
Mandelic acid-derived organogelators: applications of their solid form in rapid and efficient remediation of marine oil spills†
Xin
Zhang‡
 ,
Ran
Dai‡
,
Helue
Sun‡
,
Yuteng
Zhang
,
Di
Liu
,
Min
Wang
,
Mengmeng
Sun
and
Haitao
Yu
*
,
Ran
Dai‡
,
Helue
Sun‡
,
Yuteng
Zhang
,
Di
Liu
,
Min
Wang
,
Mengmeng
Sun
and
Haitao
Yu
*
Hebei Key Laboratory of Organic Functional Molecules, College of Chemistry and Materials Science, Hebei Normal University, Shijiazhuang, 050024, China. E-mail: haitaoyu@hebtu.edu.cn; Fax: +86-311-8078-9891; Tel: +86-311-8078-9891
First published on 19th November 2019
Abstract
Rapid industrialization has dramatically increased the probability of chemical-induced water pollution, especially the frequent spillage of organic liquids and fossil oils, which has driven the development of powerful methods to deal with these problems. Direct application of phase-selective organogelators (PSOGs) in solid form is a practical and promising alternative for cleanup of organic liquid pollutants from contaminated water. However, reports on such materials are not many in number. Herein, two mandelic acid-derived organogelators have been developed to show phase-selective gelation behaviours towards organic liquids, product oils, and a type of Chinese crude oil via a heating-and-cooling process, stirring or resting operation in biphasic mixtures of water. Particularly, gelator 1b could selectively congeal all the tested organic solvents and oils in powder form from the oil/water mixtures. Moreover, 1b could gelate the crude oil within 30 s under a certain condition. The benefits of instant gelation performance, easy collection of the resulting gels, easy recycling of oils, and reusability of the gelator make 1b ideal for real-world emergency remediation of oil spills. Additionally, 1b was compounded with filter papers to fabricate gelator–matrix composites, providing another way to apply the gelator solid in efficient elimination of crude oil from water.
Introduction
With the rapid development of industry and increasing material needs of people in everyday life, pollution of fresh water arising from spilling or release of hazardous waste such as toxic dyes, heavy metals, organic liquids, and fossil oils, etc. into various water bodies has become a serious environmental and economical issue in the 21st century especially for developing countries, driving people to develop advanced and powerful materials to deal with the current situation.1 In the past few decades, substantial attention has been given to the applications of supramolecular gels, which can be formed by the self-assembly of low-molecular-weight organogelators (LMWOGs) in organic liquids via weak intermolecular interactions including hydrogen bonding, van der Waals forces, π–π stacking forces, and so forth, in the treatment of organic pollutants in wastewater,2 especially in oil-spill remediation since the first report of phase-selective organogelators (PSOGs) in 2001.3 PSOGs that are capable of selectively congealing the oil layer from an oil/water mixture are promising and more effective oil-scavenging materials, as compared with the usual methods such as bioremediation, skimming, burning, the use of dispersants and polymer solidifiers, etc. for various spilling liquids involving organic solvents and refined oils.4 Nevertheless, studies on marine crude oil spill remediation with PSOGs are not many in number,5 even though they are of more significance because of the non-renewability of the precious resource and the irrecoverable damage caused by oil spills to the marine ecosystem.6 And how to realize the efficient gelation of the toxic oil layer from the surface of seawater is a major challenge in the real-world application research.In general, the methods that can allow a uniform dispersion of a gelator in the oil phase to form a gel include heating–cooling cycles, sonication, and mechanical force such as shaking and stirring. It is obvious that heating–cooling and sonication operations are impracticable in real situations from the technical viewpoint. In addition, there is also an option for the uniform distribution of gelators depending on the use of a carrier solvent. However, this method always involves the introduction of a toxic water-miscible solvent (e.g. methanol or THF), which may cause a secondary pollution to seawater, or hot carrier solvents and a large amount of co-solvents, limiting the application of gelators in practice. Thus, it is desirable to develop a more effective strategy to address the problems mentioned above. Direct employment of PSOGs in solid form can be an ideal alternative.
Some PSOGs that could be used to congeal crude oils in powder form have been reported recently by Sureshan, as well as Zeng and coworkers.5f,g However, quite slow gelation time (from several hours to even days) for crude oils with these PSOGs, which is undesirable for oil collection, limited their real-world application values. Later, Zeng and coworkers developed a wetting method, in which a given toxic water-miscible solvent, acetonitrile, was involved in the pre-treatment of a gelator. Although the acetonitrile-containing wetted gelator powders could congeal crude oils rapidly, the toxic water-miscible solvents ensconcing in the powders would leak into the water body if they were employed on a large scale, leading to a secondary pollution. Even though other non-toxic solvents could be used for the pre-treatment of the gelators, this method was still subjected to the consumption of a large volume of organic solvents, and long-time storage of such solvent-containing powders may inevitably give rise to the volatilization of the solvent, leading to a loss of effectiveness in the gelation of oils with the gelator. Moreover, fabrication of the solvent-containing powders on site is inconvenient due to the requirement of transportation of the solvent, which is not beneficial to instant remediation of the oil spillage and is uneconomical.5g,7 In addition, a phase-selective organogelator based on naphthalene diimide was reported very recently by Debangshu et al., and they claimed that this PSOG could show an instantaneous gelation behavior for crude oil, which was considered to be the best among all the reported results from the viewpoint of fastest gelation time.8 However, the development of more ideal and eco-friendly PSOGs that can be applied directly in solid form with excellent performances still remains a major challenge and is in great requirement.
In 2015, we reported a glucose-based PSOG which could gel aniline or nitrobenzene in powder form from contaminated water only within 1 min after a simple shaking at room temperature.9 This encouraged us to develop new excellent gelation systems for other important liquid pollutions, especially for crude oil which is a very complicated mixture, even though such PSOGs are difficult to invent. To this end, we herein devised and synthesized two new compounds incorporating a naphthyl ring, a L-valine fragment, a phenyl group or a long alkyl chain with L-mandelic acid as the scaffold based on the connection via an ester group and an amido link (Scheme 1). They are expected to serve as effective organoglators for organic solvent elimination and the remediation of oil spills from the water surface in their solid form. Therefore, their gelation performances on organic solvents, commercial fuels, and crude oil were tested. And gelator 1b, which shows better gelation behaviors than 1a, has been employed as the representative to be studied.
