
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceK2S2O8 mediated C-3 arylation of quinoxalin-2(1H)-ones under metal-, photocatalyst- and light-free conditions†
Nibedita Baruah Duttaabc,
Mayurakhi Bhuyanab and
Gakul Baishya *ab
*ab
aChemical Science & Technology Division, CSIR-North East Institute of Science and Technology, Jorhat-785006, India. E-mail: b.gakul@gmail.com; Fax: +91 3762370011; Tel: +91 3762372948
bAcademy of Scientific and Innovative Research, Ghaziabad, Uttar Pradesh-201002, India
cRain Forest Research Institute, Jorhat-785001, India
First published on 22nd January 2020
Abstract
Two facile and effective C-3 arylation protocols of quinoxalin-2(1H)-ones with arylhydrazines and aryl boronic acids respectively via free radical cross-coupling reactions under metal-, photocatalyst- and light-free conditions have been unveiled. K2S2O8 has been used as an efficient oxidant to generate aryl radicals from arylhydrazines and aryl boronic acids under two different reaction conditions. The generated aryl radicals undergo a free radical coupling reaction at the C-3 position of quinoxalin-2(1H)-ones producing 3-arylquinoxalin-2(1H)-ones in good to excellent yields. The involvement of radicals in the course of the reaction has been demonstrated by radical trapping experiments with 2,2,6,6-tetramethylpiperidine-1-oxyl.
Introduction
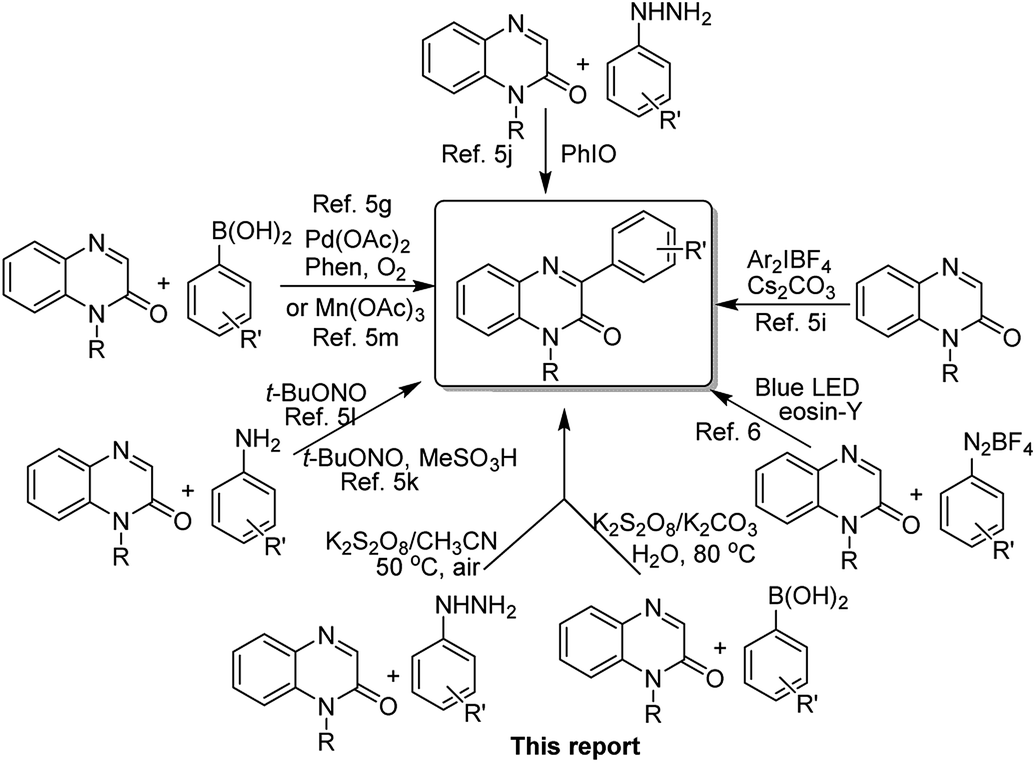
Quinoxalin-2(1H)-ones have emerged as an important class of heterocyclic compounds because of their broad biological activities and other chemical properties.1 More specifically, the skeleton of 3-arylquinoxalin-2(1H)-one is found in a variety of potential biological entities, and they exhibit a broad spectrum of pharmaceutical properties.2 Anti-tryposomal,3a anti-plasmodial,3b anti-histamine,3c anticancer,3d anti-viral,3e,f antitumor and antimicrobial,3g CFTR activators,3h FXa coagulation inhibitors2b and PDGF-βR inhibitor2c are a few of them reported in the literature. Besides, polymers obtained from these compounds can be used as semiconductors which also sets up their importance in the field of materials science.4Owing to their broad biological properties, synthetic and medicinal chemists all over the world are putting effort into developing easy-to-handle and practical methods for the synthesis of 3-arylquinoxalin-2(1H)-ones. Generally, the arylation of quinoxalin-2(1H)-ones with versatile aryl source under suitable reaction conditions produces 3-arylquinoxalin-2(1H)-ones in a single step chemical process. Recently, several research groups have highlighted the importance of this skeleton and also reported a handsome number of synthetic methodologies.5 Most of these methods undergo the radical C–C bond formation reaction at C-3 position of quinoxalin-2(1H)-ones by involving different aryl radical sources such as arylboronic acids, diaryliodonium salts, arylhydrazine/iodosobenzene, and aniline/alkyl nitrite. Very recently, Kim's group used the technique of visible light-mediated redox catalysis to obtain 3-arylquinoxalin-2(1H)-ones from arylquinoxalin-2(1H)-ones by using arenediazonium salt as an aryl radical source.6 Although these methods have their own merits, there is a need for further improvement concerning the practical and operational simplicity as well as the use of non-toxic oxidant (previous reports, Scheme 1).
 | ||
| Scheme 1 C-3 arylation of quinoxalin-2(1H)-ones. | ||
It has been well established that compared to the classical cross-coupling methods to form carbon–carbon and carbon-heteroatom bonds, the coupling reactions involving free-radicals have emerged as modern synthetic methods in organic and medicinal chemistry.7 Generally, bromo- and iodoarenes are used as traditional aryl radicals sources for various arylation reaction under different metal-catalyzed conditions.8 In recent years, back to back publications establish arylhydrazine as a readily available aryl radical source in the presence of either an oxidizing agent or a base which undergoes C–H functionalization reaction at an active C-centre of different organic compounds.9 However, the use of excess bases and toxic transition metals, longer reaction hours and low yield of the desired products are a few major drawbacks of these reported methods. Recently, Akamanchi's group has published a series of articles where they used the mixture of arylhydrazine and iodoxybenzoic acid as an aryl radical source for the arylation reaction of different organic compounds.10 Similarly, arylboronic acids are also used as aryl radical sources under different metal as well as metal-free reaction conditions for the arylation of different organic compounds such as naphthoquinone, benzoquinone, quinazoline, quinoline, quinoxalin-2(1H)-ones, imidazopyridines, etc.11 Additionally, stability, as well as easy availability in different substituted forms, makes aryl boronic acids more convenient and practical to use as aryl radical counterparts for synthesizing versatile building blocks and structural scaffolds. Even, several important organic transformations involving aryl boronic acids can be performed in water which proves their water stability as well.12 Among different organic and inorganic oxidants, potassium persulfate (K2S2O8) has come out as a suitable oxidant for many oxidative transformations since the discovery of Minisci reaction.13,14 Because of inexpensive, readily available and easy-to-handling properties it has been widely used as an oxidant in both laboratory experiments and industrial processes.14 Although several reports on the synthesis of 3-arylquinoxalin-2(1H)-ones are known in the literature, there is no such report on the use of arylhydrazine and arylboronic acid in the presence of the oxidizing agent K2S2O8 as aryl radical source for the C-3 arylation reaction of quinoxalin-2(1H)-ones. Again, the reagent system arylhydrazine/K2S2O8 has been utilized for the first time for any arylation reaction.
In continuation of our ongoing research program on the synthesis of various heterocycles,15 we, herein, report two new methods for the synthesis of 3-arylquinoxalin-2(1H)-ones via the radical coupling reaction of quinoxalin-2(1H)-ones with arylhydrazines and arylboronic acids as radical counterparts in the presence of sole oxidant K2S2O8 (this report, Scheme 1).
