
Additive manufacture of lightly crosslinked semicrystalline thiol–enes for enhanced mechanical performance†
Kimberly K.
Childress
a,
Marvin D.
Alim
 b,
Juan J.
Hernandez
a,
Jeffrey W.
Stansbury
*ac and
Christopher N.
Bowman
*ab
b,
Juan J.
Hernandez
a,
Jeffrey W.
Stansbury
*ac and
Christopher N.
Bowman
*ab
aDepartment of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Avenue, Boulder, Colorado 80303, USA. E-mail: jeffrey.stansbury@cuanschutz.edu; christopher.bowman@colorado.edu
bMaterials Science and Engineering Program, University of Colorado Boulder, 596 UCB, Boulder, Colorado 80309, USA
cDepartment of Craniofacial Biology, School of Dental Medicine, University of Colorado Denver, 12800 East 19th Avenue, Aurora, Colorado 80045, USA
First published on 3rd December 2019
Abstract
Photopolymerizable semicrystalline thermoplastics resulting from thiol–ene polymerizations were formed via fast polymerizations and achieved excellent mechanical properties. These materials have been shown to produce materials desirable for additive manufacturing (3D printing), especially for recyclable printing and investment casting. However, while well-resolved prints were previously achieved with the thiol–ene thermoplastics, the remarkable elongation at break (εmax) and toughness (T) attained in bulk were not realized for 3D printed components (εmax,bulk ∼ 790%, Tbulk ∼ 102 MJ m−3vs. εmax,print < 5%, Tprint < 0.5 MJ m−3). In this work, small concentrations (5–10 mol%) of a crosslinker were added to the original thiol–ene resin composition without sacrificing crystallization potential to achieve semicrystalline, covalently crosslinked networks with enhanced mechanical properties. Improvements in ductility and overall toughness were observed for printed crosslinked structures, and substantial mechanical augmentation was further demonstrated with post-manufacture thermal conditioning of printed materials above the melting temperature (Tm). In some instances, this thermal conditioning to reset the crystalline component of the crosslinked prints yielded mechanical properties that were comparable or superior to its bulk counterpart (εmax ∼ 790%, T ∼ 95 MJ m−3). These unique photopolymerizations and their corresponding monomer compositions exhibited concurrent polymerization and crystallization along with mechanical properties that were tunable by changes to the monomer composition, photopolymerization conditions, and post-polymerization conditioning. This is the first example of a 3D printed semicrystalline, crosslinked material with thermally tunable mechanical properties that are superior to many commercially-available resins.
While other material options exist, vat-based 3D printing reliant on photopolymerization is primarily focused on (meth)acrylate-based formulations for their combined qualities of speed of cure, range of mechanical properties, and cost.1 However, shrinkage stress,2 oxygen inhibition that generates prints with tacky surfaces,3 and incomplete cure due to early gelation hinder the performance of the final manufactured component. To circumvent these substantial disadvantages, thiol–ene based chemistries were investigated as a replacement for conventional acrylic monomers.4 Thiol–enes maintain the excellent spatiotemporal control inherent to photo-induced polymerizations while also incorporating the stereospecificity and oxygen insensitivity of a “click” reaction.5 Compared to chain-growth acrylate polymerizations, thiol–enes polymerize via a step-growth mechanism, which affords many advantages including delayed gelation, higher monomer conversion, and a reduction in shrinkage stress.6 Despite the numerous advantages of thiol–ene polymerizations, its implementation for 3D printing is in its infancy.7–13
Recently, a thermoplastic thiol–ene system composed of commercially-available 1,6-hexanedithiol (HDT) and diallyl terephthalate (DAT) monomers with previously unprecedented mechanical properties was reported.14 High molecular weight linear polymers (Mn ∼ 104 g mol−1) were achieved at low intensities in a matter of seconds, thus encouraging its consideration as a printable material. Structures with fine resolution and complex features were successfully printed and readily reformed above a relatively low melting temperature (Tm) (80 °C). This behavior was demonstrative of its potential as a reprocessable printing material for a variety of applications ranging from investment casting to recyclable consumer goods. Unfortunately, the impressive mechanical properties observed in bulk molded samples were significantly reduced when formed via layered printing. While the main advantage attributed to this printing formulation is its ease of melting, the additional resilience of the mechanics demonstrated in bulk would expand its potential applications. With some exceptions, the mechanical properties of 3D printed parts tend to fall short of their bulk cured counterparts. However, the discrepancy observed here between bulk and printed thiol–ene thermoplastic parts was substantial (e.g. εmax,bulk ∼ 790% vs. εmax,print < 5%). Many viable explanations for the considerable deterioration of the printed part's integrity exist. These poor mechanical properties were initially hypothesized to stem from the printing resin itself, wherein the incorporation of a photoabsorber reduced the molecular weight and disrupted the reinforcement associated with the development of semicrystalline domains post-polymerization. The formation of residual stresses during printing15 and the development of regions with limited adhesion between the printed layers16,17 further weakened the performance of the printed structure.
In this work, small amounts of a crosslinker were incorporated into a stoichiometric thiol–ene resin formulation as a means to manipulate the polymerization-induced crystallization process, facilitate adhesion between the layers, and ultimately control the bulk mechanical property performance through alterations in the monomer composition (Fig. 1). Polymerization kinetics, crystallization rates and amounts, and modulus development post-polymerization were analyzed to assess structure–property relationships in these materials. Formulations with low crosslinker concentrations were 3D printed and mechanically contrasted with the original linear, thermoplastic thiol–ene resins. Continuous thermal treatment post-manufacture was applied to the thermoplastic and crosslinked prints and investigated as a means to further enhance the final mechanical properties. Examples of annealing in literature previously employed heat at temperatures between the glass transition temperature (Tg) and Tm,18,19 which improved yield strength and tensile modulus.12,20–22 Here, the addition of crosslinking to the resin permitted heating at temperatures well above Tm without significant physical deformation of the print. Changes in crystallinity for the crosslinked system with thermal conditioning were monitored, and potential structure–property relationships are proposed.
