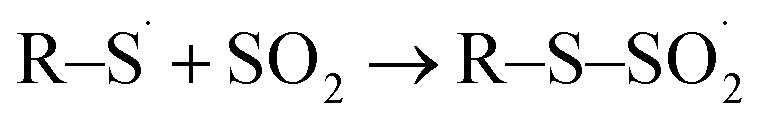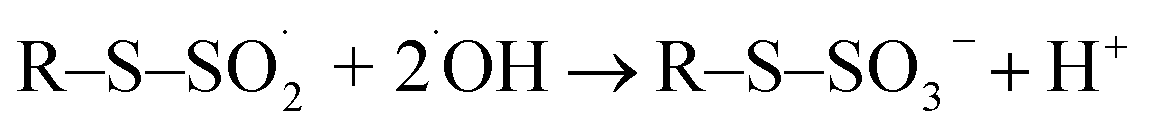DOI:
10.1039/C9RA08745A
(Paper)
RSC Adv., 2020,
10, 11598-11607
On a heavy path – determining cold plasma-derived short-lived species chemistry using isotopic labelling†
Received
24th October 2019
, Accepted 25th February 2020
First published on 17th March 2020
Abstract
Cold atmospheric plasmas (CAPs) are promising medical tools and are currently applied in dermatology and epithelial cancers. While understanding of the biomedical effects is already substantial, knowledge on the contribution of individual ROS and RNS and the mode of activation of biochemical pathways is insufficient. Especially the formation and transport of short-lived reactive species in liquids remain elusive, a situation shared with other approaches involving redox processes such as photodynamic therapy. Here, the contribution of plasma-generated reactive oxygen species (ROS) in plasma liquid chemistry was determined by labeling these via admixing heavy oxygen 18O2 to the feed gas or by using heavy water H218O as a solvent for the bait molecule. The inclusion of heavy or light oxygen atoms by the labeled ROS into the different cysteine products was determined by mass spectrometry. While products like cysteine sulfonic acid incorporated nearly exclusively gas phase-derived oxygen species (atomic oxygen and/or singlet oxygen), a significant contribution of liquid phase-derived species (OH radicals) was observed for cysteine-S-sulfonate. The role, origin, and reaction mechanisms of short-lived species, namely hydroxyl radicals, singlet oxygen, and atomic oxygen, are discussed. Interactions of these species both with the target cysteine molecule as well as the interphase and the liquid bulk are taken into consideration to shed light onto several reaction pathways resulting in observed isotopic oxygen incorporation. These studies give valuable insight into underlying plasma–liquid interaction processes and are a first step to understand these interaction processes between the gas and liquid phase on a molecular level.
Introduction
Cold atmospheric pressure plasmas (CAPs) have recently transitioned from laboratories to clinics, offering a safe and effective application directly to the patient's body.1–3 Major applications of medical plasmas are wounds, skin-derived diseases and, as off-label use, palliation in cancer patients.4–12 CAPs offer a well-documented efficacy in inactivating bacteria13 that may contribute to the stimulation of wound healing processes.14,15 Plasma treatment influences cell or tissue physiology at various levels, including metabolism, signaling, and cell fate, leading to immunomodulation, angiogenesis, tissue proliferation, or migration.16–19 High intensity treatment results in the shutdown of cellular processes and cell death by apoptosis or necrosis-like processes.20,21 These effects, especially in combination with immune cell modulation, are currently investigated for cancer treatment.22–25
Medical plasmas are multicomponent systems containing electrons, ions, electric fields, and a multiplicity of reactive oxygen and nitrogen species (ROS/RNS). Depending on plasma source, feed gas composition, and distance to the target, ROS/RNS generation varies.26–30 A major role of these plasma-derived ROS/RNS is assumed,31,32 but incongruity regarding the mode of action exists. It was argued that cell membrane-associated proteins pick up and translate the induced signals33 or that species can cross the cell membrane using specific pore proteins34 and trigger intracellular responses via cytosolic sensor proteins35 or the mitochondria.36 With regard to the lifetime of most reactive species described for CAPs, it must be acquiesced that only a small fraction can indeed diffuse into a cell or the cell's vicinity, leaving the question of the ultimate mechanism still open.37,38 With the controllability of plasma treatments in biomedical applications becoming increasingly relevant to improve safety and efficacy,39 knowledge of the relevant players in plasma–target interaction, their respective trajectories and – most importantly – their (bio) chemistry is mandatory. It can be concluded that another route of interplay between the plasma-derived species and the biological system is the covalent modification of biomolecules and the subsequent change of their activity or biological value. It has been shown that plasma-derived ROS/RNS are capable of oxidizing amino acids,40 proteins,41–45 or lipids.46,47 Thiol groups are one of the major targets for plasma-generated species.48–50 Hence, CAP impact can yield to (non-enzymatic) post-translational modifications (PTMs), with some of which transport significant influence in cellular signaling.51–54 However, so far no specific member of the various ROS or RNS could be attributed to be the major driver of these reactions. One of the reasons is the limited knowledge that is present on plasma-derived liquid chemistry. Hydrogen peroxide is a frequently reported product of the plasma liquid interaction, yet its reactivity is too low to oxidize biomolecules significantly (EO = 1.35 V, H2O2 + 2H+ + 2e− ⇌ 2H2O).55,56 The presence of other species, such as hydroxyl radicals, superoxide anion radicals, or nitric oxide can be shown in liquids by electron paramagnetic resonance,57 assumed from gas phase distributions,27,58,59 or from the detection of modified organic or inorganic targets by the plasma treatment.51,60 Given their high reactivity, the contribution of short lived ROS and RNS to the modification of biomolecules is assumingly significant.
It is a major challenge in plasma chemistry that has so far not been fully met: to determine the short lived species reactivity and to distinguish between species stemming from primary reactions in the gas phase and species created in secondary or tertiary reactions at or in the target – a liquid, a gel, or a tissue.26 Gorbanev et al.,61 and Benedikt et al.62 presented first indications by showing the activity of gas phase-derived species in aqueous model systems.
In this work, the stable oxygen isotope 18O was used in the gas (18O2) and liquid phase (H218O) to shed light onto the behavior and reactivity of reactive oxygen species (ROS) following CAP treatment. Predominantly, the argon-driven atmospheric-pressure plasma jet kINPen with shielding gas63 was utilized to treat cysteine as a chemical probe while selected experiments included the use of the helium-driven COST microplasma jet as a reference source.64 The chemical impact on cysteine was assessed using high-resolution mass spectrometry with a special focus on isotope distribution patterns. These results allow insight into the trajectories of plasma-generated ROS hitting a liquid surface and their reaction with organic tracer molecules, indicating that both gas phase-derived species and liquid phase-derived species have a biochemical potential.
Experimental
Short-lived species generation (plasma sources)
The argon-driven atmospheric-pressure plasma jet kINPen 09 (neoplas)63 was used together with a curtain gas device65 to provide defined atmospheric conditions for the experiments. The kinpen was powered by 1.1 W at a frequency of 1 MHz. Gas flux was kept constant for all conditions at 3 standard liter per minutes (slm) of pure, dry argon (5.0, Air Liquide) with the curtain gas set to 5 slm of nitrogen (5.0, Air Liquide). Besides pure argon, 1% oxygen (purity 4.8, Air Liquide, Ar/O2) was used for the experiments as these conditions offered promising oxidative thiol modification potential.53 The COST-jet64 was powered by constant 300 mW at a frequency of 13.56 MHz. Total gas flux was kept constant at 1 slm of pure dry helium (5.0, Air Liquide) with 1% oxygen admixture (purity .8, Air Liquide, He/O2). For the experiments with the kINPen, either light oxygen or heavy isotope oxygen (purity 99%, 18O2, Sigma-Aldrich) was used. Due to costs, only light oxygen was used for the control experiments with the COST-jet. All connections were flushed with nitrogen prior to switching from one oxygen variant to the other.
Sample preparation and treatment
Cysteine (L-cysteine, Sigma-Aldrich) was dissolved in double-distilled water (MilliQ) or water with isotopically labelled oxygen (H218O, 97% purity, Eurisotop) to a final concentration of 300 μM. Simultaneous treatments with 18O labeled water and 18O labeled gas were not performed. Treatments of the cysteine solutions were performed with the different gas compositions in 24-well plates using 750 μl of solution per sample and a distance between jet nozzle and liquid surface of 9 mm. All treatments were performed for 60 s and resulting samples were stored on ice and directly measured.
High-resolution mass spectrometry
Analysis. Mass spectrometry was carried out on a TripleTOF 5600 system (Sciex). Samples were diluted 1: 1 with alkaline buffer (0.3% ammonium hydroxide in methanol) and directly infused using an electronically controlled syringe pump. Each sample was acquired for 1 min using identical system settings for all samples (capillary temperature 150 °C, curtain gas: 35 psi N2, ion source gas 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 psi N2, ion source gas 2: 25 psi N2, ion spray voltage: −4 kV). To identify the structures of all observed masses, each peak of interest was isolated, fragmented, and resulting fragment masses acquired (MS/MS, collision energy −24 eV, declustering potential −10 kV) and annotated. To allow a relative quantification of observed signals, an internal standard (IS) was mixed into the sample directly in front of the mass spectrometer emitter using a mixing tee connector. Here, the amino acid valine (L-valine, Sigma-Aldrich) was used due to its mass difference to other expected signals and little interference with the rest of the spectrum. All measurements were performed in triplicates.