Results and discussion
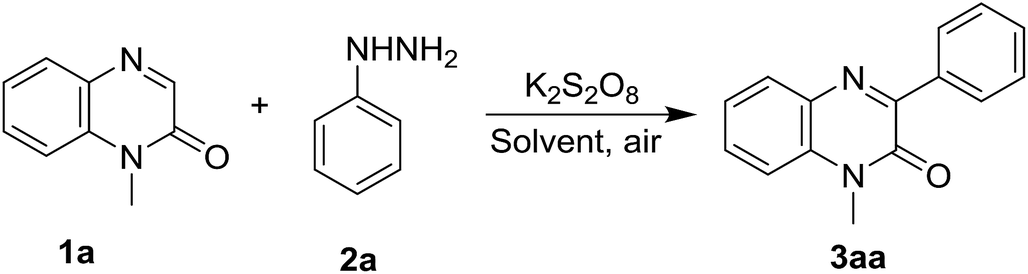
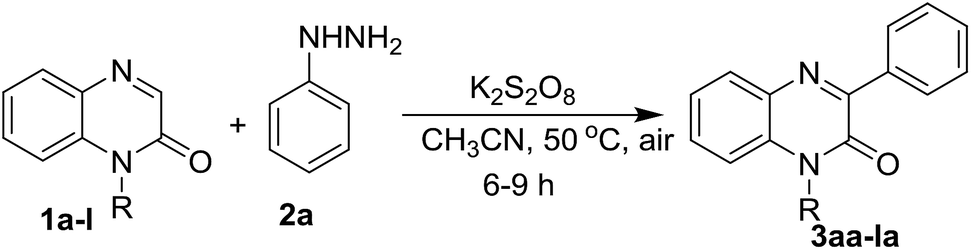
To find the optimal reaction conditions for the synthesis of 3-arylquinoxalin-2(1H)-one derivatives, we first attempted the cross-coupling reaction of quinoxalin-2(1H)-one (1a, 0.2 mmol) with phenylhydrazine (2a, 0.24 mmol) in acetonitrile at room temperature under open-air atmosphere. No reaction was perceived as both starting materials remained unchanged after 6 h of reaction time (Table 1, entry 1). We also performed the same reaction by adding 1.5 equiv. of 2a, but the same result was obtained (Table 1, entry 2). When the reaction was carried out in the presence of 1.5 equiv. of K2S2O8, after 6 hours of reaction, a new spot above the starting compound 1a was noticed in the TLC which was isolated in 49% yield and confirmed as the product 3aa by 1H and 13C NMR spectral analysis as well as by comparing with the known spectral data (Table 1, entry 3). To improve the yield of the reaction, the ratio of K2S2O8 and phenylhydrazine were increased; and accordingly, two sets of the same reaction were performed by using 1.5 equiv. of phenylhydrazine in the presence of 2.0 and 3.0 equiv. of K2S2O8. As expected, both the reactions smoothly underwent affording the product in 57% and 61% yield (Table 1, entry 4 & 5). Again, use 2.0 equiv. of 2a with 3.0 equiv. of K2S2O8 did not significantly enhance the yield of the reaction, and we obtained the product in 62% yield (Table 1, entry 6). Other solvents like DCE, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DCE: water, THF, and acetone were also employed, but none of them were successful (Table 1, entry 7, 8, 9, 10). Therefore, we selected acetonitrile as the solvent of choice. Surprisingly, when we warmed the reaction up to 50 °C, a significant enhancement of the yield was perceived and an 81% yield of the product was obtained (Table 1, entry 11). In a further improvement of the yield of 3aa, an additive like K2CO3 was also used, but we did not notice any increment (Table 1, entry 13). Instead of K2S2O8 we also used (NH4)2S2O8 as an oxidant and performed the same reaction in two different solvent systems viz. acetonitrile and 1:1 mixture of DMSO and water, but both experiments failed to give a better yield of 3aa (Table 1, entry 14 & 15). Oxidants like Mn(OAc)3·2H2O and PhI(OCOCF3)2 have also been screened for the same reaction under the optimized reaction conditions, however, they only produced a trace amount of 3aa (Table 1, entry 16 & 17). Thus, the optimized reaction conditions involve the simple stirring of quinoxalin-2(1H)-one (1a, 1.0 equiv.) with phenylhydrazine (2a, 1.5 equiv.) in the presence of K2S2O8 (3.0 equiv.) in acetonitrile at 50 °C for 6 h under open-air atmosphere (Table 1, entry 11).
:1 mixture of DCE: water, THF, and acetone were also employed, but none of them were successful (Table 1, entry 7, 8, 9, 10). Therefore, we selected acetonitrile as the solvent of choice. Surprisingly, when we warmed the reaction up to 50 °C, a significant enhancement of the yield was perceived and an 81% yield of the product was obtained (Table 1, entry 11). In a further improvement of the yield of 3aa, an additive like K2CO3 was also used, but we did not notice any increment (Table 1, entry 13). Instead of K2S2O8 we also used (NH4)2S2O8 as an oxidant and performed the same reaction in two different solvent systems viz. acetonitrile and 1:1 mixture of DMSO and water, but both experiments failed to give a better yield of 3aa (Table 1, entry 14 & 15). Oxidants like Mn(OAc)3·2H2O and PhI(OCOCF3)2 have also been screened for the same reaction under the optimized reaction conditions, however, they only produced a trace amount of 3aa (Table 1, entry 16 & 17). Thus, the optimized reaction conditions involve the simple stirring of quinoxalin-2(1H)-one (1a, 1.0 equiv.) with phenylhydrazine (2a, 1.5 equiv.) in the presence of K2S2O8 (3.0 equiv.) in acetonitrile at 50 °C for 6 h under open-air atmosphere (Table 1, entry 11).
|
|||||
|---|---|---|---|---|---|
| Entry | PhNH2NH2 (equiv.) | K2S2O8 (equiv.) | Solvent | Time (h) | Yieldsb (%) |
| a Reaction conditions: a mixture of 1a (0.2 mmol) and 2a in the presence of K2S2O8 was stirred as mentioned above in the table.b Isolated yields.c Reaction is carried out at 50 °C.d Additive K2CO3 was used.e NH4S2O8.f Mn(OAc)3·2H2O.g PhI(OCOCF3)2 were used instead of K2S2O8. | |||||
| 1 | 1.2 | — | CH3CN | 6 | n.r. |
| 2 | 1.5 | — | CH3CN | 6 | n.r. |
| 3 | 1.2 | 1.5 | CH3CN | 6 | 49 |
| 4 | 1.5 | 2.0 | CH3CN | 6 | 57 |
| 5 | 1.5 | 3.0 | CH3CN | 6 | 61 |
| 6 | 2.0 | 3.0 | CH3CN | 6 | 62 |
| 7 | 1.5 | 3.0 | ClCH2H2Cl | 6 | Complex mixture |
| 8 | 1.5 | 3.0 | ClCH2CH2Cl–H2O | 6 | Trace |
| 9 | 1.5 | 3.0 | THF | 6 | Complex mixture |
| 10 | 1.5 | 3.0 | Acetone | 6 | n.r. |
| 11c | 1.5 | 3.0 | CH3CN | 6 | 81 |
| 12c | 1.5 | 2.0 | CH3CN | 6 | 53 |
| 13d | 1.5 | 3.0 | CH3CN | 6 | 78 |
| 14e | 1.5 | 3.0 | CH3CN | 6 | 61 |
| 15e | 1.5 | 3.0 | DMSO–H2O | 6 | 61 |
| 16f | 1.5 | 3.0 | CH3CN | 6 | Trace |
| 17g | 1.5 | 3.0 | CH3CN | 6 | Trace |
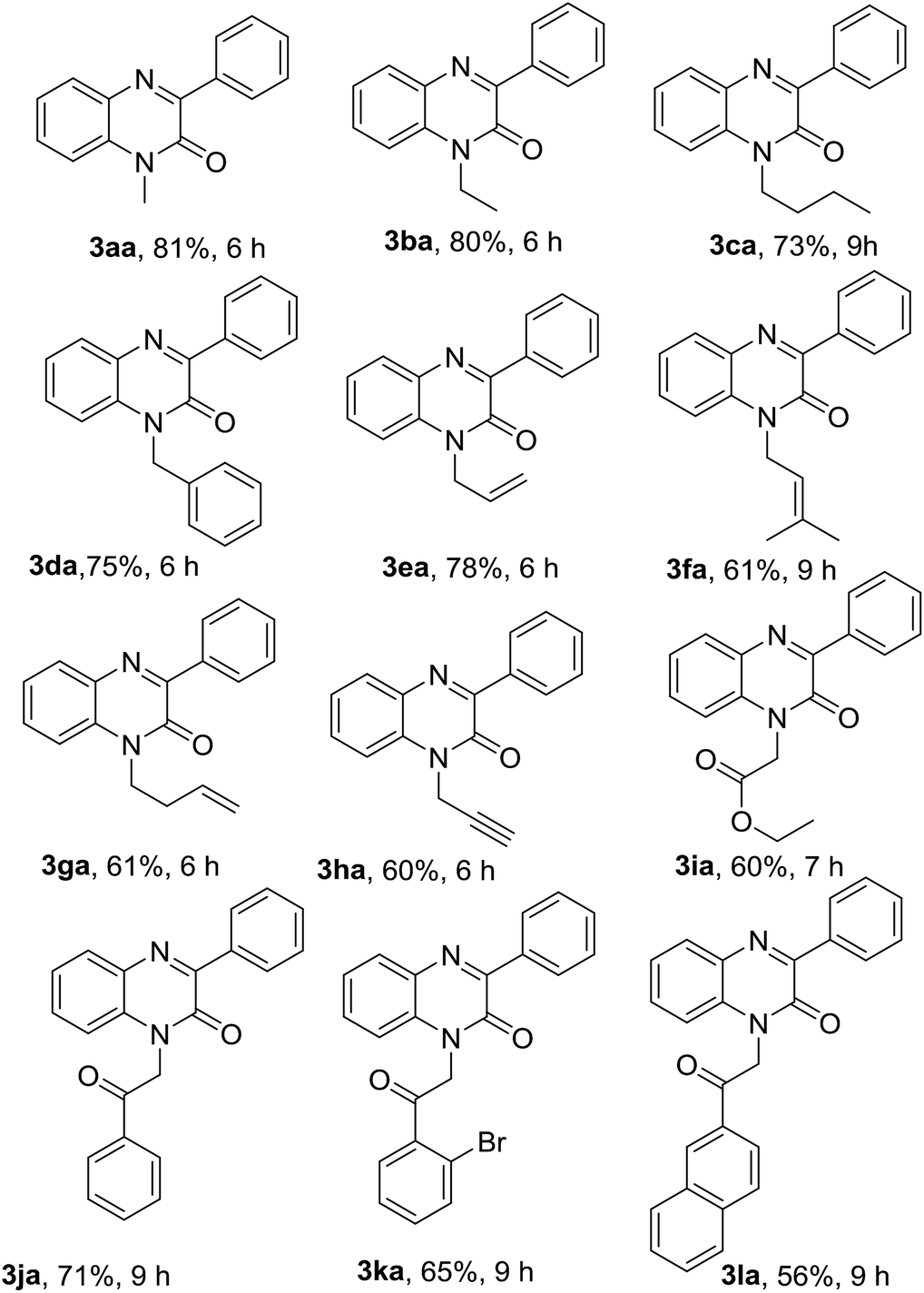
With optimized conditions in hand, we first examined the scope and general applicability of different N-substituted quinoxalin-2(1H)-one with the radical counterpart phenylhydrazine (2a). Different N-substituted quinoxalin-2(1H)-ones such as N-ethyl (1b), N-n-butyl (1c), N-benzyl (1d), N-allyl (1e), N-prenyl (1f) and N-homoallyl (1g) quinoxalin-2(1H)-ones smoothly underwent the reaction producing the respective 3-phenylquinoxalin-2(1H)-ones 3ba–3ga in very good to excellent yields. Other substituents like ethoxycarbonylmethyl and phenacyl groups at the N-1 position of quinoxalin-2(1H)-one were also well tolerated under the optimized reaction conditions and the respective N-substituted quinoxalin-2(1H)-ones (1i & 1j) produced the 3-phenylated products 3ia & 3ja in good yields. It was observed that the substituents present at the N-1 atom showed a significant variation of the yield of the product under the reaction conditions. For example, N-substituted quinoxalin-2(1H)-ones bearing N-alkyl groups like methyl, ethyl, benzyl and allyl (1a, 1b, 1d & 1e) gave more yield of the respective phenylated products (3aa, 3ba, 3da & 3ea) rather than those bearing little bulky groups namely n-butyl, prenyl, homoallyl, ethoxycarbonylmethyl and phenacyl groups (1c, 1f, 1g & 1i–j). Further, we noticed that quinoxalin-2(1H)-ones bearing more bulky groups such as 2′-bromophenacyl (1k) and naphthalene-2-acyl (1l) at the N-1 position also afforded the respective products 3ka & 3la in good yields. It would be worth mentioning that quinoxalin-2(1H)-one having N-propargyl group (1h) also took place in the reaction and produced the product 3ha in 60% yield (Table 2).
