
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn water catalyst-free synthesis of benzo[d]imidazo[2,1-b] thiazoles and novel N-alkylated 2-aminobenzo[d]oxazoles under microwave irradiation†‡
Narasimharao Mukku and
Barnali Maiti *
*
Department of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore-632014, India. E-mail: barnalimaiti.m@gmail.com
First published on 3rd January 2020
Abstract
A highly efficient unprecedented catalyst-free microwave-assisted procedure for synthesizing benzo[d]imidazo[2,1-b]thiazoles and N-alkylated 2-aminobenzo[d]oxazol in green media was developed. The transformation provided rapid access to functionalized benzo[d]imidazo[2,1-b]thiazoles from 2-aminobenzothiazole and N-alkylated 2-aminobenzo[d]oxazole from 2-aminobenzoxazole scaffolds under mild transition-metal-free conditions. This synthetic manipulation is expected to greatly expand the repertoire of reaction types in heterocyclic chemistry and pave the way for new syntheses of bioactive compounds.
Introduction
Syntheses of nitrogen-containing fused heterocyclic small molecules are attracting increasing attention in medicinal chemistry, drug design, and advanced materials due to their structural complexities.1–4 These architecturally complex molecules are frequently used as biological probes in living systems as well as drugs in medicine because their diverse heteroatoms can bind different locations of biological macromolecules. Researchers in pharmaceutical industries and academicians are highly interested in the synthesis of fused heterocyclic small molecules owing to their pharmaceutical activities and therapeutic potential. For studying cell biology and the treatment of diseases, diverse heterocyclic small molecules are extremely important as they modulate the function of enzymes and receptors.5–8 In this context, benzo[d]imidazo[2,1-b]thiazoles and benzo[d]oxazole derivatives are members of an essential class of heterocycles that have gained significant attention owing to their broad spectrum of important bioactivities. Benzo[d]imidazo[2,1-b]thiazoles have been reported to serve as a potent non-sedative anxiolytic (A),9,10 powerful anticancer agent (B),11–14 PET imaging probe of β-amyloid plaques in the brains of Alzheimer's patients (C),15–17 kinase inhibitor (D),18–20 and antimicrobially active molecule (E),21 while benzo[d]oxazole derivatives have been used to treat interval loss of nerve function (F) (Fig. 1).22,23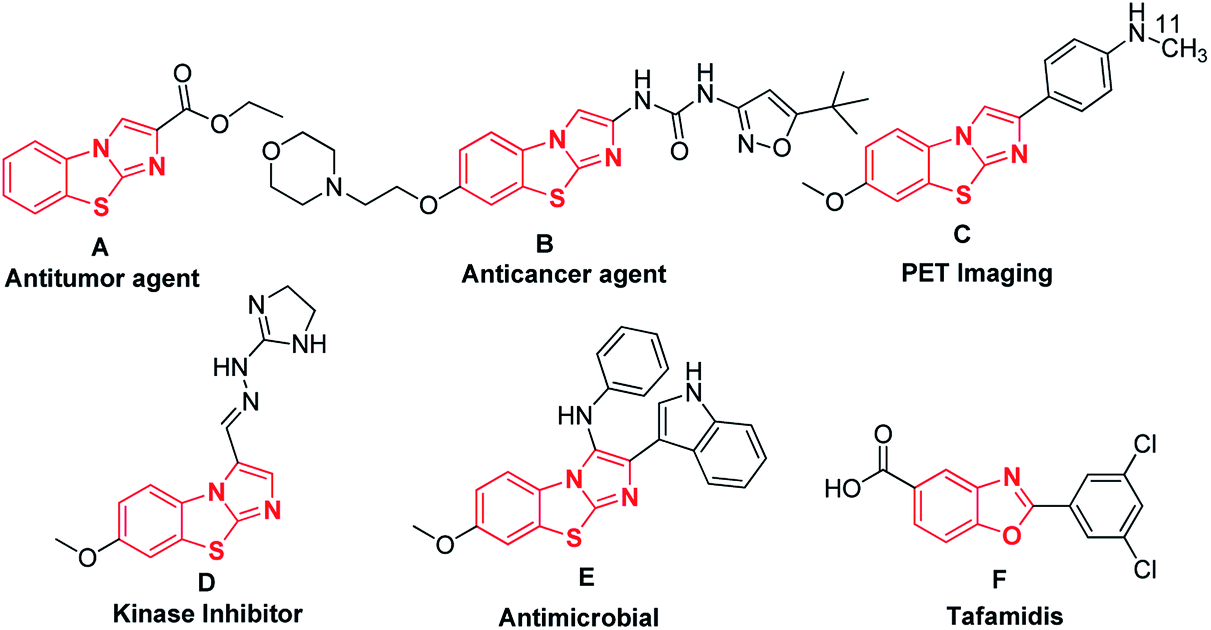 | ||
| Fig. 1 Selected biologically active benzo[d]imidazo[2,1-b]thiazoles and benzo[d]oxazole derivatives. | ||
The extraordinarily satisfying properties of the benzo[d]imidazo[2,1-b]thiazole- and benzo[d]oxazole-related drugs have motivated organic chemists to set out to synthesize, using simple methodologies, a great number of novel chemotherapeutic agents. In recent years, the syntheses of benzo[d]imidazo[2,1-b]thiazoles and benzo[d]oxazole derivatives have attracted much attention.