:20 psi N2, ion source gas 2: 25 psi N2, ion spray voltage: −4 kV). To identify the structures of all observed masses, each peak of interest was isolated, fragmented, and resulting fragment masses acquired (MS/MS, collision energy −24 eV, declustering potential −10 kV) and annotated. To allow a relative quantification of observed signals, an internal standard (IS) was mixed into the sample directly in front of the mass spectrometer emitter using a mixing tee connector. Here, the amino acid valine (L-valine, Sigma-Aldrich) was used due to its mass difference to other expected signals and little interference with the rest of the spectrum. All measurements were performed in triplicates.
Data analysis and branching calculation. After acquisition, samples were processed using the Analyst software (Analyst TF 1.7, Sciex). First, background noise was determined and 300 counts subtracted from the full spectrum (15 times background) to increase signal-to-noise quality. Afterwards, molecule structures were identified using the acquired MS/MS data and the “Formula Finder” as well as “Mass Calculator” functions of the PeakView software (PeakView 1.2.0.3, Sciex). The areas of all isotope peaks were calculated and normalized on the internal standard area to allow quantitative comparison between measuring runs or each identified structure, the theoretical isotope pattern was calculated. Intensities for all observed peaks were adjusted to remove naturally occurring 13C isotope intensities to prevent interference with isotope signals stemming from integrated 18O. Further isotope traces identified for each compound in treatments with pure argon in unlabeled water were considered as controls for impurities. Therefore, the values of isotopic masses identified in treatments with argon-only were subtracted from each corresponding isotopic mass identified in experiments with labeled oxygen. The error estimation was done by considering biological triplicates for each condition and technical duplicates for each sample, for six measurements total. Other several potential systematic errors were considered in the presented analyses. First, both isotopic labeled gas and water were not of 100% purity. Therefore, an additional error of 1% had been taken into account for all quantifications using 18O2 as well as 3% using H218O. Furthermore, evaporation had to be considered when working with isotopically labeled water. After treatment of 60 s, 20 μl of the 750 μl were evaporated. Evaporated heavy water might be dissociated in the discharge, thereby becoming a primary species while erroneous considered as a tertiary species. Therefore, an additional systematic error of 4% has to be considered. In total, expected systematic error were 1% for treatments with 18O2 and 7% for treatments with H218O. However, errors regarding 18O2 are underestimated due to imperfection of the shield gas device (see Results and discussion). The corresponding values were incorporated into all acquired standard deviations. To allow a quick overview about differences in data sets, principal component analysis (PCA) was performed with the normalized spectra using Perseus.66
Results and discussion
Cysteine as model compound
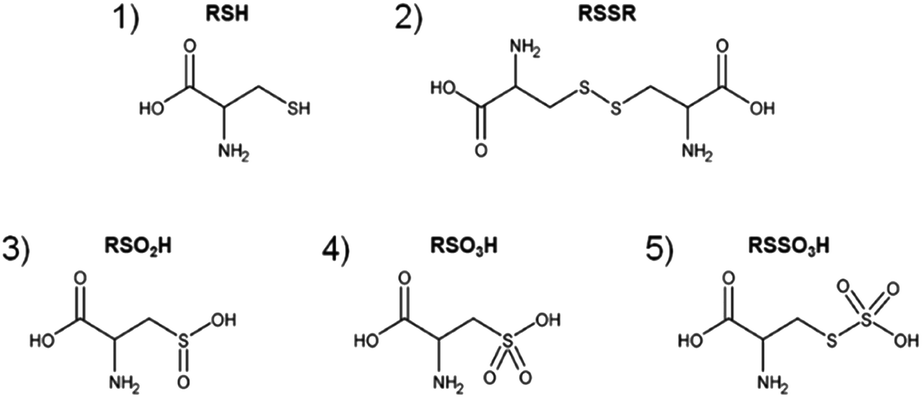
Cysteine and derivatives have been suggested as model systems to estimate the liquid phase chemistry of plasma sources, to compare the impact of discharge parameter variations such as working gas composition, and to facilitate the standardization of treatment procedure of plasma discharges for biomedical applications.48,50 The products resulting from the reaction between the CAP-derived species and cysteine were analyzed by mass spectrometry (Fig. 1). As reported previously, covalent changes to the cysteine (structure 1) by the plasma-derived species were observed, with cystine (RSSR, structure 2), cysteine sulfinic acid (RSO2H, structure 3), cysteine sulfonic acid (RSO3H, structure 4), and cysteine-S-sulfonate (RSSO3H, structure 5) and the sulfite (SO32−) and sulfate (SO42−) ions as dominant products. These compounds were chosen for their relevance in the transformation pathway of cysteine under certain redox conditions. The presence of RSO3H or SO42− indicates a strongly oxidizing environment (oxygen in the feed gas, long treatments), whereas the presence of RSSR, RSO2H, or RSSO3H reveals weakly oxidizing conditions (short treatments, nitrogen shielded feed gas).53 Here, this model was used to investigate transport processes at the interface between the gas phase (the effluent) and the target (cysteine solutions). To monitor such reactions, the chosen cysteine model with its multiple oxidation states of the thiol moiety seemed to be superior to the phenol model.
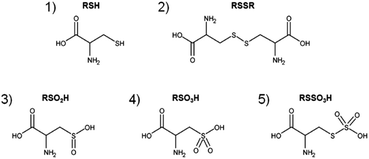 |
| | Fig. 1 Major products after direct CAP treatment of cysteine solutions. Cysteine (1, m = 121.0197u, RSH), cystine (2, m = 240.0238u, RSSR), cysteine sulfinic acid (3, m = 153.0096u, RSO2H) and cysteine sulfonic acid (4, m = 169.0045u, RSO3H) as well as cysteine-S-sulfonate (5, m = 200.9766u, RSSO3H). | |
Gas phase and liquid phase-derived species contribute to plasma liquid chemistry
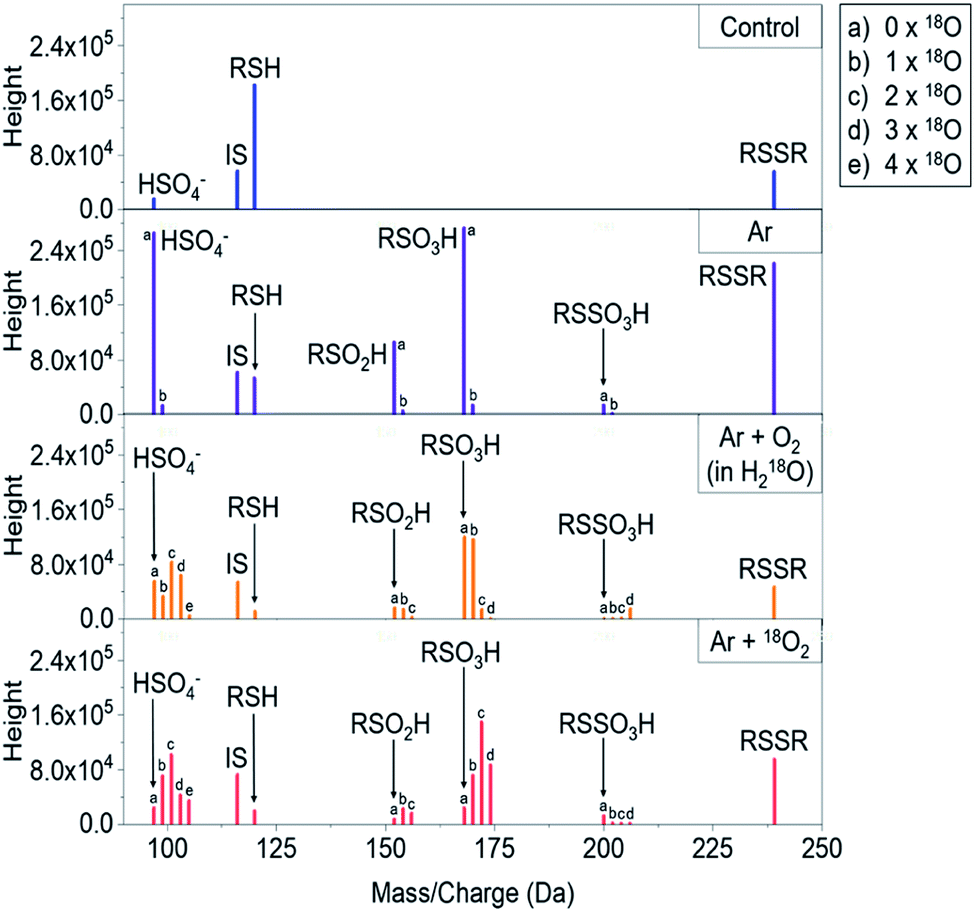
To trace the reactive species, the 18O oxygen isotope (u = 17.9992), either located in the feed gas (18O2) or in the solvent (H218O), was used during different plasma treatments. In comparison, the normal oxygen isotope 16O has a mass of u = 15.9949, generating a mass shift of 2.0043 m/z for each included 18O atom that could easily be followed by mass spectrometry (examples in Fig. 2). Hence, a differentiation between gas phase and liquid phase-derived reactive species could be made. Indeed, the transfer of gas phase 18O species into the liquid was observed for a number of products (e.g. RSO2H and RSO3H). These observations were in agreement with data published by Benedikt et al. for a phenol model investigating a micro atmospheric plasma jet (APPJ).62 Additionally, a strong role of the plasma treated target (cysteine solution in water) as an additional source of reactive species was identified. The compound cysteine-S-sulfonate (RSSO3H) almost exclusively contained liquid phase-derived oxygen. Most other products did not show such a clear-cut oxygen incorporation, indicating a mixed attack of gas and liquid phase derived species.
