
Elucidating the effect of sequence and degree of polymerization on antimicrobial properties for block copolymers†
Stephanie M.
Barbon‡
a,
Nghia P.
Truong‡
 b,
Alysha G.
Elliott
c,
Matthew A.
Cooper
c,
Thomas P.
Davis
bd,
Michael R.
Whittaker
*b,
Craig J.
Hawker
*a and
Athina
Anastasaki
*a
b,
Alysha G.
Elliott
c,
Matthew A.
Cooper
c,
Thomas P.
Davis
bd,
Michael R.
Whittaker
*b,
Craig J.
Hawker
*a and
Athina
Anastasaki
*a
aMaterials Research Laboratory, University of California, Santa Barbara, Santa Barbara, CA 93106, USA. E-mail: hawker@mrl.ucsb.edu; athina.anastasaki@mat.ethz.ch
bMonash Institute of Pharmaceutical Sciences, ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash University, Parkville, Melbourne, Victoria 3052, Australia
cInstitute of Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia
dAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia
First published on 19th November 2019
Abstract
Sequence-controlled copolymers have recently attracted great interest in a variety of applications, including antimicrobial materials. However, owing to the nature of radical polymerization, targeting multiblocks with low degree of polymerization is complicated due to the possibility of defective chains, significantly affecting the purity of the targeted materials. In addition, the effect of optimum DP and defective chains on the antimicrobial properties of sequence-controlled copolymers remains elusive. Herein, we report the quantitative synthesis of low molecular weight copolymers via photo-induced ATRP aiming to identify the influence of degree of polymerization, block order and the defects on antimicrobial properties of sequence-controlled materials. We demonstrate that sequence-controlled copolymers with shorter amine blocks increase the antimicrobial efficacy of the resulting material towards Gram-negative bacteria while shorter hydrophobic blocks improve the efficacy towards Gram-positive bacteria. Importantly, we also demonstrate that sequence-controlled materials with very low degree of polymerization (DP = 3) exhibit the highest antimicrobial activity, despite the presence of defective chains. This work offers new insights into the structure/property relationship and highlights the promise of low DP, sequence-controlled block copolymers prepared by controlled polymerizations.
Introduction
Recent developments in controlled radical polymerization techniques such as atom transfer radical polymerization (ATRP), reversible addition fragmentation chain transfer (RAFT) and nitroxide mediated polymerization (NMP), have significantly increased the degree of structural control available to polymer chemists.1–6 Specifically, in recent years, advances in these techniques have resulted in the ability to synthesize sequence-controlled block copolymers with controlled molecular weight distributions, architecture and functionality.7–12 While these techniques do not generally have single monomer level control over the sequence (due to chain dispersity), the process is significantly more scalable over solid-phase synthesis approaches and allows for the incorporation of a range of functionalities along the polymer backbone.Sequence-controlled polymers have the potential to find application in a variety of research areas including self-assembly, molecular recognition, information storage/computing,13,14 and catalysis.15,16 One exemplary application where sequence-controlled polymers offers significant promise is in antimicrobial materials. The groups of Perrier and Alabi were the first to exploit sequence control copolymers as a way to improve the selectivity of antimicrobial polymers.17 Using acrylamide-based monomers, they elegantly showed that sequence-controlled polymers outperformed statistical (or random) copolymers and demonstrated increased selectivity towards certain bacteria strains. In this pioneering study, the authors focused on comparing statistical copolymers versus multiblocks and did not address the effect of sequence or block order. In addition, the shortest block length targeted was DP = 10 in order to avoid defective chains where one or more blocks do not form. As previously described by Harrisson and co-workers, defective chains become significant at low DP (<6).18 Boyer's group also synthesized an impressive range of multiblock copolymers (consisting of acrylamide moieties) and varied the block order to tune the antimicrobial and haemolytic activities.19 In this system, the targeted DP was also above 10 (to avoid defective chains) and the purity of the studied materials was compromised by low monomer conversion (∼90% in some cases). Of particular note, these prior studies do not address the important role of the initiator/RAFT agent and monomer sequence on biological performance.20 As a result, the importance of defective chains, sequence and optimal DP remains unclear for these synthetic, polymer-based antimicrobial systems. In this work, we aim to elucidate the effect of defective chains and sequence by comparing antibacterial efficiency of copolymers having DP above and below the threshold for polymers with defective blocks.