 | ||
| Scheme 1 (i) Pyridine, acetic anhydride, r. t., overnight, 90%; (ii) thionyl chloride, DCM, 0 °C to r. t., 6 h, DCM, β-naphthylamine, r. t., overnight, 72%, then sodium hydroxide, dioxane/H2O, r. t., 30 min, 63.6%; (iii) palmitoyl-L-valine or N-benzoyl-L-valine, DMAP, DCC, r. t., DCM, 24 h, 43.0–67.0%. | ||
Results and discussion
Synthesis and gelation behaviours of the target compounds
In the present work, the materials for the synthesis of the target compounds were commercially and readily available. The synthetic procedure is outlined in Scheme 1. Firstly, L-mandelic acid as the scaffold was protected by a formyl group according to the procedure to afford precursor 2.10 Compound 2 was subjected to a reaction with 2-naphthylamine in the presence of thionyl chloride, followed by the deprotection of the formyl group in NaOH/1,4-dioxane/H2O to provide intermediate 3. Finally, the target compound 1a or 1b was obtained by the esterification of precursor 3 with premade palmitoyl-L-valine or N-benzoyl-L-valine in moderate yields.The gelation behaviors of compounds 1 were examined by the heating-and-cooling process first in eleven organic liquids, including various alcohols (isopropanol, tert-butanol, isoamyl alcohol, 2-amyl alcohol, and ethylene glycol), aliphatic (cyclohexane and 1-bromobutane) and aromatic (benzene, toluene, and 1,2-dimethylbenzene) hydrocarbons, and a carboxylic acid (acetic acid). The gel formation can be identified by an “inversion-tube” method and the results are listed in Table S1 (ESI†). It is found that 1b can gel all the tested liquids, but 1a displayed gelation behaviours just in four of them. Then most of the obtained gels can remain stable for several months at room temperature. And the thermostability of the gels can be enhanced by the increase of the gelator concentration, which is testified by the fact that the gel-to-solution transition temperature (Tgel) increases upon increasing the gelator concentration (Fig. S1, ESI†).
Characterization of the formed gels
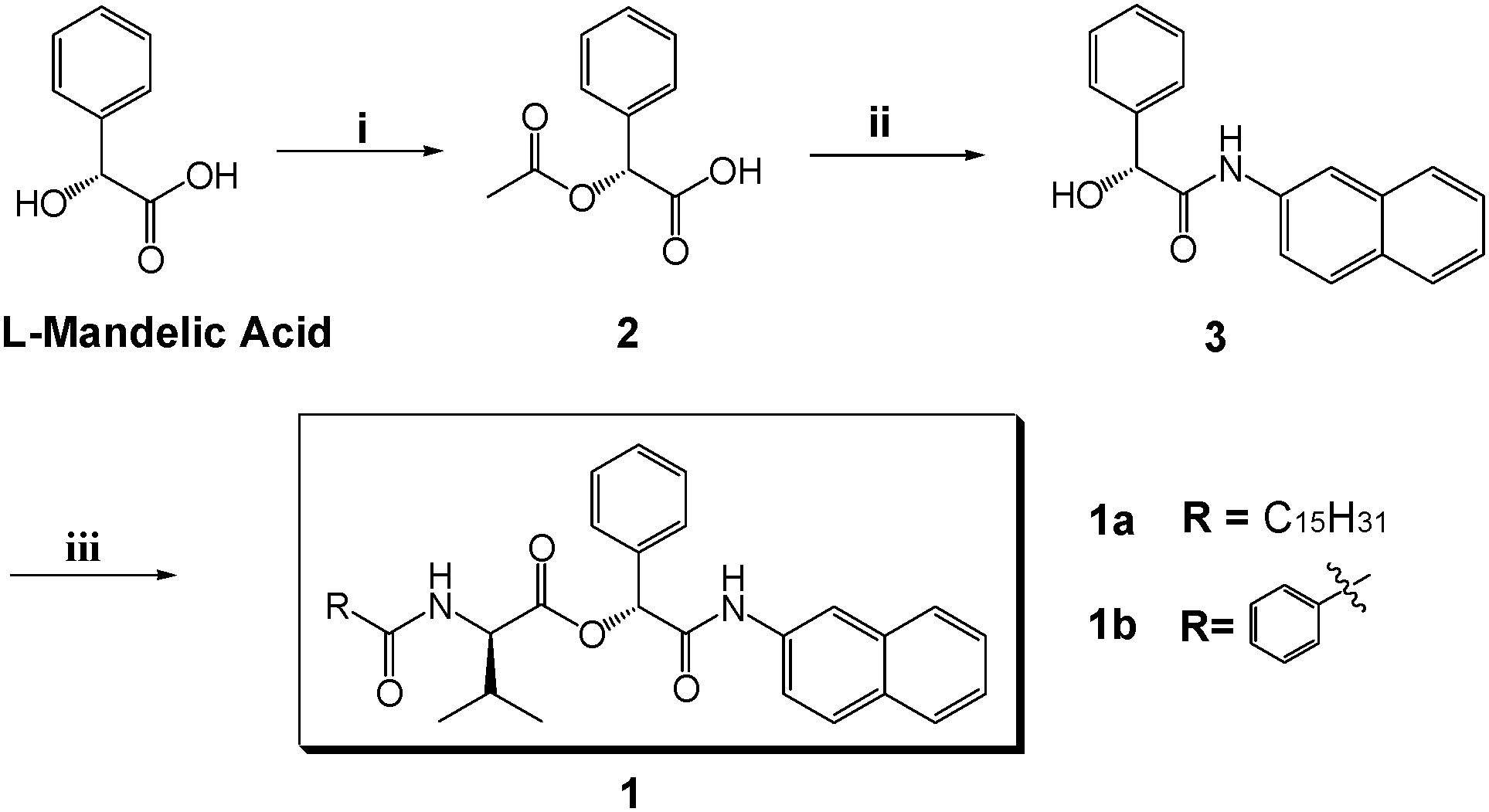
Scanning electron microscopy (SEM), temperature-dependent 1H NMR spectroscopy, powder X-ray diffraction (PXRD), and UV-vis absorption spectroscopy were utilized to characterize the obtained gels. The SEM images as shown in Fig. 1 reveal the morphologies of the xerogels prepared from gels 1a and 1b in some organic liquids, and well-defined three-dimensional (3D) networks intertwined by various aggregates can be observed. It is worth noting that xerogels 1a are found to have a sinuous rope-like aggregation structure in isopropanol and acetic acid, whereas 1b tends to form batten-like aggregates in four liquids, indicating that the aggregation mode of the gelator in the gel state is strongly dependent on the molecular structure of the gelator; here, it should be noted that 1a contains a flexible alkyl chain, instead 1b attaches a phenyl group in the same position.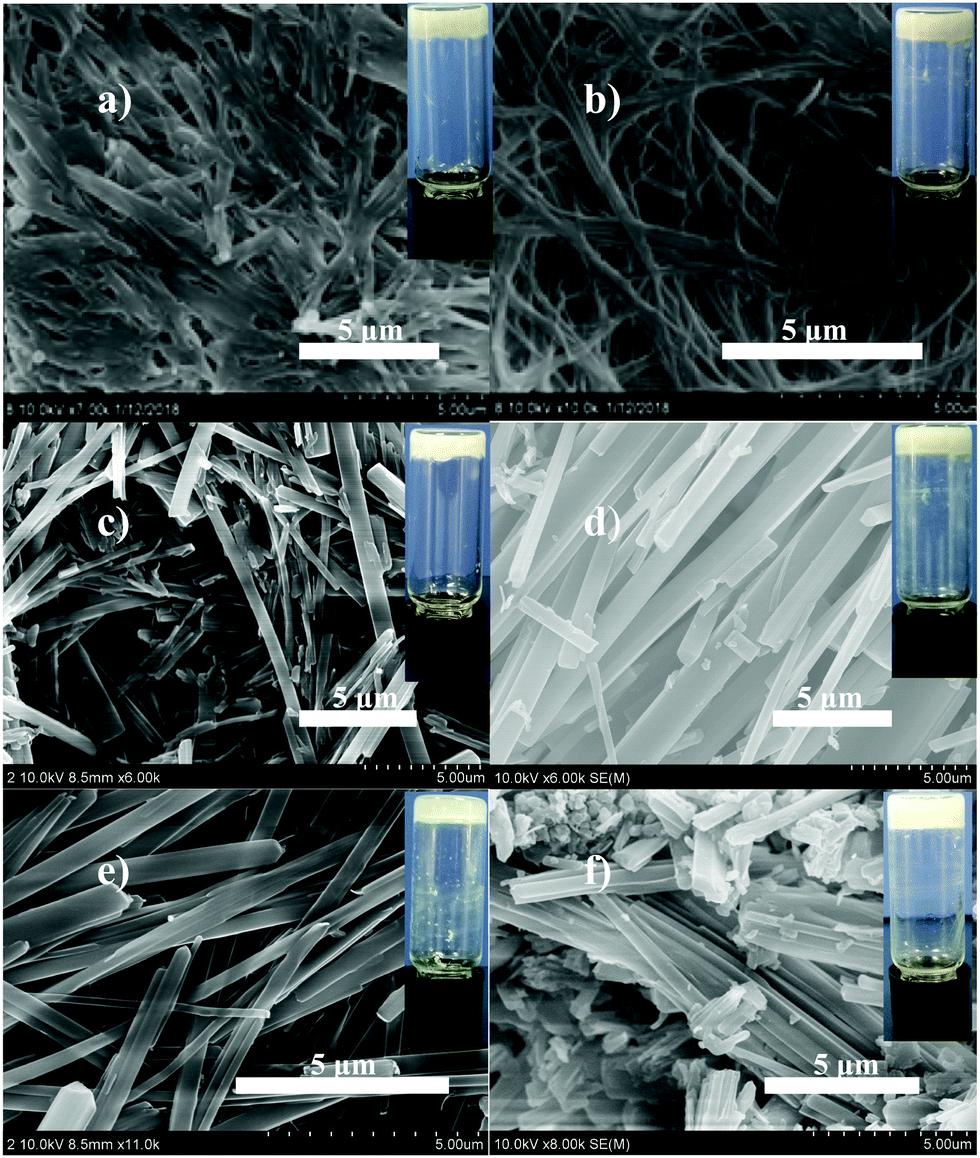 | ||
| Fig. 1 SEM images of xerogels obtained from 1a-isopropanol gel (a), 1a-acetic acid gel (b), 1b-isopropanol gel (c), 1b-acetic acid gel (d), 1b-toluene gel (e), and 1b-benzene gel (f) at their MGCs. | ||