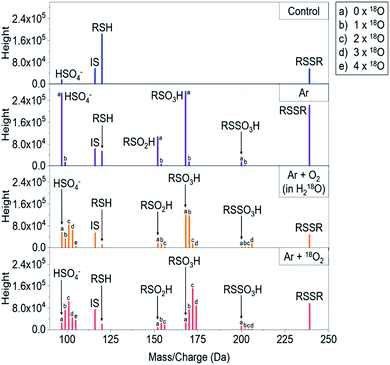 |
| | Fig. 2 Raw mass spectra showing the inclusion of 16O or 18O oxygen atoms under different labeling strategies into cysteine after kINPen treatment. RSH = cysteine, RSSR = cystine, RSO2H = cysteine sulfinic acid, RSO3H = cysteine sulfonic acid, RSSO3H = cysteine-S-sulfonate, IS = internal standard (valine), SO42− = sulfate ion. “b”-labeled signals in Ar-only samples are stemming from 13C isotopes and their normalized intensities were subtracted from corresponding signals in oxygen-containing conditions to handle isotope shadowing effects, see Experimental section for details. | |
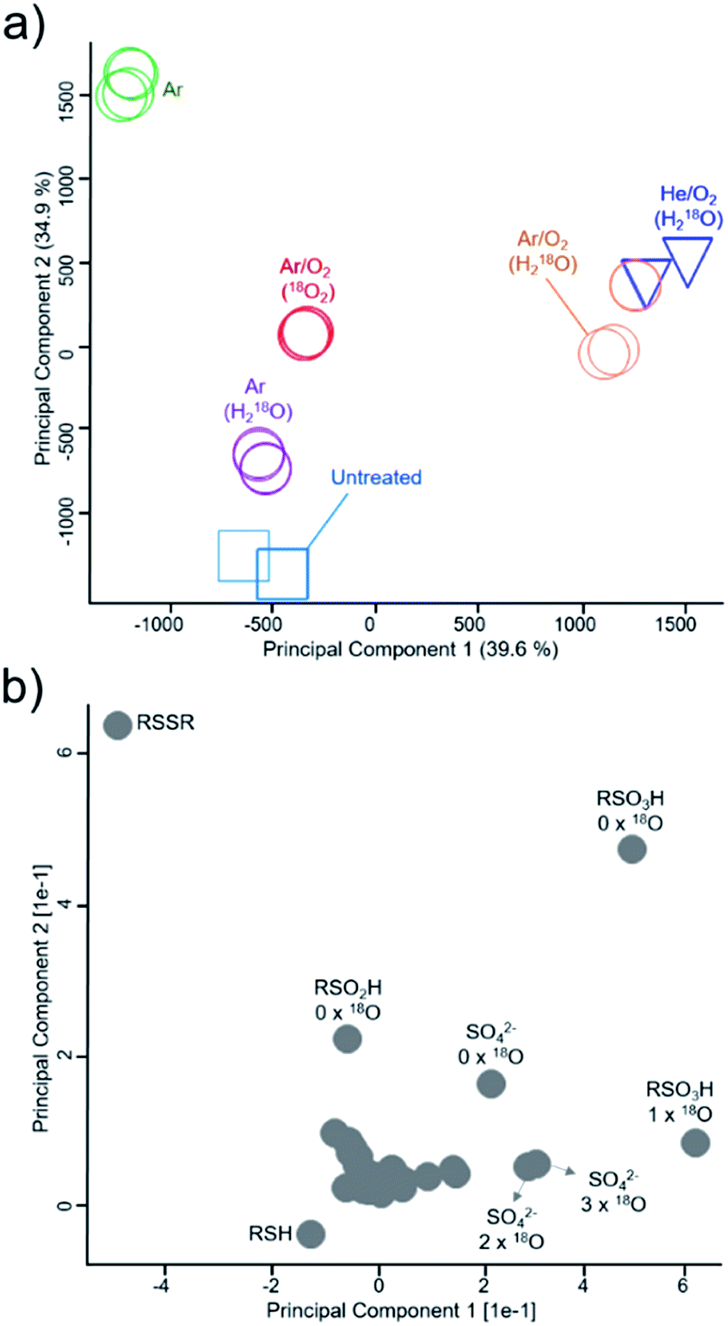
Using principal component analysis (Fig. 3), general differences in product formation and oxygen incorporation due to the various treatment conditions were easily observable in the two principal components explaining the largest differences between samples (39.6% and 34.9%, respectively). Ar/O2 (kINPen) and He/O2 (COST-jet) treatment were found in close proximity to each other, indicating similar products and isotope distributions after treatment, which was in good agreement with previous works. The loadings of the principal components (Fig. 3b) indicated a significant impact due to the presence or absence of incorporated 18O.
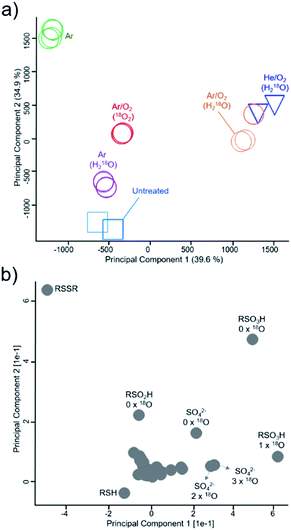 |
| | Fig. 3 Mapping of inclusion of gas phase and liquid phase-derived species in major cysteine derivatives. PCA analysis (a) and corresponding loadings ((b), loadings in relation to isotope incorporations are marked) indicate general differences in species distribution. Samples were treated with the kINPen (circles, colors indicate treatment conditions) or as control experiments with the COST-jet (triangles, He/O2 mixture in H218O) or left untreated (squares). 18O2 enriched feed gas or H218O as solvent was used to determine species origin. RSO2H = cysteine sulfinic acid, RSO3H = cysteine sulfonic acid, RSSO3H = cysteine-S-sulfonate SO42− = sulfate. All treatments (represented by different symbols/colors) were performed in triplicates and plotted individually to check for reproducibility. | |
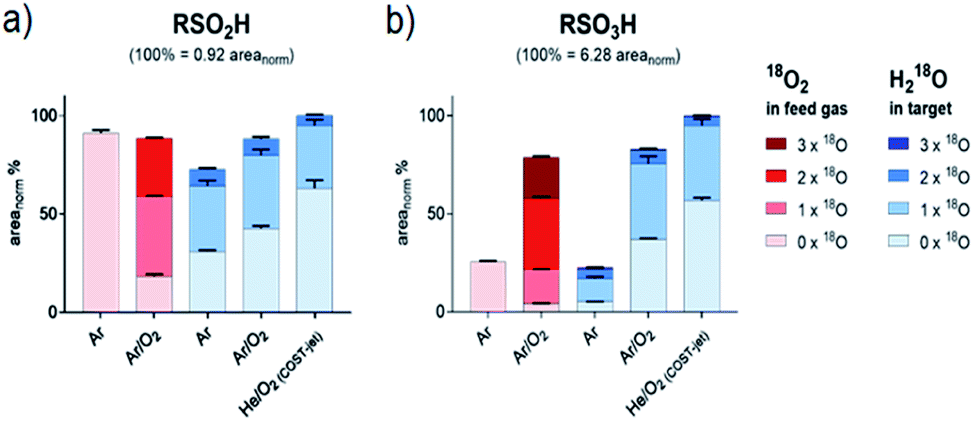
The products cysteine sulfinic and sulfonic acid. Both molecules are created by CAP treatment due to the stepwise oxidation of the thiol moiety,40,50 and were observed for all direct plasma treatment conditions. Without molecular oxygen, admixture (kINPen Ar-only) small yields of sulfonic acid indicate the strong role of gas phase-derived oxygen species for its formation (Fig. 4). Additionally, a limited incorporation of liquid phase species was observed that might stem from the initiation of the oxidative chain:| | |
H218O(aq) + hν/e−/Ar* → ˙18OH + ˙H
| (1) |
| | |
H218O(aq) + 16O(3P)(aq, from gas phase) → ˙18OH(aq) + ˙16OH(aq),
| (2) |
(18O water used as solvent, 16O molecular oxygen in the gas phase).