Results and discussion
To accurately study the effect of degree of polymerization and sequence on the antibacterial properties of polymers, the design was based on the systematic variation of block copolymers consisting of multiple hydrophobic and cationic domains. The hydrophobic domains are essential to aide in bacterial membrane perturbation which leads to cell death while the cationic domains interact with the negatively charged bacterial membranes. In particular, methyl acrylate was chosen as the hydrophobic monomer and 2-amino ethyl acrylate, protected with a tert-butyloxycarbonyl (BOC) protecting group for polymerization was selected as the comonomer, which upon deprotection gives a charged, primary amine repeat unit. It should be noted that unlike most previous reports that focused on the polymerization of acrylamide based monomers, our study is based on acrylic monomers owing to the excellent compatibility of ATRP with this monomer type.21 Photo-induced ATRP was the chosen polymerization method due to the high end group fidelity and significant control over molecular weight distributions.22–24In comparison with prior literature focused on high molecular weight, sequence-controlled materials, our interest was directed to the synthesis of low DP systems. This focus allows the effect of sequence and structure to be magnified and more easily identified. An overall degree of polymerization of 18 was therefore chosen, with 12 repeat units being derived from hydrophobic methyl acrylate monomers, and the remaining 6 units derived from protected 2-amino ethyl acrylate, which under physiological conditions would be charged after deprotection. Finally, ethyl α-bromoisobutyrate (EBiB, C2) was initially chosen as an initiator due to its ubiquity with ATRP polymerizations, and its moderate hydrophobicity. A second initiator, dodecyl α-bromoisobutyrate (DBiB, C12), was also utilized, to study the effect of the hydrophobicity of initiator structure on the antimicrobial activity of sequence-controlled polymers as increasing the hydrophobicity of the initiating group has previously been demonstrated to increase antimicrobial activity of diblock copolymers.25
Initially, a library of eight sequence-controlled polymers were synthesized, based on the C2 and C12 initiators (see Fig. 1, Table S1†). The overall DP was designed to be 18 with 6 amine and 12 methyl acrylate repeat units. When the C2 initiator was used (Fig. 1a), the hydrophobic domain (MA) follows the initiator moiety with the initial block consisting of 12 repeat units followed by an amine block of 6 repeat units (C2-MA12-b-Amine6, 1a). In an isomeric system, the hydrophobic MA domain is divided in two with blocks of lower DP (DP = 6 each) with hydrophobic segments being placed on either side of the amine block (C2-MA6-b-Amine6-b-MA6, 3a). Similarly, the amine monomer could be initially grown from the C2 initiator followed by the hydrophobic MA block (C2-Amine6-b-MA12, 2a). In the final isomeric design, the amine content was split into two blocks (DP = 3) while the hydrophobic MA block was placed in between the two amine blocks (C2-Amine3-b-MA12-b-Amine3, 4a). A similar rational was followed for the C12 initiator with four additional polymers being synthesized, as schematically represented in Fig. 1b.