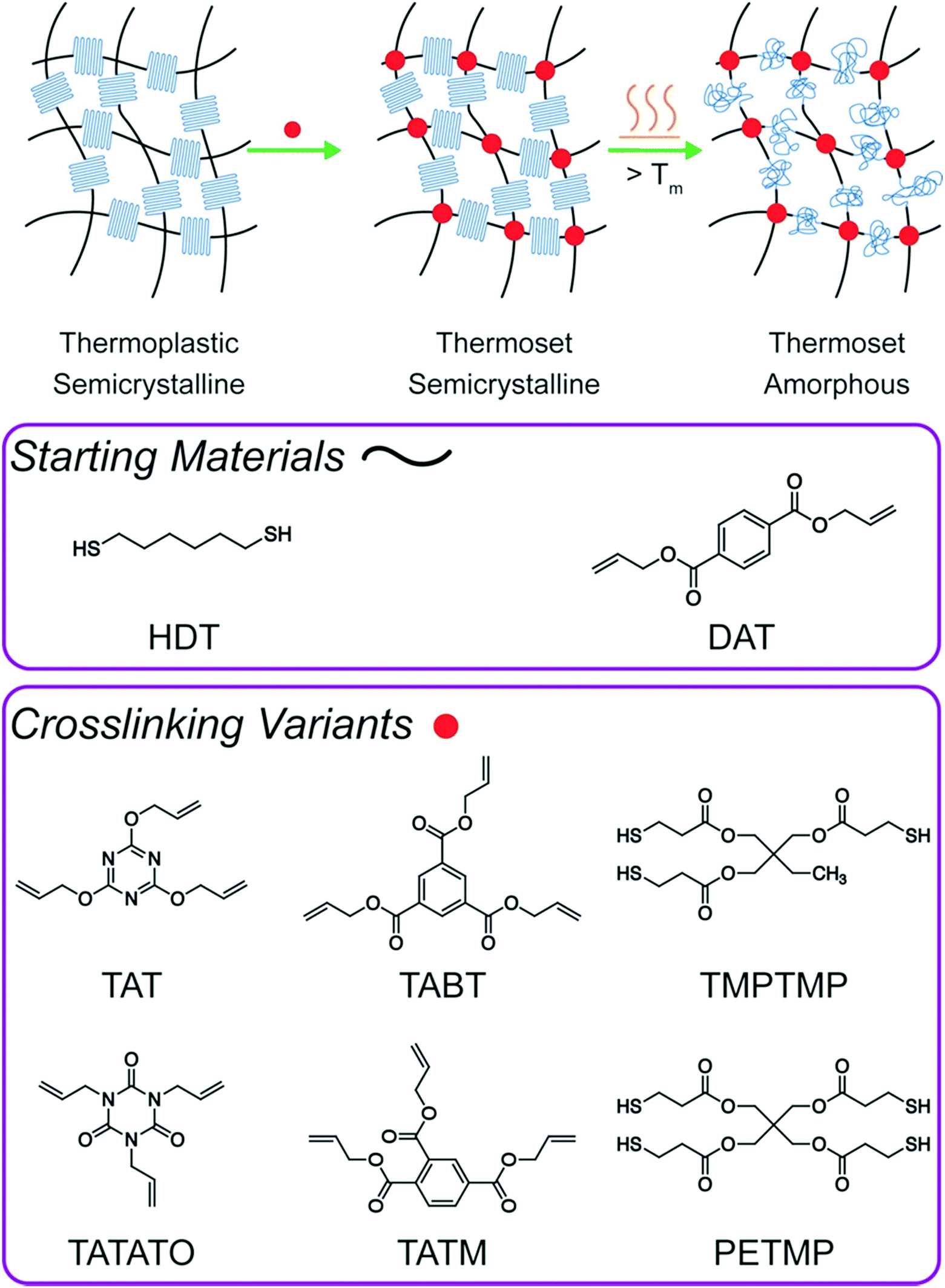 | ||
| Fig. 1 Schematic of the photopolymerization approach used produce semicrystalline, crosslinked thiol–ene materials to improve the mechanical performance of printed components. A stoichiometric amount of crosslinker was included in the original HDT–DAT formulation at concentrations low enough to maintain crystallinity. Several prints were processed post-manufacture by heating above Tm and conditioning at that temperature until the crystallites melted to form an amorphous network. | ||
An initial survey of various thiol or alkene chemical crosslinkers added to the stoichiometric linear thiol–ene formulation14 of HDT and DAT was used to establish which crosslinkers were suitable from a kinetics perspective while still enabling sufficient crystallization to occur as needed for achievement of the desired mechanical behavior. Trimethylolpropane tris(3-mercaptopropionate) (TMPTMP), pentaerythritol tetrakis(3-mercaptopropionate) (PETMP), 1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATATO), 2,4,6-triallyloxy-1,3,5-triazine (TAT), triallyl trimellitate (TATM), and triallyl 1,3,5-benzenetricarboxylate (TABT) were incorporated at 5 and 10 mol% relative to its thiol or alkene counterpart. ASTM D638 Type V dogbones cut from bulk films approximately 500 μm thick were uniaxially strained at 5 mm min−1 until fracture. Opacity remained apparent for all crosslinkers investigated but to a lesser degree than bulk HDT–DAT. This outcome is typically observed as increasing the crosslinking density reduces a sample's degree of crystallinity and crystallite thickness,23–25 with concomitant reductions for several mechanical properties, including Young's modulus, yield stress, elongation at yield, and elongation at break.26 This trend of reduced mechanical properties was consistently observed for all crosslinkers studied here (Table S1†). Additionally, the appearance of the characteristic clear and opaque striations observed for HDT–DAT and the coinciding periodic dips in tensile stress during strain decreased both in frequency and amplitude with increasing crosslinker content (Fig. S1 and S2†). From approximately 160% strain until failure, large dips in the stress–strain profile were observed for linear HDT–DAT with an average drop in stress of approximately 1.4 MPa. Addition of either 5 or 10 mol% of the trifunctional crosslinker TAT reduced the number of dips in the stress, and the average stress drop was reduced to 0.47 MPa and 0.09 MPa, respectively. These dips, which are believed to correlate to the opaque striations in the sample, intermittently develop during molecular chain orientation (i.e. strain hardening). In this regime of plastic deformation, the individual crystalline segments are separated from the lamellae after lamellae rotation in the load direction. Continued loading may cause the crystalline regions to separate further and form individual, though still interconnected, fibrils.27 It is hypothesized that the extreme plastic deformation of these materials is afforded by its ability to form these fibrils, and the coinciding lamellae slippage results in a recurring release of stress. TAT was selected as the printing monomer due its similar kinetic and crystalline properties (detailed below) and the relatively minor mechanical reductions observed in bulk. Formulations with functional molar group ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.95:0.05 HDT–DAT–TAT (5 mol% TAT) and 1:0.9:0.1 HDT–DAT–TAT (10 mol% TAT) were investigated and compared to the original 1:1 HDT–DAT (0 mol% TAT) thermoplastic.