After exemplification of our protocol with different N-substituted quinoxalin-2(1H)-one, we explored the scope of N-unsubstitued quinoxalin-2(1H)-one with a variety of phenylhydrazines. We first performed the reaction of quinoxalin-2(1H)-one (1m) with phenylhydrazine (2a) under the standard reaction conditions. Unexpectedly, the reaction smoothly underwent affording the corresponding product 3ma in very good yield (71%). To prove the efficacy of the protocol, a variety of substituted phenylhydrazines were also reacted with 1m under the optimized reaction conditions, and all the reactions underwent smoothly affording the respective products 3mb–3mf in very good yields. In contrary to the earlier reports, this protocol is equally suitable for N-unsubstitued quinoxalin-2(1H)-one and we obtained 3-arylated products in very good yields. The results were presented in Table 3.
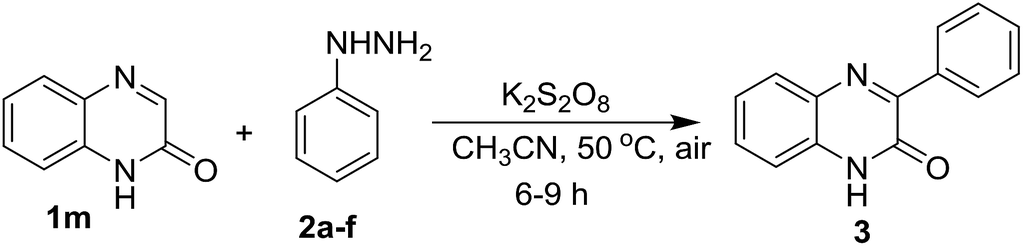

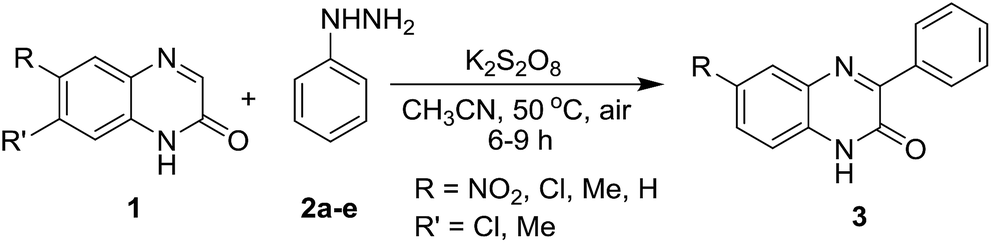
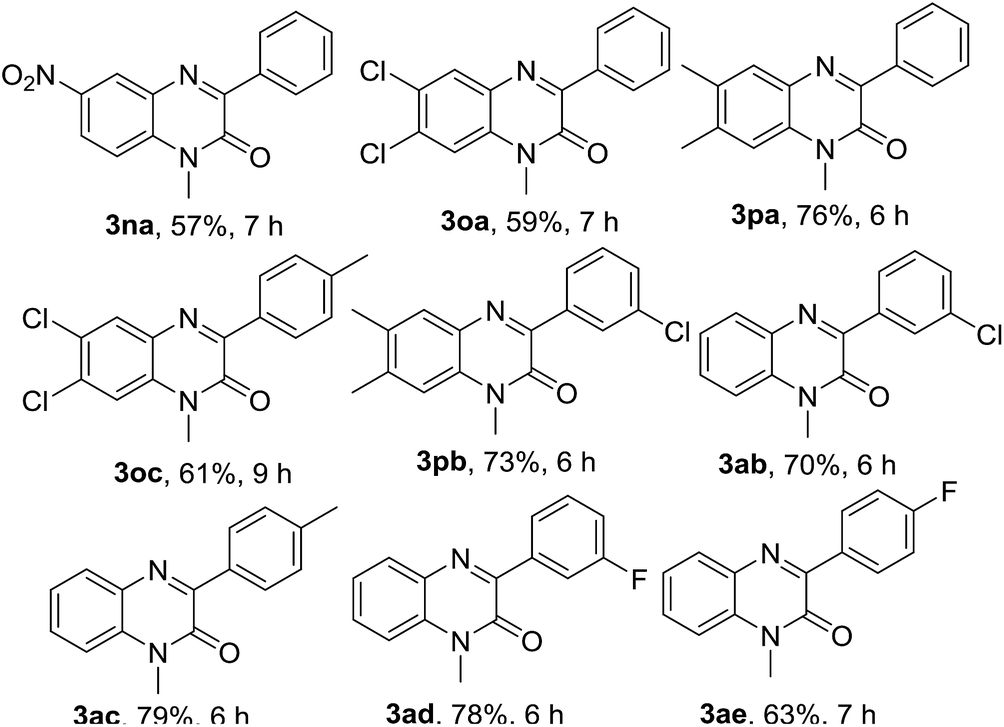
Next, we checked the compatibility of our developed protocol with a variety of quinoxalin-2(1H)-one with unsubstituted and substituted phenylhydrazines (Table 4). Different arene ring-substituted 1-methylquinoxalin-2(1H)-one for example 6-nitro-1-methylquinoxalin-2(1H)-one (1n), 6,7-dichloro-1-methylquinoxalin-2(1H)-one (1o), 6,7-dimethyl-1-methylquinoxalin-2(1H)-one (1p) also underwent the reaction with 2a producing the respective phenylated products 3na, 3oa & 3pa in very good yields. Quinoxalin-2(1H)-one bearing electron-donating group (1p) gave higher yields than those bearing electron-withdrawing groups (1n & 1o). In further exploration, a variety of phenylhydrazines bearing electron-withdrawing and electron-donating groups in the arene ring (2b, 2c, 2d & 2e) were reacted with different 1-methylquinoxalin-2(1H)-one 1a, 1o & 1p. As expected, all the reactions proceeded well and afforded the respective products 3oc, 3pb, 3ab, 3ac, 3ad & 3ae in good to very good yields (Table 4). Arylhydrazines having an electron-donating group at para-position (2c) and electron-withdrawing groups at meta-position (2b & 2d) worked better and produced higher yields than that having an electron-withdrawing group at para-position (2e).
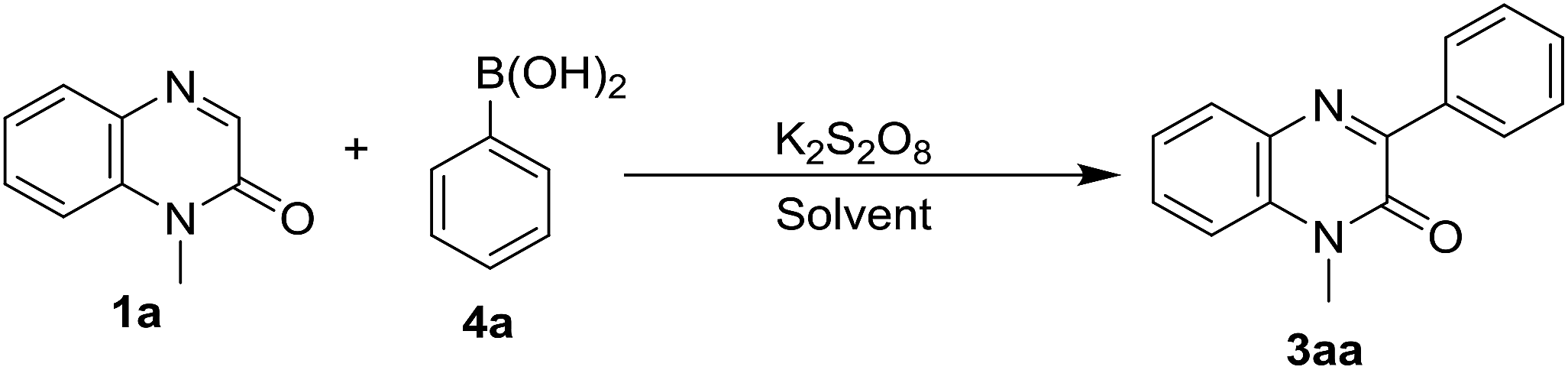
Once the exploration of our protocol with a variety of phenylhydrazines was completed, we next examined the efficacy of the arylation reaction of quinoxalin-2(1H)-ones with different aryl boronic acid. Accordingly, we performed the reaction of 1a with phenyl boronic acid (4a) in acetonitrile at room temperature under the optimized reaction conditions of Table 1. The reaction did not yield the product 3aa and both the starting materials 1a & 4a remained intact in the reaction mixture (Table 5, entry 1). As is known from the literature that aryl boronic acids form aryl radicals in the presence of excess K2S2O8 at higher temperature, hence we checked the feasibility of the reaction at a little higher temperature.16 Accordingly, we carried out the same reaction at 80 °C. This time, a very little conversion was observed in TLC, but we were not able to isolate the product 3aa (Table 5, entry 2). In further optimization, the effect of solvent on the yield of the product was also examined and accordingly, we performed the same reaction in different solvents like dichloroethane and a 1:1 mixture of dichloroethane and water at 80 °C. Surprisingly, a significant improvement of the yield was observed in the 1:1 mixture of dichloroethane and water (Table 5, entry 4). To further improve the yield, we also added a base K2CO3 in different amounts. To our delight, an addition of 1.0 equiv. of K2CO3 to the reaction mixture considerably enhanced the yield (Table 5, entry 6). Given curiosity, we also performed the same reaction in pure water keeping the other conditions unchanged and surprisingly, the reaction underwent very smoothly producing 80% yield of 3aa (Table 5, entry 8). Thus the optimized reaction conditions involve the stirring of the reaction mixture of N-methylquinoxalin-2(1H)-ones (1a, 0.5 mmol) and phenylboronic acid (4a, 0.75 mmol) in the presence of 2.0 equiv. of K2S2O8 and 1.0 equiv. of K2CO3 in water (5 mL) at 80 °C for 2 h (Table 5, entry 8). However, no reaction took place in the absence of K2S2O8 under standard reaction conditions and the starting compounds remained unchanged (Table 5, entry 9).