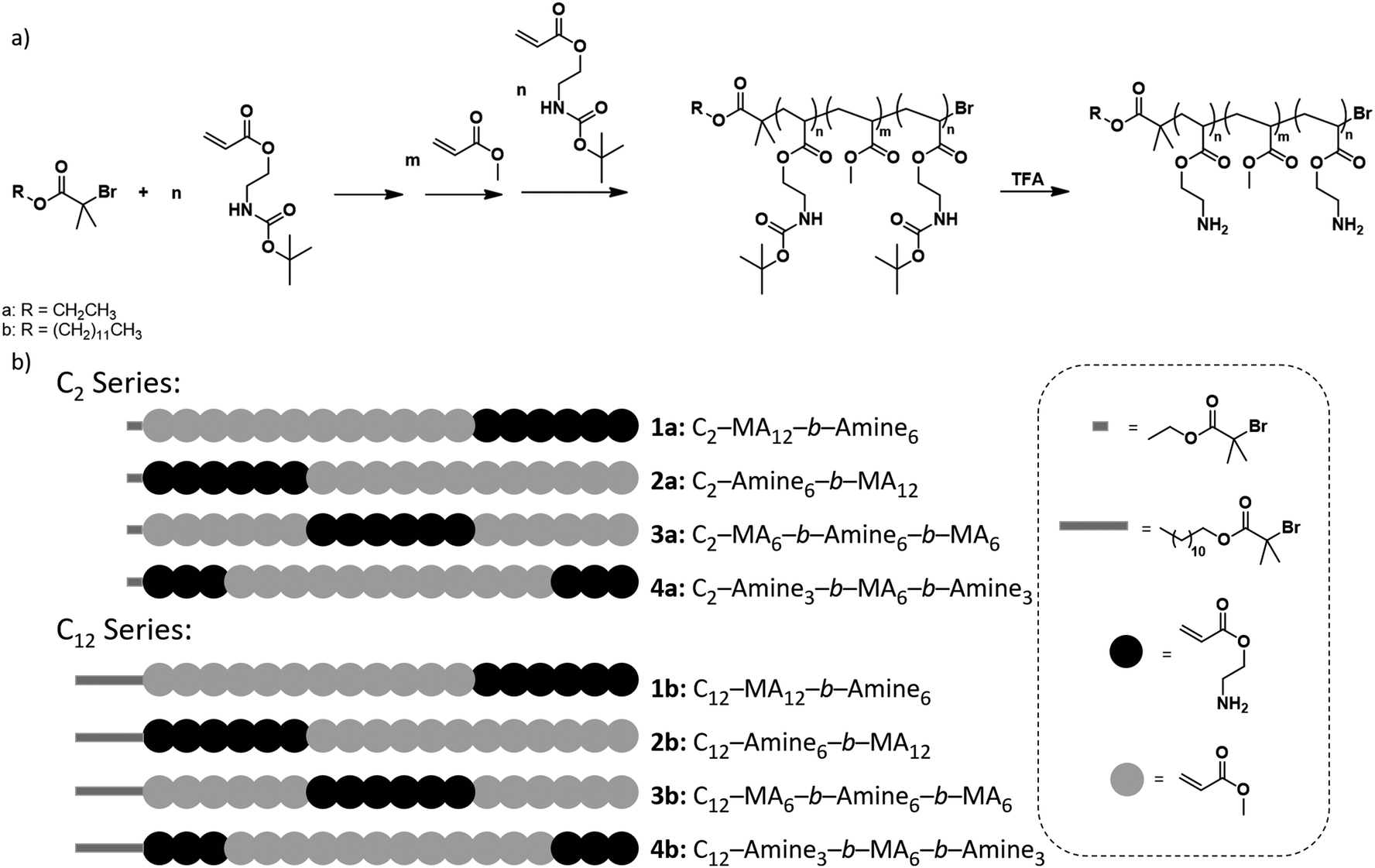 | ||
| Fig. 1 (a) General representation of the polymerization procedure for an in situ generated triblock copolymer and (b) schematic representation of the C2 and C12 series of polymers studied for their antimicrobial activity. | ||
The utility of photoinduced ATRP for the in situ synthesis of multiblock copolymers was a driver in the synthesis of these materials with trifluoroethanol being selected as solvent, due to its ability to solubilize both the Cu/Me6Tren catalyst, methyl acrylate and BOC-protected amine monomers and corresponding polymers. In addition, this solvent has previously been shown to maintain high end group functionality allowing for in situ chain extensions and high-order multiblock copolymers.9 For each chain extension, polymerization of the first block was monitored by NMR until >98% monomer conversion had been achieved. A degassed mixture of the subsequent monomer, with additional solvent and Cu/Me6Tren was then injected directly into the reaction vial in order to maintain a high reaction rate and efficient chain extension (Fig. 2a and S1–S13†). This allowed all polymers prepared for this study to be synthesized by in situ chain extension in one pot with dispersities remaining low (≤1.15), and high conversions (>98%) being obtained (Fig. S14–S26). This level of fidelity guarantees block integrity with minimal contamination from previous monomer additions. Following polymerization and purification, BOC protecting groups were removed by treatment with trifluoroacetic acid, and the deprotected block copolymers further purified before antimicrobial studies (Fig. 2b). It should also be noted that for block copolymers with low DP domains (i.e. DP = 3), there is a statistical possibility that no monomer addition occurs leading to defective chains with one or more missing blocks.18
