
The blocky structure of Ziegler–Natta “random” copolymers: myths and experimental evidence†
Claudio
De Rosa
 *,
Odda
Ruiz de Ballesteros
,
Rocco
Di Girolamo
,
Anna
Malafronte
,
Finizia
Auriemma
,
Giovanni
Talarico
and
Miriam
Scoti
*,
Odda
Ruiz de Ballesteros
,
Rocco
Di Girolamo
,
Anna
Malafronte
,
Finizia
Auriemma
,
Giovanni
Talarico
and
Miriam
Scoti
Dipartimento di Scienze Chimiche, Università di Napoli “Federico II”, Complesso Monte S. Angelo, via Cintia, I-80126 Napoli, Italy. E-mail: claudio.derosa@unina.it
First published on 20th November 2019
Abstract
The crystallization behavior of butene–ethylene random isotactic copolymers prepared with a Ziegler–Natta catalyst, in particular the surprising crystallization of form II of isotactic polybutene in samples with a high ethylene concentration, provides novel evidence of the long-debated blocky molecular structure of these “random” copolymers.
Ziegler–Natta (ZN) catalysts for the polymerization of α-olefins to isotactic polymers consist of solid particles of activated TiCl3,1,2 or of active Ti compounds (generally TiCl4) supported on magnesium chloride, in combination with an aluminum alkyl as a co-catalyst and suitable Lewis bases added to the co-catalyst (external donors) and/or to the solid catalyst (internal donors).2–4 The function of donors has been the subject of deep investigations and their structure has been designed to increase catalyst activity and stereoselectivity.2,3
The intrinsic multi-site nature of Ziegler–Natta catalysts, that is, the presence of different active catalytic sites on the surfaces of catalyst particles, produces heterogeneous polyolefin samples characterized by complicated mixtures of macromolecules that feature, in the case of homopolymers, different stereoregularities and molecular masses and a non-random distribution of stereodefects,5,6 and, in the case of copolymers of olefins, a non-random distribution of the monomeric units along the polymer chain and non-uniform composition. The heterogeneous samples are generally separable, by extraction in solvents having different boiling temperatures, into fractions containing chains grown from different sites having different molecular masses and stereoregularities and, in the case of copolymers, different comonomer concentrations.
However, a more complex structure of the macromolecules produced by the different sites with the presence of chemically bound blocks of different tacticities (stereoblocks) has been hypothesized in the case of the polypropylene homopolymer.6–8 In particular, detailed 13CNMR studies have shown that highly isotactic fractions insoluble in boiling heptane or xylene at room temperature also contain short syndiotactic blocks whose insolubility is attributed to the fact that they are chemically bound to the isotactic part.6,7 On the other hand, the atactic soluble fraction contains poorly isotactic or syndiotactic sequences5,6,8–12 or more regular isotactic sequences occurring in the form of short stereoblocks.13–15
The hypothesis of a blocky molecular structure of polyolefins (stereoblocks in the case of homopolymers) produced with Ziegler–Natta catalysts has been the subject of a long debate, giving insights into the mechanism of the Ziegler–Natta catalysts,6 where evidence from the NMR spectra of block junctions has been sought for a long time.
In this communication we present a study of the crystallization behavior of butene–ethylene (BE–ZN) random isotactic copolymers prepared with a classic Ziegler–Natta catalyst and demonstrate that the crystallization properties, and the comparison with model polymers prepared with homogeneous metallocene catalysts, provide novel evidence of the blocky molecular structure of the “random” copolymers induced by Ziegler–Natta catalysts. Our model reference systems are based on samples of isotactic polybutene (iPB) and isotactic butene–ethylene copolymers prepared with homogeneous metallocene catalysts (BE–MET) and are characterized by a controlled molecular structure with a truly random distribution of microstructural defects and comonomeric units.16–22 This ideal molecular structure allowed the proposal of a model of the crystallization behavior of iPB based on the length of regular butene sequences in model chains interrupted by the presence of randomly distributed stereodefects or comonomeric units.16,17,19,22 According to this model, when the length of the regular butene sequences is long enough, the usual crystallization of the kinetically favored form II occurs, whereas crystallization of the more stable form I from the melt or from the amorphous phase is favored when the length of the regular butene sequences is short for the presence of stereo- or constitutional defects (stereodefective iPBs or butene–ethylene copolymers).16,17,19,22 In this paper we show that “random” BE–ZN copolymer samples crystallize in form II of iPB even at high ethylene concentrations (16–20 mol%), indicating the presence of chains with long butene sequences. This is the hallmark of the non-random distribution of comonomers and their segregation in blocks forming chains with a multiblock architecture. While 13C-NMR is recognized as the most important technique for the characterization of the polyolefin molecular structure, we show that the analysis of the crystallization properties may be an adequate tool for unravelling subtle aspects of the molecular architecture.