:0.95:0.05 HDT–DAT–TAT (5 mol% TAT) and 1:0.9:0.1 HDT–DAT–TAT (10 mol% TAT) were investigated and compared to the original 1:1 HDT–DAT (0 mol% TAT) thermoplastic.
Polymerization kinetics for samples with varying TAT concentrations were monitored in real-time with Fourier-Transform Infrared Spectroscopy (FTIR). Rapid kinetics and near quantitative conversion of both the thiol and ene groups were observed for all samples after brief (10 s) irradiation with a 405 nm LED (1 mW cm−2). Despite having almost indistinguishable rates of polymerization, the final conversion of the thiol group slightly decreased (from >99.5% conversion with 0 mol% TAT to approximately 99 and 98% conversion with 5 and 10 mol% TAT, respectively) with increasing TAT concentration, which likely results from decreased thiol mobility as gelation occurs toward the end of the reaction. After polymerization, crystallization of 1:1 HDT–DAT was observed via the optical transition of the bulk film from clear to opaque. This temporal development remained visible with crosslinker addition, albeit to a lesser degree. Differential scanning calorimetry (DSC) confirmed the presence of crystalline domains in the crosslinked samples via the appearance of distinct Tm peaks (Fig. 2B). Relative degrees of crystallinity were determined by normalizing to the heat of melting for 0 mol% TAT, and approximately 86% and 64% crystallinity remained after the addition of 5 and 10 mol% TAT, respectively. Thermogram inspection revealed an extensive change in the Tm profile for increasing TAT concentrations. Initially, two nearly separate Tm regimes were present for samples without crosslinker, which is indicative of the development of domains with different crystallite sizes.12 The addition of a crosslinker remarkably altered both the crystallite size distribution and the maximum Tm,28,29 with the thermogram revealing the homogenization of crystallite size with increasing crosslinking content. The increasingly uniform transition temperatures exhibited a decrease in Tm (reduced from 62 and 79 °C for samples with 0 mol% TAT to 52 and 46 °C for samples with 5 and 10 mol% TAT, respectively), which suggests that crystal growth in a crosslinked network yielded smaller crystallites.30 Polarized optical microscopy (POM) was used to further confirm the development of crystalline domains within the crosslinked networks. POM images of bulk samples were taken several hours after brief (2 s) irradiation with a 405 nm LED bar at ∼7 mW cm−2 (Fig. 2C). In agreement with that previously observed for the thiol–ene thermoplastic, characteristic crossed spherulites were visible for all crosslinking densities. Spherulite growth was also imaged in situ with cross polarizers upon brief (1 s) irradiation with the microscope's 405 nm LED source (Fig. S3†). Shortly after irradiation and subsequent polymerization, spherulite growth for the non-crosslinked sample ensued for several minutes until impingement with other spherulites prevented further development. A considerable reduction in the rate of crystallization occurred with higher concentrations of TAT due to molecular motion restrictions imposed by the crosslinked network.
