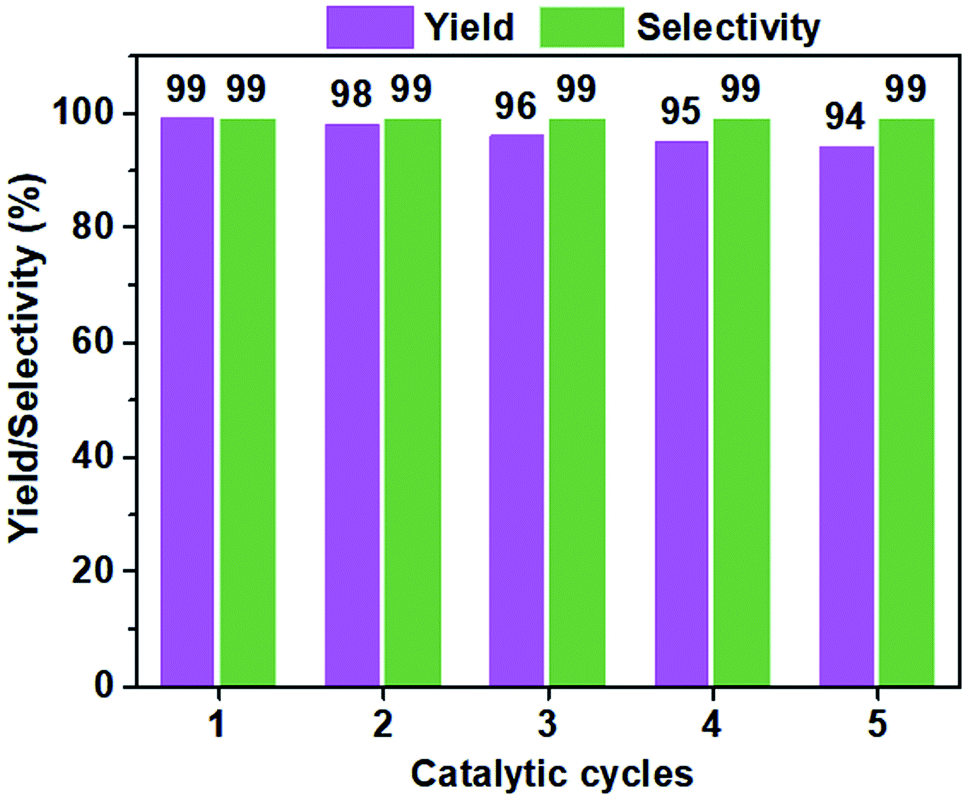 | ||
| Fig. 5 A five-cycle assessment in the catalytic reusability of VIP-Br for the CO2 conversion with ECH. Reaction conditions: ECH (2 mmol), CO2 pressure (1 bar), the catalyst (0.05 g), 60 °C, 48 h. | ||
Encouraged by the good catalytic activity and stability of VIP-Br, the substrate compatibility of various epoxides was further explored under atmospheric pressure (1 bar CO2). As shown in Fig. 6, many epoxides (1a–1h) could be smoothly converted into the corresponding cyclic carbonates (2a–2h) with high yields that determined by 1H NMR analyses of the crude products (see Fig. S9–S16†). In detail, the epibromohydrin (1a) could be easily converted into the product 2a with a high yield of 99% under very mild conditions (i.e., 60 °C, 48 h). The conversion of styrene oxide (2b), glycidyl phenyl ether (2c) and allyl glycidyl ether (2d) could be well accomplished with high yields of 93–99% at 80 °C for 48 h, which were required high temperatures (100–140 °C) using various other heterogeneous catalysts.15,16,24 Remarkably, the extreme inert long carbon-chain alkyl epoxides could be also converted into the corresponding cyclic carbonates (2e–2h) with satisfactory yields (90–94%) under atmospheric pressure using different temperatures and times. We noted that the longer carbon-chain alkyl epoxides (2g and 2h) required a relatively high temperature of 120 °C and longer time (72 h) than the shorter ones (2e and 2f), which ascribed to the increased mass-transfer resistance of these large-sized hydrophobic substrates. In a word, the present heterogeneous catalyst VIP-Br behaves favourable substrate compatibility under mild conditions.