Two samples of BE–ZN copolymers with total concentrations of ethylene of 9.1 and 14.3 mol% were prepared with a MgCl2-supported Ziegler–Natta catalyst modified using di(isobutyl) phthalate as an internal donor, combined with tri-isobutylaluminum (TIBA) as a cocatalyst and (2,3-dimethyl-butan-2-yl)trimethoxysilane as an external donor. Three samples of BE–MET copolymers of similar ethylene concentrations (8.3, 13.8 and 16.7 mol%) were also prepared with a soluble metallocene catalyst19 (Table 1).
| Sample | Eta (mol%) | M v | M w/Mnc |
|---|---|---|---|
| a From 13C-NMR analysis. b From the values of intrinsic viscosity (ESI†). c From GPC. | |||
| BE1 whole | 9.1 | 378![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
|
| BE1, sEE | 16.3 | 57800 |
6.2 |
| BE1, iEE | 7.6 | 418300 |
2.7 |
| BE2 whole | 14.3 | 279200 |
|
| BE2, sEE | 16.7 | 297300 |
5.1 |
| BE2, iEE | 12.8 | 391500 |
3.0 |
| BE–MET1 | 8.3 | 480000 |
2.2 |
| BE–MET2 | 13.8 | 366800 |
2.3 |
| BE–MET3 | 16.7 | 388819 |
2.1 |
The BE–ZN samples have been fractionated by exhaustive Kumagawa extraction using sequentially boiling diethyl ether (EE) and hexane (HE) and two fractions, soluble in diethyl ether (sEE) and insoluble in diethylether/soluble in hexane (iEE), have been separated (Table 1). For both samples, the sEE fraction presents a higher ethylene concentration and polydispersity than the insoluble iEE fraction. Moreover, 13C-NMR analysis reveals a high concentration of the constitutional triad BBB (B = butene) in both fractions, in particular in the iEE fraction of the two samples, notwithstanding the high concentration of ethylene, resulting in high average lengths of butene sequences (Table S1†). This indicates a non-random distribution of comonomers and their segregation.
The X-ray diffraction profiles of samples of the unfractionated BE–ZN copolymers and of the corresponding sEE and iEE fractions and of BE–MET copolymers crystallized by cooling from the melt at 10 °C min−1 and after aging at room temperature for a long period of time are shown in Fig. 1. The whole BE–ZN samples are crystallized in mixtures of form I and form II of iPB, as indicated by the presence of the (200)II, (220)II and (213)II + (311)II reflections of form II at 2θ = 11.9, 16.9 and 18.3°, respectively, and the (110)I, (300)I and (220)I + (211)I reflections of form I at 2θ = 9.9, 17.3 and 20.5° in the diffraction profiles a and d of Fig. 1A. The sEE fractions are almost amorphous, and the fraction sEE of the sample BE1 with 16.3 mol% of ethylene presents very small amounts of crystals of forms I and II (profile b of Fig. 1A). The iEE fractions are, instead, crystallized in form II of iPB (profiles c and f of Fig. 1A). Therefore, crystals of forms I and II in the unfractionated samples are clearly due to the crystallization of the iEE fractions (Fig. 1A) and, for the BE1 sample, also marginally due to the crystallization of the sEE fraction (Fig. 1A).