A careful literature survey revealed many reports on the designs of heteroannulation reactions of 2-aminobenzothiazoles with α-bromoketones in organic solvents.24–27 For instance, Hajra et al. developed an elegant protocol for iron-catalyzed syntheses of benzo[d]imidazo[2,1-b]thiazoles from reactions of 2-aminobenzothiazoles with ketones/chalcones.28 Furthermore, in 2016, the research groups of Jeong developed FeCl3-catalyzed reactions of 2-aminobenzothiazoles with aldehydes and nitro alkane to produce fused heterocycles.29 In 2015, Zhu et al. established Cu(OAc)2-promoted cycloaddition reactions of α-methylenyl isocyanides with 2-methyl benzothiazole to produce benzo[d]imidazo[2,1-b]thiazoles.30 Similarly, Meshram et al. developed Cu(II) and ionic liquid co-catalysed syntheses of fused heterocycles.31 In 2017, Volkova et al. synthesized diverse benzo[d]imidazo[2,1-b]thiazoles by performing copper (I, II)-catalysed reactions of 2-aminobenzothiazoles with aldehydes and alkynes in a continuous-flow reactor.32 Subsequently, Deng et al. developed metal-free syntheses of benzo[d]imidazo[2,1-b]thiazoles from 2-aminobenzothiazoles with cyclic ketones and elemental sulfur and oxygen.33 In the same year, Zhang et al. developed a catalytic oxidative cyclisation of 2-aminobenzothiazole with 2-phenoxyacetophenone under oxygen using a CuI catalyst.34 Recently, a one-pot Groebke−Blackburn−Bienaymé synthetic protocol was employed by Sharada et al. and the Gámez-Montañ group to synthesize these privileged heterocycles (Scheme 1).35–37
 | ||
| Scheme 1 Heterocyclisation of 2-aminobenothiazole: previous and present work. | ||
Although the above methodologies are impressive, the development of an efficient and transition-metal-free approach for the diversity-oriented synthesis of both benzo[d]imidazo[2,1-b]thiazoles and novel N-alkylated 2-aminobenzo[d]oxazoles derivatives using a simple methodology remains a challenging yet attractive task. Most of the syntheses developed so far suffer serious drawbacks such as the use of transition metals, harsh reaction conditions, use of toxic solvents, long reaction times and limited substrate scope.38–42 Therefore, it is warranted to devise a new strategy to construct these privileged scaffolds in a sustainable pathway, which forms the core of modern organic synthesis. The application of microwave (MW) irradiation together with green solvents has gained significant attention in recent years because of the extremely rapid growth in the generation of products in non-flammable, nontoxic environments of green solvents. MW irradiation offers several advantages such as rapid and instantaneous heating, high homogeneity of temperature, and decreased formation of side products. By choosing appropriate MW irradiation parameters, reaction selectivity can be attained and the rates of reactions can be increased. Compounds in polar solvents can more quickly absorb MW energy than can compounds in relatively apolar solvents.43–46 In an effort to solubilize organic compounds in water, isopropyl alcohol (IPA) as a polar protic organic co-solvent is used as a reaction medium that can easily absorb microwave power for efficient product generation. Moreover IPA is a powerful, nontoxic, unusual ‘green’ solvent that can easily solubilize organic compounds and efficiently absorb MW power. In our research, we set out to develop expedient catalyst-free green chemical syntheses of benzo[d]imidazo[2,1-b]thiazoles and novel N-alkylated 2-aminobenzo[d]oxazoles using H2O-IPA as the preferred reaction medium under MW irradiation. This synthetic methodology was found to offer several advantages in comparison to the already reported synthesis protocols, such as excellent yields with high purity, avoidance of toxic transition-metal catalysts, a broad substrate scope, and green and economical process resulting from the use of a combination of H2O-IPA with MW irradiation. In a continuation of our quest to develop new synthetic methodologies using sustainable chemistry,47–50 we developed in the currently described work a simple and facile procedure for the transition-metal-free synthesis of benzo[d]imidazo[2,1-b]thiazoles and novel N-alkylated 2-aminobenzo[d]oxazoles in H2O-IPA under microwave irradiation.
Results and discussion
At the outset, we employed 2-aminobenzothiazole 1 with 2-bromoacetophenone 2 as a suitable substrate, for reaction discovery and optimization, results of which are shown in Table 1. Carrying out the reaction in a room-temperature solvent-free condition for 6 h did not yield any benzo[d]imidazo[2,1-b]thiazole product 3 (Table 1, entry 1). However, upon increasing the reaction temperature to 100 °C, the product yield of compound 3 improved to 50% (Table 1, entry 2). Next we tested performing the reaction in polar aprotic solvents such as CH2Cl2 and CH3CN at their refluxing temperatures for 6 h; little improvement in the product yield of 3 was found (Table 1, entry 3, 4). Even performing the heterocyclisation reaction in a high-boiling-point solvent like DMSO at 100 °C for 6 h did not have much of an impact on the product yield of 3 (Table 1, entry 5). Interestingly, the product yield of 3 dramatically increased to 65% when we performed the reaction in a polar protic solvent such as refluxing ethanol (Table 1, entry 6). And when we performed the same set of reactions in an H2O-IPA medium as a green solvent at 100 °C for 2 h, the yield of the product 3 was observed to be 90% (Table 1, entry 7). And when subsequently applying microwave irradiation at a temperature of 100 °C and pressure of 15 bar, the yield of product 3 increased dramatically to 95% in 15 minutes of reaction time (Table 1, entry 8). The advantages of microwave irradiation combined with H2O-IPA as a reaction medium included a reduction of the reaction time along with the elimination of side product formation, showing the process to be environmentally benign and economical. Next we evaluated the scope of the reaction by employing substituted 2-aminobenzothiazoles 1 and phenacyl bromides 2. Interestingly, for both 1 and 2, the product yield was not affected by the substituent group. Whether electron-withdrawing or electron-donating substituents were used, efficient heterocyclisation in the microwave irradiation conditions occurred smoothly to result in the corresponding benzo[d]imidazo[2,1-b]thiazole products 3 in excellent yields.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temperature | Time (h) | Yieldc (%) |