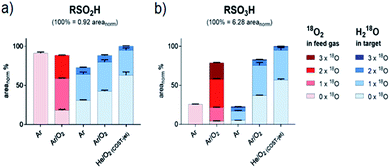 |
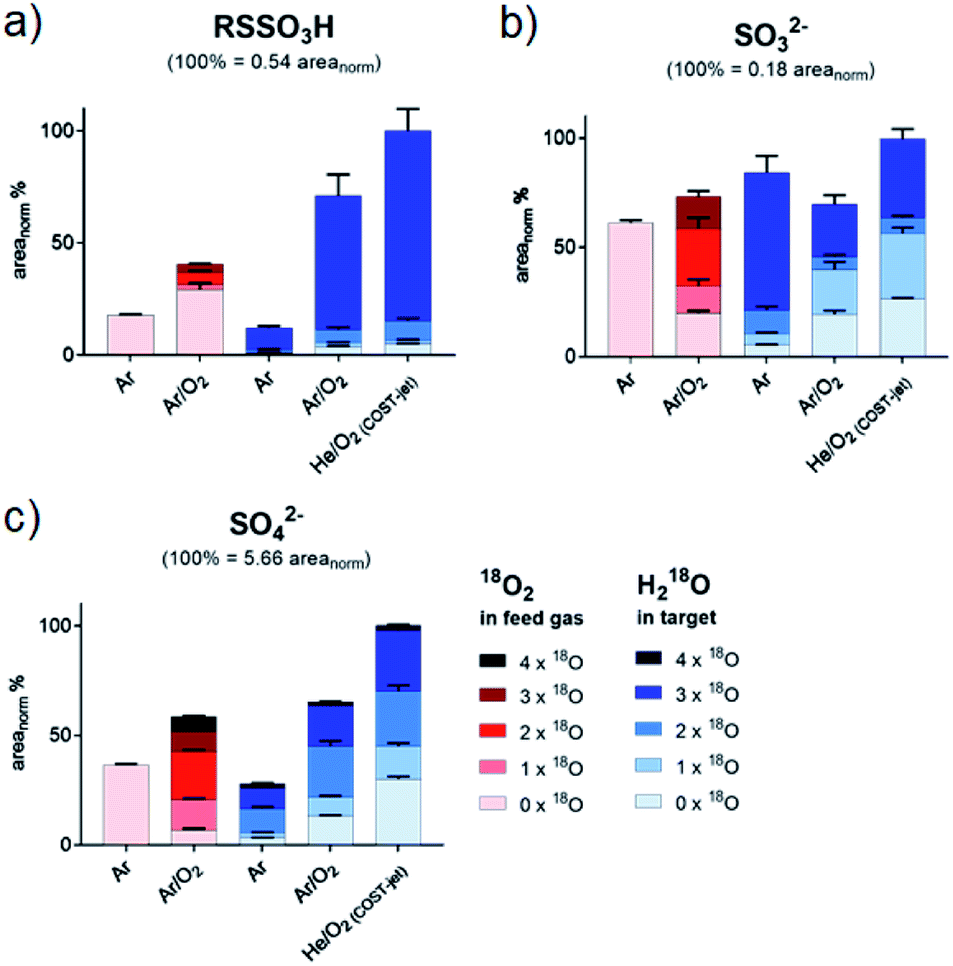
| | Fig. 4 Isotopic distribution of oxygen in sulfinic (a) and sulfonic (b) acid after CAP treatment areas for each signal were normalized on total intensities of each species (including all isotope variations). Isotope distributions were observed with labeled oxygen present in the feed gas (red) or in the target liquid (blue). Cysteine was treated by the kINPen using Ar-only (Ar) plasma or Ar with 1% O2 admixture (Ar/O2). Alternatively, treatment was performed using the COST-jet running with a He plasma with 1% O2 admixture (He/O2). | |
Here, water is cleaved either by the impact of electrons, UV photons, or energy-rich (metastable) noble gas species (reaction (1)). Atomic oxygen (O(3P)) has been suggested as another potential reactant for water cleavage (reaction (2)),62,67 however in conditions with the highest levels of O(3P) present (COST jet He/O2),68 the lowest overall inclusion of aqueous 18O species into RSO2H and RSO3H was found (Fig. 4 and Table 1). With that, reaction (2) is not a major pathway in the existing conditions and O(3P) predominantly reacts directly with the present organic molecules without a detour via liquid phase derived OH radicals. A similar formation rate of cysteine sulfinic acid was observed for the kINPen Ar only in comparison to Ar/O2, while RSO3H yields were only 25% of that of Ar/O2. Either this suggest a low conversion rate of RSO2H into RSO3H in Ar-only treatment, or that the formation of RSO3H follows a different reaction path than RSO2H. The incorporation of 18O(water) was increased in the argon-only case, indicating that a proportional larger number of 18OH radicals from the solvent were created when limited amounts of gas phase ROS (O(3P), O2(1Δg)) were available. Hence, the lysis of water is achieved according to reaction (1) by electrons (in the case of the kINPen), Ar or He higher energy states (discussed in ref. 26), and (V)UV photons which are highest if no molecular gas admixture is made.69
Table 1 Distribution of oxygen isotopes 16O and 18O in major products after plasma treatment of cysteine in labeled double-distilled water (H218O). Atom%, mean of 3 experiments
| H218O label |
RSO2H (cysteine sulfinic acid) |
RSO3H (cysteine sulfonic acid) |
RSSO3H (cysteine-S-sulfonate) |
| Ar |
Ar/O2 |
COST He/O2 |
Ar |
Ar/O2 |
COST He/O2 |
Ar |
Ar/O2 |
COST He/O2 |
| 16O/% |
65.6 |
69.4 |
79.0 |
65.0 |
78.5 |
83.8 |
11.9 |
9.7 |
8.9 |
| 18O/% |
34.4 |
30.6 |
21.0 |
34.9 |
21.7 |
16.2 |
87.9 |
90.3 |
91.1 |
| H218O label |
SO32− (sulfite)a |
SO42− (sulfate)a |
| Ar |
Ar/O2 |
COST He/O2 |
Ar |
Ar/O2 |
COST He/O2 |
| Detected as singly charged bisulfite/bisulfate ion. |
| 16O/% |
15.0 |
50.2 |
49.2 |
45.9 |
54.9 |
60.7 |
| 18O/% |
85.0 |
49.8 |
50.8 |
54.1 |
45.1 |
39.3 |
Taken together, a mixture of gas phase-derived and liquid phase secondary species attacks cysteine. Concordantly, reactive molecular dynamics simulations indicate that a proton abstraction from the thiol moiety by one hydroxyl radical followed by addition of another hydroxyl radical yielding cysteine sulfenic acid paved the way for all further modifications.49 A liquid phase-localized hydroxyl radical seems to perform this initial attack predominantly:
| | |
R–SH + ˙OH → R–S˙ + H2O
| (3) |
(R = C
3H
6NO
2).
Further reaction to sulfonic acid seem to be dominated by gas phase-derived oxygen species. For the kINPen, argon-only treatment of cysteine in H218O, the 16O/18O ratio of 2:1 indicated that precisely one oxygen atom out of the three included in the RSO3H stems from the liquid as suggested by the reactions (3) and (4). In contrast, the ratios for kINPen Ar/O2 and COST He/O2 (16O/18O 4:1) showed that on average less than one atom derived from the water lysis, fostering the relevance of gas phase-derived species for the production of the products and provide evidence that O(3P) plays an important role in introducing observed modifications. O(3P) interacts with a rate constant of about k = 1 × 1012–13 cm3 mol−1 s−1 with free thiols,70 indicating high modification efficacies. It is capable of oxidizing a thiol moiety directly to sulfenic acid.71 Especially the final oxidation step from sulfinic to sulfonic acid requires a gas phase-derived oxygen species, with O(3P) and O2(1Δg) as potential candidates as shown from gas phase measurements and 0D/2D model simulation (reactions (5)–(9)).27,72–75
| | |
R–S˙ + O(3P) → R–S–O˙
| (5) |
| | |
R–S˙ + O2(1Δg) → R–SO–O˙
| (6a) |
| | |
R–SH + O2(1Δg) → R–SO–O− + H+
| (6b) |
| | |
R–S–OO˙/ R–S–O˙ + H2O → R–S–OH + ˙OH
| (7) |
| | |
R–S–OH + [O(3P), O2(1Δg), OH] → R–SO–OH, R–SO2–OH
| (8) |
| | |
R–SO–OH + [O, O2(1Δg)] → R–SO2–O− + H+
| (9) |
(R = C
3H
6NO
2).
Treatment with the COST-jet, known for high fluxes of O(3P),68 resulted in the largest proportion of gas phase-derived oxygen in RSO2H/RSO3H (16Ogas/18Oliquid 5:1 for RSO3H). This further emphasized the transport of gas phase O(3P) into the interphase region, and presumably, the upper layers of the bulk liquid and its direct chemical reactivity with thiols and oxidized thiol moieties. Omlid et al. showed that O(3P) can migrate to a limited extent in an aqueous system.76 Besides O(3P), O2(1Δg) is the other species of interest, since it is capable of producing many of the observed products on its own (reactions (6)–(9)).77,78 While it reacts with a rate constant of k = 8.3 × 106 M−1 s−1 with free thiols, it can penetrate significantly further into the liquid bulk due to its much longer half-life79 as compared to O(3P), thereby offsetting its lower reaction rate. In the light of its biological impact in cell models and therapy, both O(3P)51,80 and O2(1Δg)81 must receive a significant attention when interpreting CAP affect in biomedical research or (re)design plasma sources for the application.