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Additive | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: a mixture of 1a (0.5 mmol), 4a (0.75 mmol) and K2S2O8 (1.0 mmol) in solvent was stirred as mentioned in the table above.b Isolated yield. n.r.: no reaction.c Product 3aa could not be isolated.d Reaction was performed without K2S2O8. | |||||
| 1 | CH3CN | — | 30 | 12 | n.r. |
| 2 | CH3CN | — | 80 | 12 | —c |
| 3 | ClCH2CH2Cl | — | 80 | 6 | —c |
| 4 | ClCH2CH2C–H2O | — | 80 | 6 | 40 |
| 5 | ClCH2CH2Cl–H2O | K2CO3 (0.25 mmol) | 80 | 6 | 57 |
| 6 | ClCH2CH2Cl–H2O | K2CO3 (0.50 mmol) | 80 | 6 | 66 |
| 7 | ClCH2CH2C–H2O | K2CO3 (0.75 mmol) | 80 | 6 | 67 |
| 8 | H2O | K2CO3 (0.50 mmol) | 80 | 2 | 80 |
| 9d | H2O | K2CO3 (0.50 mmol) | 80 | 6 | n.r. |
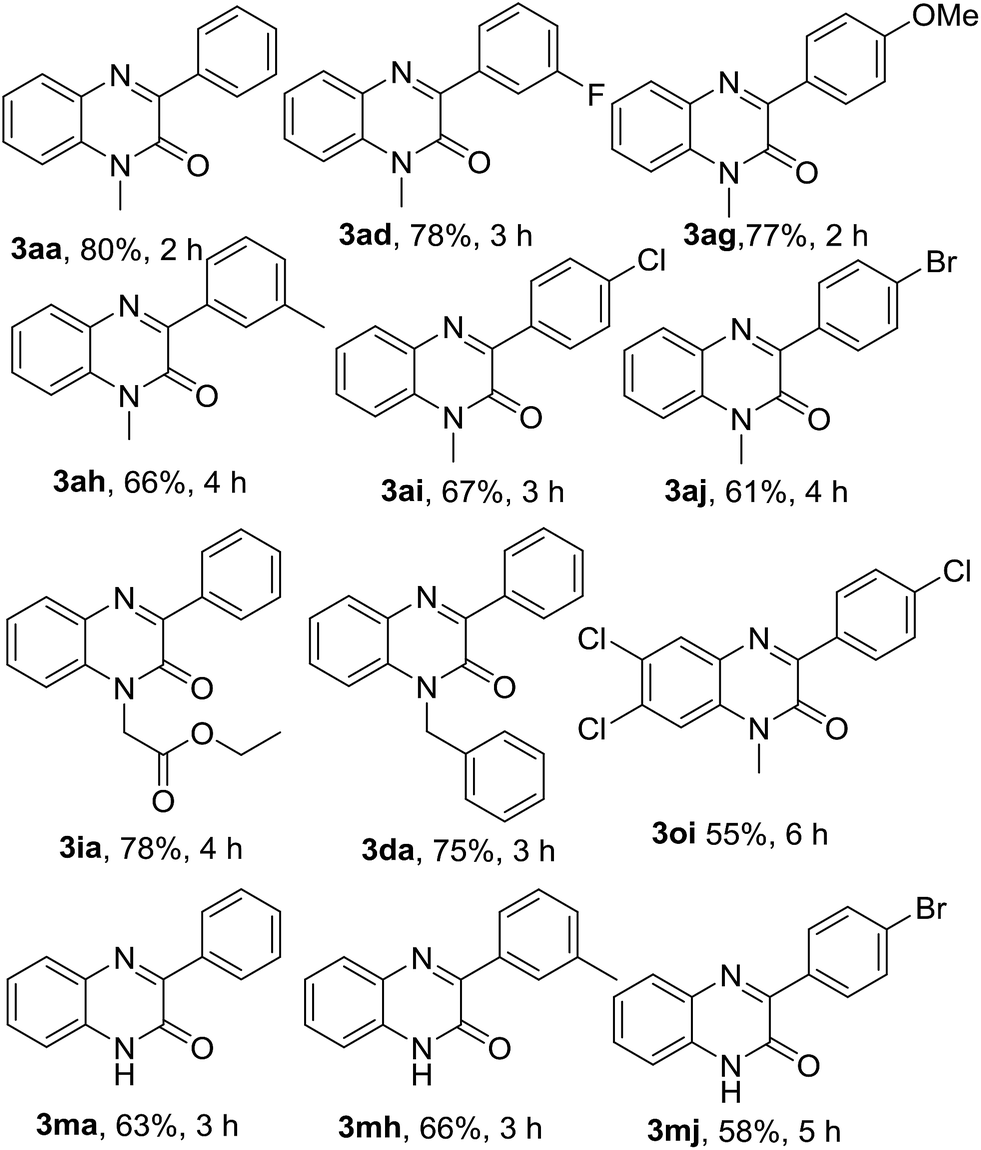
After optimization of the reaction conditions with phenylboronic acid, we engaged different aryl boronic acids bearing both electron-donating and -withdrawing substituents in the arene ring to react with quinoxalin-2(1H)-ones (Table 6). Accordingly, a series of experiments were conducted by reacting 1a with a variety of aryl boronic acids such as 3-fluorophenylboronic (4d), 4-methoxylphenylboronic acid (4g), 3-methylphenylboronic acid (4h), 4-chlorophenylboronic acid (4i) and 4-bromophenylboronic acid (4j); and all the reaction underwent smoothly producing the respective arylated quinoxalin-2(1H)-ones 3aa, 3ad, 3ag, 3ah, 3ai & 3aj in very good to excellent yields. Aryl boronic acid bearing electron-withdrawing group in meta-position (4d) gave a better yield than those of para-position (4i & 4j). Similarly, aryl boronic acid with electron-donating group in para-position (4g) afforded a better yield than that of meta-position (4h). N-Benzyl and N-ethylcarboxymethyl quinoxalin-2(1H)-ones (1d & 1i) were also reacted with 4a under the optimized reaction conditions and both substrates produced the corresponding 3-phenylquinoxalin-2(1H)-ones 3da & 3ia in very good yields. In the next couple of experiments, we also examined the efficacy of N-unsubstituted quinoxalin-2(1H)-one 3m and performed the reaction of 3m with 4a, 4h & 4j. Surprisingly, we obtained very good yields of the respective products 3ma, 3mh & 3mj.
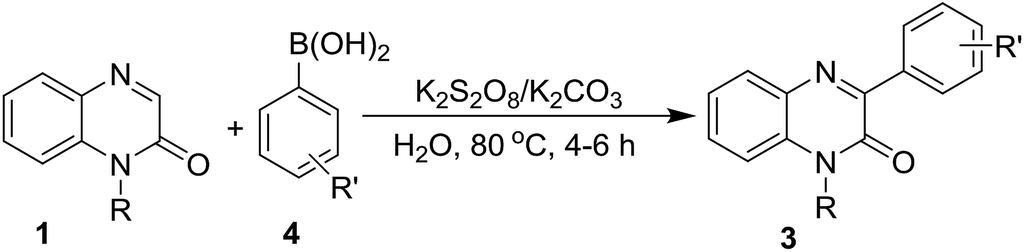
In another experiment, the robustness of the above protocol was proved by performing a gram-scale reaction with quinoxalin-2(1H)-one (3m) and phenylboronic acid (4a) under optimal reaction conditions. As expected, the product 3ma was obtained in 51% yield which is comparable to the yield obtained in the small scale preparation (Scheme 2).
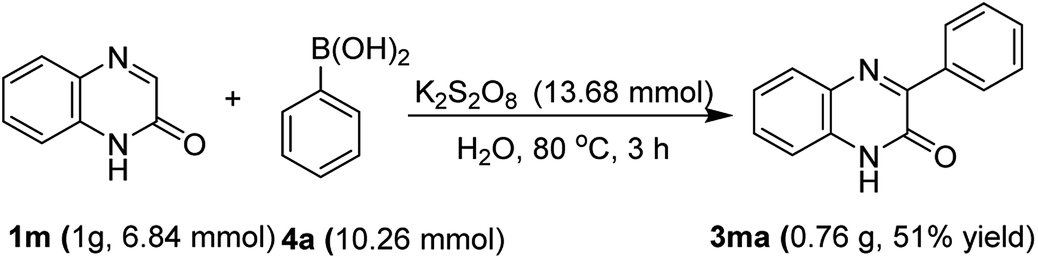 | ||
| Scheme 2 Gram-scale synthesis of 3ma. | ||
To find out a plausible reaction mechanism, a radical scavenger effect was explored by performing the arylation reaction of quinoxalin-2(1H)-one (1a) with phenylhydrazine (2a) in the presence of a radical trapping reagent 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) under the standard reaction conditions. As expected, the yield of the product 3aa sharply reduced from 81% to 19% on the addition of 2.0 equiv. of TEMPO. The same reaction was also repeated at room temperature to trap the radical, but we could not isolate the radical trapping product, however, the yield of 3aa was drastically affected affording only a 10% yield. Another radical trapping experiment was performed by reacting TEMPO with 2,4-dinitrophenylhydrazine (2g) under the optimized reaction conditions, this furnished the TEMPO adduct 1-(2,4-dinitrophenoxy)-2,2,6,6-tetramethylpiperidine (5) in 67% yield. The above experiments proved the involvement of radicals in the course of the reaction (Scheme 3).
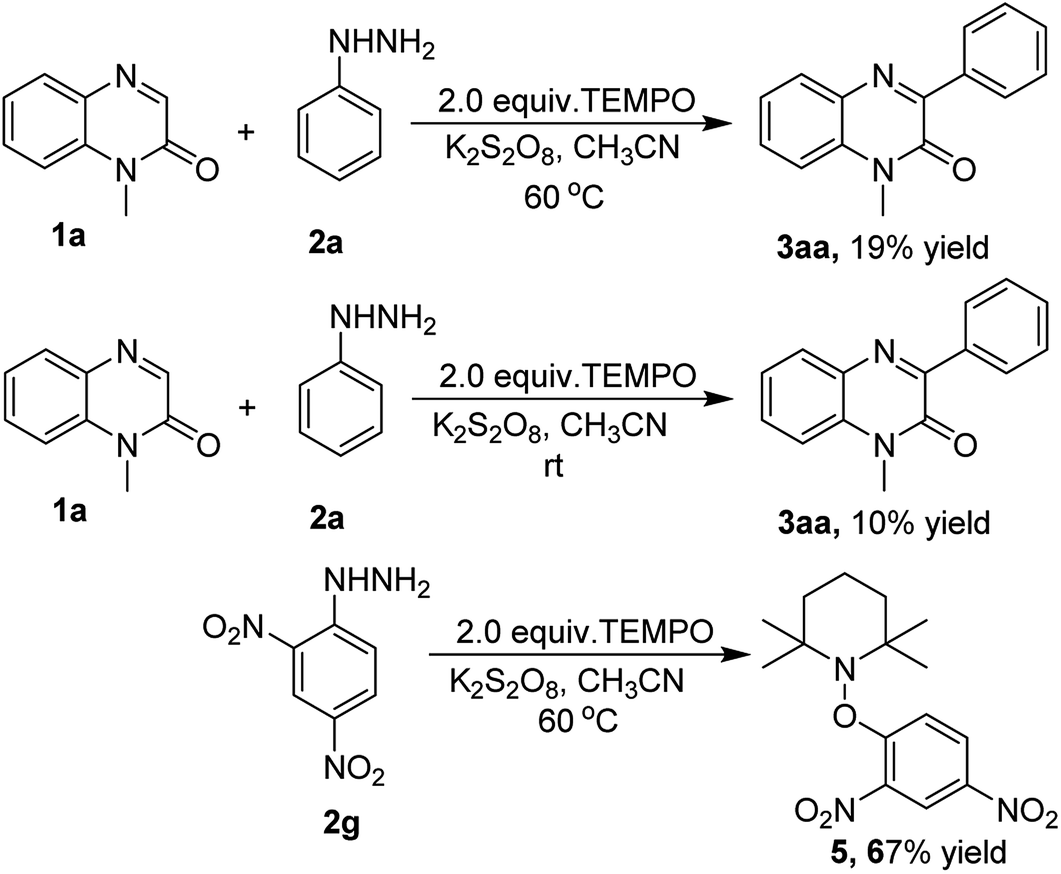 | ||
| Scheme 3 Radical scavenger effect. | ||
Based on the above result as well as literature's supports,17 a plausible mechanism has been proposed in Scheme 4. The reaction commences with the decomposition of persulfate S2O82− to the sulfate radical anion SO4˙− which reacts with phenylhydrazine to form the phenylhydrazine radical I. Again, one more sulfate anion radical reacts with I to give phenyldiazene II, which in turn reacts with another sulfate anion radical and forms III. The removal N2 gas from III produces the phenyl radical IV which undergoes Minisci-type13 radical addition reaction on to C-3 position of quinoxalin-2(1H)-one (1a), thus affording the nitrogen radical intermediate V. The formed intermediate V undergoes 1,2-hydrogen shift and produces the carbon radical VI which is again oxidized by persulfate via SET to form the carbon cation intermediate VII. Finally, the abstraction of β-H from intermediate VII by SO42− produces the desired product 3aa. In an alternative path, H˙ radical abstraction by SO4˙− from V could also furnish 3aa. Similarly, aryl boronic acid also forms aryl radical which undergoes the radical addition reaction and finally produces the 3-arylquinoxalin-2(1H)-one (Scheme 4).11f
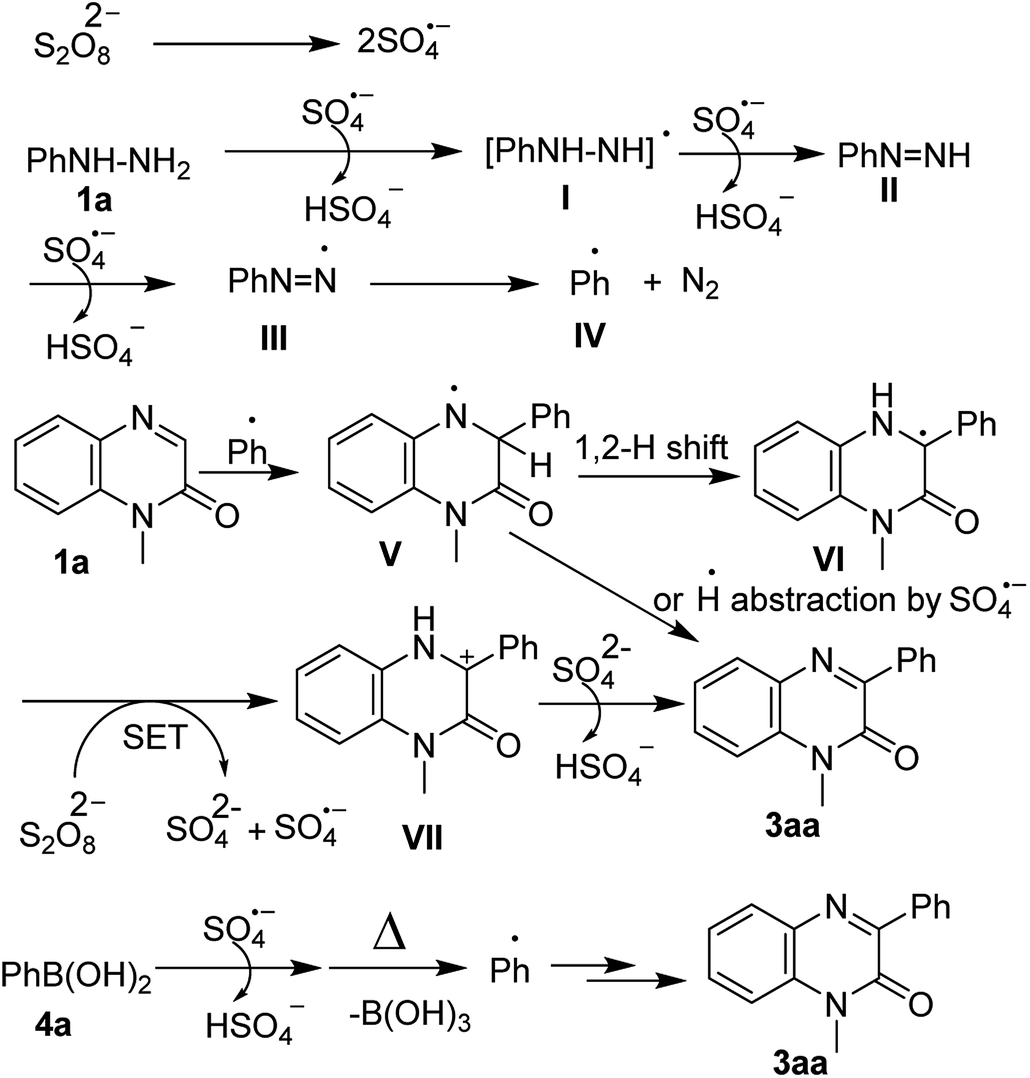 | ||
| Scheme 4 Plausible reaction mechanism. | ||
In view of the importance of 1,2,3-triazoles compounds and their uses as linkers for various applications in diverse areas of chemistry such as medicinal chemistry, agrochemistry, and material chemistry,18 we, herein, attempted to synthesize quinoxalinone linked triazole derivative and accordingly, we carried out the reaction between 3ha and commercially available 1-azidoadamantane (6) under standard reaction conditions of Huisgen [2 + 3]-cycloaddition reaction.19 The reaction smoothly underwent furnishing the product 7 in 82% yield (Scheme 5).
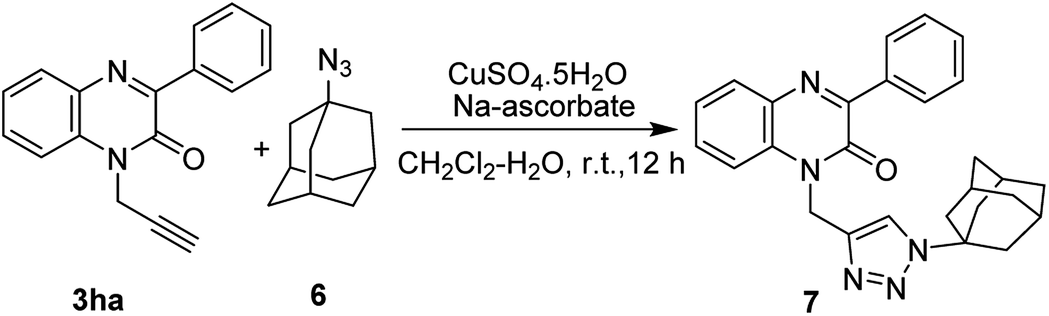 | ||
| Scheme 5 Synthesis of 3-phenylquinoxalin-2(1H)-one linked triazole. | ||
A comparison of our both protocols with the reported methods in terms of yield, reaction temperature and time has been given in Table 7. From the data presented in Table 7, it can be concluded that both methodologies show tremendous potential over earlier reported methods. These two protocols have not used any hazardous organic or inorganic oxidants like PhIO and Mn(OAc)3, transition metal catalyst viz. Pd(OAc)2, extremely corrosive and explosive reagent t-BuONO (used in 6 equiv.), strong Brønsted acid such as methane sulfonic acid and very high temperature, etc. Again, PhIO and Ar2IBF4 are not commercially available reagents. Use of cheap, readily available and non-toxic oxidant K2S2O8, mild reaction conditions (50–80 °C), shorter reaction hours (2–9 h) and environmentally benign solvents make these two transformations an alternative methodology for the construction of various 3-arylquinoxalin-2(1H)-ones from readily available starting materials.
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent system | Temp. (°C) | Time (h) | Yield (%) | Ref. |
| 1 | ArB(OH)2/Pd(OAc)2/phenanthroline/O2 | 100 | 20 | 85 | 5g |
| 2 | ArB(OH)2/Mn(OAc)2. | 120 | 12 | 80 | 5m |
| 3 | Ar2IBF4/Cs2CO3 | r. t. | 72 | 80 | 5i |
| 4 | ArNHNH2/PhIO | r. t. | 9 | 81 | 5j |
| 5 | ArNH2/t-BuONO/CH3SO3H | r. t. | 1.5 | 81 | 5k |
| 6 | ArNH2/t-BuONO | r. t. | 48 | 89 | 5l |
| 7 | ArN2BF4/Eosin-Y/blue LED | r. t. | 3 | 71 | 6 |
| 8 | ArNHNH2/K2S2O8 | 50 | 6 | 81 | This report |
| 9 | ArB(OH)2/K2S2O8/K2CO3 | 80 | 2 | 80 | This report |
Conclusions
In conclusion, we have established the reagent system comprising of arylhydrazine and K2S2O8 as a versatile reagent system for the first time to promote the free radical arylation reaction on to C-3 position of quinoxalin-2(1H)-ones. Similarly, K2S2O8/arylboronic acid has also been found to be an efficient reagent system to drive the same arylation reaction. In both the methods K2S2O8 took part in the reaction as a sole oxidant producing aryl radicals from arylhydrazines and aryl boronic acids under metal-, photocatalyst- and light-free conditions in environmentally benign solvents acetonitrile and water respectively. Both the methodologies are operationally very simple and robust, and they do not require any dry reaction conditions and special reaction set up. A wide range of N-substituted quinoxalin-2(1H)-ones bearing both electron-donating and withdrawing substituents in the arene ring have undergone the arylation reaction with various arylhydrazines and aryl boronic acids producing 3-arylquinoxalin-2(1H)-ones in very good to excellent yields. N-Unsubstituted quinoxalin-2(1H)-ones also work well under the optimized conditions of both protocols. Huisgen [2 + 3]-cycloaddition reaction on 3-arylquinoxalin-2(1H)-one derived alkyne was also accomplished to reveal its synthetic application. Further applications of these two reactions are under investigation in our laboratory.Experimental section
General experimental details
1H NMR and 13C NMR spectra were recorded in CDCl3, DMSO-d6 on 500, 400 and (125, 100) MHz NMR spectrometer, respectively, using TMS as internal standard. HRMS (ESI) data were recorded on Mass spectrometer. All the commercially available reagents were used without further purification. All experiments were monitored by thin layer chromatography using aluminium pre-coated silica gel TLC plates. After elution, plate was visualized under UV illumination at 254 nm for UV active materials. Column chromatography was performed on silica gel (100–200 mesh) eluting appropriate ethyl acetate–hexane mixture.Preparation of starting materials
The starting compounds quinoxalin-2(1H)-one (1a–1p) were prepared in accordance with literature methods.2c,5g:8 ethyl acetate–hexane as the eluent to get the pure 3-aryl quinoxalin-2(1H)-ones 3. Products 3aa, 3ba, 3da, 3ea, 3ia, 3ma, 3mc, 3me, 3nd, 3oa, 3pa, 3ab, 3ac, 3ad & 3ae are known in the literature5,6 and the spectral data of unknown compounds 3ca, 3fa, 3ga, 3ha, 3ja, 3ka, 3la, 3mb, 3md, 3mf, 3oc, 3pb, 5 & 7 have been given here.
1-Butyl-3-phenylquinoxalin-2(1H)-one (3ca). Orange liquid, 73% yield. IR (CHCl3): 3057.5, 2959.1, 1652.0, 1603.5, 1465.3, 1295.3, 1180.7, 763.5, 692.2, 426.7 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.30 (dd, 2H, J = 7.0, 3.5 Hz), 7.95 (dd, 1H, J = 8.0, 1.5 Hz), 7.56 (t, 1H, J = 8.5 Hz), 7.47–7.46 (m, 3H), 7.36–7.32 (m, 2H), 4.32 (t, 2H, J = 8.0 Hz), 1.81 (p, 2H, J = 7.5 Hz), 1.55 (sextet, 2H, J = 7.5 Hz), 1.02 (t, 3H, J = 7.5 Hz). 13C NMR (CDCl3, 125 MHz): δ 154.3, 154.0, 135.9, 133.2, 132.4, 130.5, 130.16, 130.12, 129.4, 127.9, 123.4, 113.4, 42.3, 29.2, 20.2, 13.7. HRMS (ESI): calculated m/z for C18H18N2O [M + H]+: 279.1492, found: 279.1493.
1-(3-Methylbut-2-enyl)-3-phenylquinoxalin-2(1H)-one (3fa). Orange solid, mp 97.9–115.2 °C, 61% yield. IR (CHCl3): 3295.8, 2916.7, 1652.0, 1604.4, 1466.3, 1293.8, 1178.5, 762.3, 692.1, 425.2 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.31–8.29 (m, 2H), 7.95 (dd, 1H, J = 7.5, 1.0 Hz), 7.55–7.52 (m, 1H), 7.48–7.45 (m, 3H), 7.36–7.29 (m, 2H), 5.23–5.20 (m, 1H), 4.96 (d, 2H, J = 6.5 Hz), 1.92 (s, 3H), 1.74 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ 154.2, 154.1, 137.2, 136.0, 133.2, 132.5, 130.4, 130.1, 130.0, 129.4, 127.9, 123.4, 117.8, 113.8, 40.8, 25.5, 18.3.
1-(But-3-enyl)-3-phenylquinoxalin-2(1H)-one (3ga). Yellow liquid, 61% yield. IR (CHCl3): 3075.0, 2976.6, 1651.9, 1602.8, 1465.3, 1295.7, 919.1, 762.9, 692.1, 428.4 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.31–8.29 (m, 2H), 7.96 (dd, 1H, J = 8.0, 1.5 Hz), 7.57–7.54 (m, 1H), 7.49–7.46 (m, 3H), 7.37–7.32 (m, 2H), 5.96–5.87 (m, 1H), 5.17–5.09 (m, 2H), 4.40 (t, 2H, J = 8.0 Hz), 2.58 (q, 2H, J = 7.0 Hz). 13C NMR (CDCl3, 125 MHz): δ 154.2, 154.0, 135.9, 133.8, 133.2, 132.3, 130.6, 130.2, 130.1, 129.4, 127.9, 123.5, 117.6, 113.4, 41.7, 31.4. HRMS (ESI): calculated m/z for C18H16N2O [M + H]+: 277.1335, found: 277.1346.
3-Phenyl-1-(prop-2-ynyl) quinoxalin-2(1H)-one (3ha). Yellow solid, mp 139.9–143.2 °C, 60% yield. IR (CHCl3): 3282.6, 1651.3, 1603.4, 1464.3, 1173.9, 1029.8, 753.8, 427.0 cm−1. 1H NMR (CDCl3, 400 MHz): δ 8.31 (dd, 2H, J = 6.4, 2.0 Hz), 7.96 (dd, 1H, J = 8.4, 1.6 Hz), 7.61–7.57 (m, 1H), 7.48–7.46 (m, 4H), 7.41–7.37 (m, 1H), 5.12 (d, 2H, J = 2.8 Hz), 2.30 (t, 1H, J = 2.4 Hz). 13C NMR (CDCl3, 100 MHz): δ 154.1, 153.8, 135.8, 133.9, 131.9, 130.7, 130.6, 130.5, 129.6, 128.2, 124.2, 114.1, 76.9, 73.2, 31.7. HRMS (ESI): calculated m/z for C17H12N2O [M + H]+: 261.1022, found: 261.1032.
1-(2-Oxo-2-phenylethyl)-3-phenylquinoxalin-2(1H)-one (3ja). Brownish yellow solid, mp 198.1–201.3 °C, 71% yield. IR (CHCl3): 3058.5, 1691.3, 1648.6, 1446.9, 1227.4, 989.1, 754.6, 423.5 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.33–8.30 (m, 2H), 8.10 (dd, 2H, J = 8.0, 0.5 Hz), 7.99 (dd, 1H, J = 8.0, 1.0 Hz), 7.69 (tt, 1H, J = 7.5, 2.0 Hz), 7.57 (t, 2H, J = 8.0 Hz), 7.49–7.44 (m, 4H), 7.37–7.33 (m, 1H), 6.99 (dd, 1H, J = 8.5, 1.0 Hz), 5.79 (s, 2H). 13C NMR (CDCl3, 125 MHz): δ 191.2, 154.4, 153.8, 135.8, 134.6, 134.3, 133.3, 132.8, 130.7, 130.43, 130.4, 129.6, 129.0, 128.2, 128.1, 123.9, 113.4, 48.6. HRMS (ESI): calculated m/z for C22H16N2O2 [M + H]+: 341.1292, found: 341.1285.
1-(2-(2-Bromophenyl)-2-oxoethyl)-3-phenylquinoxalin-2(1H)-one (3ka). Yellow solid, mp 168.6–172.5 °C, 65% yield. IR (CHCl3): 3059.5, 2949.1, 1960.7, 1711.9, 1648.6, 1440.2, 1217.1, 986.5, 763.6, 430.9 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.31–8.29 (m, 2H), 7.99 (dd, 1H, J = 8.0, 1.5 Hz), 7.72–7.67 (m, 2H), 7.56–7.52 (m, 1H), 7.49–7.44 (m, 4H), 7.41–7.37 (m, 2H), 7.23 (dd, 1H, J = 8.5, 1.0 Hz), 5.67 (s, 2H). 13C NMR (CDCl3, 125 MHz): δ 195.1, 154.2, 153.7, 138.6, 135.6, 133.8, 133.2, 132.68, 132.62, 130.6, 130.4, 130.3, 129.4, 128.0, 127.7, 123.9, 118.9, 113.4, 51.2. HRMS (ESI): calculated m/z for C22H15BrN2O2 [M + H]+: 419.0390, found: 419.0392.
1-(2-(Naphthalen-2-yl)-2-oxoethyl)-3-phenylquinoxalin-2(1H)-one (3la). Brown solid, mp 207.0–219.4 °C, 56% yield. IR (KBr): 3245.5, 2938.3, 2220.9, 1650.5, 1601.5, 1464.1, 1241.0, 1176.6, 750.5, 693.7, 534.2 cm−1.1H NMR (CDCl3, 500 MHz): δ 8.67 (s, 1H), 8.33 (dd, 2H, J = 6.0, 2.5 Hz), 8.10 (dd, 1H, J = 9.0, 2.0 Hz), 8.03–7.98 (m, 3H), 7.93 (d, 1H, J = 8.0 Hz), 7.68–7.59 (m, 2H),7.49–7.45 (m, 4H), 7.38 (t, 1H, J = 7.5 Hz), 7.05 (dd, 1H, J = 8.5,1.0 Hz), 5.94 (s, 2H). 13C NMR (CDCl3, 125 MHz): δ 191.1, 154.4, 153.7, 135.9, 135.7, 133.2, 132.7, 132.3, 131.8, 130.6, 130.3, 130.0, 129.6, 129.4, 129.3, 129.0, 128.9, 127.9, 127.8, 127.1, 123.8, 123.3, 123.0, 113.7, 113.4, 48.6. HRMS (ESI): calculated m/z for C26H18N2O2 [M + H]+: 391.1441, found: 391.1450.