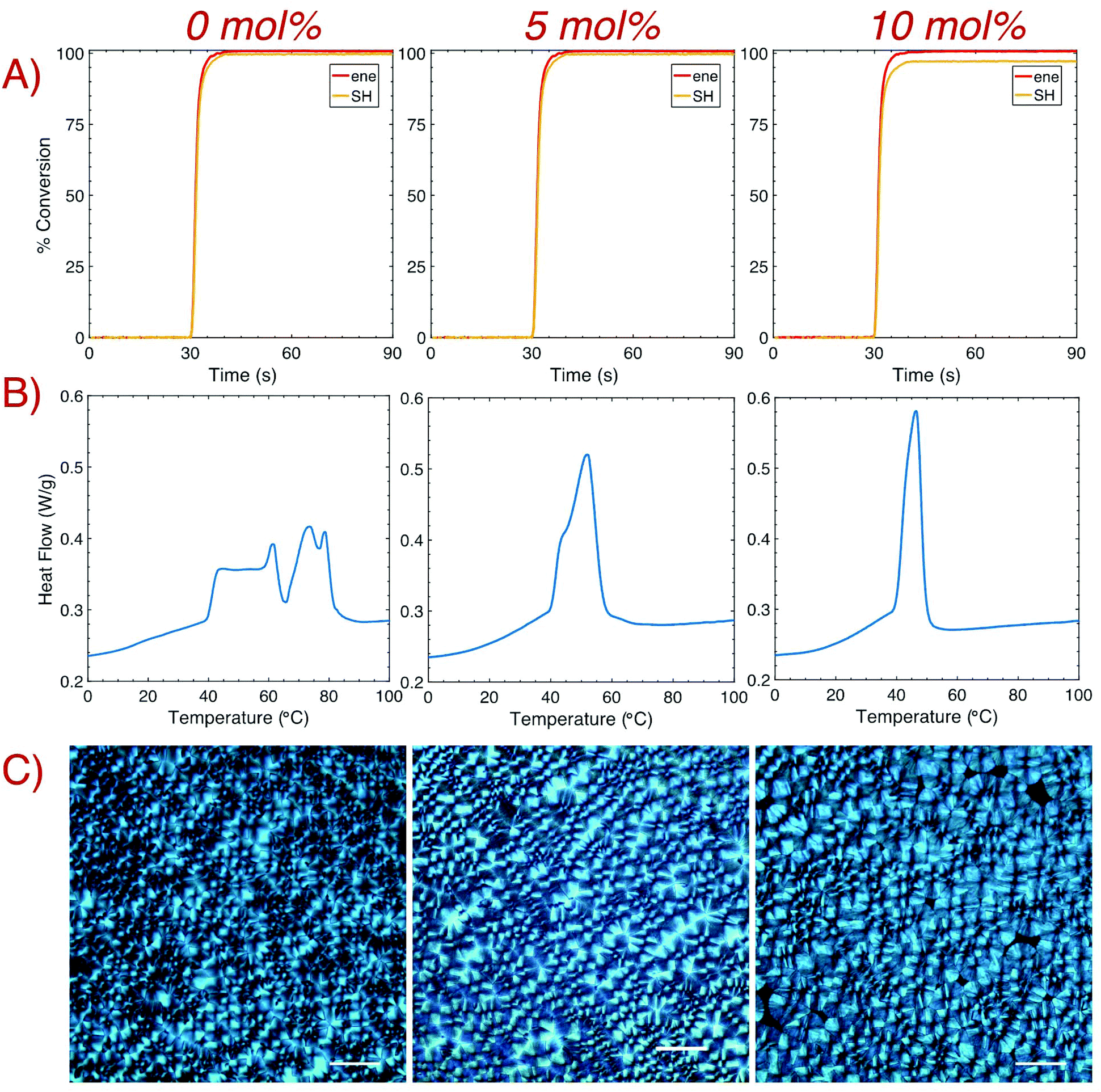 | ||
| Fig. 2 Characterization of polymerization and subsequent crystallization with the addition of the crosslinker TAT. (A) Polymerization kinetics were obtained via real-time Fourier-transform infrared spectroscopy (FTIR) after irradiation with a 405 nm LED for 10 s (1 mW cm−2). No significant change in the polymerization rate was observed with increasing crosslinker content, although the final SH conversion slightly decreased from >99% conversion without TAT to ∼97% with 10 mol% TAT. (B) Dynamic scanning calorimetry (DSC) was used to confirm the presence of crystalline domains and to determine Tm for each sample (exo down). The degree of crystallinity and the Tm,max decreased as the concentration of TAT increased. (C) Polarized optical microscopy (POM) was used to confirm crystallization of bulk samples with 0, 5, and 10 mol% TAT. Images were taken after crystallization ceased, and areas devoid of any crystals are visible for 10 mol% TAT (scale bar: 50 μm). | ||
Hindered crystallization rates with increasing TAT concentration were further confirmed indirectly through modulus development via real-time photo-rheology (Fig. 3). Samples both prepared in bulk and as printing resins were irradiated for 10 seconds with a 405 nm LED at 25 mW cm−2. A sharp increase in both the storage (G′) and loss (G′′) moduli upon irradiation was observed for all samples and is in agreement with the rapid polymerization rates obtained with FTIR. Consistent with previous findings,14G′ increased again for bulk 0 mol% TAT shortly after polymerization and eventually plateaued once crystallization was complete, whereas G′′ readings ceased soon after falling below the instrument detection limits. Complex viscosity (η*) was simultaneously measured and increased in a manner similar to G′ before also plateauing upon cessation of crystallization. The rate of crystallization was considerably reduced for bulk crosslinked samples as secondary modulus development was delayed and neither G′ nor η* plateaued in an hour. Faster rates of crystallization were observed for the printing resin due to the presence of additional nucleation sites provided by the dispersed photoabsorber carbon black (CB). The non-crosslinked printing material finished crystallizing in a few minutes, and crystallization for the 5 mol% and 10 mol% crosslinked printing materials was complete within approximately 30 and 60 minutes, respectively. Crystallization was greatly expedited with the addition of a small amount of CB (0.1 wt%), but complete crystallization of the printed layer would not occur before the next layer polymerized when implementing reasonable exposure times. This is especially true for crosslinked printing resins in which the half-life for the maximum degree of crystallinity increased from only approximately 3 minutes for the linear system to 13 minutes for 5 mol% TAT and more than 30 minutes for 10 mol% TAT (G′ and η* did not plateau within 60 minutes). While complete crystallization would not occur for any of the printing resins when implementing reasonable layer exposure times (<30 s), initial crystallite formation may harden print interfaces enough to substantially hinder interlayer adhesion, especially for systems without crosslinker. It is worth noting that, while CB is typically loaded into rubbers and elastomeric matrices at greater than 10 wt% to increase mechanical properties such as tensile strength and ductility,31 it is not expected that the relatively small amount of CB incorporated here (0.1 wt%) would improve the mechanical performance of these materials. However, due to optical attenuation from the CB during film irradiation, it is difficult to obtain a direct mechanical comparison between the bulk materials with and without CB.