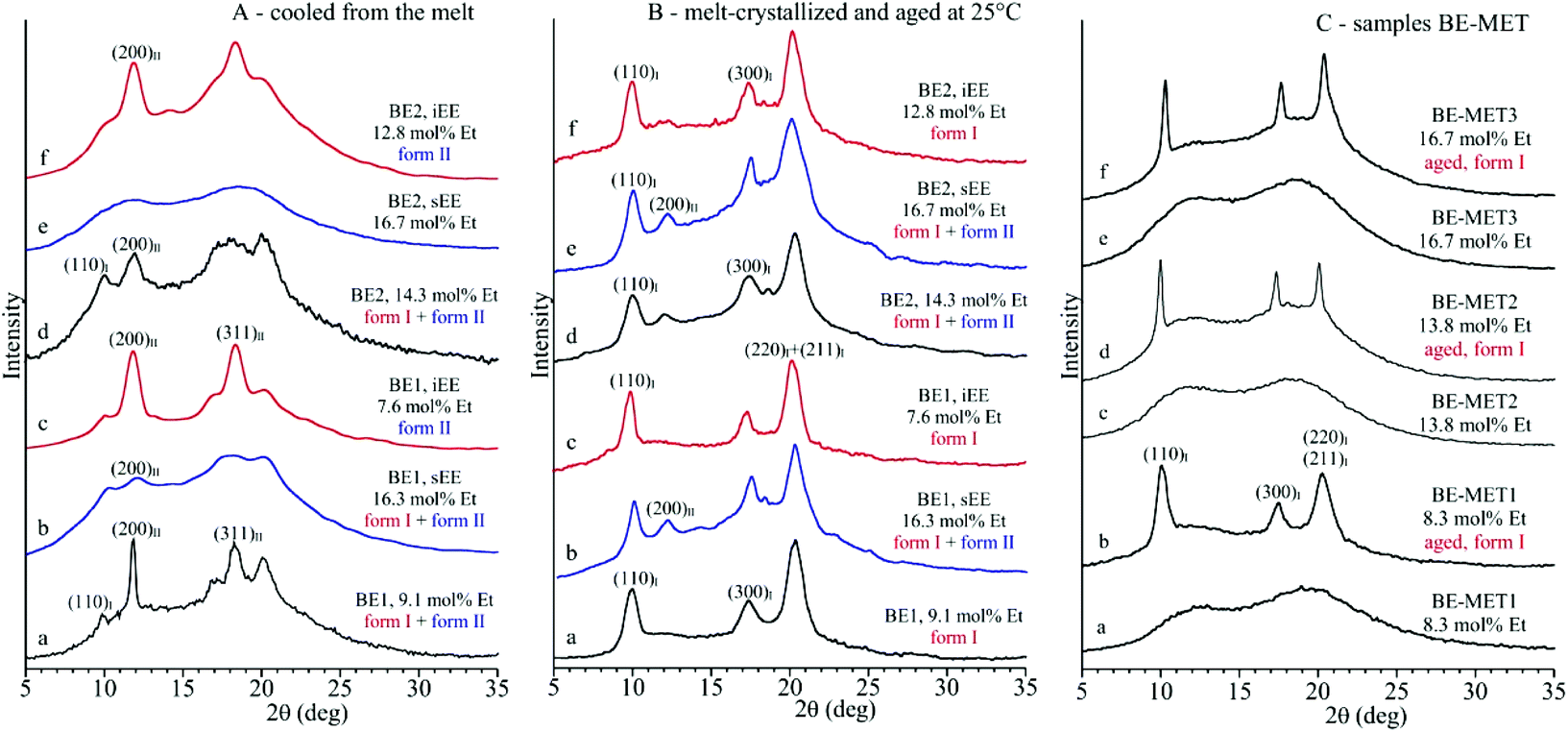 | ||
| Fig. 1 X-ray powder diffraction profiles of samples of BE–ZN and their sEE and iEE fractions (A, B) and BE–MET (C) copolymers cooled from the melt (A, C) and aged at room temperature for a long period of time (B, C) to allow transformation of form II into form I or crystallization of amorphous samples. The (110)I, (300)I and (220)I + (211)I reflections of form I of iPB at 2θ = 9.9°, 17.3° and 20.5° and the (200)II and (311)II reflections of form II of iPB at 2θ = 11.9° and 18.3°, respectively, are indicated. | ||
The three BE-MET samples cooled from the melt are, instead, amorphous because they do not crystallize from the melt (profiles a, c, and e of Fig. 1C),19 although the ethylene concentrations are similar to those of the BE–ZN sample. The fact that BE–MET samples are amorphous, whereas the samples of BE–ZN copolymers with similar ethylene concentrations are still crystalline (Fig. 1), confirms the non-random distribution of comonomers in BE–ZN copolymers that produces long butene sequences able to crystallize even at a high ethylene concentration and the perfectly random and uniform distribution of comonomers in BE–MET copolymers that strongly reduces the length of regular butene sequences preventing crystallization of form II from the melt. The amorphous samples of BE–MET, however, crystallize by ageing at room temperature in form I of iPB (profiles b, d, and f of Fig. 1C).19 In these BE–MET samples the crystallization of form II has never been observed.19
We recall that highly stereoregular samples of the iPB homopolymer always crystallize from the melt in the kinetically favored tetragonal form II22 (with chains in 11/3 helical conformation)23 that, then, spontaneously transforms at room temperature into the more stable trigonal form I (with chains in 3/1 helical conformation).24 As discussed above, a model of the crystallization behavior of iPB based on the length of regular butene sequences in model chains interrupted by the presence of randomly distributed stereodefects or comonomeric units has been recently proposed.16,17,19 Accordingly, crystallization of form II needs the presence of long regular butene