
Thermal and crystalline properties of poly(2-oxazoline)s
Natalia
Oleszko-Torbus
 *,
Alicja
Utrata-Wesołek
,
Marcelina
Bochenek
,
Daria
Lipowska-Kur
,
Andrzej
Dworak
and
Wojciech
Wałach
*,
Alicja
Utrata-Wesołek
,
Marcelina
Bochenek
,
Daria
Lipowska-Kur
,
Andrzej
Dworak
and
Wojciech
Wałach
Centre of Polymer and Carbon Materials, Polish Academy of Sciences, ul. M. Curie-Skłodowskiej 34, 41-819 Zabrze, Poland. E-mail: noleszko@cmpw-pan.edu.pl
First published on 24th October 2019
Abstract
Poly(2-substituted-2-oxazoline)s (POxs), regarded as synthetic pseudo-polypeptides, are of special interest due to their relative ease of preparation, modification possibilities and biocompatibility. Due to these properties, POxs have been investigated for a wide range of applications, including biomedical. This review gathers together data on the thermal and crystalline properties of POxs that could be helpful in designing new poly(2-oxazoline)s materials with strictly defined parameters. In the first part, a comprehensive overview of the thermal and crystalline properties of POxs in the solid state is provided. Special attention has been paid to the possibility of adjusting these properties by the copolymerization of appropriate monomers. Additionally, possible polymer chain conformations during crystallization in the solid state are discussed. In the second part, the properties of POxs crystallized from the solutions are summarized, with a special emphasis on 2-isopropyl-2-oxazoline-based (co)polymers. Here, we also consider the mechanism of crystallization from solution. At the end, the use of crystalline POxs in some applications is briefly presented.
1. Introduction
Poly(2-substituted-2-oxazoline)s (POxs) are known as synthetic “pseudo-polypeptides” due to similarities in chemical structure to polypeptides. While peptide bonds link together amino acid repeating units in peptides, in the case of POxs, a pseudo-peptide bond links a substituent with the polymer main chain. Due to this specific chemical structure (Fig. 1), an amide group interacts with a nonpolar side chain (substituent); thus, POxs show interesting side-chain-dependent properties.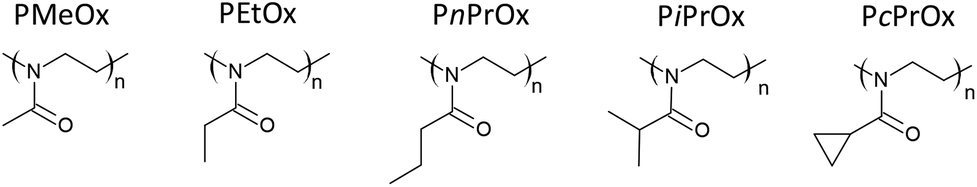 | ||
| Fig. 1 The structures of several exemplary POxs: poly(2-methyl-2-oxazoline) (PMeOx), poly(2-ethyl-2-oxazoline) (PEtOx), poly(2-n-propyl-2-oxazoline) (PnPrOx), poly(2-isopropyl-2-oxazoline) (PiPrOx) and poly(2-cyclopropyl-2-oxazoline) (PcPrOx). | ||
POxs have been known for over 50 years, as the four independent groups of Tomalia,1 Litt and Levy,2 Seeliger3 and Kagiya4 have been investigating the living cationic ring-opening polymerization mechanism of the cyclic imino ethers. Later, this issue was studied extensively by Saegusa and Kobayashi.5,6
In recent decades, an increase in the registration of patents connected with POxs (approximately 20 patents in the 1980 and over 100 in 2010)7 and an increase in the number of publications (less than 50 publications per year until the 1990s and more than 200 publications per year since the 2010s)8 has been proof of an increasing interest in these polymers both by researchers and by industry. There are a few reasons for this growth: first, no major difficulties are associated with the synthesis of POxs; second, the chemical structure and physical properties of POxs can be precisely controlled and adjusted; and finally, POxs have excellent biocompatibility.
POxs are readily obtained via cationic ring-opening polymerization (CROP). Polymerization proceeds under anhydrous conditions, and for synthesis of lower molar mass and not precisely defined polymers, strict high-vacuum techniques are not required. A wide range of different initiators (alkyl or acid halides, tosylates, triflates, Lewis acids, etc.) and terminating agents (nucleophile) are used.9–12 In the polymerization system, both cationic and covalent active centres may be present: ionic centres can be isomerized to covalent centres and vice versa (because the participation of the covalent active centres in the chain growth is negligible, the conversion of ionic pairs into a covalent centre is referred to as temporary chain termination, while the isomerization of the covalent centre into ionic ones is referred to as reinitiation).13 It was found that the kind of active centre in the system is dependent on the alkalinity of the monomer, the nucleophilicity of the counterion and the polarity of the solvent. In some cases, by the lowering the temperature and concentration of active oxazolinium cationic chain end, the rate of side reactions can be significantly reduced, approaching the polymerization close to the living system.14–16 Full conversion of the monomer, depending on the solvent and temperature, is usually reached within a few days, but this time can be significantly shortened to several hours or even minutes by applying a high temperature (above boiling point) and pressure vials as usually used in the microwave reactor.17–19 The cationic polymerization leads to well-defined 2-oxazoline homopolymers with a molar mass of up to 3 × 105 g mol−1 and a low dispersity. By copolymerization of various 2-oxazolines, linear random, gradient and block copolymers can be easily obtained.20–24 A detailed overview of the mechanism of CROP of 2-oxazolines can be found in many papers.25–28
The chemical structure and thus the properties of POxs can be tailored in a controlled manner. By an appropriate selection of an initiator and/or terminating agent, it is possible to obtain polymers with functional α- and/or ω-end groups.29 The nature of the POx side chain determines the polymer properties, both in solution and in the solid state, such as solubility, thermosensitivity (lower critical solution temperature (LCST) a upper critical solution temperature (UCST)), the glass and melting transition, and hence, the ability to crystallize. POxs containing fewer than four carbon atoms in the side chain are soluble in water. Poly(2-methyl-2-oxazoline) (PMeOx) is soluble in water regardless of the temperature, while poly(2-ethyl-2-oxazoline) (PEtOx), poly(2-isopropyl-2-oxazoline) (PiPrOx), poly(2-cyclopropyl-2-oxazoline) (PcPrOx) and poly(2-n-propyl-2-oxazoline) (PnPrOx) have limited temperature-dependent solubility and exhibit LCST-type liquid–liquid phase separation.30–35 POxs containing four or more C atoms in the side chain are not soluble in water at room temperature, but some of them exhibit UCST-type phase transitions in different solvents.36,37 In the case of POxs in the solid state, a glass transition above 0 °C is observed for 2-oxazoline homopolymers with less than four carbon atoms in the side chain, and generally, this temperature decreases linearly with the length of the side chain.38 The properties of POxs can be easily modulated by the copolymerization of appropriate monomers and the microstructure of the polymer chain.20,39 The ability of POxs to crystallize in bulk increases with increasing length of the linear side chain, starting from poly(2-n-butyl-2-oxazoline) (PnBuOx). Recently, a crystallization of some POxs both in aqueous and organic solutions, similar to that observed for proteins, was observed and described.40–42 As the crystalline precipitate is generally not soluble, this disqualifies such systems from applications where thermoresponsive behaviour is necessary. On the other hand, the crystallization of POxs is desired for some purposes.43,44
POxs are known from their excellent biocompatibility. In vitro toxicity was studied for many POx-based systems and was found to be generally rather low for many cell lines in a wide range of concentrations.45–48 With respect to in vivo toxicity, PEtOx and PMeOx have been studied most thoroughly to date. Rapid blood clearance and remarkably low uptake by the organs have been demonstrated for these POxs.49,50
Many interesting reviews on the synthesis, structure and properties of a library of POxs51–53 as well as on the possibility of their modifications54–56 were published in the last few years. Excellent literature research on POxs in the aspect of widely understood biomedical applications has also been published by Schubert,57 de la Rosa,7 Schlaad,58 Luxenhofer,8 Jordan59 and Hoogenboom.60 More detailed, comprehensive overviews of POx-based micro- and nanoparticles,61 hydrogels,62 and POxs at interfaces and on surfaces,63,64 are also available. Among these reviews, it is also obligatory to mention the issue on 50 years of poly(2-oxazoline)s, which was devoted to covering recent advances in POx chemistry and applications.65
Although many excellent literature studies are known, comprehensive data on the thermal and crystalline properties of POxs, which are helpful in POx applications, have not yet been gathered together. This wide field will be covered here. This review will first examine the thermal and crystalline properties of POxs in the solid state and adjust these properties by copolymerization of appropriate monomers. Studies on the changes in chain conformations during crystallization in the solid state will also be presented. Then, the properties of POxs crystallized both from aqueous and organic solutions will be reviewed, with special consideration of PiPrOx. Additionally, the conformation of polymer chains during crystallization from water will be discussed. Finally, the crystalline properties of POxs in some applications will be briefly presented.
It should be noted that many different abbreviations for POxs can be found in the literature. To unify, within this review, we will use abbreviations that are given in the list of abbreviations below, which might not always be consistent with the original literature.
2. Thermal and crystalline properties of poly(2-oxazoline)s in the solid state
The first systematic study on the thermal and crystalline properties of poly(2-oxazoline)s was reported by Litt et al.,66 and focused on POxs in the solid state. The authors considered POxs to be highly crystalline due to the chemical structure of the polymers. As the side chains of POxs are attached to nitrogen atoms within amide groups, this produces a high degree of symmetry. Because of the presence of three atoms in each repeating unit of the polymer main chain, it was established that the side chains must lie on alternate sides of the backbone to prevent substituent crowding. This allows for easy and symmetric packing along the chain. Further X-ray diffraction (XRD) experiments of extruded and oriented by stretching polymer fibres and calculations of the periodicity of the side chains suggested that the main chain of POx is not fully extended. On the basis of the most likely spatial structure derived from models and the X-ray analysis (Fig. 2), it was established that at every CH2CH2 bond, the chain is twisted.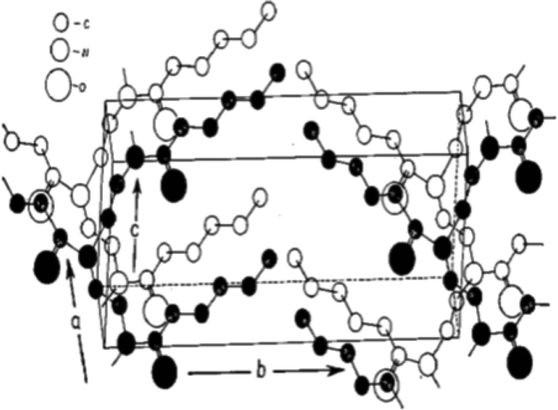 | ||
| Fig. 2 Approximate packing of the polymer molecules in the unit cell of poly(2-pentyl-2-oxazoline). Reproduced with permission from ref. 66. Copyright 1969 WILEY-VCH. | ||
Additionally, for the first time, Litt66 analysed the ability to crystallize a library of POxs (with linear and branched alkyl side chains, with cycloalkyl side chains and with aromatic phenyl ring).66 The d-spacing values, that is the distance between the backbones of the polymer chains, were determined. Although poly(2-ethyl-2-oxazoline) was found to be amorphous, POxs with longer side chains were found to be crystalline with triclinic unit cells and exhibited melting points in the vicinity of 160 °C.
Further studies on the crystallinity of POxs in the solid state focused mainly on PiPrOx. Its ability to crystallize was confirmed by X-ray diffraction analysis, and the peak positions, intensities and indexing were assigned in the obtained diffractograms.40 Two major peaks at 2θ = 7.85° (d = 11.25 Å) and 18.25° (4.85 Å) and smaller peaks at 21.50° (4.13 Å) and 24.00 (3.70 Å) were observed (Fig. 3).
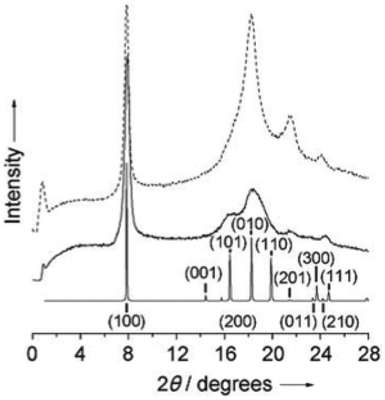 | ||
| Fig. 3 XRD data of isothermally crystallized PiPrOx. Reproduced with permission from ref. 40. Copyright 2007 WILEY-VCH. | ||
The mechanism of poly(2-oxazoline) crystallization in the solid state, involving all group motions and conformational changes, is still the subject of discussion. Recently, Sun and Wu67 studied this issue in detail on an example of PiPrOx, using spectroscopic methods for this purpose. Based on the FTIR analysis of the annealed poly(2-isopropyl-2-oxazoline) films, they found that the asymmetric and symmetric stretching bands of C–H from CH3 groups of the side chains were shifted to higher wavenumbers with increasing annealing time, while C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands were shifted to lower wavenumbers. The authors ascribed these changes to the chain ordering process during crystallization. However, the position and intensity of C–H stretching bands from CH2 of the main chain were almost the same. These results provided the somewhat unusual observation that the conformation of the polymer backbone did not change during crystallization. Raman spectroscopic studies supported this conclusion. It was noticed that stretching bands attributed to C–C from the backbone, corresponding to a trans conformation, were not present in either the amorphous or the crystalline state, but the peak attributed to C–C stretching from the gauche conformation was dominant (ν(C–C)gauche at 1090 cm−1). Thus, it was concluded that the gauche conformations of methylene groups dominate, regardless of whether the polymer is in an amorphous or crystalline state. This provided important information that the PiPrOx backbone is not subjected to significant C–C skeleton conformational changes during crystallization. However, the mathematical analysis based on two-dimensional correlation spectroscopy (2DCOS) gave another look at this thesis. It was considered that during crystallization, the backbone of PiPrOx is distorted laterally, followed by longitudinal chain adjustment. The lateral distortion of the backbone was caused by the motions of the side chains. These findings led to the conclusion that the crystallization of PiPrOx is driven mainly by the structural arrangement of side chains, which was unexpected, as in the case of crystalline polymers, an extension of the polymer backbone is normally predominant to achieve highly ordered chain alignment. Molecular dynamics simulations were employed to investigate the possible conformations of PiPrOx in amorphous and crystalline states. A proposed model of crystalline PiPrOx chain with alternating side chains and a slightly distorted backbone is shown in Fig. 4.