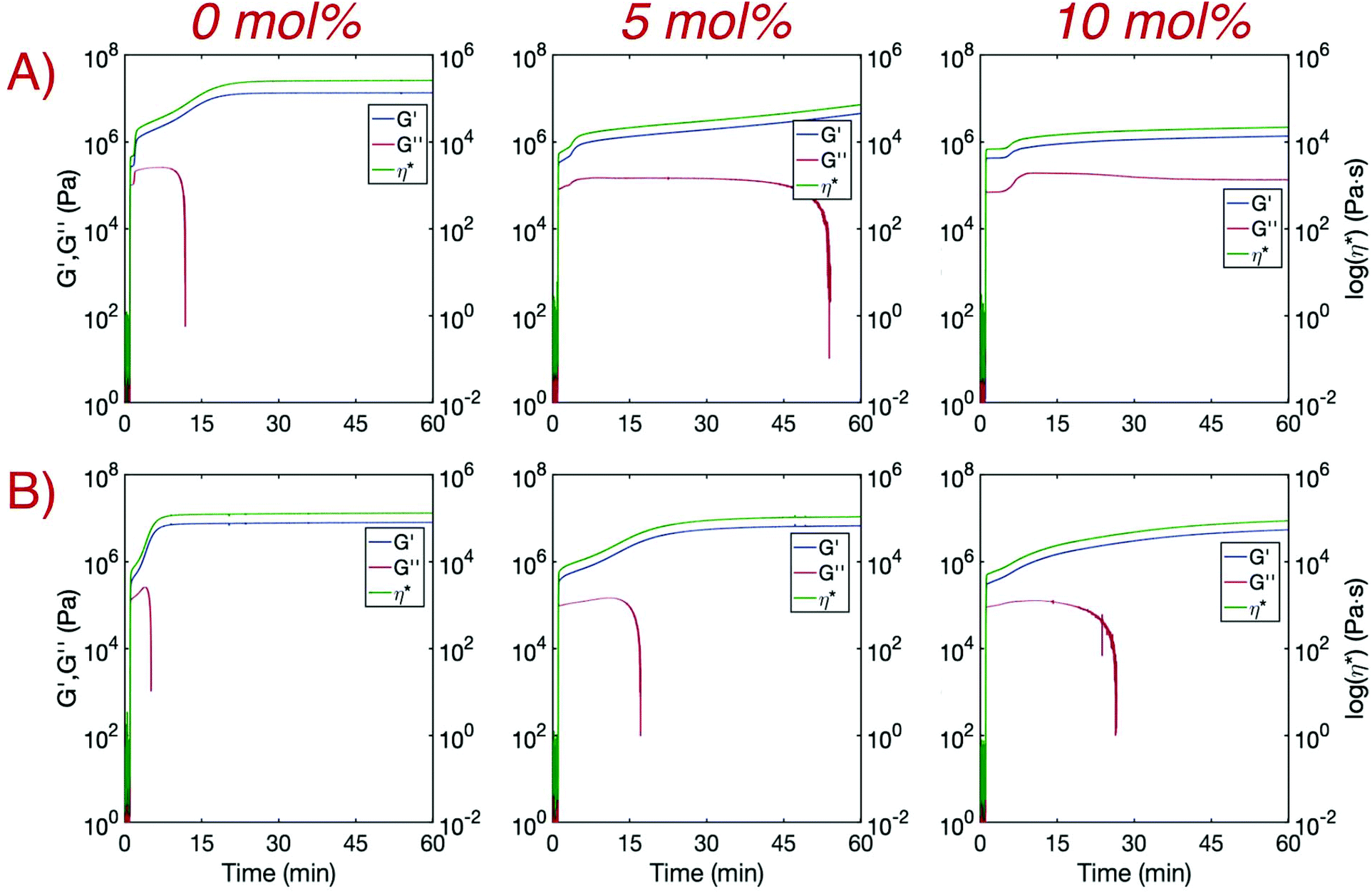 | ||
| Fig. 3 Real-time photo-rheology of (A) bulk and (B) printing resin samples consisting of 0, 5, and 10 mol% TAT, and printing samples also contained 0.1 wt% CB. After 405 nm irradiation at 25 mW cm−2 for 10 s, a rapid increase in storage (G′) and loss (G′′) moduli occurs, and a successive secondary development of G′ occurred during crystallization before plateauing. Similar behavior was concurrently observed for the complex viscosity (η*). Subsequent G′ and η* growth post-polymerization slowed with increasing crosslinking density but significantly increased with the addition of CB. | ||
A commercial LCD-based DLP 3D printer was used to print multiple ASTM D638 Type V dogbones with 0.1 wt% CB flat onto the build head (Fig. 4), and high-resolution features were obtained using SLA printing (Fig. S4†). While strain at break occurred at <5% for 1:1 HDT-DAT prints (Fig. S5†), the addition of TAT moderately increased the strain at break to approximately 36 and 50% for 5 and 10 mol% TAT, respectively (Table S2†). The Young's modulus (E) however decreased with increasing crosslinking density, corresponding with that observed in bulk. Despite the marginal improvement in ductility and toughness, the mechanical properties of the printed components still remained significantly inferior to those obtained in bulk. To address this discrepancy, continuous thermal conditioning was performed on samples with and without TAT in an attempt to further recover the diminished mechanics. This approach has been explored as a post-manufacturing technique to increase mechanical properties of the printed component by (i) increasing interfilament/interlayer adhesion and (ii) promoting recrystallization. Unlike previous work where annealing was conducted between Tg and Tm for thermoplastics, conditioning in this work was performed not only above Tg (−30 °C for all samples) but also above Tm for 2 hours before cooling at room temperature overnight. Samples without crosslinker were conditioned at 70 and 80 °C, and at these temperatures, no improvements were observed. As materials without crosslinker are thermoplastics and melt above their respective Tm, heating was not conducted above these temperatures. Samples with 5 mol% TAT were conditioned at 100 and 150 °C for 2 hours, while samples with 10 mol% TAT were also heated at 200 °C. The conditioning temperatures utilized here were well below that of the onset temperature (To ∼ 350 °C for all crosslinked prints), and minimal print deformation was observed (Fig. S6†). To test deformation, the dimensions of printed cubes (1 cm3) with 10 mol% TAT were recorded before and after conditioning at a maximum temperature of 200 °C for 2 hours. Less than 5% deformation was observed for the cube height, while a deformation of approximately 2% was observed for both the width and thickness. It is worth noting that when the temperature was progressively heated at intervals of 50 °C (100, 150, and 200 °C) and held at each temperature for half an hour, vertical and horizontal cube deformation was less than 1% and 0.5%, respectively. Therefore, thermal ramping as opposed to vigorously conditioning at high temperatures may be beneficial for some printed structures. For the tensile tests performed here, dogbones were not heated above the respective maximum conditioning temperature for each crosslinker concentration due to the onset of severe print deformation. Elongation improved by an order of magnitude for the conditioned 5 mol% TAT samples, increasing to approximately 280 ± 70 and 530 ± 30% when conditioned at 100 and 150 °C, respectively, with no substantial effect on E observed. Further mechanical improvements were attained for 10 mol% TAT samples, with εmax