Temperature-dependent 1H NMR spectroscopic studies (Fig. S2 and S3, ESI†) reveal the existence of intermolecular hydrogen bonding as evidenced by the fact that a downfield shift of the signals of amide NH protons of 1a (from δ = 10.8 to 10.5 ppm) and 1b (from δ = 8.49 to 8.41 ppm) is observed along with temperature increment. Besides, a shift of some aromatic proton signals assigned to phenyl or naphthyl rings is discovered, especially upon heating the deuterated cyclohexane-derived gel produced from 1b, suggesting the presence of π–π interactions involving the aromatic moieties within the gel phase. The proposed π–π interactions (J-aggregation) can lead to the red shift in the absorption features of both 1a and 1b seen in solutions with high gelator concentrations as compared with those with low concentrations (Fig. S4 and S5, ESI†). Furthermore, PXRD analysis reveals that xerogels 1a from isopropanol and acetic acid possess similar X-ray diffraction (XRD) patterns, which indicates their similar packing mode in the gel state. However, the packing mode of gelator 1b, as shown in Fig. S6c–e (ESI†), is different from that shown in xerogels 1a, which is also revealed by the morphologies of aggregates 1a and 1b in the gel state observed in the SEM images (Fig. 1). In addition, distinct diffraction peaks at 0.41 nm observed remarkably in the XRD patterns of xerogels 1a are attributed to the disordered packing of the long alkyl chains in the aggregates.11 Besides, the reflections with the d spacing values of 0.36 nm seen in both xerogels 1a and 1b can be assigned to the π–π stacking interactions of the aromatic moieties of the gelator molecules.12
Phase-selective gelation performances in gelator powder form
Subsequently, the abilities of compounds 1 to gel crude and commercial refined oils were also investigated. And both 1a and 1b have been found to be capable of congealing a type of Chinese crude oil (density of 0.87 g cm−3), and some commercial fuels and product oils including petrol, diesel, kerosene, pump oil, lubricating oil, and paraffin oil, either in pure oils or selectively from biphasic mixtures of water and oils through heating-and-cooling processes (Table S2, ESI†). And it should be mentioned that the gelators experimentally exhibited the same gelation performances in the biphasic systems with fresh water and seawater, thus, if it is not otherwise indicated, all the phase-selective gelation experiments were carried out in mixtures of oils with simulated seawater (the formula of the simulated seawater is shown in Table S3, ESI†). As shown in Table S2 (ESI†), both 1a and 1b can congeal the tested oils by the heating-and-cooling method. Particularly, 1b manifests more excellent gelation performances in the tested oils than 1a, which is reflected by the higher thermostability in terms of higher gel-to-solution transition temperatures (Tgel) and lower MGCs (minimum gelation concentrations) of gels 1b. And the higher gelation ability of 1b may be due to the presence of an additional phenyl group, which can engage in enhanced π–π interactions.Moreover, gelator 1b has been delightedly found to be able to congeal selectively all the tested oils, as well as benzene and toluene, from their water mixtures with or without a gentle agitation, whereas 1a fails to have such behaviours. Table 1 summarizes the gelation behaviours of 1b in the biphasic mixtures of oils and seawater with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (0.1 mL/1 mL). For example, when the fine powder of 1b (8.5 mg) was uniformly sprinkled at the surface of the crude oil, after being gently stirred at room temperature for 0.5 h, a gel was formed which could be easily separated out of the water mixture by scooping it with a special colander or by simple filtration. Next, crude oil could break away from the gelator by extraction from the isolated gel with petroleum ether that could be distilled off in a round bottom flask under vacuum (−0.08 MPa, 45 °C) after which the crude oil was left in the flask with a recovery of 82% by weight, and the gelator precipitate was filtered and dried with a recovery of 86% which could also be reused.