O stretching bands were shifted to lower wavenumbers. The authors ascribed these changes to the chain ordering process during crystallization. However, the position and intensity of C–H stretching bands from CH2 of the main chain were almost the same. These results provided the somewhat unusual observation that the conformation of the polymer backbone did not change during crystallization. Raman spectroscopic studies supported this conclusion. It was noticed that stretching bands attributed to C–C from the backbone, corresponding to a trans conformation, were not present in either the amorphous or the crystalline state, but the peak attributed to C–C stretching from the gauche conformation was dominant (ν(C–C)gauche at 1090 cm−1). Thus, it was concluded that the gauche conformations of methylene groups dominate, regardless of whether the polymer is in an amorphous or crystalline state. This provided important information that the PiPrOx backbone is not subjected to significant C–C skeleton conformational changes during crystallization. However, the mathematical analysis based on two-dimensional correlation spectroscopy (2DCOS) gave another look at this thesis. It was considered that during crystallization, the backbone of PiPrOx is distorted laterally, followed by longitudinal chain adjustment. The lateral distortion of the backbone was caused by the motions of the side chains. These findings led to the conclusion that the crystallization of PiPrOx is driven mainly by the structural arrangement of side chains, which was unexpected, as in the case of crystalline polymers, an extension of the polymer backbone is normally predominant to achieve highly ordered chain alignment. Molecular dynamics simulations were employed to investigate the possible conformations of PiPrOx in amorphous and crystalline states. A proposed model of crystalline PiPrOx chain with alternating side chains and a slightly distorted backbone is shown in Fig. 4.
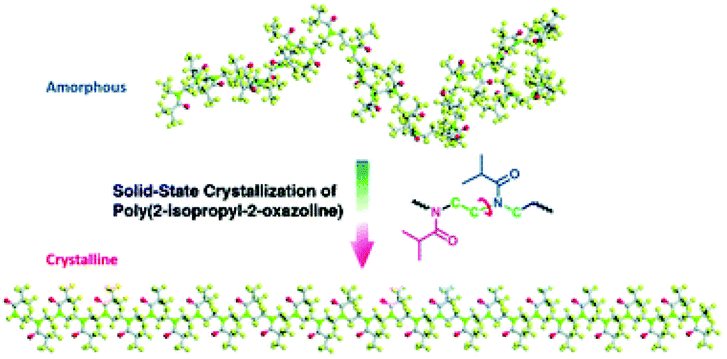 | ||
| Fig. 4 Possible conformations of PiPrOx chains both in amorphous and crystalline states. Reproduces with permission from 67. Copyright 2015 The Royal Society of Chemistry. | ||
Despite many papers considering research studies on the thermal and crystalline properties of POxs, to the best of our knowledge, the abovementioned study regarding the mechanism of crystallization and conformation of chains during ordering of these polymers in the solid state is the only one in recent years.67
The following chapters focus on the thermal and crystalline properties of 2-oxazoline homopolymers with linear and non-linear alkyl substituents as well as how these properties can be modulated by copolymerization.
2.1. Homopolymers of 2-oxazolines with linear alkyl substituents
A number of groups have studied the thermal properties of a library of poly(2-oxazoline)s with linear alkyl substituents. In 1994, Rodriguez-Parada et al.68 and Beck et al.69 independently studied the properties of POxs with 6 to 17 carbon atoms attached to the amide group in the side chains. Later, Hoogenboom and Schubert et al.17,70,71 described the thermal properties of a series of poly(2-n-alkyl-2-oxazoline)s with varying lengths of the side chains, with 1 to 11 carbon atoms.Generally, the thermal and crystalline properties of homopolymers of 2-oxazolines with linear alkyl substituents were found to be related to the length of the aliphatic side chains. Fig. 5 shows Tg and Tm values depending on the side chain length for 2-oxazoline homopolymers with 1 to 17 carbon atoms attached to the amide groups.38
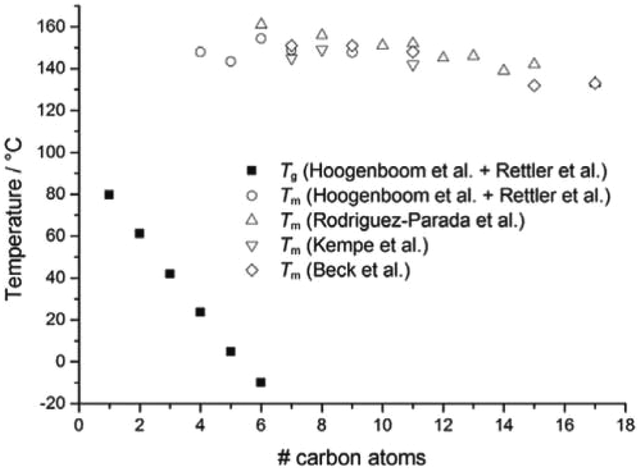 | ||
| Fig. 5 Glass transition and melting temperatures for homopolymers of 2-oxazolines with linear alkyl substituents. Reproduced with permission from ref. 38. Copyright 2017 WILEY-VCH. | ||
POxs with up to 5 carbon atoms attached to the amide groups in the side chains show a glass transition temperature (Tg) above 0 °C, which decreases with increasing side-chain length.38 When an alkyl substituent of POxs is short, the backbones of polymer chains can become closer to each other, and the amide dipoles interact more strongly, which decelerates the relaxation of the backbones and increases Tg. In the case of POxs with longer (and more flexible) alkyl substituents, the distance between the amide dipoles is larger, which consequently leads to the increase in d-spacing. This enables faster relaxation of the chain backbones; thus, Tg is observed at lower temperatures.72 For homopolymers of 2-methyl-, 2-ethyl and 2-n-propyl-2-oxazoline, no melting is generally observed during DSC measurements.17,71 However, recently, a pronounced, broad, endothermic peak in the range of 120–140 °C and of an energy of ΔH = 7 J g−1, assigned to the melting, was reported for poly(2-n-propyl-2-oxazoline).73 Moreover, when the polymer was annealed at 90 °C for merely 1 hour and then slowly cooled to room temperature (with a rate of approximately 10 °C per minute), an endothermic peak with two maxima could be observed in the DSC thermogram, with a higher enthalpy value of 23 J g−1. The degree of crystallinity (χc) measured by X-ray diffraction for this polymer was approximately 55%. Thus, the frequently encountered assumption that PnPrOx is completely amorphous and does not exhibit a tendency to crystallization seems to be unjustified, which was already reported by Litt.66 For poly(2-n-butyl-2-oxazoline) (PnBuOx), poly(2-pentyl-2-oxazoline) (PPentOx) and poly(2-hexyl-2-oxazoline) (PHexOx), both Tg and Tm are observed;17,71 however, PHexOx shows a glass transition below 0 °C.71 Additionally, for PnBuOx and PPentOx, an exothermic peak was observed during DSC measurement with the maximum at 90 and 60 °C, respectively. This was attributed to so-called cold crystallization (as this occurs during heating).72
The ability to crystallize increases with an increasing length of the substituent of POxs. Surprisingly, the length of the side chain does not significantly influence the value of the melting temperature, which is approximately 150 °C.17,52,70 However, some small deviations can be observed, depending on the molar mass. For example, in the case of poly(2-heptyl-2-oxazoline) (PHeptOx), Tm was detected at 145 and 160 °C when the molar mass was 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 21800 g mol−1, respectively.70,74
000 and 21800 g mol−1, respectively.70,74
2.2. Homopolymers of 2-oxazolines with other substituents
In addition to the POxs with linear alkyl substituents, there is also a large group of 2-oxazoline homopolymers containing different side chains attached to amide groups. These POxs are interesting because of their functionalization possibilities and further tailoring of properties for specific applications. Generally, the thermal and crystalline properties of these POxs are dependent on the nature of the substituent.75 with a significantly higher glass transition temperature compared to its linear analogue, poly(2-n-propyl-2-oxazoline). For PcPrOx with DP = 38, Tg was observed at 79 °C, and for the polymer with a slightly lower molar mass (DP = 26), Tg was 75 °C. Homopolymers containing larger cycloalkyl rings were semi-crystalline, and for these POxs, Tm of highest values among all POxs were reported thus far. For poly(2-cyclobutyl-2-oxazoline) (PcBuOx), poly(2-cyclopentyl-2-oxazoline) (PcPentOx) and poly(2-cyclohexyl-2-oxazoline) (PcHexOx), no glass transition temperature could be detected using DSC.76 A high melting temperature was registered for these homopolymers in the following order: 243, 306 and 251 °C. The melting peaks recorded in the DSC traces for all samples were not symmetric, which was explained by the formation of imperfect crystals of various morphologies. For PcBuOx, PcPentOx and PcHexOx, diffraction peaks together with a broad amorphous peak were observed in the WAXS graphs. The degree of crystallinity followed the same trend as Tm (PcPentOx (∼37%) > PcHexOx (∼25%) > PcBuOx (∼23%)).
 | ||
| Fig. 6 The structures of the selected POxs. | ||
High melting temperature values were also reported for POxs containing an adamantyl substituent (Fig. 6a).77 Poly(2-(1-adamantyl)-2-oxazoline) (PAdamOx) showed Tm at 269 °C, while poly(2-(1-adamantylmethyl)-2-oxazoline) (PMeAdamOx) had a melting peak with two maxima at 304 and 320 °C.77 After quenching, PAdamOx showed only Tg at 123 °C. For PMeAdamOx, in addition to Tg at 139 °C, an exothermic peak at 215 °C, ascribed to cold crystallization, and a melting peak at 312 °C were observed. Such differences in crystallinity between PAdamOx and PMeAdamOx were interpreted in terms of the structure of substituents that influence their mobility. With one CH2 unit in the side chain, the adamantyl group in PMeAdamOx has more freedom, and the side groups can be packed very well. As a result, PMeAdamOx has a very high tendency to crystallize. This finding was supported by the molecular model.
Interestingly, 2-oxazoline oligomers with cubane substituent (PCubOx) did not show a melting transition during calorimetric studies, but only Tg at approximately 70 °C.78 Further X-ray experiments provided diffraction peaks what confirmed however that PCubOx is semi-crystalline. Thus it was concluded that melting transition was higher than the degradation temperature which is above 250 °C.
Within this group, the thermal and crystalline properties of homopolymers are dependent on the length of the fatty acid-based unsaturated side chain. For poly[2-(3-butenyl)-2-oxazoline]s (PButenOx), a low value of Tg was observed, which was dependent on the molar mass: 9 °C for PButenOx with an Mn of 2700 g mol−1 (Đ = 1.19), 15 °C for PButenOx with an Mn of 6900 and 8800 g mol−1 (Đ = 1.22 and 1.19, respectively) and 17 °C for PButenOx with an Mn of 11500 g mol−1 (Đ = 1.16).79 Upon increasing the length of the unsaturated side chain, the melting point was detected. For example, the homopolymer of 2-decenyl-2-oxazoline (PDecenOx) (Mn = 20000 g mol−1, Đ = 1.16) melted at 130 °C (ΔH ∼ 35 J g−1), as observed in the DSC trace.74
However, POxs containing extremely long, unsaturated side chains exhibit lower Tm values than homopolymers with fatty acid-based saturated side chains. 2-“Soy alkyl”-2-oxazoline (SoyOx) is a monomer that is based on soy-bean fatty acids and has an average of 17 carbon atoms in the side chain and an average of 1.5 double bonds per monomer unit.80 For the homopolymer of SoyOx (Mn = 15000 g mol−1, Đ = 1.75), no glass transition could be detected by DSC, while Tm was observed in the vicinity of 88 °C. This is much lower than in the case of the saturated fatty acid-based homopolymers, for example, poly(2-nonyl-2-oxazoline) (Tm = 150 °C). The authors ascribed this behaviour to the presence of cis-double bonds in the fatty acid side chains that disturb crystallization. After UV curing, the homopolymer of SoyOx no longer showed a melting transition. It was concluded that this treatment partially prevented the crystallization of PSoyOx due to the lowering of the chain mobility.