occurring at approximately 780 ± 10% when conditioned at 200 °C for 2 hours. Untreated 10 mol% TAT samples achieved an εmax that was also an order of magnitude higher than the printed material without crosslinker, while no appreciable changes in E and yield stress were observed with thermal treatment. Significant improvements in print mechanics were obtained for conditioned crosslinked samples, with εmax and toughness increasing a couple orders of magnitude from the original printed thermoplastic. Not only did the ductility of 10 mol% TAT samples conditioned at 200 °C increase beyond that observed for the untreated print, but a marked increase over that of the equivalent bulk sample (∼520 ± 30%) was also obtained. Remarkably, εmax and toughness (95 ± 1 MJ m−3) determined for printed 10 mol% TAT samples conditioned at 200 °C were statistically comparable with those obtained for bulk 1:1 HDT–DAT (∼790 ± 20%, 102 ± 9 MPa). While the prints were not elastic, the substantial ductility achieved after heating was superior to many commercially-available elastomeric photopolymer resins, including Formlab Elastic,32 Formlab Flexible,33 Stratasys TangoPlus,34 and Carbon EPU 40.35 The highest εmax reported for these commercial materials was ∼270% with an ultimate tensile stress (UTS) of 8 MPa (TangoPlus), falling well below the uppermost εmax and UTS (19.8 ± 0.3 MPa) achieved for the aggressively conditioned crosslinked thiol–ene prints. It is worth mentioning that the hardness of the thiol–ene prints (∼40 shore D) was well above that of the commercial resins (TangoPlus, 68 shore A) with the exception of Formlab Flexible (85 shore A). Even amongst the reported set of photopolymerized manufactured elastomers and thermosets emphasizing mechanical performance,12,36,37 only one elastomer exhibited superior ductility,38 albeit at a much lower E and UTS. Interestingly, the distinctive, periodic striations that appeared for the bulk materials reemerged after thermal conditioning for both the 5 and 10 mol% TAT samples.
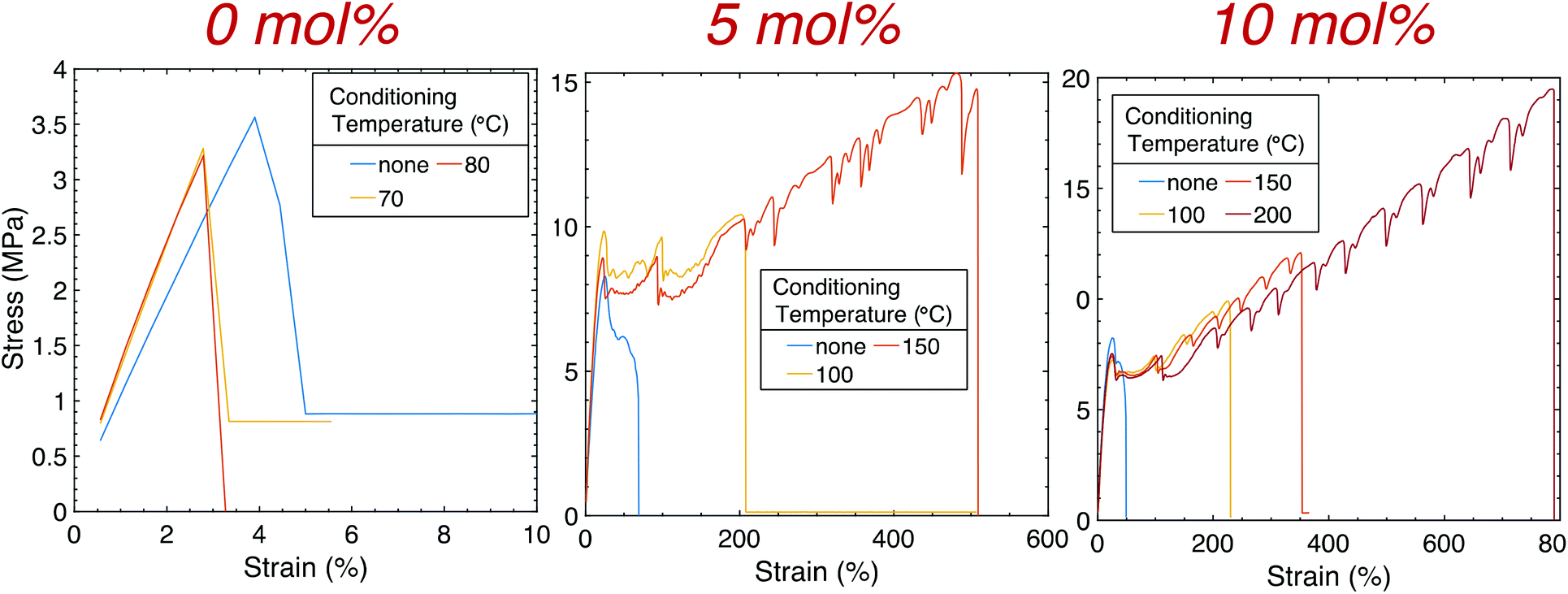 | ||
| Fig. 4 Tensile stress–strain plots of samples printed with 0, 5, and 10 mol% TAT. Thermally conditioning samples without crosslinker showed no improvement in mechanical properties, whereas conditioning crosslinked samples yielded immense enhancement of print ductility. ASTM D638 Type V dogbones were printed and uniaxially strained at 5 mm min−1. | ||
The substantial mechanical improvements observed after thermally conditioning the crosslinked prints was initially attributed to a combination of recrystallization post-heating and improved adhesion between the individual printed layers. Enhanced crystallization is known to improve mechanical properties,15,19 and thus DSC was used to approximate the extent of crystallization for each temperature explored for printed samples with 10 mol% TAT. While annealing at or below Tm previously increased the degree of crystallinity, no significant change (∼1–3% more crystalline after conditioning relative to equivalent untreated sample) in crystallinity occurred and thus recrystallization was not responsible for the improved mechanical performance. Rather, the macroscopic fusion of the individual layers into a continuous part was the primary source of the enhancement. At the low crosslinking densities explored herein, the material remained semicrystalline, thus facilitating a reciprocal relationship between the chemically crosslinked network and the crystalline scaffold. Initially, rapid polymerization and subsequent crystallization of the individual layers prevented sufficient interlayer adhesion, which was further exacerbated by the oxygen insensitivity that is inherent to thiol–enes.39 However, melting the crystalline domains promoted fusion between the printed layers, while the crosslinked network provided a permanent skeleton for recrystallization post-heating. In doing so, the interfacial stresses between layers were also removed, and the conditioned printed samples were able to withstand the stresses induced during cold-drawing.