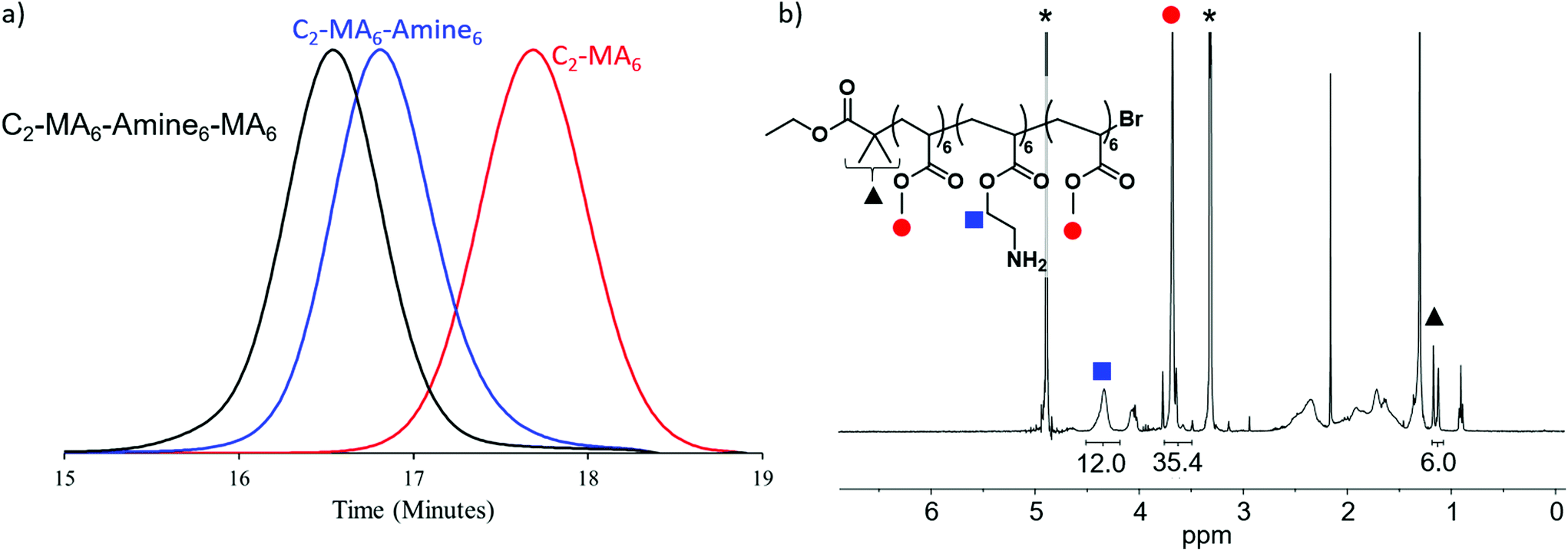 | ||
| Fig. 2 Synthesis of polymer 3a: C2-MA6-Amine6-MA6. (a) SEC data for 3a block 1 (red), block 2 (blue) and block 3 (black), showing successful chain extension. (b) Final purified NMR spectra of polymer 3a in its deprotected form in methanol-d4 (denoted by asterisks). The black triangle denotes the two CH3 groups on the EBiB initiator, the red circle denotes the protons from the methoxy group in the methyl acrylate repeat units, and the blue square denotes the protons on the methylene unit next to the oxygen in the amine acrylates. | ||
The synthesized polymers were then tested for their antimicrobial activitiy against both Gram-negative and Gram-positive bacteria, as well as for their cytotoxicity against human cells. The bacterial cell lines used in this study were: Staphylococcus aureus (Gram positive), Escherichia coli (Gram negative), Klebsiella pneumoniae (Gram negative), Acinetobacter baumannii (Gram negative), and Pseudomonas aeruginosa (Gram negative). For cytotoxicity studies, human embryonic kidney cells (HEK293) were used with the initial results shown below (see Tables 1 and S2†). For antimicrobial tests, the minimum inhibitory concentration (i.e. concentration of polymer at which 100% of bacteria growth was inhibited compared to control) is listed in μg mL−1. For cytotoxicity tests, the CC50 (i.e. concentration of polymer at which cell death of 50% of cells occurred) is also listed in μg mL−1.