000 g mol−1 (ref. 82) and 103 °C at ∼14000 g mol−1.69
POxs with a phenyl ring substituted in para position were also amorphous. A glass transition near 100 °C was determined for POxs with a thioether functional group or chlorine atom as the para substituent, while POx with a trifluoromethyl group in the para position has Tg equal to 75 °C.70 However, for POx with a tert-butyl group in the para position of the phenyl ring, neither the glass transition nor melting point were recorded in the DSC trace in the investigated temperature range from −100 to 200 °C.70
Among POxs containing condensed aromatic rings, poly(2-[4-(2,7-dimethoxycarbazoI-9-yI)butyl]-2-oxazoline) (PC4CarbOx) shows interesting thermal and crystalline properties.83 PC4CarbOx was found to crystallize during the polymerization process. The DSC traces of PC4CarbOx annealed at 160 °C displayed a sharp melting peak at an unexpectedly high temperature of 232 °C, similar to the case of POxs with cycloalkyl side chains. A barely seen glass transition was simultaneously visible. The Tg at 89 °C became prominent in the quenched sample. The fact that PC4CarbOx did not crystallize during cooling after being heated above the melting point suggested that the crystallization rate was slow. This was ascribed to large side groups that might restrict the mobility of the polymer chains and make packing difficult.
Semi-crystalline POxs with linear side chains that have a two methylene group between the amide group and the perfluorocarbon chain (consisting of 4, 6, 8, 10 or 12 carbon atoms) were extensively studied by Rodriguez-Parada et al. (Mn values from 21500 to 42100 g mol−1).68 The melting of these POxs occurred at much higher temperatures than did those of the corresponding homopolymers with hydrocarbon side chains of the same length (Tm up to 236 °C). The melting point temperature values of POxs with fluorinated linear groups increased in parallel with the side-chain length. This behaviour reflected the significant role of the fluorocarbon chains, which are more rigid than hydrocarbons and melt at higher temperatures.
A detailed, systematic study on POxs with fluorinated phenyl rings was conducted by Schubert.84 A broad library of poly(2-oxazoline)s with monofluorophenyl-(fluorine atom in the phenyl ring at ortho, meta, or para position), difluorophenyl-, trifluorophenyl-, tetrafluorophenyl- and pentafluorophenyl-substituents (Mn ∼ 8000–15000 g mol−1, Đ less than 1.4) was obtained. All polymers were amorphous with a Tg in the vicinity of that observed for PhOx (in the range from 97 to 135 °C). The POxs with two ortho-fluoro substituents on the phenyl ring revealed significantly higher Tg values than others. This was ascribed to the limited rotational freedom of the phenyl rings carrying two ortho-fluoro substituents and the presence of attractive C–F⋯CO interactions, which decrease the chain mobility and thus increase the Tg value.
Among the first group, poly(2-isopropyl-2-oxazoline) (PiPrOx), poly(2-isobutyl-2-oxazoline) (PiBuOx) and poly(2-tertbutyl-2-oxazoline) (PtBuOx) were investigated in detail. These homopolymers were found to exhibit melting at higher temperatures and had a higher tendency to crystallize than their linear analogues. PiPrOx showed a glass transition at 70 °C and melting at ∼200 °C, which is significantly higher than that of PnPrOx. During DSC analysis of PiPrOx at a standard heating rate (10 °C min−1), an exothermic peak with the maximum at approximately 150 °C, attributed to cold crystallization and a subsequent endothermic peak attributed to melting (ΔH = 11 J g−1), could be observed.42 Moreover, when the polymer was annealed at 190 °C for merely 1 hour and then slowly cooled to room temperature, an endothermic peak of high energy (ΔH = 38 J g−1) was observed. This indicated the high tendency to crystallize of this polymer. Additionally, the degree of crystallinity (χc) of isothermally crystallized PiPrOx, estimated by wide angle X-ray scattering (WAXS), was approximately 80%, which is the highest value of χc among all POxs.85 Similarly, poly(2-isobutyl-2-oxazline) (PiBuOx) exhibited a melting point at approximately 200 °C, which is considerably higher than that of PnBuOx (∼150 °C).70 This was ascribed to the lower flexibility of the isobutyl side chain than of the linear analogue, which was associated with a lower change in the entropy during the transition. Schacher et al.86 provided interesting facts regarding thermal properties of PtBuOx. Tg value of 38 °C and decomposition temperature starting from 240 °C were designated, what was not fully consistent with data obtained by Litt66,82 (lack of Tg and Tm of ∼320 °C), however similar diffraction peaks and a degree of crystallinity of approximately 65% were obtained.
Among the group with long branched alkyl substituents, poly(2-(1-ethylpentyl)-2-oxazoline) (PEPOx), poly(2-(3-ethylpentyl)-2-oxazoline) (P3EPOx) and poly(2-(3-ethylheptyl)-2-oxazoline) (PEHOx) were studied in detail. PEPOx and P3EPOx contain the same number of carbon atoms in the side chain (7 carbon atoms) and the same branching group but differ in the branching positions. Both polymers were found to be amorphous, with Tg dependent on the position of branching.87 PEPOx with the ethyl branch in the 1-position of the side chain exhibited a glass transition at approximately 30 °C, whereas P3EPOx with the branch in the 3-position showed a much lower Tg at approximately 5 °C, suggesting a lower packing density of the polymer. Moreover, branching in the 1-position was found to hinder backbone movement due to stronger steric hindrance, which resulted in a higher Tg. Increasing the length of the branched alkyl substituent to 9 carbon atoms (PEHOx) resulted in a decrease in a glass transition to −6 °C.88 The ability to crystallize these polymers was found to be suppressed compared to their linear analogues with the same number of carbon atoms in the side chain. The lack of melting was detected by DSC in the investigated temperature range, while in the case of linear poly(2-heptyl-2-oxazoline) and poly(2-nonyl-2-oxazoline), Tm was approximately 150 °C.
2.3. Control of the thermal and crystalline properties by copolymerization
The thermal and crystalline properties of POxs can be well controlled by the type and content of comonomers. By selecting appropriate comonomers both qualitatively (copolymerization of comonomers of different substituents) and quantitatively (content of comonomers in chains), it is possible to obtain POxs with different properties and the ability to crystallize. The distribution of comonomers in the chain (microstructure) also influences the final properties of POx.Generally, in the case of random and gradient copolymers, a relationship between Tg and copolymer composition approximately describes the Fox or Gordon–Taylor equation. In many cases, an increasing amount of more flexible comonomers in the copolymer causes a decrease in the glass transition value.
For example, copolymerization of 2-nonyl-2-oxazoline (NonOx) with 2-ethyl-2-oxazoline (EtOx) leads to random copolymers. As the homopolymer of NonOx is semi-crystalline with a Tm of approximately 147 °C17 and the homopolymer of EtOx is amorphous, the thermal properties of the resulting copolymers are dependent on the comonomer content.20,39 Copolymer with 80% (wt) of EtOx was amorphous with a glass transition value of approximately 50 °C. Incorporation of 60 and 45% of EtOx led to a decrease of Tg of the copolymer to approximately 40 and 30 °C, respectively. Subsequent decreasing the amount of EtOx to 15% led to copolymer where, in addition to the glass transition of 30 °C, the melting point was also observed near 105 °C. The lack of glass transition and an increase in Tm to approximately 120 °C was observed for copolymers with EtOx:NonOx weight ratio of 10:90. A similar tendency was observed for random copolymers of EtOx and NonOx described in ref. 39; however, here, the reported values of Tg and Tm were lower. The glass transition for copolymers with 80, 60 and 45% (wt) of EtOx were found to be 40, 35 and 25 °C, respectively. Additionally, the melting point was observed for the copolymers with 15% or less EtOx. Such slight discrepancies in the thermal properties of the series of copolymers described in ref. 39 and 20 could probably arise from differences in the molar masses of the studied copolymers.
In turn, copolymerization of NonOx with 2-methyl-2-oxazoline led to the copolymers with a gradient microstructure.39 Similar to the case of random copolymers, here, changes in thermal properties under the variations of the comonomers content were also observed. As the content of NonOx within copolymer increased up to 80% (wt), the glass transition decreased to approximately 20 °C. A further increase in the NonOx content led to semi-crystalline copolymers with Tm values from 100 to 140 °C.
Changes in the thermal properties with the composition of the random copolymers of NonOx with EtOx and gradient copolymers of NonOx with MeOx are compared in Fig. 7A.39
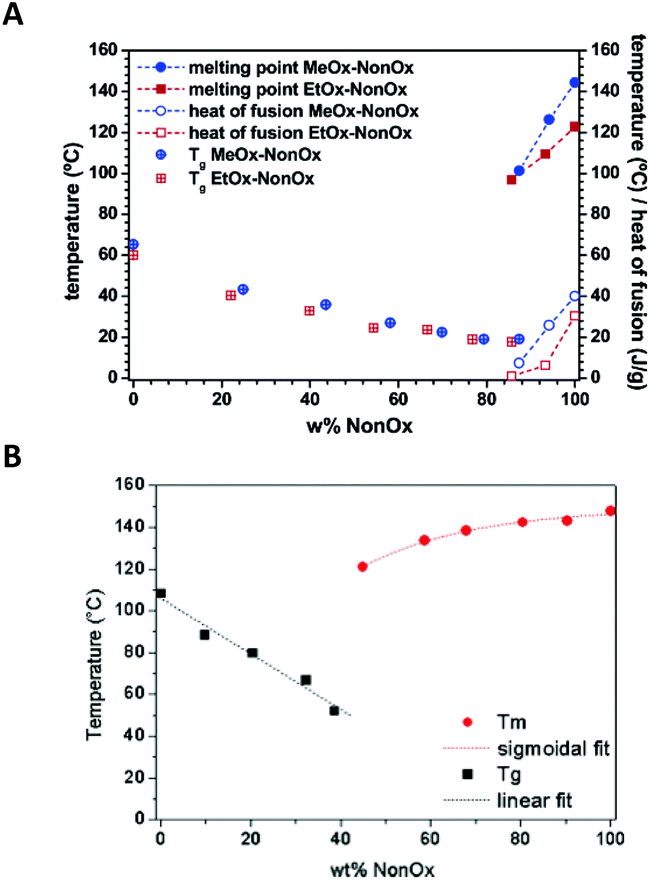 | ||
| Fig. 7 Glass transition and melting temperatures of random copolymers of NonOx with EtOx and gradient copolymers of NonOx with MeOx (A) and gradient copolymers of NonOx with PhOx (B) obtained from DSC. Reproduced with permission from ref. 39. Copyright 2006 American Chemical Society. And from ref. 89. Copyright 2009 WILEY-VCH. | ||
NonOx also forms gradient copolymers with 2-phenyl-2-oxazoline. In this case, the composition-thermal property relationship was also described (Fig. 7B).89 However, as the homopolymer of PhOx showed a glass transition at 107 °C,90Tg values of NonOx/PhOx copolymers were much higher than in the case of NonOx/MeOx copolymers. Additionally, because of different mobilities of the side chains, copolymers of NonOx/PhOx were semi-crystalline already at a NonOx content of 40% (in the case of NonOx/MeOx copolymers, Tm was observed when the content of NonOx was above 85%).39
Thermal and crystalline properties were also studied for many other random and gradient copoly(2-oxazoline)s. For example, the relationship between Tg and copolymer composition was described for a series of thermoresponsive copolymers based on 2-n-propyl-2-oxazoline with 2-methyl-, 2-ethyl- or 2-isopropyl-2-oxazoline as the 2nd comonomer (Mn from 9400 to 13100 g mol−1, Đ less than 1.4).91 The calculations of the comonomer consumption rate during polymerization indicated the gradient structure of nPrOx/iPrOx and nPrOx/MeOx and random structure for nPrOx/EtOx copolymers. DSC data demonstrated that upon increasing the content of nPrOx in each series of copolymers, Tg decreased regardless of the microstructure of the chain. It was concluded that nPrOx comonomer units acted as a plasticizer in obtained POx copolymers: it reduced Tg by increasing the mobility of the polymer chains and providing higher flexibility of the side chains compared with MeOx, EtOx or iPrOx homopolymers.
A similar structure–property relationship was observed for a library of random copolymers of EtOx and SoyOx (Mn from 6000 to 13000, Đ less than 1.54).80 With an increasing amount of SoyOx comonomer, the Tg of the copolymers was decreased from 44 °C (5 wt% of SoyOx) to 11 °C (70 wt% of SoyOx). The copolymers with more than 70 wt% of SoyOx had no glass transition and revealed a Tm between 88 and 90 °C.
The decrease in Tg due to the incorporation of comonomers of higher mobility (so-called “plasticizing” effect) was also observed for the following random copolymers: MestOx/EtOx, MestOx/nPrOx, C3MestOx/EtOx, C3MestOx/nPrOx81 and gradient copolymers: MeOx/PhOx and EtOx/PhOx.92
However, no relationship between Tg and copolymer composition was observed in the case of the gradient copolymers of 2-cyclopropyl- (cPrOx) and 2-ethyl-2-oxazoline (Mn from 14100 to 20500, Đ less than 1.16).93 In these systems, the temperature of the glass transition varied randomly between 47 and 55 °C, and it did not change upon increasing the amount of the more flexible comonomer (EtOx). All copolymers were amorphous, and surprisingly, they revealed lower Tg values than the corresponding homopolymers (apart from the one copolymer with the highest cPrOx content). Such behaviour was not reported previously for any other copoly(2-oxazoline). The authors concluded that the combination of these two monomers leads to copolymers that cannot adopt dense packing.
Not only the glass transition but also the melting temperature of random and gradient copolymers can be controlled by the appropriate selection of comonomers and their ratio.
In the case of the gradient copolymers of iPrOx and nPrOx (Mn from 11500 to 17200 g mol−1, Đ less than 1.2), the melting temperature value decreased, as well as the ΔH of melting, with an increasing amount of nPrOx.73 Copolymers did not exhibit melting when the amount of nPrOx was at least 50% mol. Thus, it was concluded that the crystallization ability of PiPrOx may be significantly reduced by gradient copolymerization of iPrOx with nPrOx. However, annealing of this copolymer at 120 °C and then cooling to room temperature at a slow rate (of 10 °C per minute) gave rise to ordering of the polymer chains and the formation of a crystalline fraction. The authors concluded that crystallization of seemingly amorphous polymer could be forced by exposure to high temperature for a sufficiently extended time.
The influence of copolymer composition on Tm was also described for a series of random copolymers based on 2-decenyl- and 2-heptyl-2-oxazoline with a degree of polymerization of 100 (Đ less than 1.2).74 The melting points of copolymers decreased with an increasing amount of DecenOx from 147 °C (at 20 mol% of DecenOx) to 135 °C (at 90 mol% of DecenOx). Similarly, ΔH decreased from 37 to 32 J g−1, respectively. The authors highlighted a significant role of the allyl groups from DecenOx during the crystallization process. It was concluded that in the case of copolymers with less than 30% mol of DecenOx, the crystallization was disorganized by the small amount of allyl groups that could rotate freely. This resulted in broad melting peaks. When the amount of DecenOx increased to over 30% mol, the allyl groups were closer to each other and their rotation was more difficult. The X-ray diffraction analysis revealed also broad reflection peaks in the copolymers with 20–60% mol of DecenOx. The authors concluded that the allyl pendant groups interfered strongly during the crystallization of the copolymers, which resulted in disordered crystals. Above 80% mol of DecenOx, the polymer crystal was dominated by the DecenOx monomer unit and had a uniform crystalline domain.
A similar trend was observed for a series of random copolymers based on cPentOx and cBuOx (Mn from 12200 to 19000 g mol−1, Đ less than 1.24).76 In the case of these copolymers, the melting points increased with an increasing amount of cPentOx from 253 °C (at 24 wt% of cPentOx) to 298 °C (at 85 mol% of cPentOx).
In contrast to random or gradient copolymers, in the case of block copoly(2-oxazoline) systems, it is difficult to find one specific relationship between thermal or crystalline properties and the copolymer composition. Generally, the thermal properties of diblock copolymer “AB” should be similar to those of homopolymer “A” when the length of block “A” in the copolymer is predominant and, conversely, similar to homopolymer “B” with the predominant length of block “B” in the copolymer.
Such a situation was observed, for example, for the diblock copolymers of NonOx with EtOx (Mn from ∼6000 to 12000 g mol−1, Đ less than 1.5).20 Copolymers containing 33 to 82% NonOx showed both a glass transition and a melting point. The Tg values (∼60 °C) were close to the Tg of the EtOx homopolymer, while Tm (∼140–150 °C) was similar to that of pure poly(2-nonyl-2-oxazoline). For the copolymers with a NonOx content of up to 33%, only Tg was observed, while only Tm was present when copolymers of above 82% of NonOx were obtained (Fig. 8).
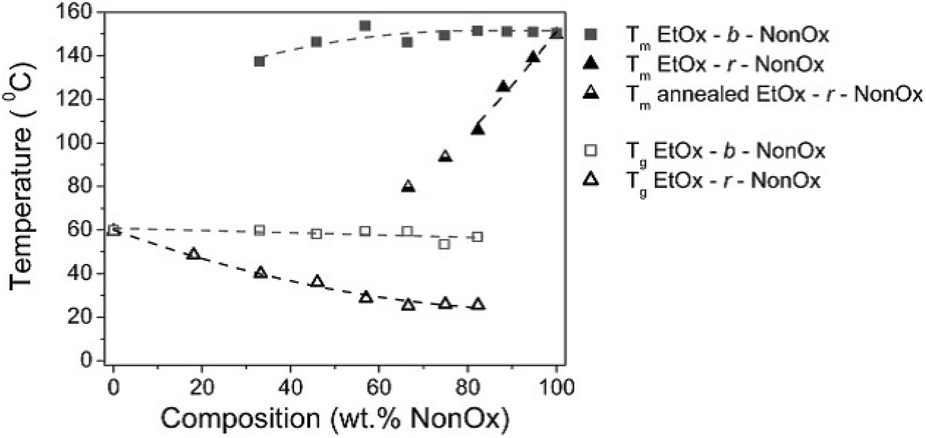 | ||
| Fig. 8 Glass transition and melting temperatures of block and random copolymers of NonOx and EtOx obtained from DSC. Reproduced with permission from ref. 20. Copyright 2007 American Chemical Society. | ||
A similar trend was observed by Litt et al.94 for semi-crystalline diblock copolymers based on 2-phenyl- and 2-undecyl-2-oxazoline (UndOx) (Mn from ∼2800 to 8100 g mol−1, Đ less than 1.3). The amount of UndOx was predominant; thus, all copolymers showed melting at a temperature range similar to that of pure poly(2-undecyl-2-oxazoline) (∼130–140 °C). Additionally, for copolymers with a constant length of the PhOx block, the melting temperatures and ΔH increased with increasing chain length of the UndOx segment. However, when the length of the UndOx block was shorter than 25 monomer units, two broad melting peaks were present in the DSC thermogram. A single and sharp melting peak was observed only when the length of the UndOx segment was equal to or longer than 25 monomer units.
The thermal properties also changed with the chain composition in the case of amorphous, amphiphilic diblock copolymers based on EtOx and 2-(4-aminophenyl)-2-oxazoline (APOx) (Mn from ∼4600 to 10800 g mol−1, Đ from ∼2 to 4).95 The glass transition of copolymers ranged from 59 °C (close to Tg of EtOx homopolymer) when the amount of EtOx was 97% mol to 119 °C (close to the Tg of the APOx homopolymer) upon increasing the APOx content (to 70 mol%). Despite the wide dispersity of molar masses of copolymers, which might suggest side reactions during copolymerization, the authors suggested that the presence of one glass transition in DSC thermograms was evidence that the copolymers were homogeneous.
An unexpected trend was described for a series of diblock copolymers of 2-phenyl-2-oxazoline with 2-methyl- and 2-ethyl-2-oxazoline, with a molar mass from 2900 to 8200 g mol−1 and a molar mass dispersity less than 1.30.90 All copolymers were amorphous. In the case of PhOx/EtOx copolymers, the glass transition was comparable to that of the homopolymer of EtOx, independent of the PhOx content. In the case of PhOx/MeOx copolymers, when the amount of 2-phenyl-2-oxazoline was 5%, Tg was decreased to 76 °C, which is less than that of pure PMeOx (79 °C). A further increase in the PhOx content in the copolymer caused a decrease in the glass transition to 69 °C (at 29% of PhOx). This is unexpected as the increasing amount of PPhOx in the copolymer should cause an increase in Tg.
The relationship between thermal and crystalline properties and the copolymer composition was also analysed for triblock 2-oxazoline systems.
For example, Litt et al.94 obtained semi-crystalline triblock copolymers of ABA type, based on UndOx and PhOx, with molar masses from 6100 to 10400 g mol−1 and a molar mass dispersity of less than 1.09. When the mole ratio of UndOx:PhOx:UndOx was 1:1:1, two melting peaks were observed with maxima at ∼110 and ∼120 °C. The small amount of amorphous PhOx decreased the melting temperature of the copolymer compared with that of the UndOx homopolymer. When the mole ratio of UndOx:PhOx:UndOx was approximately 2:1:2, only one sharp melting peak was visible near 140 °C, which is similar to that of the UndOx homopolymer.
Additionally, thermal dependencies were analysed for a library of different triblock copolymers (ABA/BAB/ABC-type) based on 2-methyl-, 2-ethyl-, 2-nonyl-, and 2-phenyl-2-oxazoline with a targeted comonomer content of 33:33:33.96 These comonomers were chosen as building blocks because they differ in the flexibility of the side groups: MeOx and PhOx have rather rigid substituents, whereas EtOx and NonOx have more flexible side chains. No Tg was detected for copolymers having both a PhOx and a NonOx in the polymer chain. The Tg ranged from 48 °C for copolymers with a high content of flexible monomers to 98 °C for copolymers with a high content of rigid comonomers. The authors concluded that as none of the triblock copolymers exhibited more than one glass transition, this indicated the lack of phase separation in the bulk. Similar value of the glass transition (60 °C) was observed for ABA-type triblock copolymers based on MeOx and NonOx of lowered amount of the central hydrophobic block.97,98 The glass transition values for a library of ABA-type triblock copolymers, all comprising the same hydrophilic PMeOx block, was determined very recently by Luxenhofer et al.98
To summarize, for 2-oxazoline copolymers with random, gradient or block microstructures, thermal and crystalline properties in bulk can be carefully controlled and are dependent on the type and content of comonomers. Increasing the amount of rigid comonomers with relatively short substituents decelerates the relaxation of the copolymer backbones and increases Tg, simultaneously decreasing the amount of crystalline phase. The plasticizing effect and lowering of the value of Tg can be observed when the presence of a comonomer with a more elastic substituent is predominant within the chain, but in many cases, this increases the ability to crystallize.
3. Properties of poly(2-oxazoline)s crystallized from solution
Beyond the crystallization of poly(2-oxazoline)s in the solid state, crystallization of POxs from solutions also occurs and has been widely described. Generally, the crystallization of proteins and other polymers from solution can be induced by liquid–liquid phase separation, driven, for instance, by the change of temperature or the addition of salt (the salting out effect).99–101 The thermodynamic driving force for crystal nucleation and growth is the concentration excess (supersaturation) of the solution above equilibrium.In the case of the aqueous solution of thermoresponsive poly(2-oxazoline)s, the liquid–liquid phase separation is driven by temperature changes (above LCST or below UCST).102 In some cases, this change increases the local density of crystallizable macromolecules, thus leading to supersaturation followed by crystallization. Additionally, the crystallization of POxs from organic and water/organic solutions was demonstrated. However, in this case, the change of temperature does not increase the local density of polymer chains (in such solvents, no thermoresponsive behaviour was detected); thus, crystallization of POxs in these conditions proceeded at a much longer time than from aqueous solution.42
Generally, crystallization from the solution was described in the most detail for PiPrOx and its copolymers41–43,85,103 but also for PEtOx,104 PiBuOx105 and PNonOx.105
3.1. Crystallization of PiPrOx from water
The crystallization of POxs from solution, driven by the LCST-type liquid–liquid phase separation, was first reported by Schlaad et al. for poly(2-isopropyl-2-oxazoline).40,41,85 In the first report, PiPrOx with 4-(N-Boc-amino)-piperidine or with two ammonium groups at the ω-chain end (Mn 5000–16000 g mol−1, Đ < 1.09) dissolved in a physiological saline solution were studied in terms of particle size by monitoring the static light scattering intensity as a function of temperature.41 The scattering intensity was observed to increase steeply at 36 °C, obviously indicating LCST-type liquid–liquid phase separation. The situation was different when PiPrOx solutions were kept above LCST for 24 h. Then, a cloudy precipitate remained stable even after cooling the solution to room temperature. Microscopic analysis of dried coagulate (PiPrOx 5000 g mol−1) revealed densely packed micron-sized spherical particles, uniform in size and shape, together with fibrils with diameters of approximately 30–50 nm and lengths of several microns. The coagulate obtained from PiPrOx with a molar mass of 13000 g mol−1 had a similar structure, but the fibrils were much shorter, less than 1 μm. On the basis of the microscopic observations, it was concluded that during prolonged heating, the fibrils were first formed, followed by their assembly into larger aggregates and finally into microspheres. The authors concluded that they resembled a decoration ball made of rattan and were thus named “cotton balls” (Fig. 9).
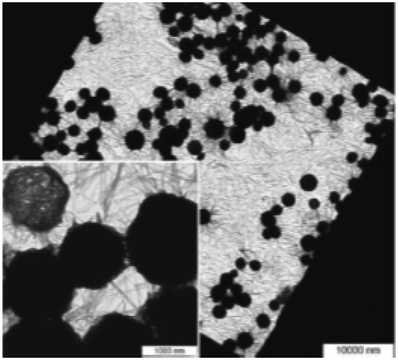 | ||
| Fig. 9 TEM micrograph of PiPrOx (Mn = 5000 g mol−1) aqueous solution annealed for 24 h at 65 °C. Reproduced with permission from ref. 41. Copyright 2007 The Royal Society of Chemistry. | ||
It was suggested that the process of formation of the fibrils and microspheres was directly coupled to self-organization or crystallization; however, no evidence for this hypothesis was provided. Further studies of PiPrOx with a methyl group at the one chain end and two ammonium groups at the ω-chain end (Mn = 5000 g mol−1, Đ = 1.08) proved that fibrils and microspheres are produced through a slow directional crystallization process.40 The crystallinity of a dried PiPrOx coagulate, formed in water at 65 °C within 24 hours, was confirmed by DSC and XRD measurements. For PiPrOx crystallized from water, Tg and Tm values in the DSC curve, together with peak positions and intensities in the XRD curve, were found similar as for PiPrOx crystallized in the solid state. The scanning force microscopy analysis of the films obtained by spin coating an aqueous solution of PiPrOx kept at 65 °C for 7 hours revealed that microspheres were made of fibrils of diameter approximately 57–90 nm and a length of several microns. As the crystallization did not occur at temperatures below the LCST, it was concluded that the phase separation seems to be a precondition for PiPrOx crystallization.40 The authors argue that crystallization of PiPrOx is driven by hydrophobic and oriented dipolar interactions and a subsequent fusion process. However, the mechanism of this process remains unclear. Detailed insight into the mechanism of crystallization of PiPrOx from solution was further studied by many researchers.106–109 In this review, it is described in the next chapter. Additionally, the kinetics of the crystallization of α-methyl-ω-hydroxy-PiPrOx (Mn = 15000 g mol−1, Đ = 1.05) and the time-dependent evolution of the morphology of crystalline spheres were extensively studied.85 Based on in situ WAXS measurements, the intensity and sharpness of the diffraction peaks increased with the annealing time of the aqueous solution of PiPrOx at 60 °C. At the same time, the degree of crystallinity (χc) of the obtained coagulate increased initially during solution annealing to remain constant (of approximately 80%) after 10 hours of annealing. Scanning electron microscopy (SEM) analysis revealed that after 4 hours of annealing, spherical particles of a diameter of ∼2 μm, built of a framework of fibre mesh, were formed. With increasing time of annealing and crystallinity of the sample, the cavities of the microspheres were filled up, while the size of the particles remained unchanged. Compact, isolated, rounded particles of regular size and shape were obtained after 24 hours of incubation (Fig. 10).