:10 (0.1 mL/1 mL). For example, when the fine powder of 1b (8.5 mg) was uniformly sprinkled at the surface of the crude oil, after being gently stirred at room temperature for 0.5 h, a gel was formed which could be easily separated out of the water mixture by scooping it with a special colander or by simple filtration. Next, crude oil could break away from the gelator by extraction from the isolated gel with petroleum ether that could be distilled off in a round bottom flask under vacuum (−0.08 MPa, 45 °C) after which the crude oil was left in the flask with a recovery of 82% by weight, and the gelator precipitate was filtered and dried with a recovery of 86% which could also be reused.
| Solvent | 1b | |
|---|---|---|
| SMGC/SBMGCa in wt% | Gelation timeb in h | |
|
a The SMGC (stirring minimum gelation concentration) and SBMGC (stirring biphasic minimum gelation concentration) values were determined using a biphasic system containing oil on top and water at the bottom with a volume ratio between oil and water of 1:10 (0.1 mL/1 mL).
b Gelation time was tested for each liquid under mild stirring at room temperature.
|
||
| Crude oil | 8.4/8.9 | 0.5 |
| Petrol | 4.6/4.8 | 0.25 |
| Diesel | 6.7/7.3 | 0.6 |
| Kerosene | 7.1/7.5 | 0.6 |
| Pump oil | 5.5/6.0 | 0.3 |
| Lubricating oil | 9.8/10.4 | 1 |
| Paraffin oil | 7.6/7.9 | 1.3 |
| Benzene | 5.9/6.3 | 0.5 |
| Toluene | 3.9/4.5 | 0.3 |
Given that the volume ratios between spilled oils and seawater are absolutely quite large and different in the actual situation, it is necessary to study the impact of the dosage of the gelator on gelation time and oil removal rates in crude oil/seawater mixtures with different large ratios such as 1:100 and 1:50. In the experiment, 0.1 mL of crude oil was introduced into 10 mL of seawater to make a test biphasic system with an oil/water ratio of 1:100. Then, different amounts of gelator 1b were spread over the surface of the crude oil phase, and each sample was rested at room temperature. The time consumed was recorded as the gelation time when the formed semi-solid was tough enough to be taken out from the artificial seawater readily by forceps. In order to calculate the oil removal rate, which herein is also defined as the recovery rate, the separated gel was extracted by petroleum ether, and the residue was subjected to vacuum distillation to recycle the crude oil. The whole phase-selective gelation and recovery process for crude oil is depicted in Fig. 2. It has been found that the gelation time and oil removal rates undergo a decrease and enhancement, respectively, with the increase in the gelator amount, as shown in Fig. 3a and Table S4 (ESI†). And upon the addition of 30 mg of gelator 1b, the gelation time can be decreased to approximately 30 s; meanwhile, the oil removal rate can reach about 90%. Nevertheless, the addition of more than 30 mg of the gelators gives rise to no significant changes in these two parameters. More interestingly, instantaneous solidification of the crude oil was observed by mechanically agitating the oil phase with the appropriate amount of the gelator (Video S1, ESI†). It is worth noting that the obtained crude oil gels with such a gelator dosage exhibit excellent mechanical strength, which is implied by their rheological data. As shown in Fig. 4a, the elastic storage modulus (G′) displays much higher values than the elastic loss modulus (G′′) in the frequency sweep experiment with a constant strain of 0.01 throughout the entire tested frequency region, suggesting the elasticity of the resulting organogel. Besides, the exceptionally high storage modulus values between 105 and 106 Pa explain its incomparable stiffness (stability). Furthermore, the yield stress can be determined to be around 5000 Pa from the crossing value of G′ and G′′ in plots of G′ and G′′ versus the stress amplitude with a constant frequency of shear stress of 1 rad s−1 (Fig. 4b), which favours the ability of the gel to resist high pressure. It is worth mentioning that the rheological data obtained above are better than those of most of the reported crude oil gels. On the other hand, similar trends can also be observed in the case of the oil/water ratio of 1:50 (0.1 mL:5 mL), as shown in Fig. 3b and Table S5 (ESI†), indicating that altering the oil/water ratios has no significant impact on the phase-selective gelation performances of gelator 1b for the crude oils. Therefore, the results obtained above demonstrate that mandelic acid-based organogelator 1b can be a rapid, efficient, and sustainable material in powder form for the remediation of marine oil spills.
 | ||
| Fig. 2 Demonstration of oil-spill recovery. (a) Biphasic system was prepared by adding 1 mL of crude oil into 100 mL of seawater (oil/water ratio = 1:100). (b) Powders of gelator 1b. (c) 300 mg of gelator powders were dispersed homogeneously over the oil surface. (d) The oil shrunk under the action of the gelator. (e) Formation of a stable gel. (f) Crude oil gel could be clamped by tweezers after 30 s. (g) Distillation of the crude oil extracted from the gel. (h) Crude oils were recycled from seawater by gelator 1b. | ||
 | ||
| Fig. 3 Changes in the gelation time and the oil removal rates of gelator 1b with crude oil/seawater ratios of (a) 1:100 (0.1 mL:10 mL) and (b) 1:50 (0.1 mL:5 mL) upon the addition of different amounts of 1b. | ||
 | ||
| Fig. 4 Rheological studies of gels formed by spreading 30 mg of gelator 1b powders over the crude oil surface (0.1 mL) in 10 mL of seawater (oil/water ratio = 1:100). (a) Frequency sweep. (b) Oscillatory stress sweep; G′ = storage modulus, G′′ = loss modulus. | ||
Importantly, tests for the repeated utilization and toxicity of the gelator are demanded for consideration of its economic and safe usage in practice. The reusability of the recovered gelators was evaluated by determination of oil removal rates using gelator 1b to repeatedly eliminate the crude oil from the oil/seawater mixtures for five cycles. It is found in Fig. S7 (ESI†) that gelator 1b can be used repeatedly at least five times and the oil removal rate can remain at almost 80% after the fifth repetition, indicating the excellent reusable feature and improved economic value of the gelator. On the other hand, gelator 1b is water-insoluble with a solubility of less than 10−5 g in 100 g of water at 25 °C, which avoids the leakage of the gelator into the water body. Besides, gelator 1b shows good stability, which is confirmed by the fact that after the gelator was soaked in artificial seawater for 15 days at room temperature, there was no toxic substance released, as shown in Fig. S8 (ESI†). A toxicology test on adult zebrafish with zero mortality indicates that gelator 1b is non-toxic to marine life (Fig. S9, ESI†). These results indicate that gelator 1b can be a reusable and eco-friendly material for liquid pollutant remediation.