The products S-sulfonate and sulfate. RSSO3H and SO42− were produced in significant amounts, with the COST-jet being more effective than the kINPen (Fig. 5). Different pathways may generate RSSO3H: the photolytic or radical driven cleavage of a C–S bond of the intermittently formed cystine (RSSR) and subsequent oxidation of the outer sulfur moiety, the oxidative cleavage of oxidized cystine derivatives, or RSSO3H is formed from the attack of a ˙SH-derived species, e.g. sulfite (SO32−) on cysteine, cystine, or the thiyl radical R–S˙.82–86 While a single reaction pathway cannot be determined with the current data, and several reactions ultimately yield the same product, the following proposed reactions will contribute to the formation of RSSO3H. Further oxidation leads to the destruction of the S–S bond of RSSO3H, yielding SO42− again:87–89| | |
R–SH → R˙ + ˙SH or R–S˙ + ˙H and R–S–S–R → R–S–S˙ + ˙R
| (10) |
| | |
˙SH + O2 → SO2˙− + H+
| (11) |
| | |
SO2˙− + O2 → SO2 + O2˙−
| (12) |
| | |
SO2 + H2O → HSO3− + H+
| (13) |
| | |
R–S–S–R + HSO3− → R–S–SO3− + R–SH
| (14a) |
| | |
R–SH + HSO3− → R–S–SO3− + H2
| (14b) |
| | |
2HSO3− + O2 → 2SO42− + 2H+
| (15) |
| |
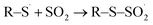 | (16) |
| |
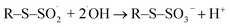 | (17) |
| | |
R–S–SO3− + 2˙OH → R–S–OH + SO42− + H+
| (18) |
(R = C3H6NO2).
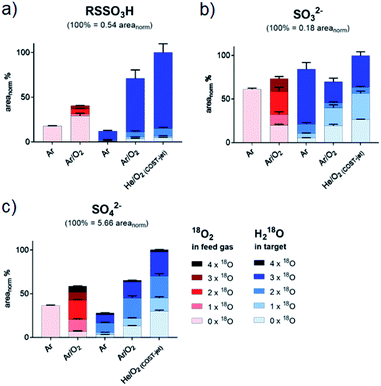 |
| | Fig. 5 Isotopic distribution of oxygen in cysteine S-sulfonate (a), sulfate (b) and sulfide (c) after CAP treatment. Areas for each signal were normalized on total intensities of each species (including all isotope variations). Isotope distributions were observed with labeled oxygen present in the feed gas (red) or in the target liquid (blue). Cysteine was treated by the kINPen using Ar-only (Ar) plasma or Ar with 1% O2 admixture (Ar/O2). Alternatively, treatment was performed using the COST-jet running with a He plasma with 1% O2 admixture (He/O2). Sulfite (b) and sulfate (c) were detected as bisulfite/bisulfate. | |
While the cleavage of disulfides by sulfite ions is well-described,89,90 its contribution must be debated. In contrast to RSO2H and RSO3H, between 80% and 90% of oxygen added to RSSO3H stemmed from liquid phase species (Fig. 5 and Table 1). The gas phase-derived ROS are not directly involved in its formation, precluding the formation of sulfite ions from SH radicals via reactions (11) and (12), or from the cleavage of a C–S bond in cysteine sulfinic or sulfonic acid (R–SO–O−/R–SO2–O−, reactions (5)–(9)). However, sulfite ions found after the plasma treatment contain a 50% mix of gas and liquid derived oxygen, indicating that also reactions precluding gas phase-derived oxygen lead to its formation. While no information is available on the reaction mechanisms of hydroxyl radicals with sulfhydryl radicals in liquids, their impact is the key to the observed isotope pattern of RSSO3H. Following reaction (1), the photolysis of H218O yields 18OH. However, RSSO3H yields are highest when oxygen is present in the gas phase (Ar/O2, He/O2 conditions). Due to the absorption of molecular O2 below 180 nm this leads to an attenuation of the VUV, and such photolysis.91 Additionally, the COST jet emits negligible VUV radiation,64 yet yielded the highest levels of RSSO3H. In contrast, when water photolysis is strongest (kINPen Ar plasma) by far the lowest amounts of RSSO3H were detected (Fig. 5a).86 This suggests, that (a) the local production of OH radicals from photolysis in the interface zone does not favour the production of RSSO3H or (b), that RSSO3H generated in the interfacial zone is immediately decayed again. With that, it must be assumed that RSSO3H is produced in the bulk by the action of OH radicals generated from the liquid. The question is; how do they get there – given their short live time they cannot penetrate that far. Interestingly, it is the O(3P) atom that may act as an intermediate carrier of chemical energy: according to recent experiments, the atom can penetrate a measurable distance in aqueous solutions,76 and following reaction (2) can lead to the formation of OH radicals distant from the interface region.67 Such, the number of accessible precursors increases while the local OH radical density is comparably low, reducing the decay of RSSO3H once it formed via reaction (18) and other short lived gas phase species. However, the cleavage of water according to (2) yields a 1:1 mixture of 16OH and 18OH. That is clearly not reflected in the product (>90% 18O). Beside a potential isotope effect, the “migration” of O–H single bonds between a 16OH radical and a neighbouring H218O water molecule results in a switch from a 16OH to an 18OH.92 Given the magnitudes higher density of water compared with the plasma generated 16O(3P) atoms, most 16OH radicals are lost resulting in the observed dominance of 18OH. This is an interesting result indicating that the impact of highly reactive species is not confined to the interface layer but can extent into the liquid bulk, and corroborates computational work done by Yusupov et al.93
Compared to the transient RSSO3H, the stable sulfate ion SO42− is the final product of thiol moiety oxidation:
| | |
R–SO3− + 2˙OH → R–OH + SO42− + H+
| (19) |
| | |
R–SO3− + O(3P) → R–O–SO3−
| (20) |
| | |
R–O–SO3− + H2O → R–OH + SO42− + H+
| (21) |
| | |
R–SO3− + O2(1Δg) → [R–O–O–SO3−] → R–OH + SO42− + H+
| (22) |
(R = C
3H
6NO
2).
It can be formed from numerous precursors by breaking the C–S bond both before and after oxidation events (reactions (19)–(22)). Beside the chemical breakage, VUV radiation may contribute as the binding energy of C–S bonds (272 kJ mol−1, 2.8 eV) lies well within the range of the emitted photons.94 In addition, a chemical cleavage via three or four-atom transition states is possible, potentially with the contribution of radical species.95 It was observed that all cysteine products decay with further treatment, with RSO3H as a final product.49 Ultimately, its accumulation also stops, indicating that consumption processes leading to the formation of SO42− counteract further build-up. The sulfate ion SO42− showed a very heterogeneous origin of its oxygen atoms, confirming the end product character (Table 1). In contrast to the kINPen, the SO42− ion's oxygen isotopes for COST jet treatment showed a significant larger proportion of gas phase-derived species that can be attributed to the higher density of O(3P) generated. Besides SO42−, small amounts of SO32− were observed. Interestingly, the 16O/18O composition indicated a higher contribution of the liquid phase species than seen for SO42−, implying bulk generated ˙OH radical oxidation of ˙SH as major chemical route to its formation. The ion plays a contributing role in the formation of RSSO3H (see Tables 1 and 2).
Table 2 Distribution of oxygen isotopes 16O and 18O in major products after gas-phase labeled plasma treatment of cysteine in normal double-distilled water (H216O). Working gas Ar/0.5% 18O2, atom%, mean of 3 experiments
| Gaseous 18O2 label |
Ar/18O2 (kINPen) |
| SO32−a (sulfite) |
SO42−a (sulfate) |
RSO2H (cysteine sulfinic acid) |
RSO3H (cysteine sulfonic acid) |
RSSO3H (cysteine-S-sulfonate) |
| Detected as singly charged bisulfite/bisulfate ion. |
| 16O/% |
50.4 |
54.9 |
43.6 |
35.6 |
80.4 |
| 18O/% |
49.6 |
45.1 |
56.4 |
64.4 |
19.6 |
Origin of oxygen in oxidized cysteine derivatives: gas phase versus liquid phase species. Using the measured abundances of each product and its respective isotope pattern, the distribution of 18O and 16O in all major cysteine oxidation products were determined. Adding the areas of all variants (e.g. RS16O3H, RS16O218OH, RS16O18O2H and RS18O3H) the fraction of each single variant was then used to calculate the atomic fraction the two oxygen isotopes (see Tables 1 and 2). The overall pattern indicated that there is no 100% contribution of either gas-phase-derived species or liquid-phase derived species. However, there a clear indications for diametric origins of the incorporated oxygen atoms in some products. The extremes were RSO3H, that incorporated a majority of gas phase-derived oxygen (up to 83.8%), and RSSO3H, that predominantly included liquid phase-derived oxygen (maximum 91.1%). The other products showed a more equally shared origin of the oxygen atoms from gas-phase and liquid-phase, especially the sulfite SO32− ion with almost 50:50 distribution in all experiments. The assumed oxidation end product SO42− also shows a commensurate isotope distribution, but with a significant tilt towards gas-phase derived oxygen.