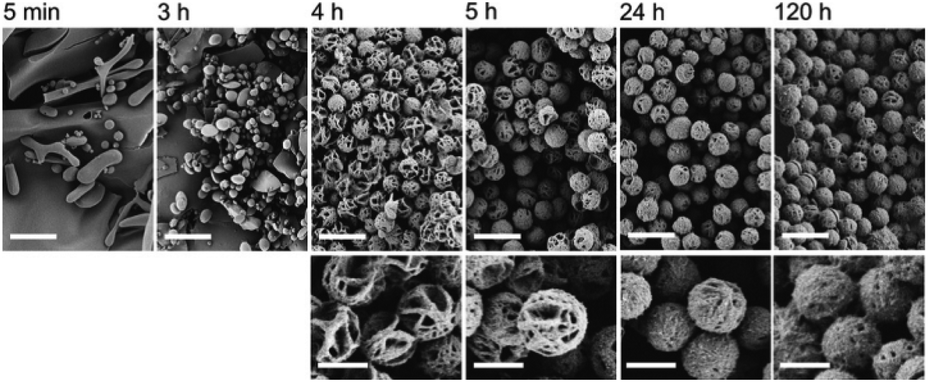 | ||
| Fig. 10 SEM micrographs showing time-dependent evolution of the morphology of PiPrOx (Mn = 15000 g mol−1) aqueous solution annealed at 60 °C. Reproduced with permission from ref. 41. Copyright 2007 The Royal Society of Chemistry. | ||
It was observed that different groups attached to PiPrOx influence the morphology of crystalline coagulates. For example, the morphology of crystallites of PiPrOx grafted with the polysaccharide pullulan was significantly different than that of non-modified PiPrOx. Winnik and Akiyoshi et al.103 grafted pullulan to PiPrOx with an Mn of 3200 g mol−1 (Đ = 1.16) and 7400 g mol−1 (Đ = 1.08) via carbonate linkages formed by reacting 1,1-carbonyldiimidazole-activated PiPrOx with the pullulan hydroxyl groups (the polymers were named PiPrOx3K-Pul and PiPrOx7K-Pul, respectively). Incubation of a PiPrOx3K-Pul aqueous solution at 70 °C (above the LCST) for 10 min resulted in the formation of objects with a hydrodynamic radius (Rh) of 470 nm and a dispersity (PDI) of 0.26. Importantly, the particles disintegrated upon cooling. A PiPrOx7K-Pul solution incubated for the same time and temperature formed smaller particles of Rh = 240 nm with a PDI of 0.18. However, after cooling, particles with Rh of 90 nm (PDI = 0.31) were still present in the solution. X-ray diffraction analysis revealed the crystalline nature of the obtained particles. The authors assumed that this short heat treatment was sufficient to induce permanent self-association of PiPrOx7K-Pul due to partial and irreversible crystallization of the PiPrOx chains. PiPrOx7K-Pul was heated for longer time periods, and the intensity of the diffraction peaks of the crystallites increased significantly. Additionally, it was observed that the patterns display a remarkable similarity to the XRD traces reported for crystalline PiPrOx obtained either by crystallization from hot water or via bulk crystallization. The crystallites were found to adopt structures of loop morphology, from 750 to 1200 nm in diameter. A few short fibrils emanating from the ring circumference could be observed (Fig. 11a–c). It is interesting to note that PiPrOx grafted with pullulan crystallized significantly faster than pure PiPrOx from aqueous solution.
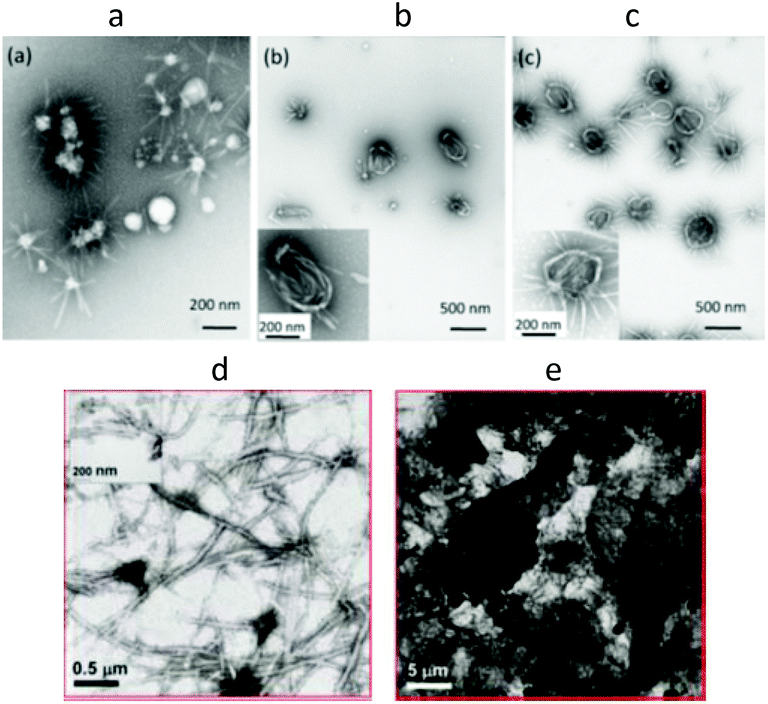 | ||
| Fig. 11 TEM images of PiPrOx7K-Pul incubated in water for 10 min (a), 1 h (b) and 3 h (c) and C18-PiPrOx-OH for 5 h (d) and 24 h (e) at 70 °C. Reproduced with permission from ref. 103. Copyright 2009 The Royal Society of Chemistry. And from ref. 110. Copyright 2009 American Chemical Society. | ||
Changes in the morphology of coagulate crystallized from aqueous solution were also monitored for PiPrOx modified with octadecyl end groups.110 Semitelechelic PiPrOx (C18-PiPrOx-OH) incubated at 70 °C for 5 and 24 h was found to form a coagulate that remained stable even after cooling of the solution to room temperature, as in the case of the unmodified PiPrOx. As revealed by TEM analysis (Fig. 11d and e), specimens obtained after 5 h of incubation formed long interconnected fibres of approximately 40 nm in diameter. With increasing heating time, the fibres assembled into large bundles that created an extensive network. Different situations were observed for telechelic PiPrOx (C18-PiPrOx-C18). The aqueous solutions of telechelic PiPrOx incubated at 70 °C for 24 h recovered their transparency upon cooling below the LCST, and no crystalline coagulate was observed. The authors concluded that the reluctance to crystallize the telechelic polymer must originate from its assembly in cold water to form flower-like micelles. It was stated that the dehydrated chains (above LCST) adopted a “crumbled loop” conformation within the polymer-rich phase. In such an environment, the motion of the chains was restricted, and reorganization towards chain alignment and crystallization could not occur.
However, a slightly different situation was described for copolymers of iPrOx with 2-(3-butenyl)-2-oxazoline (Mn of 16100 g mol−1, Đ = 1.05) dissolved in D2O.43 Based on SEM and DSC analysis, after incubation of these solutions at 60 °C for 3 days, no significant difference in the coagulate morphology and thermal properties could be observed compared to the homopolymer of iPrOx. Spherical particles of 2–3 μm in diameter, composed of nanofibres, were obtained, similar to the case of PiPrOx. Additionally, the same melting point and the degree of crystallinity as PiPrOx were designated. Moreover, the glycosylation of an aqueous dispersion of crystalline microparticles through direct photoaddition of either 1-thio-β-D-glucose or 1-thio-β-D-galactose (the thiol–ene radical addition) did not influence the morphology of crystalline coagulate.
Briefly, the prolonged incubation of PiPrOx in water above the LCST leads to crystalline coagulate which is insoluble after cooling to room temperature. Different coagulate morphologies can be obtained, depending on the incubation time, temperature and end groups of PiPrOx. The presence of insoluble material seems to be unfavourable for applications where thermoresponsive behaviour of the polymer is used, and its full dissolution after cooling is needed.
3.2. Conformation of PiPrOx chains during crystallization from water
Detailed insight into the mechanism of POx crystallization from solution for the example of PiPrOx was provided by Katsumoto and Winnik et al.,107 Sun and Wu et al.108,109 and Atılgan et al.106 and is still much debated.Initially, Katsumoto and Winnik et al.107 employed vibrational spectroscopy combined with molecular orbital calculations and spectral measurements of model compounds to monitor changes in the PiPrOx conformation during phase separation and crystallization from water. They observed that below the LCST, PiPrOx chains are hydrated and adopt a conformation in which the trans and gauche conformers coexist. As the temperature increases, the hydration layer breaks gradually; however, the polymer chains still adopt both trans and gauche conformations at this stage. When the temperature is close to or above the LCST, the solution undergoes liquid–liquid phase separation, and within the polymer-rich phase, the polymer chain adopts a mostly trans conformation. Upon prolonged heating, this conformation is stabilized and remains predominant even upon cooling the solution to below the LCST. This promotes a partial organization of the chains in trans conformation in a dense liquid phase, leading to the crystallite growth stage. A schematic representation of the changes in the conformation of PiPrOx in water as a function of temperature is shown in Fig. 12.
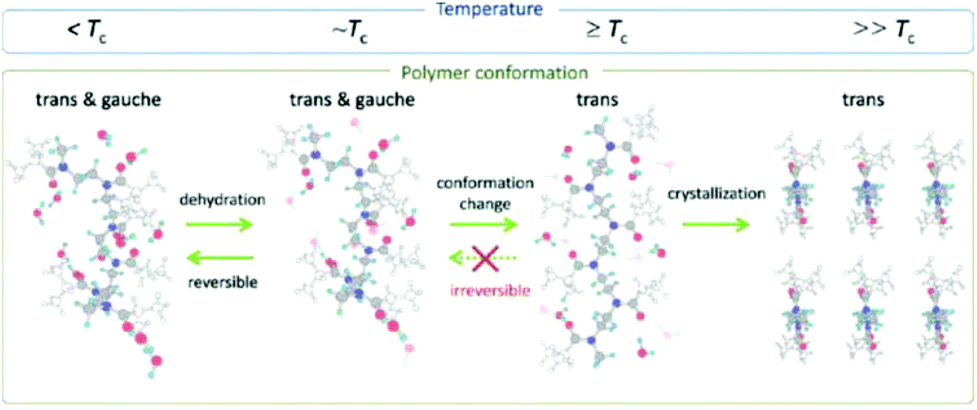 | ||
| Fig. 12 Schematic representation of the changes in the conformation of PiPrOx in water as a function of temperature. Reproduced with permission from ref. 107. Copyright 2012 American Chemical Society. | ||
These findings, showing that all trans conformations of PiPrOx chains occur prior to crystallization from water, are contrary to the conclusions proposed by Sun and Wu, who stated the impossibility for the presence of all trans conformations during crystallization of PiPrOx in the solid state.67
However, the same authors, using the FT-IR technique, studied the motions of PiPrOx functional groups and showed the importance of hydrogen bonds during nucleation and crystallization from D2O.109 The intensity of the asymmetric and symmetric stretching bands of C–H from the methyl groups of the side chains increased during annealing, while the frequencies did not change. These findings suggested that the majority of CH3 groups in the side chains remained in the dehydrated state shortly before and after crystallization. Bands attributed to the C–H bonds in methylene groups showed a slight frequency shift to lower wavenumbers, which confirmed that the backbone of PiPrOx exhibited minor dehydration during annealing at the temperature above the LCST. Upon annealing, the peaks assigned to stretching of CO being a part of the weak hydrogen interchain bonds (CO⋯D–O–D⋯OC), and the CO⋯D2O hydrogen bonds gradually disappeared, while a peak attributed to ordered CO in crystalline PiPrOx chains appeared. This led to the conclusion that the crystallization of PiPrOx in hot water is accompanied by the cleavage of CO-related hydrogen bonds and ordering of carbonyl groups. Additionally, the peak variations of the bands arising from CH3⋯OC (not easy to discern in conventional IR) were recognized using perturbation correlation moving window spectra. Thus, the cleavage of two kinds of hydrogen bonds, CH3⋯OC and CO⋯water (both CO⋯D–O–D⋯OC and CO⋯D2O), formed during liquid–liquid phase separation, was explained to be the driving force for the nucleation of POxs in hot water. Additionally, based on the 2D correlation spectroscopic analysis, it was confirmed that the crystallization of PiPrOx during liquid–liquid phase separation started from cleavage of hydrogen bonds followed by side chain reordering due to amide dipolar orientation. Induced by these movements, the backbone of PiPrOx was then distorted, and the side chains became more closely pack. During crystal growth, further chain ordering occurs, resulting in the final formation of crystalline PiPrOx chains with partial trans conformation of backbones and side chains lying alternatively on the two sides.
Further examination of the conformation of PiPrOx chains during crystallization was reported by Atılgan et al.,106 who stated that a helical PiPrOx chain conformation is a precursor of crystallization from water. Computational simulations were used in these studies to estimate the conformations of PiPrOx chains in water at different temperatures. Molecular dynamics simulations were performed to analyse single chain behaviour, while dissipative particle dynamics simulations were utilized to explain the relation of the polymer–solvent systems. A statistical analysis of the backbone torsional angles below the LCST revealed that the main chain of PiPrOx is elongated with the isopropyl side groups located away from each other so that the steric effects are decreased, and the solvation sphere is maximized. Upon increasing the temperature, the number of hydrogen bonds decreased by 12 ± 4%, and isopropyl groups are pointed to the same side, providing partial shielding from the solvent; thus, a single PiPrOx chain is more compact. The most likely conformation of the chain after molecular dynamics simulations was proposed (Fig. 13). The chain backbone was assumed to have a helical conformation with a pitch length of 15 monomers with disordered side chains.
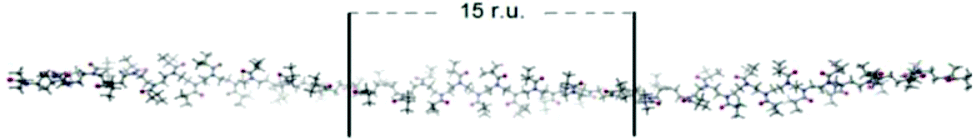 | ||
| Fig. 13 Helical PiPrOx chain conformation with 15 repeating units (r. u.) observed in water above the LCST. Reproduced with permission from ref. 106. Copyright 2016 Elsevier Ltd. | ||
The mechanism of crystallization of PiPrOx from the solution and the conformation of polymer chains during this process are constantly the subject of theoretical considerations and experimental studies. To date, a few aspects must be taken into account: polymer chains are assumed to adopt a mostly trans conformation prior to crystallization,107,108 and a helical conformation of the polymer chain is considered before crystallization.106 On the other hand, the impossibility of the trans conformation of the polymer chain as preferential to crystallization is also discussed.67
3.3. Crystallization of PiPrOx from water/organic and organic solvents
PiPrOx was widely described to crystallize from aqueous solutions but also from organic solvents. This was confirmed for PiPrOx dissolved both in water with the addition of organic solvent, such as tetrahydrofuran (THF) or ethylene glycol,40 and in pure organic solvents, including acetonitrile (ACN), dimethyl sulfoxide (DMSO), and propylene carbonate (PC).42For PiPrOx dissolved in water with the addition of organic solvent, crystallization is induced by the liquid–liquid phase separation above the LCST. In the case of PiPrOx dissolved in organic solvents, the liquid–liquid phase separation does not occur at elevated temperatures due to the lack of thermosensitivity in such solutions. The increase in the local density of the polymer cannot be induced by increased temperature; thus, crystallization occurs after prolonged heating compared to aqueous solution.
Generally, it was found that the properties of PiPrOx crystallized from water with organic solvents or from pure organic solvents were similar to the properties of PiPrOx crystallized from the aqueous solution. However, significant differences in the morphology of the crystalline phase were found.
Schlaad et al.40 observed that the addition of THF as a cosolvent (approximately 2 vol%) to the aqueous solution of PiPrOx hindered the formation of spherical particles, and a mesh of nanofibres was obtained after annealing at 65 °C for 24 h. Additionally, the predominant presence of fibres was observed when PiPrOx was annealed in water with 5% v/v ethylene glycol (Fig. 14). In such a case, the crystalline phase consisted mostly of fibril structures, whereas spherical particles were only a minority fraction.
 | ||
| Fig. 14 (a) Scanning force microscopy micrograph of the PiPrOx coagulate formed in the mixture of water and THF (98:2 v/v) annealed for 24 hours at 65 °C, (b) TEM micrograph of the PiPrOx coagulate formed in a mixture of water and ethylene glycol (95:5 v/v) Reproduced with permission from ref. 40. Copyright 2007 WILEY-VCH. | ||
As previously mentioned, the driving force for the crystallization of PiPrOx from water with the addition of organic solvent was the supersaturation induced by the liquid–liquid phase separation above the LCST, similar to the case of PiPrOx in pure aqueous solution; thus, crystallization occurred relatively quickly.
On the other hand, different behaviour was observed for PiPrOx dissolved in an organic solution without any addition of water. In such a case, as PiPrOx does not exhibit thermoresponsive behaviour in organic solvents, the phase transition under increased temperature does not occur and thus does not provide the supersaturation of the solution. A detailed study on crystallization under these conditions was performed using organic solvents with dipole moments higher than that of water (approximately 4 D), including ACN, PC and DMSO.42 PiPrOx dissolved in these solvents (at different concentrations) was incubated at elevated temperatures for a prolonged time (20 days), and the formation of crystallites was observed. DSC analysis of coagulate obtained after this time revealed unsymmetrical endothermic peaks, possibly due to the formation of imperfect crystals of various morphologies. Both the Tm and Tg of PiPrOx crystallized from organic solutions were similar to those of PiPrOx crystallized in bulk and from water. The X-ray diffraction curves were analysed to calculate the percentage of crystalline phase in PiPrOx samples. In the case of PiPrOx solutions of 5% w/v, the highest degree of crystallinity of χc ∼ 65% was obtained for PiPrOx dissolved in DMSO. χc equal to ∼60% and ∼45% was obtained in PC and in ACN, respectively. With an increasing concentration of PiPrOx in ACN to 10 and 30% w/v, an increase in the degree of crystallinity to χc ∼ 55 and 68%, respectively, was observed. This indicates that χc depends on the polymer concentration. The morphology of PiPrOx crystallized from organic solutions was found to be different than that of PiPrOx crystallized from water (Fig. 15). In the SEM micrograph of PiPrOx crystallized from acetonitrile at a very high concentration (30% w/v), a network-like structure could be observed. Within this network, separate objects possessing a fibril-like morphology could be distinguished. The length of the fibrils ranged from a few to several microns, with a width of approximately 50 nm. A dense, fibrillar network could also be observed in the SEM micrographs of PiPrOx crystallized from acetonitrile at lower concentrations, indicating that, in this case, concentration has a minor effect on sample morphology. The PiPrOx crystalline structures formed in DMSO and PC are similar to those obtained in ACN.
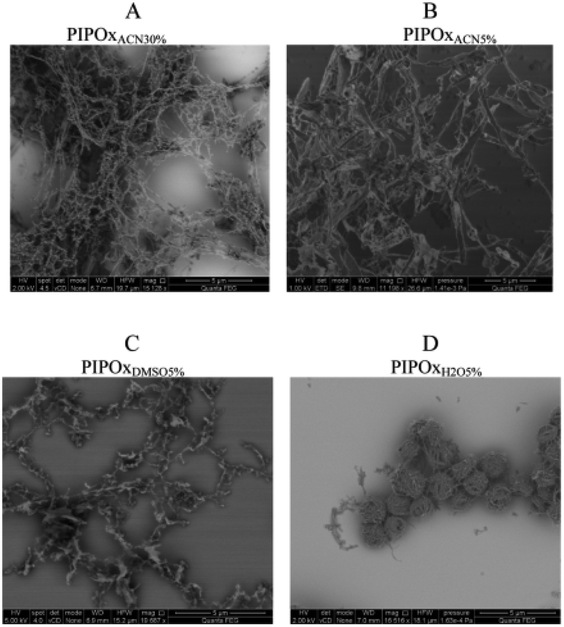 | ||
| Fig. 15 SEM micrographs of PiPrOx crystallized from different organic solutions at various concentrations. Reproduced with permission from ref. 42. Copyright 2015 American Chemical Society. | ||
Based on the studies of PiPrOx dissolved in organic solvents, it was demonstrated that the phase separation above the LCST is not a required precondition for polymer crystallization. As discussed above, the crystallization of PiPrOx from water is accelerated by the so-called solubility gap, associated with the polymer phase transition above the LCST. In the case of PiPrOx dissolved in organic solvent, the lack of this solubility gap does not prevent or disturb nucleation and crystallization. However, this process takes much longer in organic solvents. It takes several days compared with crystallization from water that occurs in several hours.
3.4. Crystallization of other POxs from solutions
In addition to the crystallization of PiPrOx, the crystallization of several other (co)poly(2-oxazolines)s from solution was described.Poly(2-ethyl-2-oxazoline) (Mw ∼ 500000 g mol−1) was found to form insoluble agglomerates in water when the solution was kept at 70 °C for 45 days.104 The X-ray diffraction analysis confirmed the crystalline character of the obtained coagulate. After the addition of salts (sodium acetate or sodium thiocyanate), the rate of agglomerate formation was significantly enhanced. The model crystal structure of PEtOx crystallized from water was proposed and is presented in Fig. 16A. The peak positions calculated by using this model were consistent with the experimentally observed peaks from X-ray diffraction analysis. In the proposed model crystal structure for PEtOx, the ethyl side groups are alternately aligned along the [100] direction to either side of the backbone. The d-spacing values were much lower compared, for example, with PiPrOx. The authors suggested that the d-spacing of crystalline POxs decreases linearly with decreasing alkyl side chain length. This is expected as the substituent opens up to either side of the backbone and shorter alkyl side chains can be more closely packed. Additionally, in the case of shorter alkyl side chains, amide dipoles can probably become closer, which gives rise to stronger intermolecular interactions. Such strong interactions can hinder the conformational relaxations needed to reach the equilibrium crystalline structure; thus, crystallization of PEtOx from water is much slower than in the case of PiPrOx. The morphology of PEtOx crystallites is much different than that of PiPrOx crystallized from water (Fig. 16B). The network-like structure consisted of entangled fibres with an average diameter of 3.8 μm that resemble PiPrOx crystallized from organic solutions.
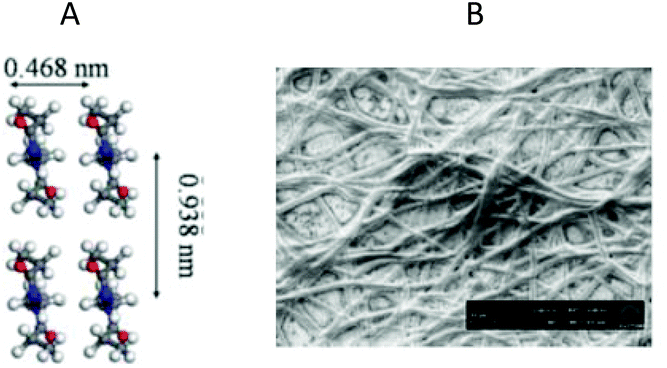 | ||
| Fig. 16 The model crystal structure of PEtOx (A) and SEM image of spin-coated PEtOX fibres from 0.2 M aqueous NaAc solution (B). Reproduced with permission from ref. 104. Copyright 2012 The Royal Society of Chemistry. | ||
Crystallization of PEtOx from solution seems to be surprising, as it is known to be amorphous in the solid state.
Next to PEtOx, crystallization of poly(2-isobutyl-2-oxazoline) (PiBuOx) and poly(2-nonyl-2-oxazoline) (PNonOx) was found to occur from an ethanol/water mixture at room temperature, which is below the upper critical solution temperature (UCST) of these polymers.105 After dissolving at elevated temperature, PiBuOx and PNonOx were left for several days at room temperature, which resulted in the formation of an insoluble precipitate. DSC analysis revealed an endothermic melting transition of PNonOx at 142 °C and in the temperature range of 134–148 °C, with a peak maximum at 141 °C for PiBuOx, indicating the semi-crystalline nature of the obtained precipitates. The large difference in melting points between the PiBuOx crystallized from solution (141 °C) and in bulk (200 °C)70 was observed, but the reason for such behaviour is not discussed. X-ray diffraction analysis confirmed the crystallinity of the obtained material. For PiBuOx, the crystalline precipitate consisted of spherical particles of ∼3 μm in diameter that were fused together into larger clusters (Fig. 17). The structures were composed of nanosheets or lamellae, not nanofibres as observed for PiPrOx crystallites.40
 | ||
| Fig. 17 SEM images of PiBuOx crystallized from ethanol/water 55:45 (w/w) at room temperature. Reproduced with permission from ref. 105. Copyright 2011 WILEY-VCH. | ||
When the concentration of PiBuOx in the ethanol/water mixture was low, the obtained crystalline spherical particles were less compact and were composed of fibres, not lamellae. Additionally, decreasing the amount of ethanol in the mixture caused changes in the crystallite morphology from spherical particles, through sunflower-like, to disk-shaped structures, exhibiting a fibrous network-like morphology. Microscopic analysis of the PNonOx crystallites revealed spherical particles of uniform size and morphology. The authors concluded that the length of the side chain does not affect the crystallization rate or the resulting morphologies.
In the case of PiBuOx and PNonOx, their ability to crystallize from solution seems justifiable because of their crystalline properties in the solid state, which is interesting and a little surprising in the case of PEtOx, which is known to be amorphous in the solid state.
3.5. The use of crystalline properties of POxs
As discussed above, during prolonged incubation of some POxs dissolved in water or organic solvents, at a certain temperature and concentration, the polymer chains arrange into elementary cells and begin to crystallize. In many cases, this process is undesirable because the precipitated crystalline phase is not soluble after changing the temperature to the initial value. This fact may potentially disqualify the POxs aqueous systems from applications where, for example, thermoresponsive behaviour is necessary. On the other hand, the ability of POxs to crystallize was used in many applications, for example, in the crystallization-driven self-assembly to obtain micelles or in substance separation systems for molecular recognition or in the polymeric solid supports for cell culture to improve the cell adhesion and proliferation.43,44,111Taton and Lecommandoux et al.111 used the crystalline properties of poly(2-isopropyl-2-oxazoline) in the crystallization-driven self-assembly of block copolymers of iPrOx and MeOx. These copolymers exhibited LCST-type liquid–liquid phase separation in aqueous solution at 57 °C. By increasing the temperature to 65 °C, the formation of well-defined micelles with Rh = 17 nm was observed, as confirmed by dynamic light scattering. This process was reversible only when the solution was kept at the elevated temperature for a short period of time (less than 90 minutes), and unimers were detected when the solution was then cooled. In contrast, when the solution was kept above the LCST for more than 90 minutes, crystallization occurred, and the initially well-defined and nearly monodisperse micelles progressively became unstable. Next to the micellar nanostructures, the fibre-like objects were then observed that were arranged into denser “fibrenodes”. After 24 hours of annealing, both micellar nanostructures of approximately 30 nm in diameter and a dense network of fibres were observed. The X-ray analysis revealed that both the intensity and the sharpness of the crystalline peaks progressively increased with time of annealing, confirming an increase in the crystallinity of the samples. The authors concluded that the self-assembled micelles acted as crystallization seeds and that the spatial restriction caused by the crystallization of the PiPrOx block could result in the formation of spherical or distorted crosslinked micelles and fibres. Crystallization-driven self-assembly was also mentioned in the case of the gradient copolymers of NonOx with PhOx containing 50 and 60 mol% of NonOx.36 The authors noticed that these copolymers formed thermoresponsive micelles in an ethanol–water mixture (80:20 wt%) below the UCST. Prolonged incubation of such systems led to large self-assembled structures, and their formation might be driven by crystallization of the NonOx segment.
The ability of POxs to crystallize was used by Schlaad for molecular recognition and cell sensing.43 In this case, glycosylated crystalline microspheres based on the copolymer of iPrOx and 2-(3-butenyl)-2-oxazoline (Mn of 16100 g mol−1, Đ = 1.05) were used. To obtain crystalline particles, the copolymer was incubated at 60 °C for 3 days. The obtained spherical particles of 2–3 μm in diameter, composed of nanofibres, were then glycosylated through direct photoaddition (thiol–ene radical addition) of either 1-thio-β-D-glucose (Glc) or 1-thio-β-D-galactose (Gal). The SEM images obtained for the dried products confirmed that the original structure of particles was preserved after glycosylation (Fig. 18).
 | ||
| Fig. 18 SEM micrographs of crystalline microspheres of copolymers of iPrOx with 2-(3-butenyl)-2-oxazoline (top) and microspheres glycosylated with Glc (bottom). Reproduced with permission from ref. 43. Copyright 2009 WILEY-VCH. | ||
The molecular recognition and cell-sensing abilities of glycosylated particles were measured by the strength of their interaction with lectins – proteins that bind glucose or galactose. It was estimated that nearly half of the Glc or Gal units attached to the crystalline particles participated in the binding of lectin. Moreover, the highly specific protein recognition of the glycosylated crystalline microspheres was observed during the separation of a mixture of both lectins. Owing to their versatile and simple synthesis as well as their biocompatibility, the authors assumed that these materials could be eligible for biological and biomedical applications such as protein isolation and separation.
The thermoresponsive nanolayers composed of POxs with different degrees of crystallinity were used as biomaterials to enhance cell adhesion and detachment.44 PiPrOx layers covalently bonded to glass or silica wafers were obtained via surface termination of the living polymer chains (Mn = 20800 g mol−1, Đ = 1.01). Fibrillar crystallites formed in the polymerization mixture (acetonitrile as a solvent) settled onto the wafers next to the amorphous polymer. The amount of crystallite adsorbed on thermoresponsive polymer layers was controlled by the time of annealing of the PiPrOx solution (Fig. 19). The incubation of PiPrOx in the polymerization mixture for 2 days caused that after grafting, only 20% of the surface was covered by the fibrils, while 70% of the entire area was covered when the polymerization mixture was incubated for 7 days before grafting. The changeable amount of crystallite on the surface influenced the nanolayer properties, such as cell adhesion and detachment. The higher content of crystallites weakened the temperature response of the layer, as evidenced by the philicity and thickness measurements. The presence of crystallites on the PiPrOx layers promoted the proliferation of human dermal fibroblasts. After cell sheet formation, the temperature was lowered. Changes in the physicochemical properties of the layer caused by the temperature response of the polymer led to cell sheet detachment.
 | ||
| Fig. 19 AFM micrographs of surfaces obtained after annealing of PiPrOx in ACN for 0, 2, 4 and 7 days and morphology of confluent fibroblast sheets cultured on PiPrOx surfaces with different crystallite contents. Reproduced with permission from ref. 44. Copyright 2015 American Chemical Society. | ||
4. Conclusions
The thermal and crystalline properties of poly(2-oxazoline)s are summarized in this review.For homopolymers of 2-oxazolines in the solid state, these properties were found to be determined by the nature of the side chain attached to an amide group. For POxs with linear alkyl side chains, the thermal and crystalline properties were dependent on the length of the aliphatic side chain. Very high melting temperatures, above 200 °C, exhibited POxs with cycloalkyl side chains as well as with condensed aromatic rings and fluorinated linear side chains. Relatively low Tm values were detected for POxs with extremely long, unsaturated side chains. A glass transition below 0 °C was exhibited by poly(2-hexyl-2-oxazoline), branched PEHOx and PC3MestOx containing methyl ester side chains. The highest value of Tg was recorded for adamantyl-containing PMeAdamOx. The majority of POxs exhibit the ability to crystallize, especially those containing linear alkyl chains, large cycloalkyl rings, condensed aromatic rings or linear fluorinated chains as a substituent. Special attention must be paid to PiPrOx, which exhibit relatively high values of Tg and Tm, both with a high ability to crystallize. The thermal and crystalline properties of POxs may be easily controlled by the copolymerization of appropriate monomers. In many cases, an increasing amount of more flexible comonomers in the copolymer causes a decrease in Tg (plasticizing effect). The melting point of POxs and their ability to crystallize can be significantly reduced by copolymerization with monomers that weaken the strong interactions between the chains. However, crystallization cannot be eliminated completely, even in the case of seemingly amorphous POxs, as it can be forced by exposure of the polymers to a high, predefined temperature for an extended time.
Many POxs exhibit a tendency to crystallize not only in the solid state but also from solution. This process was studied most extensively for PiPrOx; nevertheless, PEtOx, PiBuOx and PNonOx were also observed to crystallize from solution. In the case of aqueous solutions of thermoresponsive POxs, changing the temperature above the LCST or below the UCST causes liquid–liquid phase separation, which is a driving force to increase the local density of crystallizable macromolecules and further crystallization. When POx is dissolved in organic solvent, where no thermoresponsive behaviour is observed, supersaturation of the solution is not provided by the phase transition; however, crystallization occurs, but it proceeds much more slowly. The presence of insoluble, crystalline material seems to be unfavourable for applications where thermoresponsive behaviour is used and a return to total solubility is needed. However, the crystalline properties of POxs were used in some applications, for example, in the self-assembly of polymer chains to obtain micelles or in the polymeric solid supports for improvement of the cell culture.
These synthetic pseudo-polypeptides offer structural and functional diversity that can be used to obtain polymers with strictly defined properties.
Abbreviations
| 2DCOS | Two-dimensional correlation spectroscopy |
| ACN | Acetonitrile |
| APOx | (2-(4-Aminophenyl)-2-oxazoline |
| CROP | Cationic ring-opening polymerization |
| DMSO | Dimethyl sulfoxide |
| DSC | Differential scanning calorimetry |
| LCST | Lower critical solution temperature |
| P3EPOx | Poly(2-(3-ethylpentyl)-2-oxazoline) |
| PAdamOx | Poly(2-(1-adamantyl)-2-oxazoline) |
| PButenOx | Poly[2-(3-butenyl)-2-oxazoline] |
| PC | Propylene carbonate |
| PC3MestOx | Poly(2-methoxycarbonylpropyl-2-oxazoline) |
| PC4CarbOx | Poly(2-[4-(2,7-dimethoxycarbazoI-9-yI)butyl]-2-oxazoline) |
| PcBuOx | Poly(2-cyclobutyl-2-oxazoline) |
| PcHexOx | Poly(2-cyclohexyl-2-oxazoline) |
| PcPentOx | Poly(2-cyclopentyl-2-oxazoline) |
| PcPrOx | Poly(2-cyclopropyl-2-oxazoline) |
| PDecenOx | Poly(2-decenyl-2-oxazoline) |
| PEHOx | Poly(2-(3-ethylheptyl)-2-oxazoline) |
| PEPOx | Poly(2-(1-ethylpentyl)-2-oxazoline) |
| PEtOx | Poly(2-ethyl-2-oxazoline) |
| PHeptOx | Poly(2-heptyl-2-oxazoline) |
| PHexOx | Poly(2-hexyl-2-oxazoline) |
| PiBuOx | Poly(2-isobutyl-2-oxazoline) |
| PiPrOx | Poly(2-isopropyl-2-oxazoline) |
| PMeAdamOx | Poly(2-(1-adamantylmethyl)-2-oxazoline) |
| PMeOx | Poly(2-methyl-2-oxazoline) |
| PMestOx | Poly(2-methoxycarbonylethyl-2-oxazoline) |
| PnBuOx | Poly(2-n-butyl-2-oxazoline) |
| PNonOx | Poly(2-nonyl-2-oxazoline) |
| PnPrOx | Poly(2-n-propyl-2-oxazoline) |
| POx | Poly(2-substituted-2-oxazoline) |
| PPentOx | Poly(2-pentyl-2-oxazoline) |
| PPhOx | Poly(2-phenyl-2-oxazoline) |
| PSoyOx | Poly(2-“soy alkyl”-2-oxazoline) |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
| T g | Glass transition temperature |
| THF | Tetrahydrofuran |
| T m | Melting temperature |
| UCST | Upper critical solution temperature |
| UndOx | 2-Undecyl-2-oxazoline |
| WAXS | Wide-angle X-ray scattering |
| XRD | X-ray diffraction |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Project 2016/21/D/ST5/01951.References
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2253–2265 CrossRef CAS.
- A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 881–886 CrossRef CAS.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed. Engl., 1966, 5, 875–888 CrossRef CAS.
- T. Kagiya, S. Narisawa, T. Maeda and K. Fukui, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- T. Saegusa, H. Ikeda and H. Fujii, Macromolecules, 1972, 5, 354–358 CrossRef CAS.
- T. Saegusa, S. Kobayashi and Y. Nagura, Macromolecules, 1974, 7, 713–716 CrossRef CAS.
- V. R. de la Rosa, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS.
- M. Glassner, D. D'hooge, J. Y. Park, P. Van Steenberge, B. Monnery, M.-F. Reyniers and R. Hoogenboom, Eur. Polym. J., 2015, 65, 298–304 CrossRef CAS.
- M. Einzmann and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2821–2831 CrossRef CAS.
- M.-E. Kourti, G. C. Vougioukalakis, N. Hadjichristidis and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2520–2527 CrossRef CAS.
- J. S. Hrkach and K. Matyjaszewski, Macromolecules, 1992, 25, 2070–2075 CrossRef CAS.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Słomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. Leenen, S. F. G. M. van Nispen, M. van der Loop, C. H. Abeln, A. M. J. van den Berg and U. S. Schubert, Macromolecules, 2005, 38, 7957–7966 CrossRef CAS.
- F. Hu, S. Xie, L. Jiang and Z. Shen, RSC Adv., 2014, 4, 59917–59926 RSC.
- B. D. Monnery, V. V. Jerca, O. Sedlacek, B. Verbraeken, R. Cavill and R. Hoogenboom, Angew. Chem., Int. Ed., 2018, 57, 15400–15404 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659–671 CrossRef CAS.
- C. Guerrero-Sanchez, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2006, 3797–3799 RSC.
- R. M. Paulus, T. Erdmenger, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2007, 28, 484–491 CrossRef CAS.
- M. W. M. Fijten, J. M. Kranenburg, H. M. L. Thijs, R. M. Paulus, B. M. van Lankvelt, J. de Hullu, M. Springintveld, D. J. G. Thielen, C. A. Tweedie, R. Hoogenboom, K. J. Van Vliet and U. S. Schubert, Macromolecules, 2007, 40, 5879–5886 CrossRef CAS.