Conclusions
In summary, semicrystalline, crosslinked materials demonstrating concurrent polymerization and crystallization were successfully photopolymerized. Rapid reaction kinetics and relatively high amounts of crystallinity were obtained with light amounts of crosslinking, while the rates of crystallization significantly varied with the addition of crosslinker and photoabsorber. The addition of the crosslinker TAT in small amounts (5 and 10 mol%) allowed for the partial recovery of the excellent mechanical properties exhibited for the bulk systems. Ductility and toughness were moderately improved by the addition of the crosslinker but remained well below that obtained in bulk, while substantial mechanical recovery was achieved by thermally conditioning the prints far above Tm for a relatively short period of time (2 hours). Crosslinked materials conditioned at their respective maximum temperatures exhibited enhancements in εmax and toughness, especially for samples with 10 mol% TAT. Not only did this semicrystalline, crosslinked printed material outperform its bulk counterpart, but its mechanical properties were comparable to those of the original bulk HDT–DAT thermoplastic. The attained mechanical properties of the heated prints greatly surpassed many commercially-available printing resins and several other materials reported in literature. These significant improvements were primarily attributed to melting of the crystalline domains above Tm and the subsequent fusion of the individual printed layers. Whereas raising temperature above the Tm would melt the thermoplastic print, heating the crosslinked printed structures at elevated temperature only melted the crystalline domains while the crosslinked network provided a permanent structure to prevent significant print deformation. To our knowledge, this is the first example of a printed, crosslinked semi-crystalline material with thermally-improved mechanics above Tm. The mechanical properties achieved herein make this a promising printing material for applications requiring plastics with high impact strength and preferable deformation over shear fracture.Conflicts of interest
A provisional patent on this work has been filed by the Venture Partners at the University of Colorado Boulder.Acknowledgements
This work was supported by NIH/NIDCR (R21DE028444) and NSF DMR 1809841. Imaging work was performed at the BioFrontiers Institute Advanced Light Microscopy Core. A Nikon Ti-E Microscope was used to perform super resolution microscopy and is supported by the Howard Hughes Medical Institute. The Microscopy Core was also used to conduct data analysis and visualization work. The Analysis Workstation was supported by NIH 1S10RR026680-01A1.We would like to acknowledge the team at Colorado Photopolymer Solutions (CPS), particularly Amelia Davenport, for useful discussions regarding our experimental procedures. We would also like to thank Dr. Parag Shah for his assistance with experimental measurements obtained for this manuscript.
References
- J. W. Stansbury and M. J. Idacavage, Dent. Mater., 2015, 32, 54–64 CrossRef PubMed.
- S. Schoerpf, Y. Catel, N. Moszner, C. Gorsche and R. Liska, Polym. Chem., 2019, 10, 1357–1366 RSC.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2009, 42, 2551–2556 CrossRef CAS PubMed.
- A. Oesterreicher, J. Wiener, M. Roth, A. Moser, R. Gmeiner, M. Edler, G. Pinter and T. Griesser, Polym. Chem., 2016, 7, 5169–5180 RSC.