kINPen versus COST-jet – atomic vs. singlet oxygen?. kINPen (Ar/O2) and COST-jet (He/O2) yielded similar amounts of the major products that also share a similar oxygen isotope distribution. However, the COST-jet featured slightly higher oxygen incorporation from the gas phase compared to that of the kINPen. In addition to ubiquitous hydroxyl radicals produced by both sources under all conditions,30,57 other components of a discharge might affect radical formation in the liquid. Currently, electrons influencing the liquid surface are discussed as initiators for various liquid chemistry processes.96 However, electrons play a minor role for the two sources used here. The COST-jet features an electric field perpendicular to the gas flux,64 thereby preventing electrons from leaving the electrode area. While the kINPen is ignited using a linear electric field, the discharge is relatively remote from the treatment zone (12 mm in total taking into account liquid displacement due to the gas flux). A known difference between both plasma sources is generation of O(3P) and O2(1Δg) and the observed isotope pattern differences seem to be related to these species. The COST-jet produces high amounts of O(3P)64 with about 8 × 1014 cm−3 atoms at the working distance of 4 mm.97 In comparison, calculated O(3P) densities for the kINPen reach a similar level (5 × 1014 cm−3).58 However, TALIF spectroscopy measurements indicate a highly dynamic density of O(3P) in the kINPen's effluent. Starting as high as 3.5 × 1015 cm−3, densities quickly decrease with a rate of 0.5 × 1015 cm−3 mm−1 along the z-axis of the effluent resulting in lower O(3P) levels at the gas–liquid interphase in normal conditions (9 mm nozzle – liquid). In contrast to O(3P), O2(1Δg) densities in the effluent of both sources are comparable, with a tendency to higher production rates in the COST jet (1 × 1015 cm−3 in normal conditions, up to 6 × 1015 cm−3 at high power settings98) than in the kINPen (8 × 1014 cm−3 at standard conditions27). O2(1Δg) has a significantly longer half-life. For the kINPen, O2(1Δg) was still measured at 192 mm away from the nozzle. However, the loss starting from 100 mm was significant (2/3 of the initial value). Both jets produce ozone, especially at high oxygen admixtures and long distances from the nozzle. Due to limitations in solubility and negligible effects in control experiments using an ozonizer (data not shown), a limited role in respect to liquid chemistry can be assumed. Taken together, the higher inclusion of gas phase species in COST-jet treated cysteine might be due to higher levels of O(3P) interacting either directly with the cysteine molecule or with the liquid, yielding OH radicals.
Summary and conclusions
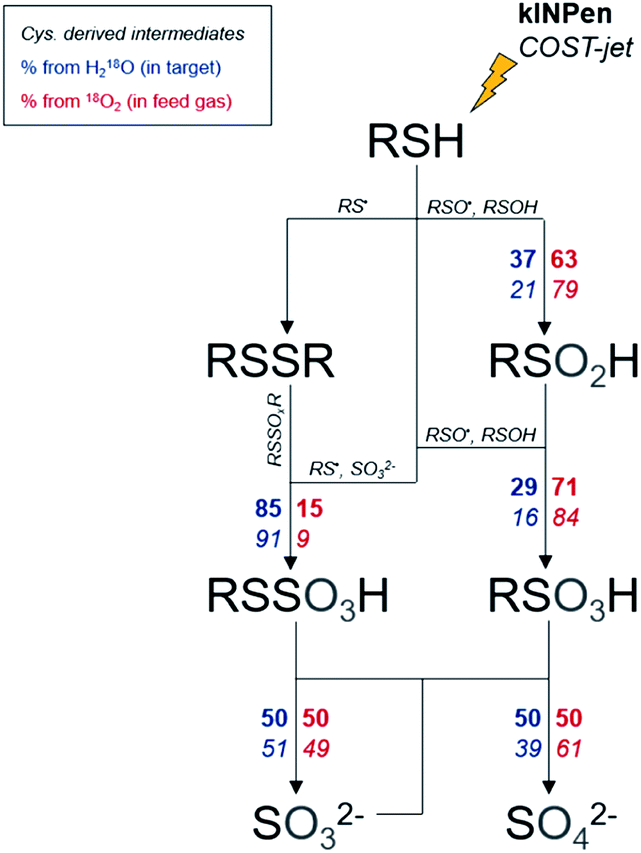
Heavy oxygen (18O2) was used to track the fate of plasma-generated ROS in a cysteine model using mass spectrometry. Furthermore, H218O was used in a reverse experiment to observe the role of liquid-derived reactive species under the same conditions. It became apparent that gas and liquid phase species play different roles: while some products are mostly driven by gas phase species, others require the presence of liquid phase species derived from the solvent system (Fig. 6). The observed isotope distribution pattern allows the assumptions that, (a) gas-phase derived short-lived reactive species are active in the gas–liquid interphase, (b) short-lived reactive species are generated in the liquid phase especially from gas-phase derived ROS (e.g. atomic oxygen), and (c) the formation of liquid phase species occurs in the interface and in deeper layers. The dominant gas phase-derived species was found to be O(3P) and OH radicals in the liquid phase.
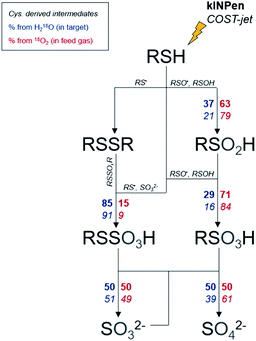 |
| | Fig. 6 Labeled oxygen branching ratios for monitored species. Branching ratios are indicated for the kINPen (bold) and COST-jet (italic). Oxygen can either stem from the water in the treated target (blue, left side of arrows) or from the plasma (red, right side of arrows). For clarity, the actual protonation/deprotonation is not reflected. | |
Concerning the application of CAP in the clinics, these results suggest a significant role of the target onto the treatment efficacy: in a humid environment such as the mucosa or during surgery, target derived species as OH radicals intensify the oxidative impact of the CAP. When treating dry tissue such as intact skin, gas phase derived species dominate and an overall milder impact of the CAP results. Recent data on the oxidation of complex lipids by CAP corroborate these conclusions.99
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Funding from the German Federal Ministry of Education and Research (grant number 03Z22DN12 to K. W., grant number 03Z22DN11 to S. B.) supported this work. The authors would like to thank Volker Schulz von-der-Gathen and Patrick Preissing for their great support with the COST-jet.
References
- S. Bekeschus, A. Schmidt, A. Kramer, H. R. Metelmann, F. Adler, T. von Woedtke, F. Niessner, K. D. Weltmann and K. Wende, Environ. Mol. Mutagen., 2018, 59, 268–277 Search PubMed.
- K. Wende, S. Bekeschus, A. Schmidt, L. Jatsch, S. Hasse, K. D. Weltmann, K. Masur and T. von Woedtke, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2016, 798–799, 48–54 CrossRef CAS PubMed.
- S. Kluge, S. Bekeschus, C. Bender, H. Benkhai, A. Sckell, H. Below, M. B. Stope and A. Kramer, PLoS One, 2016, 11, e0160667 CrossRef PubMed.
- H.-R. Metelmann, C. Seebauer, V. Miller, A. Fridman, G. Bauer, D. B. Graves, J.-M. Pouvesle, R. Rutkowski, M. Schuster, S. Bekeschus, K. Wende, K. Masur, S. Hasse, T. Gerling, M. Hori, H. Tanaka, E. H. Choi, K.-D. Weltmann, P. Metelmann, D. von Hoff and T. von Woedtke, Clin. Plasma Med., 2018, 9, 6–13 CrossRef.
- M. Keidar, D. Yan, I. I. Beilis, B. Trink and J. H. Sherman, Trends Biotechnol., 2018, 36, 586–593 CrossRef CAS PubMed.
- G. Isbary, J. Heinlin, T. Shimizu, J. Zimmermann, G. Morfill, H. U. Schmidt, R. Monetti, B. Steffes, W. Bunk and Y. Li, Br. J. Dermatol., 2012, 167, 404–410 CrossRef CAS PubMed.
- G. Isbary, G. Morfill, H. U. Schmidt, M. Georgi, K. Ramrath, J. Heinlin, S. Karrer, M. Landthaler, T. Shimizu, B. Steffes, W. Bunk, R. Monetti, J. L. Zimmermann, R. Pompl and W. Stolz, Br. J. Dermatol., 2010, 163, 78–82 CAS.
- T. Shimizu and Y. Ikehara, J. Phys. D: Appl. Phys., 2017, 50, 503001 CrossRef.
- H. Tanaka, K. Ishikawa, M. Mizuno, S. Toyokuni, H. Kajiyama, F. Kikkawa, H.-R. Metelmann and M. Hori, Rev. Mod. Plasma Phys., 2017, 1, 3 CrossRef.
- J. Gay-Mimbrera, M. C. Garcia, B. Isla-Tejera, A. Rodero-Serrano, A. V. Garcia-Nieto and J. Ruano, Adv. Ther., 2016, 33, 894–909 CrossRef CAS.
- F. Brehmer, H. Haenssle, G. Daeschlein, R. Ahmed, S. Pfeiffer, A. Görlitz, D. Simon, M. Schön, D. Wandke and S. Emmert, J. Eur. Acad. Dermatol. Venereol., 2015, 29, 148–155 CrossRef CAS PubMed.