sequences (the highly stereoregular and regioregular iPB homopolymer or iPB copolymers with low comonomer concentrations). Form II is destabilized and the direct crystallization of form I from the melt or from the amorphous phase is favored when the length of the regular butene sequences is short for the presence of stereo- or constitutional defects (stereodefective iPBs with the concentration of stereodefects higher than 2–3 mol%,16–18,22 or butene–ethylene copolymers with ethylene concentrations of 5–6 mol%),19,22 as in the case of BE–MET samples of Fig. 1C.
The DSC melting curves of the samples of Fig. 1A cooled from the melt and crystallized in form II (whole samples and iEE fractions) are shown in Fig. 2A and A′. These data indicate that crystals of form II of samples of copolymers of different ethylene concentrations melt at nearly the same temperature of 75–80 °C, which is almost independent of the ethylene concentration and lower than the melting temperature of 120 °C of form II of highly stereoregular iPB homopolymer samples.17,22
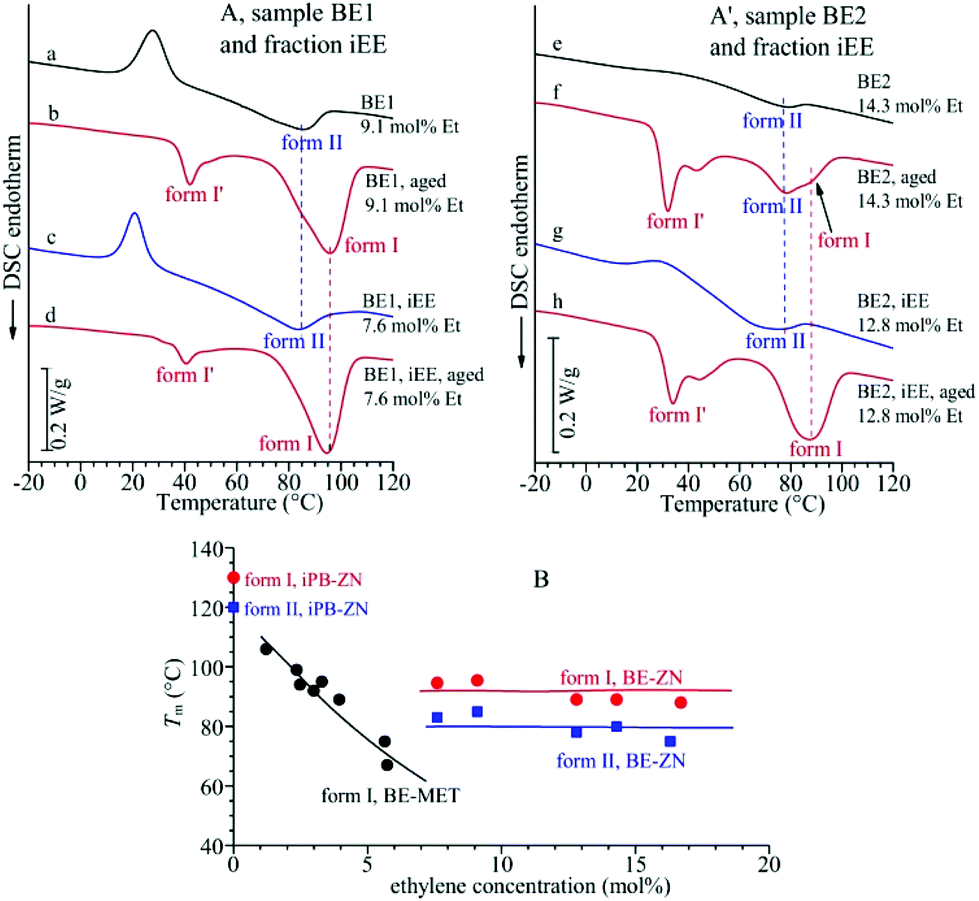 | ||
| Fig. 2 (A, A′) DSC heating curves of the unfractionated samples BE1 (a, b) and BE2 (e, f) and their iEE fractions (c, d, g, h) crystallized by cooling from the melt (a, c, e, g) and aged at room temperature (b, d, f, h). (B) Values of the melting temperature of crystals of form II and form I in melt-crystallized and aged samples of BE–ZN copolymers and their iEE fractions in comparison with the melting temperature of form I of BE–MET copolymers taken from ref. 19. | ||
The diffraction profiles of the BE–ZN samples cooled from the melt (Fig. 1A) and aged at room temperature for a long period of time (Fig. 1B) show that crystals of form II formed in the unfractionated samples (profiles a and d of Fig. 1A), in the sEE fraction of the BE1 sample (profile b of Fig. 1A) and in the iEE fractions of both samples BE1 and BE2 (profiles c and f of Fig. 1A), transform into form I by ageing at room temperature for several days (Fig. 1B). The amorphous BE–MET samples crystallize by ageing at room temperature in form I (Fig. 1C). The values of the degree of crystallinity evaluated from the diffraction profiles of Fig. 1 of samples cooled from the melt and of samples aged at room temperature are reported in Table S2 of the ESI.†