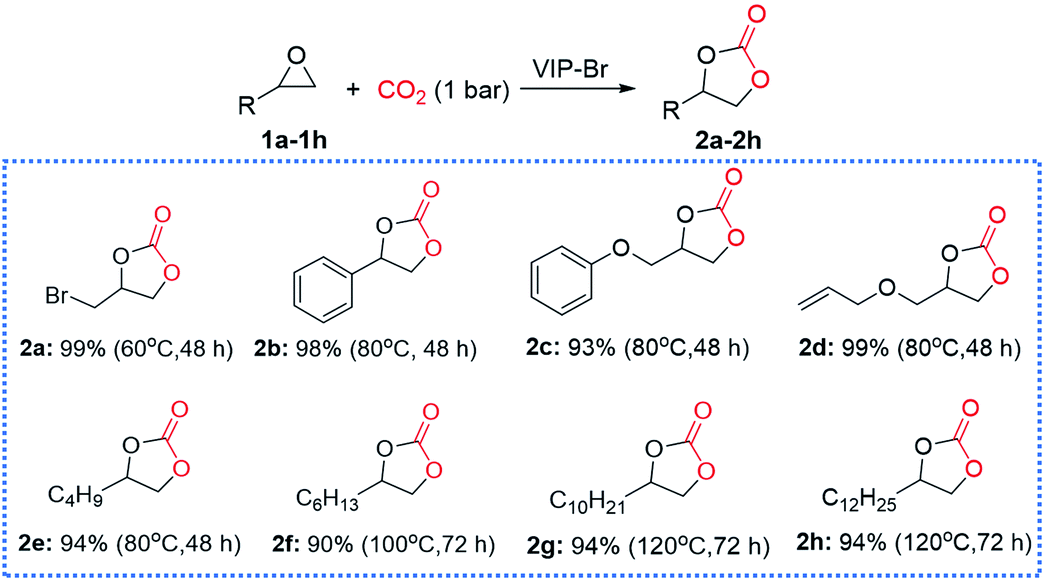 | ||
| Fig. 6 Substrate compatibility of various epoxides for the CO2 cycloaddition over the catalyst VIP-Br. Reaction conditions: substrate (2.0 mmol), catalyst (0.05 g), CO2 (1 bar), 60–120 °C, 48–72 h. | ||
Insights into the catalytic behavior
Previous works have revealed that the combination of halogen anions and hydrogen bond donors –OH groups can enhance the activation and conversion of CO2 with epoxides.14,28,57,68 Obviously, the present VIP-Br has both intrinsic viologen ionic sites with nucleophilic Br− anions and H-bonded water molecules, which makes for the good catalytic performance by the synergistic catalysis effect.64,65 Based on the above experimental results and previous reports, a plausible catalytic reaction mechanism is proposed for the cycloaddition of CO2 with epoxides over the catalyst VIP-Br (Scheme 2). First, the epoxide substrate was electrophilically activated by the hydrogen-bonding interaction between the oxygen atoms from epoxides and the free –OH groups within H-bonded water, thus inducing the ring opening of the epoxide via the C–O bond polarization.65,69 Second, the nearby nucleophilic Br− anion will attack the less hindered β-carbon atom of the activated epoxide, leading to the formation of an oxyanion intermediate that can be stabilized by the available multiple –OH groups from the H-bonded water.69,70 Subsequently, the insertion of CO2 into the oxyanion intermediate will create the OH-stabilized alkylcarbonate anion intermediate.17,32,69 At last, the intramolecular ring-closing of the above alkylcarbonate anion intermediate can afford the cyclic carbonate, along with the release of Br− anions and regeneration of the catalyst.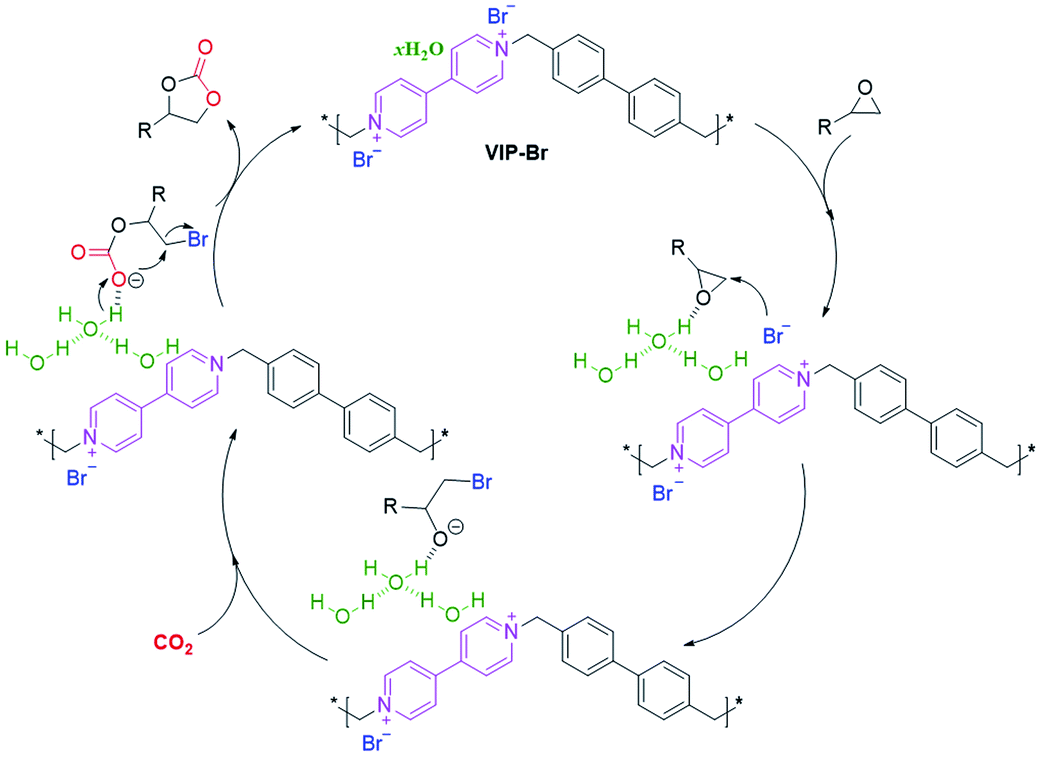 | ||
| Scheme 2 A proposed catalytic reaction mechanism for the cycloaddition of CO2 with epoxides over the catalyst VIP-Br with Br− anions and H-bonded water molecules. | ||