- C. Ulrich, F. Kluschke, A. Patzelt, S. Vandersee, V. A. Czaika, H. Richter, A. Bob, J. Hutten, C. Painsi, R. Huge, A. Kramer, O. Assadian, J. Lademann and B. Lange-Asschenfeldt, J. Wound Care, 2015, 24, 196 CrossRef CAS , 198–200, 202–193..
- T. Maisch, T. Shimizu, Y. F. Li, J. Heinlin, S. Karrer, G. Morfill and J. L. Zimmermann, PLoS One, 2012, 7, e34610 CrossRef CAS PubMed.
- A. Schmidt, S. Bekeschus, K. Wende, B. Vollmar and T. von Woedtke, Exp. Dermatol., 2017, 26, 156–162 CrossRef CAS PubMed.
- S. Arndt, A. Schmidt, S. Karrer and T. von Woedtke, Clin. Plasma Med., 2018, 9, 24–33 CrossRef.
- S. Arndt, P. Unger, M. Berneburg, A. K. Bosserhoff and S. Karrer, J. Dermatol. Sci., 2018, 89, 181–190 CrossRef CAS.
- J. R. Liu, G. M. Xu, X. M. Shi and G. J. Zhang, Sci. Rep., 2017, 7, 11698 CrossRef.
- A. Schmidt and S. Bekeschus, Antioxidants, 2018, 7, 146 CrossRef PubMed.
- N. Kaushik, N. Uddin, G. B. Sim, Y. J. Hong, K. Y. Baik, C. H. Kim, S. J. Lee, N. K. Kaushik and E. H. Choi, Sci. Rep., 2015, 5, 8587 CrossRef CAS PubMed.
- J. Xia, W. Zeng, Y. Xia, B. Wang, D. Xu, D. Liu, M. G. Kong and Y. Dong, J. Biophotonics, 2019, 12, e201800046 CrossRef PubMed.
- F. Virard, S. Cousty, J. P. Cambus, A. Valentin, P. Kemoun and F. Clement, PLoS One, 2015, 10, e0133120 CrossRef PubMed.
- K. Rödder, J. Moritz, V. Miller, K.-D. Weltmann, H.-R. Metelmann, R. Gandhirajan and S. Bekeschus, Appl. Sci., 2019, 9, 660 CrossRef.
- G. Pasqual-Melo, R. K. Gandhirajan, I. Stoffels and S. Bekeschus, Clin. Plasma Med., 2018, 10, 1–8 CrossRef.
- S. Bekeschus, C. Seebauer, K. Wende and A. Schmidt, Biol. Chem., 2018, 400, 63–75 Search PubMed.
- A. Lin, Y. Gorbanev, P. Cos, E. Smits and A. Bogaerts, Clin. Plasma Med., 2018, 9, 9 CrossRef.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham, D. B. Graves, R. C. H. M. Hofman-Caris, D. Maric, J. P. Reid, E. Ceriani, D. F. Rivas, J. E. Foster, S. C. Garrick, Y. Gorbanev, S. Hamaguchi, F. Iza, H. Jablonowski, E. Klimova, J. Kolb, F. Krcma, P. Lukes, Z. Machala, I. Marinov, D. Mariotti, S. M. Thagard, D. Minakata, E. C. Neyts, J. Pawlat, Z. L. Petrovic, R. Pflieger, S. Reuter, D. C. Schram, S. Schroter, M. Shiraiwa, B. Tarabova, P. A. Tsai, J. R. R. Verlet, T. von Woedtke, K. R. Wilson, K. Yasui and G. Zvereva, Plasma Sources Sci. Technol., 2016, 25, 053002 CrossRef.
- H. Jablonowski, J. S. Sousa, K.-D. Weltmann, K. Wende and S. Reuter, Sci. Rep., 2018, 8, 12195 CrossRef PubMed.
- H. Jablonowski, A. Schmidt-Bleker, K. D. Weltmann, T. von Woedtke and K. Wende, Phys. Chem. Chem. Phys., 2018, 20, 25387–25398 RSC.
- A. Lin, Y. Gorbanev, J. De Backer, J. Van Loenhout, W. Van Boxem, F. Lemière, P. Cos, S. Dewilde, E. Smits and A. Bogaerts, Adv. Sci, 2019, 1802062 CrossRef PubMed.
- Y. Gorbanev, C. C. W. Verlackt, S. Tinck, E. Tuenter, K. Foubert, P. Cos and A. Bogaerts, Phys. Chem. Chem. Phys., 2018, 20, 2797–2808 RSC.
- S. Mitra, L. N. Nguyen, M. Akter, G. Park, E. H. Choi and N. K. Kaushik, Cancers, 2019, 11, 1030 CrossRef PubMed.
- N. Jha, J. J. Ryu, E. H. Choi and N. K. Kaushik, Oxid. Med. Cell. Longev., 2017, 2017, 7542540 Search PubMed.
- G. Bauer and D. B. Graves, Plasma Processes Polym., 2016, 13, 1157–1178 CrossRef CAS.
- D. Yan, H. Xiao, W. Zhu, N. Nourmohammadi, L. G. Zhang, K. Bian and M. Keidar, J. Phys. D: Appl. Phys., 2017, 50, 055401 CrossRef.
- A. Schmidt, S. Dietrich, A. Steuer, K.-D. Weltmann, T. von Woedtke, K. Masur and K. Wende, J. Biol. Chem., 2015, 290, 6731–6750 CrossRef CAS PubMed.
- R. K. Gandhirajan, K. Rodder, Y. Bodnar, G. Pasqual-Melo, S. Emmert, C. E. Griguer, K. D. Weltmann and S. Bekeschus, Sci. Rep., 2018, 8, 12734 CrossRef PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- N. K. Kaushik, B. Ghimire, Y. Li, M. Adhikari, M. Veerana, N. Kaushik, N. Jha, B. Adhikari, S. J. Lee, K. Masur, T. von Woedtke, K. D. Weltmann and E. H. Choi, Biol. Chem., 2018, 400, 39–62 Search PubMed.
- A. Mesbah and D. B. Graves, J. Phys. D: Appl. Phys., 2019, 52, 30LT02 CrossRef CAS.
- E. Takai, T. Kitamura, J. Kuwabara, S. Ikawa, S. Yoshizawa, K. Shiraki, H. Kawasaki, R. Arakawa and K. Kitano, J. Phys. D: Appl. Phys., 2014, 47, 285403 CrossRef.
- J.-W. Lackmann, S. Schneider, E. Edengeiser, F. Jarzina, S. Brinckmann, E. Steinborn, M. Havenith, J. Benedikt and J. E. Bandow, J. R. Soc., Interface, 2013, 10, 20130591 CrossRef PubMed.
- M. Yusupov, J.-W. Lackmann, J. Razzokov, S. Kumar, K. Stapelmann and A. Bogaerts, Plasma Processes Polym., 2018, 15, 1800022 CrossRef.
- E. Takai, K. Kitano, J. Kuwabara and K. Shiraki, Plasma Processes Polym., 2012, 9, 77–82 CrossRef CAS.
- J. W. Lackmann, S. Baldus, E. Steinborn, E. Edengeiser, F. Kogelheide, S. Langklotz, S. Schneider, L. I. O. Leichert, J. Benedikt, P. Awakowicz and J. E. Bandow, J. Phys. D: Appl. Phys., 2015, 48, 494003 CrossRef.
- J.-W. Lackmann, E. Edengeiser, S. Schneider, J. Benedikt, M. Havenith and J. E. Bandow, Plasma Med., 2013, 3, 115–124 CrossRef.
- M. Yusupov, K. Wende, S. Kupsch, E. C. Neyts, S. Reuter and A. Bogaerts, Sci. Rep., 2017, 7, 5761 CrossRef CAS PubMed.
- S. Maheux, G. Frache, J. S. Thomann, F. Clement, C. Penny, T. Belmonte and D. Duday, J. Phys. D: Appl. Phys., 2016, 49, 344001 CrossRef.
- G. Bruno, T. Heusler, J.-W. Lackmann, T. von Woedtke, K.-D. Weltmann and K. Wende, Clin. Plasma Med., 2019, 14, 100083 CrossRef.
- J. W. Lackmann, K. Wende, C. Verlackt, J. Golda, J. Volzke, F. Kogelheide, J. Held, S. Bekeschus, A. Bogaerts, V. Schulz-von der Gathen and K. Stapelmann, Sci. Rep., 2018, 8, 7736 CrossRef PubMed.
- C. Klinkhammer, C. Verlackt, D. Smilowicz, F. Kogelheide, A. Bogaerts, N. Metzler-Nolte, K. Stapelmann, M. Havenith and J. W. Lackmann, Sci. Rep., 2017, 7, 13828 CrossRef PubMed.
- K. Wende, P. Williams, J. Dalluge, W. V. Gaens, H. Aboubakr, J. Bischof, T. von Woedtke, S. M. Goyal, K. D. Weltmann, A. Bogaerts, K. Masur and P. J. Bruggeman, Biointerphases, 2015, 10, 029518 CrossRef PubMed.