- I. A. Barker, M. P. Ablett, H. T. J. Gilbert, S. J. Leigh, J. A. Covington, J. A. Hoyland, S. M. Richardson and A. P. Dove, Biomater. Sci., 2014, 2, 3–7 RSC.
- S. Bertlein, G. Brown, K. S. Lim, T. Jungst, T. Boeck, T. Blunk, J. Tessmar, G. J. Hooper, T. B. F. Woodfield and J. Groll, Adv. Mater., 2017, 1703404, 1–6 Search PubMed.
- H. Leonards, S. Engelhardt, A. Hoffman, L. Pongratz, S. Schriever, J. Bläsius, M. M. Wehner and A. Gillner, Proc. SPIE, 2015, 93530F Search PubMed.
- L. Chen, Q. Wu, G. Wei, R. Liu and Z. Li, J. Mater. Chem. C, 2018, 6, 11561–11568 RSC.
- D. G. Sycks, T. Wu, H. S. Park and K. Gall, J. Appl. Polym. Sci., 2018, 135, 46259 CrossRef.
- T. J. Wallin, J. H. Pikul, S. Bodkhe, B. N. Peele, B. C. Mac Murray, D. Therriault, B. W. McEnerney, R. P. Dillon, E. P. Giannelis and R. F. Shepherd, J. Mater. Chem. B, 2017, 5, 6249–6255 RSC.
- M. D. Alim, K. K. Childress, N. Baugh, A. M. Martinez, M. K. McBride, B. T. Worrell, B. D. Fairbanks, J. W. Stansbury, R. R. McLeod and C. N. Bowman, Mater. Horiz. 10.1039/c9mh01336a.
- M. Ivey, G. W. Melenka, J. P. Carey and C. Ayranci, Adv. Manuf.: Polym. Compos. Sci., 2017, 3, 81–91 CAS.
- H. Prajapati, D. Chalise, D. Ravoori, R. M. Taylor and A. Jain, Addit. Manuf., 2019, 26, 242–249 CrossRef CAS.
- M. S. Stark, Improving and Understanding Inter-filament Bonding in 3D-printed Polymers, University of Tennessee Honors Thesis Projects, 2016.
- C. C. Ibeh, Thermoplastic Materials: Properties, Manufacturing Methods, and Applications, Taylor & Francis, 2011 Search PubMed.
- J. W. Chen, J. Dai, J. H. Yang, T. Huang, N. Zhang and Y. Wang, J. Mater. Res., 2013, 28, 3100–3108 CrossRef CAS.
- H. Wu, X. Li, J. Chen, L. Shao, T. Huang, Y. Shi and Y. Wang, Composites, Part B, 2013, 44, 439–445 CrossRef CAS.
- K. R. Hart, R. M. Dunn, J. M. Sietins, C. M. Hofmeister Mock, M. E. Mackay and E. D. Wetzel, Polymer, 2018, 144, 192–204 CrossRef CAS.
- S. Rangisetty and L. D. Peel, in Proceedings of the ASME 2017 Conference on Smart Materials, Adaptive Structures, and Intelligent Systems, Snowbird, UT, 2017, pp. 1–12 Search PubMed.
- A. Charlesby and N. H. Hancock, Proc. R. Soc. London, Ser. A, 1953, 218, 245–255 CrossRef CAS.
- P. J. Hendra, A. J. Peacock and H. A. Willis, Polymer, 1987, 28, 705–709 CrossRef CAS.
- P. Kurian, K. E. George and D. J. Francis, J. Elastomers Plast., 1993, 25, 12–21 CrossRef CAS.
- A. J. Peacock, J. Macromol. Sci., Polym. Rev., 2001, 41, 285–323 CrossRef.
- G. W. Ehrenstein, Polymeric Materials: Structure-Properties-Applications, Hanser/Gardner Publications, 2001 Search PubMed.
- C. Liu, S. B. Chun, P. T. Mather, L. Zheng, E. H. Haley and E. B. Coughlin, Macromolecules, 2002, 35, 9868–9874 CrossRef CAS.
- T. Chung, A. Romo-Uribe and P. T. Mather, Macromolecules, 2008, 41, 184–192 CrossRef CAS.
- Y. Xue, Q. Zhao and C. Luan, J. Colloid Interface Sci., 2001, 243, 388–390 CrossRef CAS.
- E. B. Sebok and R. L. Taylor, in Encyclopedia of Materials: Science and Technology, 2001, pp. 902–906 Search PubMed.
- Elastic Material Data Sheet, https://formlabs-media.formlabs.com/datasheets/Elastic_Resin_Technical.pdf.
- Flexible Material Data Sheet, https://formlabs-media.formlabs.com/datasheets/Flexible_Technical.pdf.
- Tango, https://www.stratasys.com/materials/search/tango.
- EPU: Elastomeric Polyurethane, https://www.carbon3d.com/materials/epu-elastomeric-polyurethane/.
- C. J. Thrasher, J. J. Schwartz and A. J. Boydston, ACS Appl. Mater. Interfaces, 2017, 9, 39708–39716 CrossRef CAS PubMed.
- Z. Ji, X. Zhang, C. Yan, X. Jia, Y. Xia, X. Wang and F. Zhou, Macromol. Rapid Commun., 2019, 1800873, 1800873 CrossRef PubMed.
- D. K. Patel, A. H. Sakhaei, M. Layani, B. Zhang, Q. Ge and S. Magdassi, Adv. Mater., 2017, 29, 1606000 CrossRef PubMed.
- Z. Zhao, X. Mu, J. Wu, H. J. Qi and D. Fang, Extreme Mech. Lett., 2016, 9, 108–118 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01452g |
| This journal is © The Royal Society of Chemistry 2020 |