From these results, the first significant trend observed is a pronounced difference in antibacterial activity upon changing the sequence or location of the amine functionality. In polymer 2b (C12-Amine6-b-MA12), the amine moiety is located directly next to the C12 initiator, followed by the hydrophobic MA block. This polymer exhibits markedly improved antimicrobial activity when compared to the diblock 1a (C12-MA12-b-amine6), where the sequence is reversed and the two hydrophobic domains are followed by the cationic amine block. This trend is also apparent for the triblocks 3b (C12-MA12-b-Amine6-b-MA6) and 4b (C12-Amine3-b-MA12-b-Amine3), where enhanced antimicrobial activity is evident upon connecting an amine domain directly to the C12 initiating unit. Identical trends can also be observed in the case of the C2 initiator for both diblock and triblock copolymers. It can therefore be concluded that the sequence of the amine moiety relative to the initiator is crucial and plays a major role in determining the antimicrobial activity. These results highlight that sequence-controlled polymers may serve as a useful tool to improve the performance of polymer-based systems as well as allowing a fundamental understanding of the mechanism behind antimicrobial activity for copolymer systems.
When comparing this library of block copolymers, an interesting observation is the high antimicrobial activity of polymer 4b (C12-Amine3-b-MA12-b-Amine3), which consists of short amine blocks with a total DP of 3 per block. As previously reported, polymer blocks where the targeted degree of polymerization is less than DP = 6 will have a significant number of defective chains, even when using controlled polymerization techniques. For polymer 4b, the inherent dispersity of the controlled radical polymerization process will dictate that a distribution of domain sizes with 0, 1, 2, 3, 4, 5, etc. units of amine functionality will be present. As schematically represented in Fig. 3, the polymers with 0 repeat units are termed defective chains, and can be considered diblock copolymers.
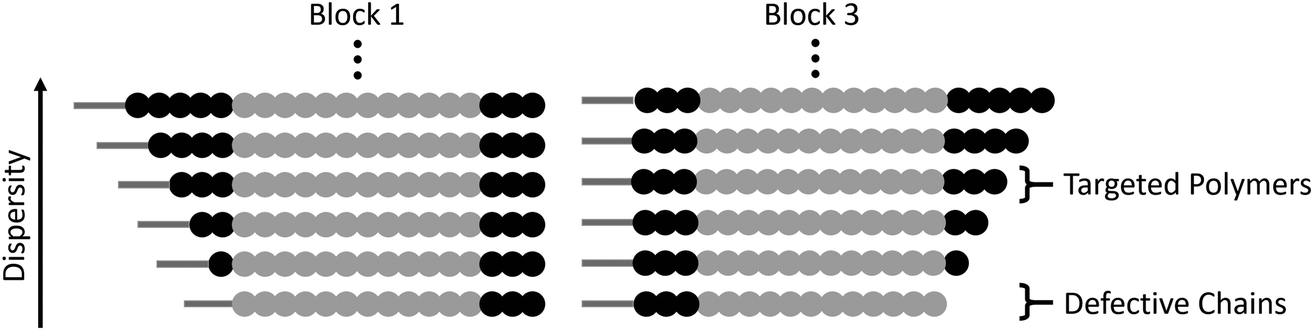 | ||
| Fig. 3 Depiction of the presence of defective chains present in polymer 4b [C12-Amine3-b-MA12-b-Amine3]. For clarity, dispersity in block 2 is ignored as no defective chains are expected, and dispersity in block 1 (left column) and block 3 (right column) are depicted separately. | ||
Using previously reported methods,18 calculations show that defective chains from block 1 of polymer 4b account for more than 5% of the polymer mixture. In the same polymer, the second block consists of 12 methyl acrylate repeat units, which theoretically has ∼0.001% domains with 0 MA units and therefore leads to an essentially pure hydrophobic block. Finally, the third block is again comprised of 3 amine monomers, introducing an additional population (∼5%) of defective chains, leading to ∼10% of chains being a mixture of different diblock copolymers in 4b. It is interesting to note that despite the presence of defective chains, polymer 4b exhibits the highest antimicrobial activity. This data suggests that targeting sequence-controlled polymers with low degrees of polymerization can still lead to high performance despite the “imperfect” backbone composition. This is particularly important as prior studies have focused on the synthesis of sequence-controlled polymers with higher DPs (typically >10 per block) designed to avoid the presence of defective chains (Table 2).