Removal of crude oil from water with gelator-attached filter papers
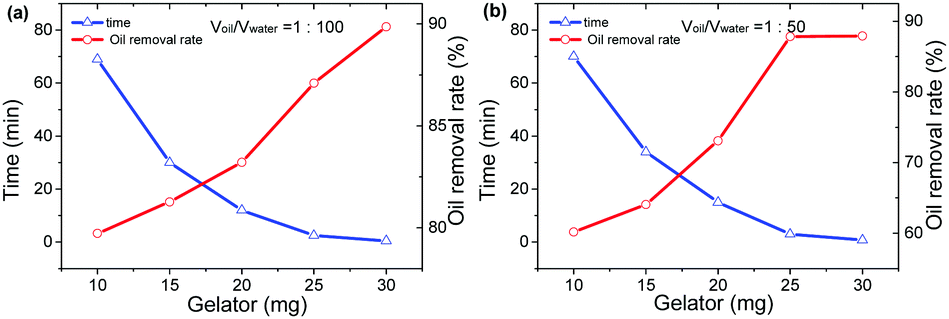
Apart from the direct employment of gelator powders, creation of a gelator–matrix composite is another way to realize the efficient removal of crude oils from the biphasic mixtures of crude oils and seawater in gelator solid form. The matrices used to fabricate the composite with a gelator can be generally those materials having a multihole structure including natural polymers and carbon.13 In the present work, we adopt inexpensive filter papers as substrates, and the gelators are attached onto them to make gelator-attached filter papers (GAFPs) which can act as sorbents for crude oils. In a typical fabricating procedure for GAFPs, circular filter papers (FPs) with a diameter of 2 cm were immersed respectively in 2 mL of ethanol solutions containing different amounts of gelator 1b (0–10 mg). Herein, low-, middle-, and high-speed FPs are involved in the attachment of the gelators to them. It is found in Fig. 5 that the attachment capability of the FPs can be enhanced by increasing the concentration of gelator 1b in ethanol, and the attachment capacity of the high-speed FPs can reach about ca. 196 mg g−1 (4.9 mg of gelators attached to a circular FP with a diameter of 2 cm which is the gelator-attachment limit) when the gelator concentration is 5 mg mL−1 in 2 mL of ethanol, which is larger than those of the other two types of FPs (180 mg g−1 for the middle-speed FP, and 124 mg g−1 for the low-speed FP) under the same conditions. Notably, the obtained gelator composites are extremely hydrophobic which can be proven by the fact that the GAFPs could always float over the water surface when they were placed on the surface of the artificial seawater in a beaker, whereas the FPs sunk to the bottom of water upon adsorbing a substantial amount of water within 30 s, as shown in Fig. 6b. Besides, upon dropping a water droplet over the surface of a GAFP, the water droplet was never adsorbed by the composite with a contact angle of 100.2° as shown in Fig. 6a and c, also indicating the hydrophobicity of the GAFP. This property is crucial for GAFPs to adsorb crude oils from their biphasic mixtures with seawater effectively.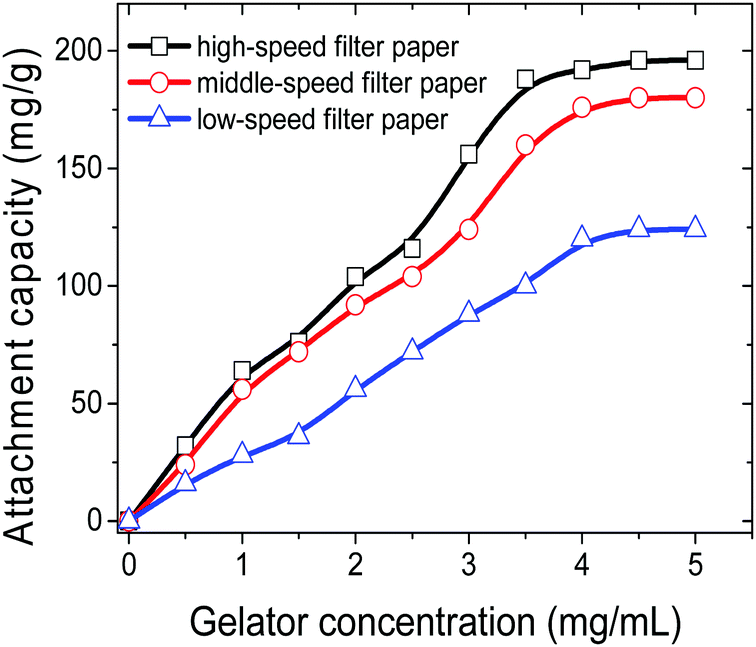 | ||
| Fig. 5 The attachment capabilities of three types of circular filter papers with a diameter of 2 cm to gelator 1b upon immersing the filter papers respectively in 2 mL of ethanol solutions containing 0–10 mg of the gelators. The unit of the attachment capacity is mg g−1. | ||
 | ||
| Fig. 6 (a) Photograph of a water droplet placed over a high-speed GAFP with a diameter of 2 cm (the attachment capacity for 1b is 196 mg g−1); (b) the FPs sank to the bottom of water in a beaker within 30 s, whereas the GAFPs was hydrophobic enough to float over the water surface; (c) image of a water droplet and the corresponding contact angle over the surface of the GAFP. | ||
In order to investigate the crude oil removal efficiency of GAFPs, the three types of circular FPs of diameter 2 cm, with different amounts of gelator 1b attached onto them, were separately placed over the surface of crude oil/seawater mixtures in some beakers, each of which contained 0.1 mL of the crude oil as well as 10 mL of artificial seawater (Fig. 7a). As shown in Fig. 7b and c, after the GAFP was rested for 5 min, the crude oil could be more or less adsorbed by the GAFP, and then the crude oil-adsorbed GAFP was taken out of the seawater by forceps and dried to calculate the oil removal rate (Fig. 7d). Moreover, the adsorbed crude oil could be separated from the GAFP by washing the crude oil-adsorbed GAFP with petroleum ether (Fig. 2g and h). Fig. 7e shows the eluted crude oil in petroleum ether which can be recovered via simple reduced pressure distillation. Furthermore, the test results of oil removal rates using GAFPs as sorbents are summarized in Fig. 8. According to Fig. 8, the oil removal rates can be increased with the increase of gelator amounts on PFs, and they can reach maximum uptake degrees of about 40%, 60%, and 80%, respectively, for low-, middle- and high-speed GAFPs within their gelator-attachment limits. However, the free PFs exhibit quite low oil removal efficiency of less than 5%. Furthermore, the oil removal efficiency with high speed GAFPs of different diameters, which were fabricated under the same conditions as those mentioned above in Fig. 5, was also investigated in the crude oil/seawater mixtures with an oil/water ratio of 1:100 (0.1 mL:10 mL). As shown in Fig. S10 (ESI†), the oil removal rate can reach almost 90% with the increase of the diameter of the GAFP to 3.0 cm which loads 6.8 mg of the gelator. It is worth mentioning that this strategy can significantly reduce the gelator usage as compared with the case of the direct utilization of gelator powders, which may be due to the enhancement of the contact extent between gelators and crude oils as the gelators are uniformly diffused over the GAFPs.