Conclusions
In summary, a series of viologen-based porous ionic polymers were constructed by a one-pot facile Menshutkin reaction. The obtained VIP-X have special crystalline ordered structures and flower-like porous structures with considerable surface area up to 56 m2 g−1, and abundant halogen anions-paired viologen ionic sites and useful H-bonded water molecules. The typical VIP-Br exhibits promising heterogeneous catalytic activities in the CO2 conversion with various epoxides into cyclic carbonates under mild conditions, which attributes to the synergistic catalysis of nucleophilic Br− anions and HBD co-catalyst H-bonded water. We predict that more task-specific porous ionic polymers will be developed based on this facile and catalyst-free strategy towards diverse targeted applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21603089, 21706106 and 21503098), the Natural Science Foundation of Jiangsu Province (BK20160209), the Natural Science Foundation of Jiangsu Higher Education Institutions of China (16KJB150014), Postgraduate Research & Practice Innovation Program of Jiangsu Province, TAPP and PAPD.References
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- J. K. Sun, M. Antonietti and J. Yuan, Chem. Soc. Rev., 2016, 45, 6627–6656 RSC.
- D. Xu, J. Guo and F. Yan, Prog. Polym. Sci., 2018, 79, 121–143 CrossRef CAS.
- Z. W. Liu and B. H. Han, Current Opinion in Green and Sustainable Chemistry, 2019, 16, 20–25 CrossRef.
- K. Huang, J. Y. Zhang, F. Liu and S. Dai, ACS Catal., 2018, 8, 9079–9102 CrossRef CAS.
- T. Yamada, Y. Tominari, S. Tanaka and M. Mizuno, J. Phys. Chem. B, 2017, 121, 3121–3129 CrossRef CAS PubMed.
- K. Kim, O. Buyukcakir and A. Coskun, RSC Adv., 2016, 6, 77406–77409 RSC.
- J. Byun, H. A. Patel, D. Thirion and C. T. Yavuz, Polymer, 2017, 126, 308–313 CrossRef CAS.