The DSC melting curves of these aged samples, reported in Fig. 2A and A′, show that these crystals of form I obtained by transformation of form II melt at nearly the same temperature of 90–95 °C, which seems to be almost independent of the ethylene concentration, whereas form I of the iPB homopolymer melts at 130 °C.17,22 The constant melting temperatures of form II and form I of BE–ZN copolymers are compared in Fig. 2B with the melting temperature of crystals of form I (obtained from transformation of form II) of aged samples of BE–MET,19 which decreases with the ethylene concentration. The values of the melting temperature and enthalpy of crystals of form II and form I and of the degree of crystallinity evaluated from DSC curves of Fig. 2 in BE–ZN and BE–MET copolymers are also reported in Table S2 of the ESI.†
The strange behavior of the BE–ZN copolymers that crystallize in form II of iPB even in samples containing very high concentrations of ethylene of 14–16 mol% (Fig. 1A), compared to metallocene samples that crystallize in form I in samples with 5–6 mol% of ethylene and do not crystallize for higher ethylene concentrations19 (Fig. 1C), is the hallmark of the presence of long crystallizable butene sequences in BE–ZN samples and much shorter butene sequences in metallocene BE–MET samples. At the same ethylene concentration, the regular crystallizable butene sequences along the chains of BE–ZN samples are always longer than those in the chains of perfectly random metallocene BE–MET copolymers. This is in agreement with the non-random and non-uniform intra- and inter-molecular distribution of comonomers and the presence of segregated defects in the BE–ZN samples. Moreover, the constant melting temperatures of crystals of form II and form I (Fig. 2B) that are nearly independent of the ethylene concentration indicate that the length of the regular crystallizable butene sequences is almost the same in the different fractions of the BE–ZN samples and give evidence of a blocky molecular structure of these Ziegler–Natta copolymers (Fig. 3).