a Reaction was performed using 1a (1 mmol), 2a (1 mmol), H2O-IPA (4 ml, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).b Microwave reaction were carried out in Biotage Initiator Microwave Synthesizer operated at 2450 MHz at 15 bar pressure.c Yield of the isolated product. :1).b Microwave reaction were carried out in Biotage Initiator Microwave Synthesizer operated at 2450 MHz at 15 bar pressure.c Yield of the isolated product. |
||||
| 1 | Neat | rt | 6 | 0 |
| 2 | Neat | 100 °C | 6 | 50 |
| 3 | CH2Cl2 | Reflux | 6 | 15 |
| 4 | CH3CN | Reflux | 6 | 10 |
| 5 | DMSO | 100 °C | 6 | 15 |
| 6 | EtOH | 80 °C | 6 | 65 |
| 7 | H2O-IPA | 100 °C | 2 | 90 |
| 8 | H2O-IPA | MW, 100b °C | 0.25 | 95 |

All of these reactions were performed under high-pressure microwave conditions in typically 12–15 minutes. Table 2 shows the substrate scope for the efficient synthesis of substituted benzo[d]imidazo[2,1-b]thiazoles in green media. After completion of the reactions, the corresponding benzo[d]imidazo[2,1-b]thiazole derivatives were obtained with excellent yields and then simple work-ups were carried out involving removal of solvents under reduced pressure, extraction, and solvent evaporation, all without the use of column chromatography.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | (C6H5)R2 | Yielda | LRMSb | H-bond donor | H-bond acceptor | clogPc |
| a Isolated yields.b LMRS was recorded by using the GC-MS method.c clog P estimated by using by ChemBioOffice 2010. | |||||||
| 3a | H | C6H5 | 95 | 250 | 0 | 2 | 4.86 |
| 3b | H | 4-(C6H5)CH3 | 94 | 264 | 0 | 2 | 5.36 |
| 3c | H | 4-(C6H5)OCH3 | 93 | 280 | 0 | 3 | 4.87 |
| 3d | H | –CO2Et | 95 | 246 | 0 | 4 | 2.94 |
| 3e | H | 4-(C6H5)CN | 96 | 275 | 0 | 3 | 4.29 |
| 3f | H | 4-(C6H5)–CF3 | 92 | 318 | 0 | 2 | 5.74 |
| 3g | H | 4-(C6H5)NO2 | 90 | 295 | 0 | 3 | 4.60 |
| 3h | 7-OEt | 4-(C6H5)CH3 | 94 | 308 | 0 | 3 | 5.90 |
| 3i | 7-OEt | 4-(C6H5)–CF3 | 94 | 362 | 0 | 3 | 6.28 |
| 3j | 7-OEt | 2,4-(C6H5)(F)2 | 92 | 330 | 0 | 3 | 5.69 |
| 3k | 7-NO2 | C6H5 | 90 | 295 | 0 | 4 | 4.61 |
| 3l | 7-NO2 | 4-(C6H5)CF3 | 92 | 363 | 0 | 4 | 5.50 |
Finally the identities of the purified products were confirmed by characterizing them spectroscopically, specifically using 1H NMR, 13C NMR, and mass spectroscopy (MS).
To further examine the efficiency of this heterocyclisation reaction and to swiftly expand our unique compound library, we extended the substrate scope to 2-aminobenzoxazoles 4 as suitable substrates. We reacted the 2-aminobenzoxazole 4 with α-bromoketone 2a in the same microwave irradiation condition as described above for 15 minutes, and this reaction only produced the N-alkylated 2-amino benzo[d]oxazoles 5a, and did so in 90% yield, as shown in Scheme 2.
 | ||
| Scheme 2 Attempted synthesis of substituted benzo[d]imidazo[2,1-b]oxazoles in green media. | ||
Furthermore, to obtain the cyclised benzo[d]imidazo[2,1-b]oxazoles 6, we tested various polar aprotic and polar protic solvents in various conditions as outlined in Table 1; here, no cyclised product 6 was found. This result was probably due to the oxygen atom of the 2-amino benzo[d]oxazole moiety being more electronegative than the sulphur atom of 2-aminobenzothiazole. Furthermore, the presumably relatively high localization of the lone pair of electrons of oxygen on the oxygen atom of the 2-amino benzo[d]oxazole moiety further deactivated the nitrogen atom of the amine group to attack the adjacent carbonyl moiety. Finally using the variously substituted α-bromoketones 2, we obtained N-alkylated 2-amino benzo[d]oxazole products 5 in good yield, as shown in Table 3.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | (C6H5)R2 | Yielda | LRMSb | H-bond donor | H-bond acceptor | clogPc |
| a Isolated yields.b LRMS was recorded by using the GC-MS method.c clogP estimated by using by ChemBioOffice 2010. |
|||||||
| 5a | H | C6H5 | 94 | 253 | 1 | 4 | −0.39 |
| 5b | H | 4-(C6H5)OCH3 | 93 | 283 | 1 | 5 | −0.17 |
| 5c | H | 4-(C6H5)CI | 94 | 287 | 1 | 4 | 0.39 |
| 5d | H | 4-(C6H5)CF3 | 94 | 321 | 1 | 4 | 0.62 |
| 5e | H | 2,4-(C6H5)(F)2 | 92 | 289 | 1 | 4 | −0.45 |
| 5f | 5-CI | 4-(C6H5)OCH3 | 93 | 317 | 1 | 5 | 0.39 |
Next, to evaluate the drug-like properties of the synthesized compounds in our libraries, we applied Lipinski's rule of five by calculating the molecular weight, clogP, the number of hydrogen bond donors and acceptors and the number of rotatable bonds of each member of the library. The rule predicts than an organic molecule can have drug-like physical properties if its molecular weight is less than 500, its clogP value is not more than 5, and it has no more than 10 and 5 hydrogen bond acceptors and donors, respectively. However, a molecule violating just one of the Lipinski rules may still be considered as a potential drug candidate. Fortunately the predicted values of drug-like properties for these synthesized compounds were found to be within the accepted limits of Lipinski's rule of five, as shown in Tables 2 and 3.