3-(3-Chlorophenyl) quinoxalin-2(1H)-one (3mb). Brown solid, mp 233.6–237.7 °C, 70% yield. IR (KBr): 3421.8, 2926.4, 1667.3, 1610.8, 1417.9, 1116.5, 902.2, 754.6, 618.5 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ 12.67 (s, 1H), 8.38 (s, 1H), 8.30 (dd, 1H, J = 8.0, 1.5 Hz), 7.87 (d, 1H, J = 7.5 Hz), 7.59–7.52 (m, 3H), 7.36 (d, 2H, J = 7.0 Hz). 13C NMR (DMSO-d6, 125 MHz): δ 154.8, 152.7, 137.8, 132.9, 132.5, 132.2, 131.1, 130.28, 130.21, 129.2, 129.1, 128.0, 123.8, 115.5. HRMS (ESI): calculated m/z for C14H9ClN2O [M + H]+: 257.0476, found: 257.0486.
3-(3-Fluorophenyl) quinoxalin-2(1H)-one (3md). Brownish yellow solid, mp 225.6–227.5 °C, 76% yield. IR (KBr): 3433.8, 3068.5, 2848.7, 1668.5, 1607.8, 1475.1, 1246.5, 896.9, 783.0, 555.4 cm−1.1H NMR (DMSO-d6, 500 MHz) δ 12.66 (s, 1H), 8.20–8.31 (m, 2H), 7.86 (dd, 1H, J = 8.0, 2.5 Hz), 7.58–7.52 (m, 2H), 7.38–7.32 (q, 3H, J = 6.5 Hz). 13C NMR (DMSO-d6, 125 MHz): δ 162.9 (d, J = 240.625 Hz), 154.8, 152.7, 138.1 (d, J = 8.125 Hz), 132.5, 132.1, 131.1, 130.2 (d, J = 8.0 Hz), 129.2, 125.5 (d, J = 2.625 Hz), 123.8, 117.4 (d, J = 21.0 Hz), 116.1(d, J = 23.375 Hz), 115.5. HRMS (ESI): calculated m/z for C14H9FN2O [M + H]+: 241.0772, found: 241.0775.
3-(4-(Trifluoromethyl) phenyl) quinoxalin-2(1H)-one (3mf). Brown solid, mp 291.7–294.7 °C, 59% yield. IR (KBr): 3434.6, 2850.5, 1663.3, 1610.8, 1330.1, 1114.8, 906.5, 760.1, 618.6 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.70 (s, 1H), 8.51 (d, 2H, J = 8.5 Hz), 7.86 (d, 3H, J = 8.5 Hz), 7.59 (t, 1H, J = 8.0 Hz), 7.36–7.33 (m, 2H). 13C NMR (DMSO-d6, 125 MHz): δ 154.8, 153.1, 139.6, 132.6, 132.2, 131.3, 130.2, 129.3, 125.1, 125.0, 123.9, 115.5. HRMS (ESI): calculated m/z for C15H9F3N2O [M + H]+: 291.0740, found: 291.0750.
6,7-Dichloro-1-methyl-3-p-tolylquinoxalin-2(1H)-one (3oc). Brown solid, mp 180.9–183.4 °C, 61% yield. IR (CHCl3): 3394.4, 2919.5 1661.6, 1417.7, 1294.6, 94.2, 883.3, 427.0 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.22 (d, 2H, J = 8.5 Hz), 7.95 (s, 1H), 7.36 (s, 1H), 7.27 (d, 2H, J = 7.5 Hz), 3.67 (s, 3H), 2.41 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ 154.7, 154.1, 141.4, 133.9, 132.6, 132.5, 132.1, 130.8, 129.6, 128.9, 127.3, 114.9, 29.5, 21.5. HRMS (ESI): calculated m/z for C16H12Cl2N2O [M + H]+: 319.0399, found: 319.0407.
3-(3-Chlorophenyl)-1,6,7-trimethylquinoxalin-2(1H)-one (3pb). Yellow solid, mp 172.0–173.8 °C, 73% yield. IR (CHCl3): 3366.3, 2919.5 1637.7, 1419.4, 1298.8, 1011.1, 677.0, 431.0 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.34 (t, 1H, J = 2.0 Hz), 8.26 (dt, 1H, J = 7.5, 1.5 Hz), 7.67 (s, 1H), 7.43–7.37 (m, 2H), 7.08 (s, 1H), 3.72 (s, 3H), 2.42 (s, 3H), 2.35(s, 3H). 13C NMR (CDCl3, 125 MHz): δ 154.5, 151.0, 140.9, 137.9, 134.0, 132.9, 131.47, 131.40, 130.5, 129.9, 129.4, 129.2, 127.6, 114.1, 29.2, 20.7, 19.2. HRMS (ESI): calculated m/z for C17H15ClN2O [M + H]+: 299.0946, found: 299.0958.
1-(2,4-Dinitrophenoxy)-2,2,6,6-tetramethylpiperidine (5). Yellow liquid, 67% yield. IR (CHCl3): 3115.7, 2977.2, 2938.4, 1602.1, 1533.0 (N–O stretch), 1470.2, 1365.5(N–O stretch), 1271.3, 426.9 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.06 (dd, 1H, J = 2.5, 2.0 Hz), 8.36 (dd, 1H, J = 9.5, 3.0 Hz), 8.09 (d, 1H, J = 9.5 Hz), 1.68–1.65 (m, 6H), 1.31 (s, 6H), 1.02 (s, 6H). 13C NMR (CDCl3, 125 MHz): δ 161.4, 139.6, 135.1, 128.6, 121.1, 118.3, 61.7, 39.4, 31.8, 20.7, 16.5.
:8 ethyl acetate–hexane as the eluent to get the pure 3-aryl quinoxalin-2(1H)-ones 3. Products 3aa, 3ad, 3ag, 3ah, 3ai, 3aj, 3ia, 3ja, 3oi, 3ma, 3mh & 3mj are known in the literature.5:1) system in a 25 mL round-bottomed flask and stirred for 5 min. CuSO4·5H2O (0.150 mmol) and sodium ascorbate (2.8 mmol) were added and the reaction mixture was stirred at r.t. for 12 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the crude mixture was partitioned between H2O (15 mL) and CH2Cl2 (20 mL). The aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic phases were dried over anhydrous sodium sulphate and concentrated under rotary evaporator and column chromatographed on silica gel (100–200 mesh) using 2:8 ethyl acetate–hexane as the eluent to get the pure compound 7. Yellow liquid, 82% yield. IR (CHCl3): 3400.8, 2911.7, 1651.8, 1604.2, 1444.4, 1101.1, 911.3, 733.2, 462.8 cm−1. 1H NMR (CDCl3, 500 MHz): δ 8.31 (dd, 2H, J = 7.0, 3.5 Hz), 8.01 (dd, 1H, J = 8.5, 1.0 Hz), 7.92 (dd, 1H, J = 8.0, 1.5 Hz), 7.75 (s, 1H), 7.60–7.56 (m, 1H), 7.49–7.48 (m, 3H), 7.36–7.33 (m, 1H), 5.58 (s, 2H), 2.20–2.16 (m, 9H), 1.76–1.70 (m, 6H). 13C NMR (CDCl3, 125 MHz): δ 154.4, 153.8, 141.3, 135.8, 133.1, 132.4, 130.5, 130.2, 130.1, 129.3, 128.0, 123.8, 120.2, 114.8, 59.7, 42.7, 38.3, 35.7, 29.2. HRMS (ESI): calculated m/z for C27H27N2O [M + H]+: 438.2288, found: 438.2293.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi, for financial support under the In-house Project OLP 2020. MB thanks DST, New Delhi for INSPIRE fellowship. Authors also thank the Director, CSIR-NEIST for his continuous support and encouragement.Notes and references
- (a) A. Carta, S. Piras, G. Loriga and G. Paglietti, Mini-Rev. Med. Chem., 2006, 6, 200 CrossRef PubMed; (b) X. Li, K. Yang, W. Li and W. Xu, Drugs Future, 2006, 31, 979 CrossRef CAS; (c) R. Liu, Z.-H. Huang, M. G. Murray, X.-Y. Guo and G. Liu, J. Med. Chem., 2011, 54, 5747 CrossRef CAS PubMed; (d) S. Hussain, S. Parveen, X. Hao, S.-Z. Zhang, W. Wang, X.-Y. Qin, Y.-C. Yang, X. Chen, S.-J. Zhu, C.-J. Zhu and B. Ma, Eur. J. Med. Chem., 2014, 80, 383 CrossRef CAS PubMed.
- (a) X. Qin, X. Hao, H. Han, S. Zhu, Y. Yang, B. Wu, S. Hussain, S. Parveen, C. Jing, B. Ma and C. Zhu, J. Med. Chem., 2015, 58, 1254 CrossRef CAS PubMed; (b) J. A. Willardsen, D. A. Dudley, W. L. Cody, L. Chi, T. B. McClanahan, T. E. Mertz, R. E. Potoczak, L. S. Narasimhan, D. R. Holland, S. T. Rapundalo and J. J. Edmunds, J. Med. Chem., 2004, 47, 4089 CrossRef CAS PubMed; (c) K. Aoki, T. Obata, Y. Yamazaki, Y. Mori, H. Hirokawa, J.-I. Koseki, T. Hattori, K. Niitsu, S. Takeda, M. Aburada and K.-I. Miyamoto, Chem. Pharm. Bull., 2007, 55, 255 CrossRef CAS PubMed; (d) K. Aoki, J.-I. Koseki, S. Takeda, M. Aburada and K.-I. Miyamoto, Chem. Pharm. Bull., 2007, 55, 922 CrossRef CAS PubMed; (e) K. Kánai, P. Arányi and Z. Böcskei, et al., J. Med. Chem., 2008, 51, 7514 CrossRef PubMed; (f) M. Weıwer, J. Spoonamore and J. Wei, et al., ACS Med. Chem. Lett., 2012, 3, 1034 CrossRef PubMed; (g) Z. Xu, A. Y. Shaw, J. Dietrich, A. P. Cappelli, G. Nichol and C. Hulme, Mol. Diversity, 2012, 16, 73 CrossRef CAS PubMed; (h) S. Hussain, S. Parveen and X. Qin, et al., Bioorg. Med. Chem. Lett., 2014, 24, 2086 CrossRef CAS PubMed; (i) L. Shi, J. Zhou and J. Wu, et al., Bioorg. Med. Chem., 2016, 24, 1840 CrossRef CAS PubMed.
- (a) C. Urquiola, M. Vieites and G. Aguirre, Bioorg. Med. Chem., 2006, 14, 5503 CrossRef CAS PubMed; (b) B. Zarranz, M. Jaso and L. M. Lima, Rev. Bras. Cienc. Farm., 2006, 42, 55 CrossRef; (c) H. M. Refaat, A. A. Moneer and O. M. Khalil, Arch. Pharmacal Res., 2004, 27, 1093 CrossRef CAS PubMed; (d) A. M. S. E. Newahie, N. S. M. Ismail, D. A. A. E. Ella and K. A. M. Abouzid, Arch. Pharm. Chem. Life Sci., 2016, 349, 309 CrossRef PubMed; (e) R. Liu, Z. Huang, M. G. Murray, X. Guo and G. Liu, J. Med. Chem., 2011, 54, 5747 CrossRef CAS PubMed; (f) L. Yang, P. Wang, J.-F. Wu, L.-M. Yang, R.-R. Wang, W. Pang, Y.-G. Li, Y.-M. Shen, Y.-T. Zheng and X. Li, Bioorg. Med. Chem., 2016, 24, 2125 CrossRef CAS PubMed; (g) S. A. M. El-Hawash, N. S. Habib and M. A. Kassem, Arch. Pharm., 2006, 339, 1 Search PubMed; (h) O. Cil, P. W. Phuan, S. Lee, J. Tan, P. M. Haggie, M. H. Levin, L. Sun, J. R. Thiagarajah, T. Ma and A. S. Verkman, Cell Mol. Gastroenterol. Hepatol., 2016, 2, 317 CrossRef PubMed.
- J. Quinn, C. Guo, L. Ko, B. Sun, Y. He and Y. Li, RSC Adv., 2016, 6, 22043 RSC.
- (a) A. Y. Shaw, C. R. Denning and C. Hulme, Synthesis, 2013, 45, 459 CrossRef CAS; (b) S. Krupkova, P. Funk, M. Soural and J. Hlava, ACS Comb. Sci., 2013, 15, 20 CrossRef CAS PubMed; (c) A. Carta, I. Briguglio, S. Piras, P. Corona, G. Boatto, M. Nieddu, P. Giunchedi, M. E. Marongiu, G. G. F. liano, S. Blois, C. Ibba, B. Busonera and P. Colla, Bioorg. Med. Chem., 2011, 19, 7070 CrossRef CAS PubMed; (d) D. Li, H. Ma and W. Yu, Adv. Synth. Catal., 2015, 357, 3696 CrossRef CAS; (e) Y. M. Yan, Y. Gao and M. W. Ding, Tetrahedron, 2016, 72, 5548 CrossRef CAS; (f) J. Areephong, B. Huo, I. I. Mbaezue and K. E. O. Ylijoki, Tetrahedron Lett., 2016, 57, 3124 CrossRef CAS; (g) A. Carrër, J.-D. Brion, S. Messaoudi and M. Alami, Org. Lett., 2013, 15, 5606 CrossRef PubMed; (h) A. Carrër, J.-D. Brion, M. Alami and S. Messaoudi, Adv. Synth. Catal., 2014, 356, 3821 CrossRef; (i) K. Yin and R. Zhang, Org. Lett., 2017, 19, 1530 CrossRef CAS PubMed; (j) S. Paul, J. H. Ha, G. E. Park and Y. R. Lee, Adv. Synth. Catal., 2017, 359, 1515 CrossRef CAS; (k) J. Yuan, S. Lim and L. Qu, Adv. Synth. Catal., 2017, 359, 4197 CrossRef CAS; (l) K. Yin and R. Zhang, Synlett, 2018, 28, 597 Search PubMed; (m) B. Ramesh, C. R. Reddy, G. R. Kumar and B. V. S. Reddy, Tetrahedron Lett., 2018, 59, 628 CrossRef CAS; (n) T. Nishio, J. Org. Chem., 1984, 49, 827 CrossRef CAS; (o) Q. Yu, Y. Zhang and J. –P. Wan, Green Chem., 2019, 21, 3436 RSC.
- S. J. Kwon, H. I. Jung and D. Y. Kim, ChemistrySelect, 2018, 3, 5824 CrossRef CAS.
- For recent reports on radical arylation reactions, see: (a) W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803 RSC; (b) A. Studer and M. Brossart, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 1st edn., 2001, vol. 2, p. 62 Search PubMed; (c) J. Fossey, D. Lefort and J. Sorba, Free Radicals in Organic Chemistry, Wiley, New York, 1995, p. 167 Search PubMed.
- (a) S. E. Vaillard and A. Studer, Radical Arylations, in: Encyclopedia of Radicals in Chemistry, Biology, and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, New York, 2012, vol. 2 Search PubMed; (b) H. Jasch and M. R. Heinrich, Tin Hydrides and Functional Group Transformations, in Encyclopedia of Radicals in Chemistry, Biology, and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, New York, 2012, vol. 2 Search PubMed; (c) G. Pratsch and M. R. Heinrich, in Topics in Current Chemistry, ed. M. R. Heinrich and A. Gans-uer, Springer, Heidelberg, 2012, vol. 320, p. 33 Search PubMed.
- A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfaredb and Z. Rahmani, RSC Adv., 2018, 8, 33828 RSC and references cited therein.
- (a) P. Patil, A. Nimonkar and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331 CrossRef CAS PubMed; (b) R. R. Jadhav, S. N. Huddar and K. G. Akamanchi, Eur. J. Org. Chem., 2013, 6779 CrossRef CAS; (c) G. Wagh, S. Autade, P. C. Patil and K. G. Akamanchi, New J. Chem., 2018, 42, 3301 RSC.
- (a) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, D. B. Matthew and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292 CrossRef CAS PubMed; (b) J. Wang, S. Wang, G. Wang, J. Zhang and X.-Q. Yu, Chem. Commun., 2012, 48, 11769 RSC; (c) D. Arghya, M. Srimanta, M. Arun, D. Uttam and D. Maiti, Eur. J. Org. Chem., 2013, 5251 Search PubMed; (d) K. Komeyama, T. Kashihara and K. Takaki, Tetrahedron Lett., 2013, 54, 1084 CrossRef CAS; (e) P. S. Parvinder, A. Sravan Kumar, Y. Mahipal, V. P. Singh and R. A. Vishwakarma, J. Org. Chem., 2013, 78, 2639 CrossRef PubMed; (f) A. Ilangovan, A. Polu and G. Satish, Org. Chem. Front., 2015, 2, 1616 RSC; (g) M. Khoshneviszadeh, M. Soheilizad, M. Fardpour and M. Mahdavi, Moatsh. Chem., 2017, 148, 1817 CrossRef CAS.
- (a) P. R. Boruah, A. A. Ali, B. Saikia and D. Sarma, Green Chem., 2015, 17, 1442 RSC; (b) L. Botella and C. Nájera, J. Organomet. Chem., 2002, 663, 46 CrossRef CAS.
- F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27 CrossRef CAS.
- S. Mandal, T. Bera, G. Dubey, J. Saha and J. K. Laha, ACS Catal., 2018, 8, 5085 CrossRef CAS and references cited therein.
- (a) N. B. Dutta, S. Sharma, R. L. Chetry and G. Baishya, ChemistrySelect, 2019, 4, 5817 CrossRef CAS; (b) N. Hazarika, B. Sarmah, M. Bordoloi, P. Phukan and G. Baishya, Org. Biomol. Chem., 2017, 15, 2003 RSC.
- (a) P. Bartlett and J. D. Cotman, J. Am. Chem. Soc., 1949, 71, 1419 CrossRef CAS; (b) I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055 CrossRef CAS.
- (a) M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A.-L. Lee, Org. Lett., 2018, 20, 6863 CrossRef PubMed; (b) M. J. Davies, B. C. Gilbert, C. B. Thomas and J. J. Young, J. Chem. Soc., Perkin Trans. 2, 1985, 1199 RSC.
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed; (b) K. Wang, M. Chen, Q. Wang, X. Shi and J. K. Lee, J. Org. Chem., 2013, 78, 7249 CrossRef CAS PubMed; (c) C.-Y. Chen, P.-H. Lee, Y.-Y. Lin, W.-T. Yu, W.-P. Hu, C.-C. Hsu, Y.-T. Lin, L.-S. Chang, C.-T. Hsiao, J.-J. Wang and M.-I. Chung, Bioorg. Med. Chem. Lett., 2013, 23, 6854 CrossRef CAS PubMed; (d) Y. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848 RSC.
- (a) T. Jin, S. Kamijo and Y. Yamamoto, Eur. J. Org. Chem., 2004, 3789 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed; (c) G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS PubMed; (d) F. Amblard, J. H. Cho and R. F. Schinaz, Chem. Rev., 2009, 109, 4207 CrossRef CAS PubMed and references cited therein; (e) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, J. Org. Chem., 2013, 78, 5031 CrossRef CAS PubMed; (f) N. V. Sokolova and V. G. Nenajdenko, RSC Adv., 2013, 3, 16212 RSC; (g) J. C. Loren, A. Krasinski, V. V. Fokin and K. B. Sharpless, Synlett, 2005, 2847 CAS; (h) A. A. Ali, M. Chetia, B. Saikia, P. J. Saikia and D. Sarma, Tetrahedron Lett., 2015, 56, 5892 CrossRef CAS; (i) B. Saikia, P. P. Saikia, A. Goswami, N. C. Barua, A. K. Saxena and N. Suri, Synthesis, 2011, 19, 3173 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data of all new compounds; 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/d0ra00013b |
| This journal is © The Royal Society of Chemistry 2020 |