 | ||
Fig. 3 Scheme of the crystallization behaviour of chains of iPB copolymers with a non-random distribution of ethylene comonomeric units ( ) that produce blocks with short or long butene ( ) that produce blocks with short or long butene ( ) sequences that crystallize in form I or form II of iPB, respectively. ) sequences that crystallize in form I or form II of iPB, respectively. | ||
This is confirmed by the similar values of 9–10 nm of the average size of crystals of form II that crystallize from the melt and of crystals of form I in melt-crystallized and aged samples of the different fractions of BE–ZN copolymers, reported in Table S3 of the ESI.† These values have been evaluated from the widths of the (200)II reflection of form II at 2θ = 11.9° and of the (110)I reflection of form I at 2θ = 9.9° in the diffraction profiles of Fig. 1. The size evaluated from the (200)II reflection of form II corresponds to the size along the a axis of the tetragonal crystals of form II, whereas the size evaluated from the (110)I reflection of form I corresponds to the size along the direction perpendicular to the (110) plane of the trigonal crystals of form I, and are representative of the average size of the crystals. The similar sizes of 9–10 nm for crystals of form II and form I in both samples BE1 and BE2 and in their fractions sEE and iEE (Table S3†), regardless of the different ethylene concentrations, is in agreement with the constant melting temperatures of crystals of form II and form I, which are independent of the ethylene concentration (Fig. 2B and Table S2†), and confirms that the length of the regular crystallizable butene sequences is almost the same in the different fractions of the BE–ZN samples. In the case of metallocene BE–MET copolymers, the size of crystals of form I, instead, is different in the three samples because the different ethylene concentrations produce very different lengths of crystallizable butene sequences (Table S3†).
These data can be explained considering the structure of the ZN catalyst and the reaction mechanism (a model of the ZN catalytic site is shown in the ESI, Fig. S1†). The presence of different active catalytic sites on the surfaces of the Ziegler–Natta catalyst produces heterogeneous samples characterized by mixtures of macromolecules each of them featuring a non-random distribution of the monomeric units along the polymer chain and non-uniform composition. The samples are separable in fractions containing chains grown from different sites having different molecular masses and polydispersities and comonomer concentrations. All fractions are composed of chains containing multiblocks where the comonomers are segregated, with blocks characterized by long butene sequences and low ethylene concentration that crystallize in form II of iPB (Fig. 3) and blocks with shorter butene sequences and higher ethylene concentration that may crystallize in form I of iPB (Fig. 3) or may not. The average length of crystallizable butene sequences is nearly the same in the different fractions of the different samples, even for large differences in ethylene concentrations (at least in the range of the analyzed composition), according to the constant melting temperatures of form II and form I that do not depend on the ethylene concentration. The main difference between the different fractions is the relative amount of the constituting blocks; the fractions soluble in ether contain prevailingly blocks with short butene sequences and the insoluble fractions contain mainly the blocks with long butene sequences. The surprising result that the length of crystallizable butene sequences is nearly the same regardless of the feed comonomer concentration indicates that for the more isospecific catalytic sites that produce long crystallizable butene sequences (Fig. S1A†) the “local” product of the reactivity ratio r1r2 is nearly the same and does not change with the feed comonomer composition. These results demonstrate that fine details of the molecular structure of polyolefins and copolymers of olefins may be obtained through the analysis of the crystallization behavior. In this case, the crystallization properties and the comparison with model polymers provide strong evidence of a blocky molecular structure of butene–ethylene copolymers induced by Ziegler–Natta catalysts. This molecular architecture and the perspectives opened by the possible control of the block lengths by a suitable choice of internal and external donors and conditions of polymerization are of particular relevance for tailoring the mechanical and elastomeric properties of these materials.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Boor, Ziegler-Natta catalysts and polymerization, Academic Press, New York, 1979 Search PubMed.
- Polypropylene Handbook, ed. N. Pasquini, Hanser Publishers, 2005 Search PubMed.