 | ||
| Fig. 7 Demonstration of oil spill remediation using a GAFP with a diameter of 2 cm (attachment capacity for gelator 1b is 196 mg g−1). (a) Biphasic system was formed by introducing 0.1 mL of the crude oil into 10 mL of artificial seawater. (b) The GAFP was placed over the crude oil surface. (c) The crude oil accumulated under the action of the gelator composite. (d) The crude oil-adsorbed GAFP could be taken out of the water phase by tweezers after 5 min. (e) The recovered crude oil in petroleum ether. | ||
 | ||
| Fig. 8 The oil removal rates of three types of GAFPs with a diameter of 2 cm and different amounts of gelator 1b attached onto them for crude oils from the crude oil/seawater mixtures with an oil/water ratio of 1:100 (0.1 mL:10 mL) at room temperature. | ||
Conclusions
In summary, we report two novel L-mandelic acid-derived compounds (1a and 1b) bearing a naphthyl group and a L-valine moiety. These two compounds can be used as phase-selective gelators for organic liquids, product oils, and a type of Chinese crude oil. Particularly, 1b can congeal the product oils, benzene and toluene, and the crude oil in powder form at room temperature under mild stirring or resting. Systematic experiments indicate that the formation of the crude oil-gels can be realized within 30 s under a certain condition. Furthermore, the advantages, including the high mechanical strength of the formed gels for convenient collection, the high oil removal efficiency, and the easy recycling of oils and gelators, lead us to propose gelator 1b to be an ideal oil-removing agent for real-life applications in the emergency remediation of marine oil spills and efficient treatment of other organic liquids. In addition, gelator 1b can be distributed onto a cheap filter paper to fabricate a gelator-attached filter paper (GAFP). It allows the GAFP to provide another option for the application of the gelator in solid form for oil cleanup from contaminated water.Experimental section
Materials and techniques
All the materials, chemical reagents, and solvents were commercially available from Sigma-Aldrich and other providers. All the solvents of analytical grade for synthesis and tests were used directly without any purification. All the reaction processes were checked by thin-layer chromatography (TLC) with the help of UV-lamp irradiation (254 nm and 365 nm). Column-chromatography was used to purify the intermediates and target compounds with Merck millipore silica gel of 300–400 mesh. A X-4 melting point apparatus was used to determine the melting points of solid compounds.NMR, mass, and absorption spectra were recorded on a Bruker Avance AVIII-500Q spectrometer, an AB SCIEX 3200Q TRAP LC/MS/MS system equipped with an Agilent 1260 Infinity HPLC, and a PerkinElmer Double Beam UV/vis Spectrometer, respectively. An Elementar Vario EL III elemental analyzer (Germany) was used for elemental analyses of compounds. A Bruker D8 ADVANCE X-ray diffractometer was employed for the powder X-ray diffraction (PXRD) measurements. SEM images were recorded on a Hitachi S4800 FE-SEM microscope. The xerogel samples for PXRD and SEM images were fabricated by freezing the obtained gels in liquid nitrogen, then drying them under vacuum. All dynamic rheological studies were conducted on a Rheometer (MCR 302, Anton Paar, Austria). The frequency sweep experiment was performed with a constant strain of 0.01 over the entire test frequency range and the Oscillatory stress sweep experiment was performed with a constant frequency of shear stress of 1 rad s−1. The fluorescence measurements were carried out on a SHIMADZU RF-6000 fluorescence spectrophotometer. The contact angle tests were carried out on a KYOWA DMo-501 contact angle goniometer.
Synthesis procedures
:10). The reaction was stirred at room temperature for 30 minutes and then filtered to give a white solid, 0.64 g, yield 63.6%. M. p. 137.5–138.7 °C.
1H NMR (500 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.38 (s, 1H), 7.82 (dt, J = 17.6, 9.0 Hz, 4H), 7.58 (d, J = 7.5 Hz, 2H), 7.47 (t, J = 7.4 Hz, 1H), 7.34–7.44 (m, 3H), 7.31 (t, J = 7.2 Hz, 1H), 6.51 (d, J = 3.9 Hz, 1H), 5.19 (d, J = 4.1 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 171.9, 141.2, 136.6, 133.7, 130.3, 128.7, 128.5, 128.1, 127.9, 127.8, 127.1, 126.8, 125.1, 120.9, 116.3, 74.5. Elem. anal. C18H15NO2: calcd C 77.96 H 5.45 N 5.05; found. C 75.57 H 5.37 N 5.13. EPI-MS C18H15NO2 ([M + H]+): calcd 278.3, found. 278.3.
:2.5) to give a white solid, 1.26 g, yield 43.0%. M. p. 77.0–78.6 °C.
1H NMR (500 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.22–8.30 (m, 2H), 7.80–7.90 (m, 3H), 7.65 (d, J = 7.6 Hz, 1H), 7.59 (d, J = 7.3 Hz, 2H), 7.36–7.50 (m, 5H), 6.10 (s, 1H), 4.33 (t, J = 7.0 Hz, 1H), 2.16 (dt, J = 14.2, 7.0 Hz, 3H), 1.43–1.51 (m, 2H), 1.16–1.24 (m, 24H), 0.93 (d, J = 6.7 Hz, 6H), 0.85 (t, J = 6.7 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 173.6, 171.6, 167.1, 136.3, 135.6, 133.7, 130.4, 129.2, 129.0, 128.9, 127.9, 127.8, 127.7, 126.9, 125.3, 120.4, 116.2, 75.8, 58.2, 35.2, 31.7, 30.2, 29.5, 29.3, 29.1, 29.0, 25.7, 22.5, 18.7, 14.4. Elem. anal. C39H54N2O4: calcd C 76.18 H 8.85 N 4.56; found. C 76.06 H 7.65 N 4.62. EPI-MS C39H54N2O4 ([M + H]+): calcd 615.8, found. 615.7.
:2) to give a white solid, 1.30 g, yield 67.0%. M. p. 152.5–153.0 °C.
1H NMR (500 MHz, DMSO-d6) δ 10.54 (d, J = 41.8 Hz, 1H), 8.73 (dd, J = 30.9, 7.6 Hz, 1H), 8.26 (d, J = 22.6 Hz, 1H), 7.81–7.90 (m, 5H), 7.64 (d, J = 7.3 Hz, 1H), 7.56 (dt, J = 9.6, 7.0 Hz, 3H), 7.38–7.49 (m, 7H), 6.12 (d, J = 24.6 Hz, 1H), 4.49 (t, J = 7.5 Hz, 1H), 2.30–2.38 (m, 1H), 1.12 (t, J = 7.0 Hz, 3H), 0.99–1.02 (m, 3H). 13C NMR (125 MHz, DMSO-d6) δ 171.5, 168.0, 167.0, 136.3, 135.6, 134.3, 133.7, 131.9, 130.4, 129.3, 129.2,129.1, 129.0, 128.6, 128.1, 128.0, 127.9, 127.8, 127.0, 125.3, 120.4, 116.3, 75.8, 59.4, 30.0,19.6, 19.5, 19.4. Elem. anal. C30H28N2O4: calcd C 74.98 H 5.87 N 5.83; found. C 73.74 H 6.27 N 6.06. EPI-MS C30H28N2O4 ([M + Na]+): calcd 503.5, found. 503.3.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21971054, 21801060, 21272054, and 21502040), the Nature Science Foundation of Hebei Province (B2019205172 and B2018205199), and the Key Program of Nature Science Foundation of Hebei Normal University (no. L2019Z04). We also gratefully acknowledge the support from the National Demonstration Center for Experimental Chemistry Education of Hebei Normal University.Notes and references
- (a) X. Luo, Z. Pan, F. Pei, Z. Jin, K. Miao, P. Yang, H. Qian, Q. Chen and G. Feng, J. Ind. Eng. Chem., 2018, 59, 410–415 CrossRef CAS; (b) J. Zhao, S. Ge, L. Liu, Q. Shao, X. Mai, C. X. Zhao, L. Hao, T. Wu, Z. Yu and Z. Guo, Ind. Eng. Chem. Res., 2018, 57, 231–241 CrossRef CAS; (c) J. Zhao, S. Ge, D. Pan, Y. Pan, V. Murugadoss, R. Li, W. Xie, Y. Lu, T. Wu, E. K. Wujcik, Q. Shao, X. Maiand and Z. Guo, J. Electrochem. Soc., 2019, 166, H3074–H3083 CrossRef CAS; (d) B. Zhao, Q. Shao, L. Hao, L. Zhang, Z. Liu, B. Zhang, S. Ge and Z. Guo, J. Colloid Interface Sci., 2018, 511, 39–47 CrossRef CAS; (e) C. Lou, T. Jing, J. Tian, Y. Zheng, J. Zhang, M. Dong, C. Wang, C. Hou, J. Fan and Z. Guo, J. Mater. Res., 2019, 34, 2964–2975 CrossRef CAS; (f) M. Zhang, J. Meng, Q. Liu, S. Gu, L. Zhao, M. Dong, J. Zhang, H. Hou and Z. Guo, J. Mater. Res., 2019, 34, 3050–3060 CrossRef CAS; (g) X. Shi, C. Wang, Y. Ma, H. Liu, S. Wu, Q. Shao, Z. He, L. Guo, T. Ding and Z. Guo, Powder Technol., 2019, 356, 726–734 CrossRef CAS; (h) D. Pan, S. Ge, J. Zhao, J. Tian, Q. Shao, L. Guo, X. Mai, T. Wu, V. Murugadoss, H. Liu, T. Ding, S. Angaiah and Z. Guo, Ind. Eng. Chem. Res., 2019, 58, 836–848 CrossRef CAS; (i) J. Tian, Q. Shao, J. Zhao, D. Pan, M. Dong, C. Jia, T. Ding, T. Wu and Z. Guo, J. Colloid Interface Sci., 2019, 541, 18–29 CrossRef CAS; (j) J. Zhao, S. Ge, D. Pan, Q. Shao, J. Lin, Z. Wang, Z. Hu, T. Wu and Z. Guo, J. Colloid Interface Sci., 2018, 529, 111–121 CrossRef CAS; (k) D. Pan, S. Ge, J. Tian, Q. Shao, L. Guo, H. Liu, S. Wu, T. Ding and Z. Guo, Chem. Rec., 2019, 19, 1–16 CrossRef CAS; (l) D. Pan, S. Ge, J. Zhao, Q. Shao, L. Guo, X. Zhang, J. Lin, G. Xu and Z. Guo, Dalton Trans., 2018, 47, 9765–9778 RSC; (m) C. Huang, X. Shi, C. Wang, L. Guo, M. Dong, G. Hu, J. Lin, T. Ding and Z. Guo, Int. J. Biol. Macromol., 2019, 140, 1167–1174 CrossRef CAS PubMed; (n) G. Li, Z. Mai, X. Shu, D. Chen, M. Liu and W. Xu, Adv. Compos. Hybrid Mater., 2019, 2, 254–265 CrossRef CAS; (o) L. Dashairya, M. Rout and P. Saha, Adv. Compos. Hybrid Mater., 2018, 1, 135–148 CrossRef CAS; (p) S. Sun, L. Zhu, X. Liu, L. Wu, K. Dai, C. Liu, C. Shen, X. Guo, G. Zheng and Z. Guo, ACS Sustainable Chem. Eng., 2018, 6, 9866–9875 CrossRef CAS; (q) H. Kang, Z. Cheng, H. Lai, H. Ma, Y. Liu, X. Mai, Y. Wang, Q. Shao, L. Xiang, X. Guo and Z. Guo, Sep. Purif. Technol., 2018, 201, 193–204 CrossRef CAS; (r) Z. Li, B. Wang, X. Qin, Y. Wang, C. Liu, Q. Shao, N. Wang, J. Zhang, C. Shen and Z. Guo, ACS Sustainable Chem. Eng., 2018, 6, 13747–13755 CrossRef CAS; (s) X. Zhang, X. Wang, X. Liu, C. Lv, Y. Wang, G. Zheng, H. Liu, C. Liu, Z. Guo and C. Shen, ACS Sustainable Chem. Eng., 2018, 6, 12580–12585 CrossRef CAS.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- (a) H. Yu, B. Liu, Y. Wang, J. Wang and Q. Hao, Soft Matter, 2011, 7, 5113–5115 RSC; (b) D. R. Trivedi and P. Dastidar, Chem. Mater., 2006, 18, 1470–1478 CrossRef CAS; (c) S. Dehnath, A. Shome, S. Dutta and P. K. Das, Chem. – Eur. J., 2008, 14, 6870–6881 CrossRef; (d) S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS; (e) S. Mukherjee and B. Mukhopadhyay, RSC Adv., 2012, 2, 2270–2273 RSC; (f) A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250–5252 RSC; (g) S. Basak, J. Nanda and A. Banerjee, J. Mater. Chem., 2012, 22, 11658–11664 RSC; (h) C. Tsai, Y. Cheng, L. Shen, K. Chang, I. Ho, J. Chu and W. Chung, Org. Lett., 2013, 15, 5830–5833 CrossRef CAS; (i) M. Konda, I. Maity, D. B. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 1482–1488 CrossRef CAS; (j) S. Mukherjee, C. Shang, X. Chen, X. Chang, K. Liu, C. Yu and Y. Fang, Chem. Commun., 2014, 50, 13940–13943 RSC; (k) J. Liu, J. Li, P. Lin, N. Zhang, X. Han, B. Zhang and J. Song, Chem. Commun., 2016, 52, 13975–13978 RSC; (l) C. Narayana, R. K. Upadhyay, R. Chaturvedi and R. Sagar, New J. Chem., 2017, 41, 2261–2267 RSC; (m) X. Ran, Y. Li, Q. Gao, W. Qiu and L. Guo, Asian J. Org. Chem., 2017, 6, 95–101 CrossRef CAS.
- (a) Rajkamal, D. Chatterjee, A. Paul, S. Banerjee and S. Yadav, Chem. Commun., 2014, 50, 12131–12134 RSC; (b) L. Yan, G. Li, Z. Ye, F. Tian and S. Zhang, Chem. Commun., 2014, 50, 14839–14842 RSC; (c) C. S. K. Raju, B. Pramanik, T. Kar, P. V. C. Rao, N. V. Choudary and R. Ravishankar, RSC Adv., 2016, 6, 53415–53420 RSC; (d) C. Ren, F. Chen, F. Zhou, J. Shen, H. Su and H. Zeng, Langmuir, 2016, 32, 13510–13516 CrossRef CAS; (e) C. Ren, G. H. B. Ng, H. Wu, K. Chan, J. Shen, C. Teh, J. Y. Ying and H. Zeng, Chem. Mater., 2016, 28, 4001–4008 CrossRef CAS; (f) A. M. Vibhute, V. Muvvala and K. M. Sureshan, Angew. Chem., Int. Ed., 2016, 55, 7782–7785 CrossRef CAS; (g) C. Ren, J. Shen, F. Chen and H. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3847–3851 CrossRef CAS; (h) H. Sun, J. Chuai, H. Wei, X. Zhang and H. Yu, J. Hazard. Mater., 2019, 366, 46–53 CrossRef CAS PubMed; (i) C. Zhuan, Y. Li, X. Yuan, J. Zhao and X. Hou, J. Appl. Polym. Sci., 2019, 136, 47052 CrossRef.
- M. A. Hubbe, O. J. Rojas, M. Fingas and B. S. Gupta, BioRes., 2013, 8, 3038–3097 Search PubMed.
- (a) J. Li, Y. Huo and H. Zeng, J. Mater. Chem. A, 2018, 6, 10196–10200 RSC; (b) J. Li, Y. Huo and H. Zeng, Langmuir, 2018, 34, 8058–8064 CrossRef CAS.
- S. Datta, S. Samanta and D. Chaudhuri, J. Mater. Chem. A, 2018, 6, 2922–2926 RSC.
- X. Zhang, J. Song, W. Ji, N. Xu, N. Gao, X. Zhang and H. Yu, J. Mater. Chem. A, 2015, 3, 18953–18962 RSC.
- S. Peukert, Y. C. Sun, R. Zhang, B. Hurley, M. Sabio, X. Y. Shen, C. Gray, J. S. Tao, R. Cebula and S. Wattanasin, Bioorg. Med. Chem. Lett., 2008, 18, 1840–1844 CrossRef CAS.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem. – Eur. J., 2001, 7, 2245–2253 CrossRef.
- H. Xu, A. K. Das, M. Horie, M. S. Shaik, A. M. Smith, Y. Luo, X. Lu, R. Collins, S. Y. Liem, A. Song, P. L. Popelier, M. L. Turner, P. Xiao, I. A. Kinloch and R. V. Ulijn, Nanoscale, 2010, 2, 960–966 RSC.
- (a) A. Prathap and K. M. Sureshan, Angew. Chem., Int. Ed., 2017, 56, 9405–9409 CrossRef CAS; (b) M. Liu, B. Li, H. Zhou, C. Chen, Y. Liu and T. Liu, Chem. Commun., 2017, 53, 2810–2813 RSC; (c) H. Zhang, S. Lyu, X. Zhou, H. Gu, C. Ma, C. Wang, T. Ding, Q. Shao, H. Liu and Z. Guo, J. Colloid Interface Sci., 2019, 536, 245–251 CrossRef CAS; (d) S. Zhang, H. Liu, S. Yang, X. Shi, D. Zhang, C. Shan, L. Mi, C. Liu, C. Shen and Z. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 10922–10932 CrossRef CAS; (e) M. Liu, Z. Yang, H. Sun, C. Lai, X. Zhao, H. Peng and T. Liu, Nano Res., 2016, 9, 3735–3746 CrossRef CAS; (f) Z. Wang, R. Wei, J. Gu, H. Liu, C. Liu, C. Luo, J. Kong, Q. Shao, N. Wang, Z. Guo and X. Liu, Carbon, 2018, 139, 1126–1135 CrossRef CAS; (g) Z. Shi, G. Xua, J. Deng, M. Dong, V. Murugadoss, C. Liu, Q. Shao, S. Wu and Z. Guo, Green Chem. Lett. Rev., 2019, 12, 235–243 CrossRef CAS; (h) G. Xu, Z. Shi, Y. Zhao, J. Deng, M. Dong, C. Liu, V. Murugadoss, X. Mai and Z. Guo, Int. J. Biol. Macromol., 2019, 126, 376–384 CrossRef CAS PubMed; (i) Z. Shi, C. Jia, D. Wang, J. Deng, G. Xu, C. Wu, M. Dong and Z. Guo, Int. J. Biol. Macromol., 2019, 133, 964–970 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional tables, figures, and videos. See DOI: 10.1039/c9qm00603f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2020 |