In addition, we have demonstrated the potential utility of this synthetic protocol on the gram scale by using the model reaction. As shown in Scheme 3, the reaction afforded 1.5 g of 3a in 95% yield, demonstrating the potential applications of the present synthetic protocol for large-scale syntheses of benzo[d]imidazo[2,1-b]thiazole scaffolds.
 | ||
| Scheme 3 Gram-scale catalyst-free synthesis of benzo[d]imidazo[2,1-b] thiazole 3a. | ||
Based on the experimental results of the reaction, a plausible mechanistic pathway for this heteroannulation reaction was derived, and this pathway is outlined in Scheme 4. According to this proposed pathway, the reaction occurred via a sequence of steps involving a nucleophilic bromo-substitution by the N-atom of 2-amino benzothiazole to lead to the N-alkylated adduct a, which on liberation of HBr formed the intermediate b followed by an intramolecular cyclization to obtain intermediate c. The final step of the reaction involved the elimination of a water molecule from intermediate c to afford benzo[d]imidazo[2,1-b] thiazole 3 derivatives. Furthermore, according to the proposed mechanism, the reactions of 2-aminobenzoxazole with α-bromoketones involved selective substitution by the secondary amine of 2-aminobenzo[d]oxazol, leading to the N-alkylated 2-aminobenzo[d]oxazol derivatives. The lack of any observed hetero annulation was attributed to the greater electronegativity of oxygen than of sulphur resulting in the electron pair on the oxygen atom being more localized and hence further deactivating the benzoxazole moiety for hetero cyclisation. In addition, an attempted reaction in a sealed tube with microwave irradiation for a long time did not facilitate the heteroannulation reactions, indicative of a relative lack of delocalization of the benzoxazole moiety oxygen electron pair for heterocyclisation compared to that of the benzothiazole moiety sulphur electron pair.
 | ||
| Scheme 4 A plausible mechanism for the syntheses of benzo[d]imidazo[2,1-b]thiazole derivatives. | ||
Conclusions
In summary, we have demonstrated a novel green methodology for the syntheses of benzo[d]imidazo[2,1-b]thiazoles and N-alkylated 2-aminobenzo[d]oxazoles from 2-amino benzothiazoles and 2-aminobenzoxazoles with α-haloketone under microwave irradiation in a green medium. The prominent features of this approach include green reaction environments, high atom economy, single synthetic operation, relatively short reaction time, high yield, and the lack of any need to perform chromatographic purification. These features coupled with the reaction being conducted under microwave irradiation and without the use of toxic transition-metal catalysts as well as being operationally simple and involving the use of inexpensive starting materials make the method potentially scalable and useful for the preparation of a wide range of analogues.Experimental section
Substituted 2-aminobenzothiazoles, 2-aminobenzoxazoles and substituted 2-bromo acetophenones purchased from Sigma Aldrich and solvents from commercial suppliers were used without further purification. 1H NMR and 13C NMR data were recorded using a Bruker DRX400 spectrometer (400 MHz). Chemical shifts are reported in ppm relative to an internal standard. Coupling constant (J) values are given in Hz. Multiplicities of peaks are given as d (doublet), m (multiplet), s (singlet), and t (triplet). Mass spectra were recorded using a PerkinElmer Clarus 600 GC MS spectrometer. IR spectra were recorded using a Bomen DA8 3 FTS spectrometer. Microwave reactions were carried out in a 2011 model Biotage Initiator Microwave Synthesizer, SW version: 4.1.3. Build 11768 using an IR sensor as internal probe for the control of temperature and compressed air system for cooling.Representative procedure for the synthesis of benzo[d]imidazo[2,1-b]thiazole 3a
In a 10 ml microwave vial, 2-aminobenzothiazole 1a (0.125 mg, 0.83 mmol, 1.0 equiv.) and 2-bromo acetophenone 2a (0.165 mg, 0.83 mmol, 1.0 equiv.) were added to 4 ml of H2O-IPA (1:1). The reaction mixture was irradiated with microwaves for 15 min at 100 °C and 15 bar pressure. After completion of the reaction as monitored using TLC, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The precipitated solid product was filtered and washed with ice-cooled ether (10 ml) and dried under vacuum to obtain benzo[d]imidazo[2,1-b]thiazole 3a in 95% yield.
Representative procedure for the synthesis of 2-amino-3-(2-(4-methoxyphenyl)-2-oxoethyl)benzo[d]oxazol-3-ium 5a
In a 10 ml microwave vial, 2-aminobenzoxazole 4a (0.134 mg, 1.00 mmol, 1.0 equiv.) and 2-bromo acetophenone 2a (0.198 mg, 1.00 mmol, 1.0 equiv.) were added to 4 ml of H2O-IPA (1:1). The reaction mixture was irradiated with microwaves for 15 min at 100 °C and 15 bar pressure. After completion of the reaction as monitored using TLC, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The precipitated solid product was filtered and washed with ice-cooled ether (10 ml) and dried under vacuum to obtain 2-amino-3-(2-(4-methoxyphenyl)-2-oxoethyl)benzo[d]oxazol-3-ium 5a in 95% yield.