- P. C. Barbè, G. Cecchin and L. Noristi, Adv. Polym. Sci., 1987, 81, 1 Search PubMed.
- J. C. Chadwick, Ziegler–Natta Catalysts, in Encyclopedia of Polymer Science and Technology, Wiley, 2003, vol. 8, p. 517 Search PubMed.
- P. Corradini, V. Busico and G. Guerra, Monoalkene Polymerization: Stereospecificity, in Comprehensive Polymer Science, ed. G. Allen and J. C. Bevington, Pergamon Press, Oxford, 1988, vol. 4, pp. 29–50 Search PubMed.
- V. Busico and R. Cipullo, Prog. Polym. Sci., 2001, 26, 443 CrossRef CAS.
- V. Busico, R. Cipullo, P. Corradini and R. De Biasio, Macromol. Chem. Phys., 1995, 196, 491 CrossRef CAS.
- Y. Doi, Makromol. Chem., Rapid Commun., 1982, 3, 635 CrossRef CAS.
- V. Busico, P. Corradini, L. De Martino, F. Graziano and A. Iadicicco, Macromol. Chem. Phys., 1991, 192, 49 CrossRef CAS.
- P. Pino and R. Mulhaupt, Angew. Chem., Int. Ed., 1980, 19, 857 CrossRef.
- V. Busico, R. Cipullo, P. Corradini, L. Landriani, M. Vacatello and A. L. Segre, Macromolecules, 1995, 28, 1887 CrossRef CAS.
- V. Busico, R. Cipullo, G. Talarico, A. L. Segre and J. C. Chadwick, Macromolecules, 1997, 30, 4786 CrossRef CAS.
- V. Busico, R. Cipullo, G. Monaco, G. Talarico, M. Vacatello, J. C. Chadwick, A. L. Segre and O. Sudmejier, Macromolecules, 1999, 32, 4173 CrossRef CAS.
- V. Busico, P. Corradini, R. De Biasio, L. Landriani and A. L. Segre, Macromolecules, 1994, 27, 4521 CrossRef CAS.
- V. Busico, R. Cipullo, G. Monaco, M. Vacatello and A. L. Segre, Macromolecules, 1997, 30, 6251 CrossRef CAS.
- C. De Rosa, F. Auriemma and L. Resconi, Angew. Chem., Int. Ed., 2009, 48, 9871 CrossRef CAS.
- C. De Rosa, F. Auriemma, O. Ruiz de Ballesteros, F. Esposito, D. Laguzza, R. Di Girolamo and L. Resconi, Macromolecules, 2009, 42, 8286 CrossRef CAS.
- C. De Rosa, F. Auriemma, M. Villani, O. Ruiz de Ballesteros, R. Di Girolamo, O. Tarallo and A. Malafronte, Macromolecules, 2014, 47, 1053 CrossRef CAS.
- C. De Rosa, O. Ruiz de Ballesteros, F. Auriemma, R. Di Girolamo, C. Scarica, G. Giusto, S. Esposito, S. Guidotti and I. Camurati, Macromolecules, 2014, 47, 4317 CrossRef CAS.
- C. De Rosa, O. Tarallo, F. Auriemma, O. Ruiz de Ballesteros, R. Di Girolamo and A. Malafronte, Polymer, 2015, 73, 156 CrossRef CAS.
- O. Tarallo, O. Ruiz de Ballesteros, A. Bellissimo, M. Scoti, A. Malafronte, F. Auriemma and C. De Rosa, Polymer, 2018, 158, 231 CrossRef CAS.
- C. De Rosa, F. Auriemma, A. Malafronte and M. Scoti, Polym. Cryst., 2018, 1, e10015 Search PubMed.
- A. Turner-Jones, J. Polym. Sci., Part B: Polym. Lett., 1963, 1, 455 CrossRef CAS.
- G. Natta, P. Corradini and I. W. Bassi, Nuovo Cimento, 1960, 15, 52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of polymer synthesis, molecular characterization and analysis of the Ziegler–Natta catalytic sites. See DOI: 10.1039/c9py01485c |
| This journal is © The Royal Society of Chemistry 2020 |