- S. Bekeschus, J. Kolata, C. Winterbourn, A. Kramer, R. Turner, K. D. Weltmann, B. Broker and K. Masur, Free Radic. Res., 2014, 48, 542–549 CrossRef CAS PubMed.
- J.-W. Lackmann, G. Bruno, H. Jablonowski, F. Kogelheide, B. Offerhaus, J. Held, V. Schulz-von der Gathen, K. Stapelmann, T. von Woedtke and K. Wende, PLoS One, 2019, 14, e0216606 CrossRef PubMed.
- C. T. Stomberski, H. L. Zhou, L. Wang, F. van den Akker and J. S. Stamler, J. Biol. Chem., 2019, 294, 1568–1578 CrossRef CAS PubMed.
- G. Merényi, J. Lind and L. Engman, J. Chem. Soc., Perkin Trans. 2, 1994, 2551–2553 RSC.
- D. T. Sawyer, Oxygen chemistry, Oxford University Press, 1991 Search PubMed.
- H. Tresp, M. U. Hammer, J. Winter, K. D. Weltmann and S. Reuter, J. Phys. D: Appl. Phys., 2013, 46, 435401 CrossRef.
- A. Schmidt-Bleker, J. Winter, A. Bösel, S. Reuter and K.-D. Weltmann, Plasma Sources Sci. Technol., 2015, 25, 015005 CrossRef.
- S. Schroter, A. Wijaikhum, A. R. Gibson, A. West, H. L. Davies, N. Minesi, J. Dedrick, E. Wagenaars, N. de Oliveira, L. Nahon, M. J. Kushner, J. P. Booth, K. Niemi, T. Gans and D. O'Connell, Phys. Chem. Chem. Phys., 2018, 20, 24263–24286 RSC.
- P. Lukes, E. Dolezalova, I. Sisrova and M. Clupek, Plasma Sources Sci. Technol., 2014, 23, 015019 CrossRef.
- Y. Gorbanev, D. O'Connell and V. Chechik, Chemistry, 2016, 22, 3496–3505 CrossRef CAS PubMed.
- J. Benedikt, M. Mokhtar Hefny, A. Shaw, B. R. Buckley, F. Iza, S. Schakermann and J. E. Bandow, Phys. Chem. Chem. Phys., 2018, 20, 12037–12042 RSC.
- S. Reuter, T. von Woedtke and K.-D. Weltmann, J. Phys. D: Appl. Phys., 2018, 51, 233001 CrossRef.
- J. Golda, J. Held, B. Redeker, M. Konkowski, P. Beijer, A. Sobota, G. Kroesen, N. S. J. Braithwaite, S. Reuter, M. M. Turner, T. Gans, D. O’Connell and V. Schulz-von der Gathen, J. Phys. D: Appl. Phys., 2016, 49, 084003 CrossRef.
- A. Schmidt-Bleker, J. Winter, S. Iseni, M. Dunnbier, K. D. Weltmann and S. Reuter, J. Phys. D: Appl. Phys., 2014, 47, 145201 CrossRef.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef CAS PubMed.
- R. G. Quiller, T. A. Baker, X. Deng, M. E. Colling, B. K. Min and C. M. Friend, J. Chem. Phys., 2008, 129, 064702 CrossRef CAS PubMed.
- D. Ellerweg, A. Von Keudell and J. Benedikt, Plasma Sources Sci. Technol., 2012, 21, 034019 CrossRef.
- S. Schneider, J.-W. Lackmann, F. Narberhaus, J. E. Bandow, B. Denis and J. Benedikt, J. Phys. D: Appl. Phys., 2011, 44, 295201 CrossRef.
- S. M. Omlid, M. Zhang, A. Isor and R. D. McCulla, J. Org. Chem., 2017, 82, 13333–13341 CrossRef CAS PubMed.
- M. Zhang, G. E. Ravilious, L. M. Hicks, J. M. Jez and R. D. McCulla, J. Am. Chem. Soc., 2012, 134, 16979–16982 CrossRef CAS PubMed.
- S. Reuter, J. Winter, A. Schmidt-Bleker, D. Schroeder, H. Lange, N. Knake, V. Schulz-von der Gathen and K. D. Weltmann, Plasma Sources Sci. Technol., 2012, 21, 024005 CrossRef.
- J. Waskoenig, K. Niemi, N. Knake, L. M. Graham, S. Reuter, V. Schulz-von der Gathen and T. Gans, Plasma Sources Sci. Technol., 2010, 19, 045018 CrossRef.
- C. C. W. Verlackt, W. Van Boxem and A. Bogaerts, Phys. Chem. Chem. Phys., 2018, 20, 6845–6859 RSC.
- G. R. Buettner and R. D. Hall, Biochim. Biophys. Acta, Gen. Subj., 1987, 923, 501–507 CrossRef CAS.
- S. M. Omlid, S. A. Dergunov, A. Isor, K. L. Sulkowski, J. T. Petroff, E. Pinkhassik and R. D. McCulla, Chem. Commun., 2019, 55, 1706–1709 RSC.
- T. P. A. Devasagayam, A. R. Sundquist, P. Di Mascio, S. Kaiser and H. Sies, J. Photochem. Photobiol., B, 1991, 9, 105–116 CrossRef CAS.
- M. Rougee, R. V. Bensasson, E. J. Land and R. Pariente, Photochem. Photobiol., 1988, 47, 485–489 CrossRef CAS PubMed.
- A. U. Khan and T. Wilson, Chem. Biol., 1995, 2, 437–445 CrossRef CAS PubMed.
- S. Bekeschus, K. Wende, M. M. Hefny, K. Rödder, H. Jablonowski, A. Schmidt, T. v. Woedtke, K.-D. Weltmann and J. Benedikt, Sci. Rep., 2017, 7, 2791 CrossRef PubMed.
- M. T. Jarvi, M. S. Patterson and B. C. Wilson, Biophys. J., 2012, 102, 661–671 CrossRef CAS PubMed.
- J. B. Kohl, A. T. Mellis and G. Schwarz, Br. J. Pharmacol., 2019, 176, 554–570 CrossRef CAS PubMed.
- L. Pecci, M. Costa, A. Antonucci, G. Montefoschi and D. Cavallini, Biochem. Biophys. Res. Commun., 2000, 270, 782–786 CrossRef CAS PubMed.
- A. Kotronarou, G. Mills and M. R. Hoffmann, Environ. Sci. Technol., 1992, 26, 2420–2428 CrossRef CAS.
- G. H. Morine and R. R. Kuntz, Photochem. Photobiol., 1981, 33, 1–5 CrossRef CAS.
- H. Jablonowski, R. Bussiahn, M. U. Hammer, K.-D. Weltmann, T. von Woedtke and S. Reuter, Phys. Plasmas, 2015, 22, 122008 CrossRef.
- T. N. Das, R. E. Huie, P. Neta and S. Padmaja, J. Phys. Chem. A, 1999, 103, 5221–5226 CrossRef CAS.
- G. Mills, K. H. Schmidt, M. S. Matheson and D. Meisel, J. Phys. Chem., 1987, 91, 1590–1596 CrossRef CAS.
- J. L. Bailey and R. D. Cole, J. Biol. Chem., 1959, 234, 1733–1739 CAS.
- T. W. Thannhauser, Y. Konishi and H. A. Scheraga, Anal. Biochem., 1984, 138, 181–188 CrossRef CAS PubMed.
- P. H. Krupenie, J. Phys. Chem. Ref. Data, 1972, 1, 423–534 CrossRef CAS.
- P. Jiang, X. Chi, Q. Zhu, M. Cheng and H. Gao, Nat. Commun., 2019, 10, 3175 CrossRef PubMed.
- M. Yusupov, E. C. Neyts, P. Simon, G. Berdiyorov, R. Snoeckx, A. C. T. van Duin and A. Bogaerts, J. Phys. D: Appl. Phys., 2014, 47, 025205 CrossRef.
- S. Soorkia, C. Dehon, S. S. Kumar, M. Pedrazzani, E. Frantzen, B. Lucas, M. Barat, J. A. Fayeton and C. Jouvet, J. Phys. Chem. Lett., 2014, 5, 1110–1116 CrossRef CAS PubMed.
- E. L. Clennan, Acc. Chem. Res., 2001, 34, 875–884 CrossRef CAS PubMed.
- P. Rumbach, D. M. Bartels, R. M. Sankaran and D. B. Go, Nat. Commun., 2015, 6, 7248 CrossRef CAS PubMed.
- D. Ellerweg, J. Benedikt, A. von Keudell, N. Knake and V. Schulz-von der Gathen, New J. Phys., 2010, 12, 013021 CrossRef.
- J. S. Sousa, K. Niemi, L. J. Cox, Q. T. Algwari, T. Gans and D. O'Connell, J. Appl. Phys., 2011, 109, 123302 CrossRef.
- J. Striesow, J.-W. Lackmann, Z. Ni, S. Wenske, K. D. Weltmann, M. Fedorova, T. von Woedtke and K. Wende, Chem. Phys. Lipids, 2019, 104786 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08745a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Giuliana Brunoa,
Michael Lalkb,
Klaus-Dieter Weltmannc,
Thomas von Woedtke
*a,
Giuliana Brunoa,
Michael Lalkb,
Klaus-Dieter Weltmannc,
Thomas von Woedtke