To further study the effect of defective chains and degree of polymerization, three additional triblock polymers were synthesized, based on polymer 4b (C12-Amine3-b-MA12-b-Amine3) as a starting point. For these materials, the middle hydrophobic block was kept constant at DP = 12, while the two amine domains, originally consisting of 3 amine units per block was replaced by on average 1, 2 and 6 amine units: 5b (C12-Amine1-b-MA12-b-Amine1), 6b (C12-Amine2-b-MA12-b-Amine2) and 7b (C12-Amine6-b-MA12-b-Amine6). Upon comparing the performance for these four triblocks it is evident that polymer 4b exhibits the highest antimicrobial activity (in particular towards Gram negative bacteria) while increasing the number of amine groups leads to decreased activity. This suggests that the degree of polymerization and number of amine units are important design features in these systems. It should also be noted that upon further decreasing the block length of the amine functionality from DP = 3 to DP = 2 (6b; >25% defective chains) and DP = 1 (5b; >60% defective chains) per block, a significant reduction in antibacterial activity was observed which could also be attributed to the increasing percentage of defective chains.
Building on these insights, the effect of degree of polymerization for the hydrophobic methyl acrylate block was then studied. From the parent polymer 4b (C12-Amine3-b-MA12-b-Amine3), where the length of the middle MA block is DP = 12, the degree of polymerization for the methyl acrylate domain was systematically varied from 12 (4b, C12-Amine3-b-MA12-b-Amine3) to 6 (8b, C12-Amine3-b-MA6-b-Amine3) and 3 (9b, C12-Amine3-b-MA3-b-Amine3). When comparing all three polymers 4b, 8b and 9b, the antimicrobial activity towards Gram negative bacteria is similar (∼16–32 μg mL−1), however, the antimicrobial efficacy towards Gram positive bacteria was significantly lower in polymers with shorter methyl acrylate blocks (8b and 9b). Additionally the cytotoxicity was reduced as the number of methyl acrylates is decreased, with polymer 9b (C12-Amine3-b-MA3-b-Amine3) having one of the lowest cytotoxicity values for polymers based on a dodecyl initiating group. These results suggest that future research in this area (in particular when targeting Gram-positive bacteria) should examine multiblock polymers with low DP domains and strategies for removing defective chains may further enhance performance.
Conclusions
In conclusion, a series of sequence-controlled copolymers with amine containing acrylate and methyl acrylate repeat units were synthesized by photoinduced ATRP, based on either a dodecyl or ethyl substituted initiator. Interestingly, both the block order, and the degree of polymerization of both the amine and methyl acrylate blocks was shown to significantly impact the antimicrobial activity – with shorter amine acrylate blocks increasing the antimicrobial efficacy of the polymer towards Gram negative bacteria, and shorter methyl acrylate blocks increasing the efficacy of the polymer as an antimicrobial agent towards Gram positive bacteria. The efficacy of materials with low degrees of polymerization (DP = 3), which is below the threshold for polymers without defective blocks, illustrates the importance of studying low DP polymers, as well as the utility of one-pot photo-ATRP procedures for facile and large scale synthesis of antimicrobial polymers.Experimental
General considerations
All reactions were carried out under an argon atmosphere commercially obtained reagents were used as received. All reactions were performed in vials, unless otherwise noted. CuBr2, 2,2,2-trifluoroethanol, ethyl α-bromoisobutyrate, 1-dodecanol, α-bromoisobutyryl bromide and methyl acrylate were purchased from Sigma-Aldrich. Tris[2-(dimethylamino)ethyl] amine (Me6Tren) and acryloyl chloride, were purchased from Alfa Aesar. 2-(Boc-amino)-ethanol-Boc-ethanolamine was purchased from Chem-Impex International Inc. Human embryonic kidney cells (HEK293) were purchased from ATCC. tert-Butyloxycarbonyl protected 2-amino ethyl acrylate,26 and dodecyl α-bromoisobutyrate27 were synthesized according to previously published procedures.Nuclear magnetic resonance (NMR) spectra were recorded on a Varian 600 MHz instrument. All 1H and 13C{1H} NMR experiments are reported in δ units, parts per million (ppm), and were measured relative to the signal for residual chloroform (7.26 ppm) or deuterated chloroform (77.16 ppm) respectively in the deuterated solvent unless otherwise stated. Size exclusion chromatography (SEC) was performed on a Waters Acquity APC System, with Acquity UPLC PDA and ACQUITY UPLC refractive index detectors. The polymerization light source (UV: λmax ≈ 360 nm) was a commercial nail curing lamp (Thermal Spa—obtained on-line from Amazon) equipped with 3 × 16 W bulbs.