2-Phenylbenzo[d]imidazo[2,1-b]thiazole (3a)28
Yield: 95%, white solid; mp: 88–90 °C, Rf: 0.5% (40% EtOAc/n-Hexane); 1H NMR (400 MHz, CDCl3) 7.87 (s, 1H), 7.79 (d, J = 7.52 Hz, 2H), 7.60 (d, J = 7.96 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.37–7.32 (m, 3H), 7.24–7.18 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 148.1, 147.7, 133.8, 132.1, 130.3, 128.7, 127.5, 126.2, 125.2, 124.9, 124.4, 112.6, 106.9. MS (GC-MS): 250; IR (KBr, cm−1): 3134, 1599, 1580, 1490, 1256.2-(p-Tolyl)benzo[d]imidazo[2,1-b]thiazole (3b)28
Yield: 94%; brown solid; mp: 105–107 °C, Rf: 0.4 (30% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 7.86 (s, 1H), 7.69 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.40–7.36 (m, 1H), 7.28–7.24 (m, 1H), 7.16 (d, J = 8.0 Hz, 1H), 2.31 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 148.0, 147.8, 137.3, 132.2, 131.1, 130.3, 129.4, 126.2, 125.1, 124.8, 124.4, 112.6, 106.4, 21.3; MS (GC-MS): 264; IR (KBr, cm−1): 3139, 1550, 1485, 1253, 821, 725.2-(4-Methoxyphenyl)benzo[d]imidazo[2,1-b]thiazole (3c)28
Yield: 93%; white solid; mp: 168–170 °C; Rf: 0.6 (40% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.69 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.33 (t, J = 7.7 Hz, 1H), 6.96 (d, J = 8.5 Hz, 2H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.4, 147.9, 147.6, 132.3, 130.2, 126.7, 126.5, 126.2, 124.7, 124.4, 114.2, 112.6, 105.9, 55.3. MS (GC-MS): 280; IR (KBr, cm−1): 2835, 1545, 1485, 1398, 1238, 831.Ethyl benzo[d]imidazo[2,1-b]thiazole-2-carboxylate (3d)25
Yield: 95%; white solid; mp: 95–97 °C, Rf: 0.6 (30% EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.57 (t, J = 7.7 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 4.30 (q, J = 7.2 Hz, 2H), 1.32 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.3, 148.0, 137.8, 131.8, 130.3, 127.3, 126.7, 125.5, 119.6, 114.8, 60.7, 14.8. MS (GC-MS): 246; IR (KBr, cm−1): 3125, 1786, 1703, 1525, 1496, 1296, 1232, 1020.4-(Benzo[d]imidazo[2,1-b]thiazol-2-yl)benzonitrile (3e)33
Yield: 96%; white solid; mp: 210–212 °C, Rf: 0.5% (40% EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.04–8.00 (m, 3H), 7.97 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.57 (t, J = 8.0 Hz, 1H), 7.44 (t, J = 8.0 Hz, 1H), 13C NMR (100 MHz, DMSO-d6) δ 148.2, 144.8, 138.7, 133.3, 132.0, 129.8, 127.7, 126.0, 125.6, 123.2, 119.5, 114.0, 118.8, 109.7; MS (GC-MS): 275; IR (KBr, cm−1): 3053, 2220, 1605, 1493, 1408, 837.2-(4-(Trifluoromethyl)phenyl)benzo[d]imidazo[2,1-b]thiazole (3f)28
Yield: 92% light-brown solid; mp: 136–138 °C; Rf: 0.6 (40% EtOAc/n-hexane) 1H NMR (400 MHz, DMSO-d6) δ 8.30 (s, 1H), 7.93 (d, J = 7.6 Hz, 2H), 7.74–7.69 (m, 2H), 7.57 (d, J = 7.6 Hz, 2H), 7.47–7.42 (m, 1H), 7.34–7.31 (t, J = 7.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 148.2, 144.8, 136.7, 131.8, 129.9, 129.2, 126.7, 125.7, 125.6, 125.2, 124.5, 113.3, 109.1. MS (GC-MS): 318; IR (KBr, cm−1): 1616, 1496, 1321, 1101, 1068, 746.2-(4-Nitrophenyl)benzo[d]imidazo[2,1-b]thiazole (3g)30
Yield: 90%; brown solid; mp: 250–252 °C, Rf: 0.6 (40% EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.28 (d, J = 8.8 Hz, 1H), 8.24 (m, 1H), 8.10 (d, J = 8.8 Hz, 2H), 8.00 (dd, J = 8.0, 1.6 Hz, 2H), 7.57 (t, J = 7.2 Hz, 1H), 7.46–7.42 (m, 1H), 13C NMR (400 MHz, DMSO-d6) δ 146.5, 145.8, 145.6, 141.9, 140.9, 137.2, 132.0, 130.0, 129.9, 127.1, 126.0, 125.6, 125.4, 114.0, 112.3; MS (GC-MS): 295; IR (KBr, cm−1): 3148, 1595, 1492, 1323, 1103, 852.7-Ethoxy-2-(p-tolyl)benzo[d]imidazo[2,1-b]thiazole (3h)
Yield: 94%; brown solid; mp: 175–177 °C; Rf: 0.6 (40% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.42–7.38 (m, 1H), 7.16 (s, 1H), 7.14–7.11 (m, 1H), 4.03–3.94 (m, 2H), 2.30 (s, 3H), 1.39 (t, J = 7.2 Hz, 3H).13C NMR (100 MHz, CDCl3) δ 156.5, 147.3, 137.2, 131.4, 131.1, 129.4, 127.0, 126.3, 125.0, 114.0, 113.0, 109.4, 106.4, 64.3, 21.3, 14.8. HRMS (ESI, m/z): calcd for C18H16N2OSH [M + H]+: 309.1062. Found: 309.1064, IR (KBr, cm−1) 2974, 1601, 1548, 1498, 1260, 1244, 1056, 825.7-Ethoxy-2-(4-(trifluoromethyl)phenyl)benzo[d]imidazo[2,1-b]thiazole (3i)
Yield: 94%; white solid; mp: 177–180 °C, Rf: 0.5 (60% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H), 7.87 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.8 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 6.95 (d, J = 8.8, 2.4 Hz, 1H), 4.00 (q, J = 7.2 Hz, 2H), 1.39 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 156.9, 147.9, 145.5, 137.2, 131.5, 128.9, 126.0, 125.7 (d, JCF = 3.8 Hz), 125.1, 114.2, 113.3, 109.4, 107.9, 64.4, 14.8. HRMS (ESI, m/z): calcd for C18H13F3N2OSH [M + H]+: 363.0779. Found: 363.0775, IR (KBr, cm−1) 1614, 1500, 1475, 1317, 1161, 1105, 1060, 833.2-(2,4-Difluorophenyl)-7-ethoxybenzo[d]imidazo[2,1-b]thiazole (3j)
Yield: 92%; white solid; mp: 164–166 °C;, Rf: 0.4 (40% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 8.20–8.14 (m, 1H), 7.97 (d, J = 3.8 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.16 (d, J = 2.4 Hz, 1H), 6.98–6.93 (m, 2H), 6.90–6.84 (m, 1H), 4.05 (q, J = 6.9 Hz, 2H), 1.44 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 163.0 (d, JCF = 12 Hz), 160.5 (q, JCF = 12 Hz), 158.2 (d, JCF = 12 Hz), 156.7, 147.1, 139.7, 131.4, 128.7 (q, JCF = 5 Hz), 126.2, 118.2 (q, JCF = 4 Hz), 113.9, 113.2, 111.6 (dd, JCF = 21.0, 4.0 Hz), 110.4 (d, JCF = 15 Hz), 109.4, 103.9 (t, JCF = 26 Hz), 64.3, 14.8. HRMS (ESI, m/z): calcd for C17H12F2N2OSH [M + H]+: 331.0717. Found: 331.0712. IR (KBr, cm−1): 2986, 1577, 1497, 1481, 1259, 1061, 1041, 845.7-Nitro-2-phenylbenzo[d]imidazo[2,1-b]thiazole (3k)32