- C. Kim, S. C. Lee, J. H. Shin, J.-S. Yoon, I. C. Kwon and S. Y. Jeong, Macromolecules, 2000, 33, 7448–7452 CrossRef CAS.
- J.-S. Park and K. Kataoka, Macromolecules, 2006, 39, 6622–6630 CrossRef CAS.
- J.-S. Park and K. Kataoka, Macromolecules, 2007, 40, 3599–3609 CrossRef CAS.
- Y. Morishima, T. Tanaka and S.-i. Nozakura, Polym. Bull., 1981, 5, 19–24 CrossRef CAS.
- K. Aoi and M. Okada, Prog. Polym. Sci., 1996, 21, 151–208 CrossRef CAS.
- S. Kobayashi and H. Uyama, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 192–209 CrossRef CAS.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- A. Dworak, B. Trzebicka, A. Kowalczuk, C. Tsvetanov and S. Rangelov, Polimery, 2014, 59, 88–94 CrossRef CAS.
- J.-S. Park, Y. Akiyama, F. M. Winnik and K. Kataoka, Macromolecules, 2004, 37, 6786–6792 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- R. Konefał, J. Spěváček and P. Černoch, Eur. Polym. J., 2018, 100, 241–252 CrossRef.
- C. Diab, Y. Akiyama, K. Kataoka and F. M. Winnik, Macromolecules, 2004, 37, 2556–2562 CrossRef CAS.
- S. Huber and R. Jordan, Colloid Polym. Sci., 2008, 286, 395–402 CrossRef CAS.
- S. Huber, N. Hutter and R. Jordan, Colloid Polym. Sci., 2008, 286, 1653–1661 CrossRef CAS.
- D. Christova, R. Velichkova, W. Loos, E. J. Goethals and F. D. Prez, Polymer, 2003, 44, 2255–2261 CrossRef CAS.
- R. Hoogenboom, H. M. L. Lambermont-Thijs, M. J. H. C. Jochems, S. Hoeppener, C. Guerlain, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Soft Matter, 2009, 5, 3590–3592 RSC.
- H. M. L. Lambermont-Thijs, H. P. C. v. Kuringen, J. P. W. v. d. Put, U. S. Schubert and R. Hoogenboom, Polymers, 2010, 2, 188–199 CrossRef CAS.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten, S. Wijnans, A. M. J. van den Berg, H. M. L. Thijs and U. S. Schubert, J. Comb. Chem., 2006, 8, 145–148 CrossRef CAS.
- A. L. Demirel, M. Meyer and H. Schlaad, Angew. Chem., Int. Ed., 2007, 46, 8622–8624 CrossRef CAS.
- M. Meyer, M. Antonietti and H. Schlaad, Soft Matter, 2007, 3, 430–431 RSC.
- N. Oleszko, A. Utrata-Wesołek, W. Wałach, M. Libera, A. Hercog, U. Szeluga, M. Domański, B. Trzebicka and A. Dworak, Macromolecules, 2015, 48, 1852–1859 CrossRef CAS.
- C. Diehl and H. Schlaad, Chem. – Eur. J., 2009, 15, 11469–11472 CrossRef CAS.
- N. Oleszko, W. Wałach, A. Utrata-Wesołek, A. Kowalczuk, B. Trzebicka, A. Klama-Baryła, D. Hoff-Lenczewska, M. Kawecki, M. Lesiak, A. L. Sieroń and A. Dworak, Biomacromolecules, 2015, 16, 2805–2813 CrossRef CAS.
- R. Donev, N. Koseva, P. Petrov, A. Kowalczuk and J. Thome, World J. Biol. Psychiatry, 2011, 12, 44–51 CrossRef.
- K. Kempe, A. Vollrath, H. W. Schaefer, T. G. Poehlmann, C. Biskup, R. Hoogenboom, S. Hornig and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 1869–1873 CrossRef CAS.
- R. Luxenhofer, G. Sahay, A. Schulz, D. Alakhova, T. K. Bronich, R. Jordan and A. V. Kabanov, J. Controlled Release, 2011, 153, 73–82 CrossRef CAS.
- J. Tong, M. C. Zimmerman, S. Li, X. Yi, R. Luxenhofer, R. Jordan and A. V. Kabanov, Biomaterials, 2011, 32, 3654–3665 CrossRef CAS.
- F. C. Gaertner, R. Luxenhofer, B. Blechert, R. Jordan and M. Essler, J. Controlled Release, 2007, 119, 291–300 CrossRef CAS.
- P. Goddard, L. E. Hutchinson, J. Brown and L. J. Brookman, J. Controlled Release, 1989, 10, 5–16 CrossRef CAS.
- R. Hoogenboom, Macromol. Chem. Phys., 2007, 208, 18–25 CrossRef CAS.
- R. Hoogenboom, Eur. J. Lipid Sci. Technol., 2011, 113, 59–71 CrossRef CAS.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- B. Guillerm, S. Monge, V. Lapinte and J.-J. Robin, Macromol. Rapid Commun., 2012, 33, 1600–1612 CrossRef CAS.
- K. Lava, B. Verbraeken and R. Hoogenboom, Eur. Polym. J., 2015, 65, 98–111 CrossRef CAS.
- E. Rossegger, V. Schenk and F. Wiesbrock, Polymers, 2013, 5, 956–1011 CrossRef.
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504–1520 CrossRef CAS.
- H. Schlaad, C. Diehl, A. Gress, M. Meyer, A. L. Demirel, Y. Nur and A. Bertin, Macromol. Rapid Commun., 2010, 31, 511–525 CrossRef CAS.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS.
- P. Wilson, P. C. Ke, T. P. Davis and K. Kempe, Eur. Polym. J., 2017, 88, 486–515 CrossRef CAS.
- A. M. Kelly and F. Wiesbrock, Macromol. Rapid Commun., 2012, 33, 1632–1647 CrossRef CAS.
- G. Morgese and E. M. Benetti, Eur. Polym. J., 2017, 88, 470–485 CrossRef CAS.
- L. Tauhardt, K. Kempe, M. Gottschaldt and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 7998–8011 RSC.
- R. Hoogenboom, Eur. Polym. J., 2017, 88, 448–450 CrossRef CAS.
- M. Litt, F. Rahl and L. G. Roldan, J. Polym. Sci., Part A-2, 1969, 7, 463–473 CrossRef CAS.
- S. Sun and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 31084–31092 RSC.
- J. M. Rodriguez-Parada, M. Kaku and D. Y. Sogah, Macromolecules, 1994, 27, 1571–1577 CrossRef CAS.
- M. Beck, P. Birnbrich, U. Eicken, H. Fischer, W. E. Fristad, B. Hase and H.-J. Krause, Angew. Makromol. Chem., 1994, 223, 217–233 CrossRef CAS.
- K. Kempe, M. Lobert, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3829–3838 CrossRef CAS.
- E. F.-J. Rettler, J. M. Kranenburg, H. M. L. Lambermont-Thijs, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2010, 211, 2443–2448 CrossRef CAS.
- A. L. Demirel, P. Tatar Güner, B. Verbraeken, H. Schlaad, U. S. Schubert and R. Hoogenboom, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 721–729 CrossRef CAS.
- N. Oleszko-Torbus, W. Wałach, A. Utrata-Wesołek and A. Dworak, Macromolecules, 2017, 50, 7636–7645 CrossRef CAS.
- G. Cai and M. H. Litt, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2679–2688 CrossRef CAS.
- M. M. Bloksma, C. Weber, I. Y. Perevyazko, A. Kuse, A. Baumgärtel, A. Vollrath, R. Hoogenboom and U. S. Schubert, Macromolecules, 2011, 44, 4057–4064 CrossRef CAS.
- V. V. Jerca, K. Lava, B. Verbraeken and R. Hoogenboom, Polym. Chem., 2016, 7, 1309–1322 RSC.
- B. R. Hsieh and M. H. Litt, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2501–2515 CrossRef CAS.
- M. Glassner, B. Verbraeken, V. V. Jerca, K. Van Hecke, J. Tsanaktsidis and R. Hoogenboom, Polym. Chem., 2018, 9, 4840–4847 RSC.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. W. M. Fijten and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5371–5379 CrossRef CAS.
- P. Bouten, K. Lava, J. van Hest and R. Hoogenboom, Polymers, 2015, 7, 1998–2008 CrossRef CAS.
- T. G. Bassiri, A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871–879 CrossRef.
- B. R. Hsieh and M. H. Litt, Macromolecules, 1985, 18, 1388–1394 CrossRef CAS.
- M. Lobert, H. M. L. Thijs, T. Erdmenger, R. Eckardt, C. Ulbricht, R. Hoogenboom and U. S. Schubert, Chem. – Eur. J., 2008, 14, 10396–10407 CrossRef CAS.
- C. Diehl, P. Černoch, I. Zenke, H. Runge, R. Pitschke, J. Hartmann, B. Tiersch and H. Schlaad, Soft Matter, 2010, 6, 3784–3788 RSC.
- T. Rudolph, K. Kempe, S. Crotty, R. M. Paulus, U. S. Schubert, I. Krossing and F. H. Schacher, Polym. Chem., 2013, 4, 495–505 RSC.
- K. Kempe, E. F. J. Rettler, R. M. Paulus, A. Kuse, R. Hoogenboom and U. S. Schubert, Polymer, 2013, 54, 2036–2042 CrossRef CAS.
- K. Kempe, S. Jacobs, H. M. L. Lambermont-Thijs, M. M. W. M. Fijten, R. Hoogenboom and U. S. Schubert, Macromolecules, 2010, 43, 4098–4104 CrossRef CAS.
- H. M. L. Lambermont-Thijs, M. J. H. C. Jochems, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6433–6440 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten, R. M. Paulus, H. M. L. Thijs, S. Hoeppener, G. Kickelbick and U. S. Schubert, Polymer, 2006, 47, 75–84 CrossRef CAS.
- N. Oleszko-Torbus, A. Utrata-Wesołek, W. Wałach and A. Dworak, Eur. Polym. J., 2017, 88, 613–622 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. W. M. Fijten, B. M. van Lankvelt and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 416–422 CrossRef CAS.
- M. Glassner, K. Lava, V. R. de la Rosa and R. Hoogenboom, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3118–3122 CrossRef CAS.
- G. Cai and M. H. Litt, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3603–3618 CrossRef CAS.
- J. Kronek, J. Lustoň, Z. Kroneková, E. Paulovičová, P. Farkaš, N. Petrenčíková, L. Paulovičová and I. Janigová, J. Mater. Sci.: Mater. Med., 2010, 21, 879–886 CrossRef CAS.
- R. Hoogenboom, F. Wiesbrock, H. Huang, M. A. M. Leenen, H. M. L. Thijs, S. F. G. M. van Nispen, M. van der Loop, C.-A. Fustin, A. M. Jonas, J.-F. Gohy and U. S. Schubert, Macromolecules, 2006, 39, 4719–4725 CrossRef CAS.
- M. M. Lübtow, L. Keßler, A. Appelt-Menzel, T. Lorson, N. Gangloff, M. Kirsch, S. Dahms and R. Luxenhofer, Macromol. Biosci., 2018, 18, 1800155 CrossRef PubMed.
- M. M. Lübtow, M. S. Haider, M. Kirsch, S. Klisch and R. Luxenhofer, Biomacromolecules, 2019, 20, 3041–3056 CrossRef PubMed.
- S. B. Larson, J. S. Day, R. Cudney and A. McPherson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 310–318 CrossRef CAS PubMed.
- S. Fermani, G. Falini, M. Minnucci and A. Ripamonti, J. Cryst. Growth, 2001, 224, 327–334 CrossRef CAS.
- J. P. K. Doye, A. A. Louis, I. C. Lin, L. R. Allen, E. G. Noya, A. W. Wilber, H. C. Kok and R. Lyus, Phys. Chem. Chem. Phys., 2007, 9, 2197–2205 RSC.
- F. Pooch, V. Teltevskij, E. Karjalainen, H. Tenhu and F. M. Winnik, Macromolecules, 2019, 52, 6361–6368 CrossRef CAS PubMed.
- N. Morimoto, R. Obeid, S. Yamane, F. M. Winnik and K. Akiyoshi, Soft Matter, 2009, 5, 1597–1600 RSC.
- P. T. Güner, A. Mikó, F. F. Schweinberger and A. L. Demirel, Polym. Chem., 2012, 3, 322–324 RSC.
- C. Diehl, I. Dambowsky, R. Hoogenboom and H. Schlaad, Macromol. Rapid Commun., 2011, 32, 1753–1758 CrossRef CAS.
- T. Furuncuoğlu Özaltın, V. Aviyente, C. Atılgan and L. Demirel, Eur. Polym. J., 2017, 88, 594–604 CrossRef.
- Y. Katsumoto, A. Tsuchiizu, X. Qiu and F. M. Winnik, Macromolecules, 2012, 45, 3531–3541 CrossRef CAS.
- T. Li, H. Tang and P. Wu, Langmuir, 2015, 31, 6870–6878 CrossRef CAS.
- S. Sun and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 32232–32240 RSC.
- R. Obeid, F. Tanaka and F. M. Winnik, Macromolecules, 2009, 42, 5818–5828 CrossRef CAS.
- C. Legros, M.-C. De Pauw-Gillet, K. C. Tam, D. Taton and S. Lecommandoux, Soft Matter, 2015, 11, 3354–3359 RSC.
| This journal is © The Royal Society of Chemistry 2020 |