General procedure for copolymer formation: polymer 4b
A stock solution of copper(II) bromide (8.2 mg, 0.037 mmol) and Me6Tren (59.2 μL, 0.221 mmol) was prepared in 1.2 mL trifluoroethanol. In a scintillation vial, BOC protected 2-amino ethyl acrylate (0.30 g, 1.4 mmol, 3.0 eq.) was dissolved in 0.3 mL of the trifluoroethanol stock solution. Dodecyl α-bromoisobutyrate (0.16 g, 0.46 mmol, 1.0 eq.) was then added, the vial was capped with a septum, and the solution was degassed with argon for 5 minutes. With stirring, the polymerization mixture was then irradiated with ∼360 nm light for 8 hours in a commercial UV-nail lamp system. Once NMR integration confirmed >98% conversion of the BOC-protected 2-aminoethyl acrylate, a second vial, charged with 0.3 mL of the trifluoroethanol stock solution, 0.2 mL trifluoroethanol, and methyl acrylate (0.48 g, 5.5 mmol, 12 eq.) was then degassed with argon for 5 minutes, and the contents were transferred by syringe to the polymerization vial. This mixture was returned to the UV-nail lamp, and irradiated with ∼360 nm light with stirring for an additional 12 hours. After NMR integration confirmed >98% conversion of the methyl acrylate, a third vial, charged with 0.3 mL of the trifluoroethanol stock solution, and BOC-protected 2-aminoethyl acrylate (0.30 g, 1.4 mmol, 3 eq.) was then degassed with argon for 5 minutes, and the contents were transferred by syringe to the polymerization vial. This mixture was returned to the UV-nail lamp, and irradiated with ∼360 nm light with stirring for an additional 12 hours. The resulting polymer was purified via filtration through a plug of neutral alumina, and then concentrated in vacuo. Finally, polymers (∼200 mg) with the BOC-protected 2-aminoethyl acrylate groups were deprotected by dissolving in dichloromethane (CH2Cl2, ∼2 mL) followed by adding trifluoroacetic acid (TFA, ∼2 mL) and stirring overnight (15 h). Subsequently, CH2Cl2 and TFA were removed by evaporation with a gentle air stream for 2 h. 5 mL of acetone was added to dissolve the products and then evaporated under a stream of air for 2 h. This process of adding and evaporating acetone was repeated 5 times to completely remove the excess TFA. Finally, the resulting deprotected polymers were dried under high vacuum (1 mbar) for 48 h.Minimum inhibitory concentration (MIC) assay
The polymers tested were dissolved in DMSO and then diluted with cation-adjusted Mueller Hinton Broth (CaMHB; BD, Cat. No. 212322) to a final solution containing 20 volume % of DMSO. The highest concentration tested for each compound was 512 μg mL−1 in the MIC assay. Bacteria (see Table 3 below) were cultured in CaMHB at 37 °C overnight, then diluted 40-fold and incubated at 37 °C for a further 2–3 h. The resultant mid-log phase cultures were diluted in CaMHB for each respective assay, and added to each well of the compound-containing 384-well plates to give a final cell density of 5 × 105 CFU mL−1. The final concentration range was 4–512 μg mL−1 for all compounds. The plates were covered and incubated at 37 °C for 20 h. Two biological replicates were conducted on the same day using separate starter cultures (final n = 4). Optical density was read at 600 nm (OD600) using a Tecan M1000 Pro Spectrophotometer. MIC was determined as the lowest concentration at which OD600 demonstrated 100% growth inhibition compared to growth control. The MICs were then pooled and analysed by multiple linear regression to identify significant (α = 0.95) physicochemical drivers of antimicrobial activity (as quantified by MIC). The R statistical computing package (3.0.2) was used to perform regressions.| ID | Species | Strain | Description |
|---|---|---|---|
| GP_020:02 | Staphylococcus aureus | ATCC 43300 | MRSA |
| GN_001:02 | Escherichia coli | ATCC 25922 | FDA control |
| GN_003:02 | Klebsiella pneumoniae | ATCC 700603 | MDR |
| GN_034:02 | Acinetobacter baumannii | ATCC 19606 | Type strain |
| GN_042:02 | Pseudomonas aeruginosa | ATCC 27853 | QC strain |
Cytotoxicity assay