Yield: 90%; white solid; mp: 218–220, Rf: 0.6 (30% EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6) δ 9.12 (d, J = 2 Hz, 1H), 8.91 (s, 1H), 8.47 (dd, J = 2.0, 8.8 Hz, 1H), 8.17 (d, J = 8.8 Hz, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.46 (t, J = 7.6 Hz, 2H), 7.33 (t, J = 7.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 149.3, 147.7, 144.6, 136.4, 133.8, 131.1, 129.3, 128.7, 125.3, 123.1, 122.1, 114.0, 110.1. MS (GC-MS): 295; IR (KBr, cm−1): 3068, 1580, 1500, 1332, 814.7-Nitro-2-(4-(trifluoromethyl)phenyl)benzo[d]imidazo[2,1-b]thiazole (3l)
Yield: 92%; brown solid; mp: 225–227 °C; Rf: 0.5 (60% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 2.0 Hz, 1H), 8.35 (dd, J = 8.8, 2.2 Hz, 1H), 8.06 (s, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 135.7, 128.1, 126.1, 126.0, 125.9, 125.8, 125.6, 122.5, 120.8, 112.6, 108.3. MS (GC-MS): 363, HRMS (ESI, m/z): calcd for C16H8F3N3O2SH [M + H]+: 364.0368. Found: 364.0369, IR (KBr, cm−1): 3421, 3377, 1681, 1321, 1107, 1064.2-Amino-3-(2-oxo-2-phenylethyl)benzo[d]oxazol-3-ium bromide (5a)
Yield: 94%; white solid; mp: 140–142 °C; Rf: 0.6 (40% EtOAc/n-hexane).1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.0 Hz, 2H), 7.76 (t, J = 7.6 Hz, 1H), 7.63 (t, J = 7.6 Hz, 2H), 7.41 (d, J = 7.2 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.20–7.13 (m, 2H), 5.60 (s, 2H). 13C NMR (100 Hz, CDCl3) δ 192.7, 154.6, 142.5, 134.8, 134.6, 132.0, 129.4, 128.8, 124.5, 122.9, 110.2, 110.1, 48.8. MS (GC-MS): 253; IR (KBr, cm−1): 2924, 1766, 1689, 1487, 1224.2-Amino-3-(2-(4-methoxyphenyl)-2-oxoethyl)benzo[d]oxazol-3-ium bromide (5b)
Yield: 93%; white solid; mp: 160–162 °C; Rf: 0.6 (40% EtOAc/n-hexane).1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.8 Hz, 2H), 7.25–7.23 (m, 1H), 7.16–7.10 (m, 2H), 7.00 (d, J = 8.8 Hz, 2H), 6.85–6.83 (m, 1H), 5.19 (s, 2H), 3.90 (s, 3H). 13C NMR (100 Hz, CDCl3) δ 189.2, 164.5, 154.7, 142.7, 130.5, 127.1, 123.9, 122.7, 114.3, 110.2, 108.7, 55.6, 47.8. MS (GC-MS): 283; IR (KBr, cm−1): 2972, 1765, 1670, 1601, 1572, 1485, 1240, 1174, 1103, 1016.2-Amino-3-(2-(4-chlorophenyl)-2-oxoethyl)benzo[d]oxazol-3-ium bromide (5c)
Yield: 94%; pale yellow solid; mp: 176–178 °C; Rf: 0.4 (40% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 8.3 Hz, 2H), 7.17–7.17 (m, 1H), 7.09–7.07 (m, 2H), 6.78–6.74 (m, 1H), 5.13 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 189.8, 154.7, 142.8, 141.1, 132.4, 130.9, 129.6, 129.5, 124.0, 122.9, 110.3, 108.6, 48.0. MS (GC-MS): 287; IR (KBr, cm−1): 2966, 1759, 1689, 1587, 1485, 1400, 1088, 1020.2-Amino-3-(2-oxo-2-(4-(trifluoromethyl)phenyl)ethyl)benzo[d]oxazol-3-ium bromide (5d)
Yield: 94%; off-white solid; mp: 146–148 °C; Rf: 0.4 (40% EtOAc/n-hexane); 1H NMR (400 MHz, DMSO-d6). 1H NMR (400 MHz, DMSO) δ 8.30 (d, J = 8.0 Hz, 2H), 8.06 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.51–7.38 (m, 2H), 6.01 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 190.4, 159.4, 144.2, 137.4, 130.9, 129.8, 126.6, 126.4, 125.5, 112.1, 111.7, 50.9. MS (GC-MS): 321; HRMS (EI, m/z): calcd for C16H11F3N2O2H [M + H]+: 321.0851. Found: 321.0852, IR (KBr, cm−1): 2907, 2652, 1689, 1494, 1178, 1132, 1063.2-Amino-3-(2-(2,4-difluorophenyl)-2-oxoethyl)benzo[d]oxazol-3-ium bromide (5e)
Yield: 92%; white solid; mp: 125–127 °C; Rf: 0.6 (40% EtOAc/n-hexane) 1H NMR (400 MHz, CDCl3) δ 8.06 (q, J = 8.4 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.17–7.12 (m, 2H), 7.07–6.97 (m, 2H), 6.81 (t, J = 3.8 Hz, 1H) 5.15 (d, J = 3.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 187.6 (d, JCF = 5.8 Hz), 168.2, 165.5 (d, JCF = 12.5 Hz), 164.7 (d, JCF = 12.5 Hz), 162.0 (d, JCF = 12.5 Hz), 154.7, 142.8, 133.2 (q, JCF = 4.6 Hz), 131.1, 123.9, 122.8, 113.2 (d, JCF = 3.0 Hz), 110.3, 108.4, 51.4 (d, JCF = 13.8 Hz), MS (GC-MS): 289; IR (KBr, cm−1): 3051, 1749, 1697, 1612, 1485, 1238, 1201, 1109, 1022.2-Amino-5-chloro-3-(2-(4-methoxyphenyl)-2-oxoethyl)benzo[d]oxazol-3-ium bromide (5f)
Yield: 93%; white solid; mp: 160–162 °C; Rf: 0.6 (40% EtOAc/n-hexane).1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 6.84 (s, 1H), 5.17 (s, 2H), 3.90 (s, 3H). 13C NMR (100 Hz, CDCl3) δ 183.9, 159.9, 136.5, 127.5, 125.8, 124.8, 122.2, 117.9, 109.6, 106.3, 104.6, 50.9, 43.1. MS (GC-MS): 317; IR (KBr, cm−1): 2933, 1766, 1674, 1486, 1240.Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
The authors thank DST-Govt of India for funding through DST-SERB-YSS/2015/00450. The authors also thank the Chancellor and Vice Chancellor of VIT for providing an opportunity to carry out this study, and VIT for providing ‘VIT SEED GRANT’ for carrying out this research work.References
- P. Das, M. D. Delost, M. H. Qureshi, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2019, 62, 4265–4311 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- L. M. Blair and J. Sperry, J. Nat. Prod., 2013, 76, 794–812 CrossRef CAS PubMed.
- D. Chen, S. J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565–9578 RSC.
- K. R. Simonetta, J. Taygerly, K. Boyle, S. E. Basham, C. Padovani, Y. Lou, T. J. Cummins, S. L. Yung, S. K. V. Soly, F. Kayser, J. Kuriyan, M. Rape, M. Cordozo, M. A. Gallop, N. F. Bence, P. A. Barsanti and A. Saha, Nat. Commun., 2019, 10(1402), 1–10 CAS.
- Z. Guo, S. Y. Hong, J. Wang, S. Rehan, W. Liu, H. Peng, M. Das, W. Li, S. Bhat, B. Peiffer, B. R. Ullman, C. M. Tse, Z. Tarmakova, C. Schiiene-Fischer, G. Fischer, I. Coe, V. O. Paaviliinen, Z. Sun and J. O. Liu, Nat. Chem., 2018, 11, 254–263 CrossRef PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discov., 2016, 15, 533–550 CrossRef CAS PubMed.
- E. E. Carlson, ACS Chem. Biol., 2010, 5, 639–653 CrossRef CAS PubMed.
- S. Clements-Jewery, G. Danswan, C. R. Gardner, S. S. Matharu, R. Murdoch, W. R. Tully and R. Westwood, J. Med. Chem., 1988, 31, 1220–1226 CrossRef CAS PubMed.
- A. M. Farag, A. S. Mayhoub, S. E. Barakat and A. H. Bayomi, Bioorg. Med. Chem., 2008, 16, 4569–4578 CrossRef CAS PubMed.
- Q. Chao, K. G. Sprankle, R. M. Grotzfeld, A. G. Lai, T. A. Carter, A. M. Velasco, R. N. Gunawardanne, M. D. Cramer, M. F. Gardener, J. James, P. P. Zarrinkar, H. K. Patel and S. S. Bhagwat, J. Med. Chem., 2009, 52, 7808–7816 CrossRef CAS PubMed.
- A. Furlan, F. Colombo, A. Kover and N. Issaly, Eur. J. Med. Chem., 2012, 47, 239–254 CrossRef CAS PubMed.
- A. Andreani, S. Burnelli, M. Granaiola and A. Leoni, J. Med. Chem., 2008, 51, 809–816 CrossRef CAS PubMed.
- G. Trapani, M. Franco, A. Latrofa, A. Reho and G. Liso, Eur. J. Pharm. Sci., 2001, 14, 209–216 CrossRef CAS PubMed.
- B. H. Yousefi, A. Manook, A. Drzezga, B. Reutern, M. Schwaiger, H. J. Wester and G. Henriksen, J. Med. Chem., 2011, 54, 949–956 CrossRef CAS PubMed.
- B. H. Yousefi, A. Drzezga, B. Reutern and A. Manook, ACS Med. Chem. Lett., 2011, 2, 673–677 CrossRef CAS PubMed.
- D. Alagille, H. DaCosta, R. M. Baldwin and G. D. Tamagnan, Bioorg. Med. Chem. Lett., 2011, 21, 2966–2968 CrossRef CAS PubMed.
- A. Andreani, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, L. I. Varoli, D. Lannigan, J. Smith, D. Scudiero, S. Kondapaka and R. H. Shoemaker, Eur. J. Med. Chem., 2011, 46, 4311–4323 CrossRef CAS PubMed.
- A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, S. Locatelli, R. Morigi, M. Rambaldi, L. Varoli, N. Calonghi, C. Cappadone, M. Valtattorni, M. Zini, C. Stefanelli, L. Masotti and R. H. Shoemaker, J. Med. Chem., 2008, 51, 7508–7513 CrossRef CAS PubMed.
- H. C. Shen, F. X. Ding, Q. Deng and L. C. Wilsie, J. Med. Chem., 2009, 52, 2587–2602 CrossRef CAS PubMed.
- T. H. Al-Tel, R. A. Al-Qawasmeh and R. Zaarour, Eur. J. Med. Chem., 2011, 46, 1874–1881 CrossRef CAS PubMed.
- C. E. Bulawaa, S. Connelly, M. DeVit, L. Wang, C. Weigel, J. A. Fleminga, J. Packman, E. T. Powerse, R. L. Wisemang, T. R. Fossh, I. A. Wilson, J. W. Kellye and R. Labaudinière, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9629–9634 CrossRef PubMed.
- R. Sant’anna, P. Gallego, L. Z. Robinson, A. Pereira-Henriques, N. Ferreira, F. Pinheiro, S. Esperante, I. Pallares, O. Huertas, M. R. Almeida, N. Reixach, R. Insa, A. Velazquez-campoy, D. Reverter, N. Reig and S. Ventura, Nat. Commun., 2016, 10787, 1–13 Search PubMed.
- M. S. Christodoulou, F. Colombo, D. Passarella, G. Leronimo, V. Zuco, M. D. Cesare and F. Zunino, Bioorg. Med. Chem., 2011, 19, 1649–1657 CrossRef CAS PubMed.
- S. P. Shaik, M. V. P. S. Vishnuvardhan, F. Sultana, S. Rao, C. Bagul, D. Bhattacharjee, J. S. Kapure, N. Jain and A. Kamal, Bioorg. Med. Chem., 2011, 19, 1649–1657 CrossRef PubMed.
- F. Sultana, S. R. Bonam, V. G. Reddy, V. L. Nayak, R. Akunuri, S. R. Routhu, A. Alarifi, M. S. K. Halmuthur and A. Kamal, Bioorg. Chem., 2018, 76, 1–12 CrossRef CAS PubMed.
- S. D. Barchechath, I. Tawatao, M. Corr, A. D. Carson and H. B. Cottom, J. Med. Chem., 2005, 48, 6409–6422 CrossRef CAS PubMed.
- S. Mishra, K. Monir, S. Mitra and A. Hajra, Org. Lett., 2014, 16, 6084–6087 CrossRef CAS PubMed.
- S. G. Balwe and Y. T. Jeong, RSC Adv., 2016, 6, 107225–107232 RSC.
- J. Wang, J. Li and Q. Zhu, Org. Lett., 2015, 17, 5336–5339 CrossRef CAS PubMed.
- S. G. Kumar, S. P. Ragini, A. S. Kumar and H. M. Meshram, RSC Adv., 2015, 5, 51576–51580 RSC.
- I. V. Rassokhina, T. A. Tikhonova, S. G. Kobylskoy, I. Y. Babkin, V. Z. Shirinian, V. Gevorgyan, I. V. Zavarzin and Y. A. Volkova, J. Org. Chem., 2017, 82, 9682–9692 CrossRef CAS PubMed.
- Y. Xie, X. Chen, Z. Wang, H. Huang, B. Yi and G. J. Deng, Green Chem., 2017, 19, 4294–4295 RSC.
- J. Zhang, X. Lu, T. Li, S. Wang and G. Zhong, J. Org. Chem., 2017, 82, 5222–5229 CrossRef CAS PubMed.
- S. Vidyacharan, A. H. Shinde, B. Satpathi and D. S. Sharada, Green Chem., 2014, 16, 1168–1175 RSC.
- K. G. Kishore, A. I. Jacome, A. R. Gomez, A. S. Conejo, U. M. V. Basavanag, K. Wrobel and R. G. Montano, Tetrahedron Lett., 2016, 57, 3556–3560 CrossRef CAS.
- M. A. Claudio-Catalan, S. G. Pharande, A. Quezada-Soto, K. G. Kishore, A. Renteria-Gomez, F. Padilla-Vaca and R. Gamez-Montan, ACS Omega, 2018, 3, 5177–5186 CrossRef CAS PubMed.
- X. Zhang, J. Jia and C. A. Ma, Org. Biomol. Chem., 2012, 10, 7944–7948 RSC.
- S. G. Balwe, T. K. Lim, B. G. Cho and Y. T. Jeong, Tetrahedron, 2017, 73, 3564–3570 CrossRef CAS.
- I. I. Roslan, K. H. Ng and G. Chuah, Beilstein J. Org. Chem., 2017, 13, 2739–2750 CrossRef CAS PubMed.
- W. Hao, X. Sang, J. Jiang and M. Cai, Tetrahedron Lett., 2016, 57, 1511–1514 CrossRef CAS.
- J. K. Malik, M. N. Noolvi, F. V. Manvi, B. K. Nanjwade, H. M. Patel, S. N. Manjula, M. C. Rao and A. Barve, Lett. Drug Des. Discovery, 2011, 8, 717–724 CrossRef CAS.
- S. Dhole, M. Selvaraju, B. Maiti, K. Chanda and C. M. Sun, ACS Comb. Sci., 2015, 17, 310–316 CrossRef CAS PubMed.
- M. Rodriguez, P. Prieto, A. de. la. Hoz, A. Diaz-Ortiz and J. I. Garcia, Org. Biomol. Chem., 2014, 12, 2436–2445 RSC.
- A. T. Garrison, E. S. Childress, D. C. Davis and C. W. Lidsley, J. Org. Chem., 2019, 84(9), 5855–5862 CrossRef CAS PubMed; C. Bordoni, C. M. Cima, E. Azzali, G. Costantino and A. Brancale, RSC Adv., 2019, 9, 20113–20117 RSC.
- A. Kokel and B. Torok, Green Chem., 2017, 19, 2515–2519 RSC; B. Maiti, K. Chanda, M. Selvaraju, C. C. Tseng and C. M. Sun, ACS Comb. Sci., 2013, 15, 291–297 CrossRef CAS PubMed.
- J. Chen, L. Mei, J. Liu, C. Zhong, B. Yuan and Q. Li, RSC Adv., 2019, 9, 28576–28580 RSC; R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110(10), 6302–6337 CrossRef CAS PubMed.
- B. Maiti, K. Chanda and C. M. Sun, Org. Lett., 2009, 11, 4826–4829 CrossRef CAS PubMed.
- R. D. Padmaja, V. C. Devi, N. Mukku, K. Chanda and B. Maiti, ACS Omega, 2018, 3, 4583–4590 CrossRef CAS PubMed.
- R. N. Rao, M. M. Balamurali, B. Maiti, R. Thakuria and K. Chanda, ACS Comb. Sci., 2018, 20, 164–171 CrossRef CAS PubMed.
Footnotes |
| † The article is dedicated to Prof. Chung Ming Sun for his contributions to diversity-oriented synthesis. |
| ‡ Electronic supplementary information (ESI) available: 1H and 13CNMR, GCMS, IR, HRMS spectra of compounds 3a–3l, and 5a–5d ESI page no 2–33. See DOI: 10.1039/c9ra08929b |
| This journal is © The Royal Society of Chemistry 2020 |