- Y. Byun, S. H. Je, S. N. Talapaneni and A. Coskun, Chem.–Eur. J., 2019, 25, 10262–10283 CrossRef CAS PubMed.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev.: Sci. Eng., 2019, 61, 214–269 CrossRef CAS.
- X. Wang, Y. Zhou, Z. Guo, G. Chen, J. Li, Y. Shi, Y. Liu and J. Wang, Chem. Sci., 2015, 6, 6916–6924 RSC.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- G. Chen, Y. Zhang, J. Xu, X. Liu, K. Liu, M. Tong and Z. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS.
- M. A. Ziaee, Y. Tang, H. Zhong, D. Tian and R. Wang, ACS Sustainable Chem. Eng., 2019, 7, 2380–2387 CrossRef CAS.
- Q. Wang, X. Ling, T. Ye, Y. Zhou and J. Wang, J. Mater. Chem. A, 2019, 7, 19140–19151 RSC.
- L. Liu, F. Guo, J. Xu, J. Hu, H. Wang, H. Liu and M. Wang, Fuel, 2019, 244, 439–446 CrossRef CAS.
- S. Ding, C. Tian, X. Zhu, H. Wang, H. Wang, C. W. Abney, N. Zhang and S. Dai, Chem. Commun., 2018, 54, 5058–5061 RSC.
- T. Škorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705–6716 CrossRef PubMed.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- Y.-P. Zhao, Y. Li, C.-Y. Cui, Y. Xiao, R. Li, S.-H. Wang, F.-K. Zheng and G.-C. Guo, Inorg. Chem., 2016, 55, 7335–7340 CrossRef CAS PubMed.
- Y. Xiao, S.-H. Wang, Y.-P. Zhao, F.-K. Zheng and G.-C. Guo, CrystEngComm, 2016, 18, 2524–2531 RSC.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- Q. Sun, Y. Jin, B. Aguila, X. Meng, S. Ma and F.-S. Xiao, ChemSusChem, 2017, 10, 1160–1165 CrossRef CAS.
- K. Hu, Y. Tang, J. Cui, Q. Gong, C. Hu, S. Wang, K. Dong, X. Meng, Q. Sun and F.-S. Xiao, Chem. Commun., 2019, 55, 9180–9183 RSC.
- M. Tong, Y. Lan, Q. Yang and C. Zhong, Chem. Eng. Sci., 2017, 168, 456–464 CrossRef CAS.
- S. Fischer, J. Schmidt, P. Strauch and A. Thomas, Angew. Chem., Int. Ed., 2013, 52, 12174–12178 CrossRef CAS.
- K. Jie, H. Chen, P. Zhang, W. Guo, M. Li, Z. Yang and S. Dai, Chem. Commun., 2018, 54, 12706–12709 RSC.
- Y. Zhang, K. Liu, L. Wu, H. Zhong, N. Luo, Y. Zhu, M. Tong, Z. Long and G. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16907–16916 CrossRef CAS.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulouc and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- S.-B. Yu, H. Lyu, J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Polym. Chem., 2016, 7, 3392–3397 RSC.
- L. Wang, C. Zeng, H. Xu, P. Yin, D. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. Ma, Chem. Sci., 2019, 10, 1023–1028 RSC.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gandará, M. Lusi, D. S. S. Rao, S. Vimala, S. K. Prasad, J. Raya, D. S. Han, R. Jagannathan, J.-C. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- G. Chen, X. Huang, Y. Zhang, M. Sun, J. Shen, R. Huang, M. Tong, Z. Long and X. Wang, Chem. Commun., 2018, 54, 12174–12177 RSC.
- L.-Z. Peng, P. Liu, Q.-Q. Cheng, W.-J. Hu, Y. A. Liu, J.-S. Li, B. Jiang, X.-S. Jia, H. Yang and K. Wen, Chem. Commun., 2018, 54, 4433–4436 RSC.
- P. Samanta, P. Chandra, S. Dutta, A. V. Desai and S. K. Ghosh, Chem. Sci., 2018, 9, 7874–7881 RSC.
- G. Chen, Y. Zhou, X. Wang, J. Li, S. Xue, Y. Liu, Q. Wang and J. Wang, Sci. Rep., 2015, 5, 11236 CrossRef.
- P. Zhang, Z.-A. Qiao, X. Jiang, G. M. Veith and S. Dai, Nano Lett., 2015, 15, 823–828 CrossRef CAS PubMed.
- P. Zhang, M. Li, B. Yang, Y. Fang, X. Jiang, G. M. Veith, X.-G. Sun and S. Dai, Adv. Mater., 2015, 27, 8088–8094 CrossRef CAS PubMed.
- A. A. Rajaa and C. T. Yavuz, RSC Adv., 2014, 4, 59779–59784 RSC.
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 RSC.
- S. Hou, N. Chen, P. Zhang and S. Dai, Green Chem., 2019, 21, 1455–1460 RSC.
- J.-K. Tang, S.-B. Yu, C.-Z. Liu, H. Wang, D.-W. Zhang and Z.-T. Li, Asian J. Org. Chem., 2019, 8, 1912–1918 CrossRef CAS.
- G. Chen, L. Zhang, Y. Zhang, K. Liu, Z. Long and Y. Wang, J. Mater. Chem. A, 2019, 7, 7194–7201 RSC.
- H. D. Correia, S. Chowdhury, A. P. Ramos, L. Guy, G. J.-F. Demetsb and C. Bucherc, Polym. Int., 2019, 68, 572–588 CrossRef CAS.
- G. Chen, W. Hou, J. Li, X. Wang, Y. Zhou and J. Wang, Dalton Trans., 2016, 45, 4504–4508 RSC.
- Y. Jiang, J. Li, P. Jiang, Y. Li and Y. Leng, Catal. Commun., 2018, 111, 1–5 CrossRef CAS.
- G. Das, S. K. Sharma, T. Prakasam, F. Gándara, R. Mathew, N. Alkhatib, N. Saleh, R. Pasricha, J.-C. Olsen, M. Baias, S. Kirmizialtin, R. Jagannathan and A. Trabolsi, Commun. Chem., 2019, 2, 1–9 CrossRef CAS.
- J. Mohan, Organic Spectroscopy: Principles and Applications, Alpha Science International Ltd., Harrow, 2004, p. 37 Search PubMed.
- L. Schottner, R. Ovcharenko, A. Nefedov, E. Voloshina, Y. Wang, J. Sauer and C. Woll, J. Phys. Chem. C, 2019, 123, 8324–8335 CrossRef.
- Y. Wang, J. Nie, C. Lu, F. Wang, C. Ma, Z. Chen and G. Yang, Microporous Mesoporous Mater., 2020, 292, 109751 CrossRef.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- Y. Leng, D. Lu, C. Zhang, P. Jiang, W. Zhang and J. Wang, Chem.–Eur. J., 2016, 22, 8368–8375 CrossRef CAS PubMed.
- Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 2534–2541 CrossRef CAS PubMed.
- P. Puthiaraj, S. Ravi, K. Yu and W.-S. Ahn, Appl. Catal., B, 2019, 251, 195–205 CrossRef CAS.
- J. Liu, G. Zhao, O. Cheung, L. Jia, Z. Sun and S. Zhang, Chem.–Eur. J., 2019, 25, 9052–9059 CrossRef CAS PubMed.
- R. A. Watile, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 1051–1055 RSC.
- D. Ma, K. Liu, J. Li and Z. Shi, ACS Sustainable Chem. Eng., 2018, 6, 15050–15055 CrossRef CAS.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- Y. A. Alassmy and P. P. Pescarmona, ChemSusChem, 2019, 12, 3856–3863 CrossRef CAS.
- G. Chen, Y. Zhou, P. Zhao, Z. Long and J. Wang, ChemPlusChem, 2013, 78, 561–569 CrossRef CAS.
- G. Chen, Y. Zhou, Z. Long, X. Wang, J. Li and J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 4438–4446 CrossRef CAS.
- P. Yingcharoen, C. Kongtes, S. Arayachukiat, K. Suvarnapunya, S. V. C. Vummaleti, S. Wannakao, L. Cavallo, A. Poater and V. D'Elia, Adv. Synth. Catal., 2019, 361, 366–373 CrossRef CAS.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS.
- M. Liu, X. Li, L. Liang and J. Sun, J. CO2 Util., 2016, 16, 384–390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09088f |
| This journal is © The Royal Society of Chemistry 2020 |