HEK293 cells suspended in DMEM medium supplemented with 10% FBS were seeded at 5000 cells per well in a volume of 20 μL. The compound was serially diluted two-fold for concentrations ranging from 4 to 512 μg mL−1 and then plated in duplicate (20 μL). The cell plates were incubated for 20 h at 37 °C, 5% CO2. After the incubation, 5 μL of 100 μM resazurin in PBS was added to each well for a final concentration of approximately 11 μM. The plates were then incubated for 3–4 h at 37 °C, 5% CO2. The fluorescence intensity was read using the TECAN Infinite M1000 PRO with excitation/emission 560/590 nm. Two biological replicates were conducted on separate days (final n = 4). The data was analysed using Microsoft Excel and GraphPad Prism software. Cytotoxicity or cell viability were calculated using the following equation:
Using nonlinear regression analysis of log(concentration) vs. normalised cytotoxicity, using variable fitting, CC50 (concentration at 50% cytotoxicity) were calculated. In addition, the maximum percentage of cytotoxicity is reported. Any value with >, indicates a sample with no cytotoxicity or CC50 above the maximum tested concentration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the National Science Foundation through the MRSEC Program of the National Science Foundation through Grant No. DMR-1720256 (IRG-3). S. B. and A. A. acknowledge the California NanoSystems Institute for an Elings Prize Fellowship in Experimental Science. A. A. is grateful to the European Union's Horizon 2020 research and innovation program for a Marie Curie Global Postdoctoral Fellowship, and S. B. is grateful to the National Science and Engineering Research Council of Canada for the Banting Postdoctoral Fellowship. N. P. T. acknowledges the award of a DECRA Fellowship from the Australian Research Council (DE180100076).References
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2016, 9, 171 CrossRef.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406 RSC.
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Macromolecules, 2014, 47, 5051 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef.
- G. M. Church, Y. Gao and S. Kosuri, Science, 2012, 337, 1628 CrossRef CAS PubMed.
- L. Qian, E. Winfree and J. Bruck, Nature, 2011, 475, 368 CrossRef CAS.
- A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083 RSC.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196 CrossRef CAS.
- (a) A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, ACS Appl. Mater. Interfaces, 2017, 9, 40117 CrossRef CAS; (b) J. S. Brown, Z. J. Mohamed, C. M. Artim, D. N. Thornlow, J. F. Hassler, V. P. Rigoglioso, S. Daniel and C. A. Alabi, Commun. Biol., 2018, 1, 220 CrossRef PubMed; (c) M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590 CrossRef CAS.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef . Note that this reference assumes a Poisson distribution, which may affect the value of the defective chains calculated slightly.
- P. R. Judzewitsch, T.-K. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 4559 CrossRef CAS.
- T. D. Michl, K. E. S. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Travern, M. Haeussler, L. Meagher and H. J. Griesser, Polym. Chem., 2014, 5, 5813 RSC.
- J. L. Grace, J. X. Huang, S.-E. Cheah, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, RSC Adv., 2016, 6, 15469 RSC.
- A. Anastasaki, V. Nikolaou, A. Simula, J. Godfrey, M. Li, G. Nurumbetov, P. Wilson and D. M. Haddleton, Macromolecules, 2014, 47, 3852 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141 CrossRef CAS.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219 CrossRef CAS.
- J. L. Grace, A. G. Elliott, J. X. Huang, E. K. Schneider, N. P. Truong, M. A. Cooper, J. Li, T. P. Davis, J. F. Quinn, T. Velkov and M. R. Whittaker, J. Mater. Chem. B, 2017, 5, 531 RSC.
- K. A. Berchtold, J. Nie, J. W. Stansbury and C. N. Bowman, Macromolecules, 2008, 41, 9035 CrossRef CAS.
- L.-W. Zhu, B.-H. Wu, L.-S. Wan and Z.-K. Xu, Polym. Chem., 2014, 5, 4311 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01435g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |