
Celebrating 100 years of “polymer science”: Hermann Staudinger's 1920 manifesto
Holger
Frey
 * and
Tobias
Johann
* and
Tobias
Johann
Johannes Gutenberg University Mainz, Germany. E-mail: hfrey@uni-mainz.de
“Der Kautschuk ist danach ein sehr hochmolekularer Kohlenwasserstoff mit vielen Äthylenbindungen, und das chemische Verhalten entspricht auch völlig dieser Auffassung. Die Äthylenbindungen können teilweise oder ganz […] abgesättigt werden, ohne dass sich die kolloidalen Eigenschaften ändern, also ohne dass die “Makromolekel” zerfällt”.
Translation: “Natural rubber is therefore a high molecular weight hydrocarbon with many ethylene bonds, and its chemical behaviour is also completely in line with this hypothesis. The ethylene bonds can be saturated partially or fully without impact on the colloidal properties – i.e. without degradation of this “Macromolecule””.
More than 30 years later, the Nobel Prize in Chemistry 1953 was awarded to Hermann Staudinger “for his discoveries in the field of macromolecular chemistry”. This was the reward and culmination for an uncomfortable and strong character who, as one may express it today, did not rest in a “comfort zone”, even though he was a highly regarded scientist already in 1920 in the area of organic chemistry.
We hope that this translation will help many readers to comprehend the disruptive character of this work 100 years ago. One may understand its immense impetus, leading eventually to the new field of “macromolecular Science” (Staudinger himself preferred “macromolecular”) or “polymer science”.

125. H. Staudinger: on polymerisation
Some time ago G. Schroeter3 expressed interesting views regarding the composition of polymerisation products, especially pertaining to the constitution of polymeric ketenes. According to him these structures are molecular compounds and not cyclobutane derivatives, as previously assumed,4 since these polymeric ketenes differ from cyclobutane derivatives accessible from acetone-dicarboxylic ester derivatives in essential points.The same views regarding the composition of polymerisation products were already expressed in the year 1909 in a dissertation carried out in the laboratory of Thiele,5 following an investigation on the polymerisation of the asymmetric diphenylethylene. The bimolecular polymerisation product was not thought to represent the tetraphenylcyclobutane, but a molecular compound, in which partial valences lead to the cohesion of the unsaturated molecules.
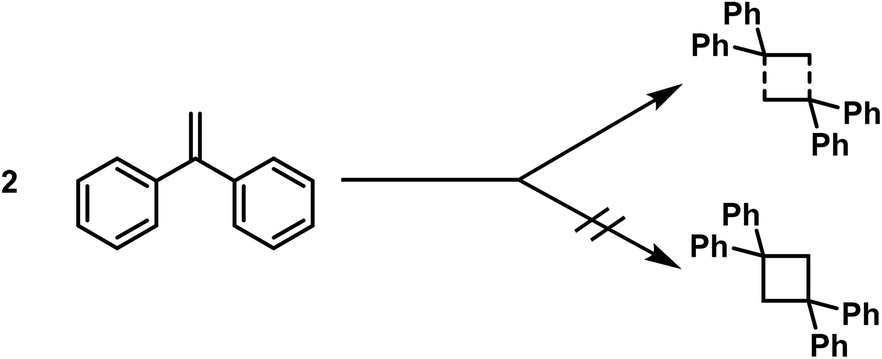
Before turning to the specific question of the constitution of the polymeric ketenes, I would like to share some of the views I have developed based on a series of works in recent years, and as it is already partly described in the works of L. Lautenschläger7 and E. Suter.8
General considerations regarding polymerisation processes
The terms “polymerisation process” and “polymerisation products” are not used evenly in chemistry, as Franke's remarks in Weyl's “Methods of Organic Chemistry” show.9 Holleman referred to polymerisation processes as such processes in which two or more molecules of a compound are linked in such a way that they can be regenerated from it again.10 As will be shown later in detail, just this latter criterion is not essential for polymerisation processes. The polymerisation products may exhibit a rather different degree of degradability, depending on their constitution.Polymerisation processes in a broader sense are all processes in which two or more molecules combine to form a product of equal composition, but higher molecular weight. However, these processes can be divided into two subgroups:
1. On the one hand, we find polymerisation processes in which the polymerisation product still has the same bonding type of the atoms as the monomolecular compound.
Examples for this are: the formation of hexaphenylethane from triphenylmethyl, the formation of paraldehyde from acetaldehyde and generation of the cyclobutanedione derivative from the ketenes. Furthermore, the transformation of styrene to metastyrene [polystyrene], of isoprene to [polyisoprene] rubber and the reaction of formaldehyde to paraformaldehyde. We suggest that these processes should be called “true polymerisation processes”, and the resulting products “true polymerisation products”.
2. On the other hand, one often finds polymerisation processes that occur under more or less pronounced rearrangement of the atoms, and consequently the polymerisation product does not show the same bonding type of the atoms as the monomolecular precursor.
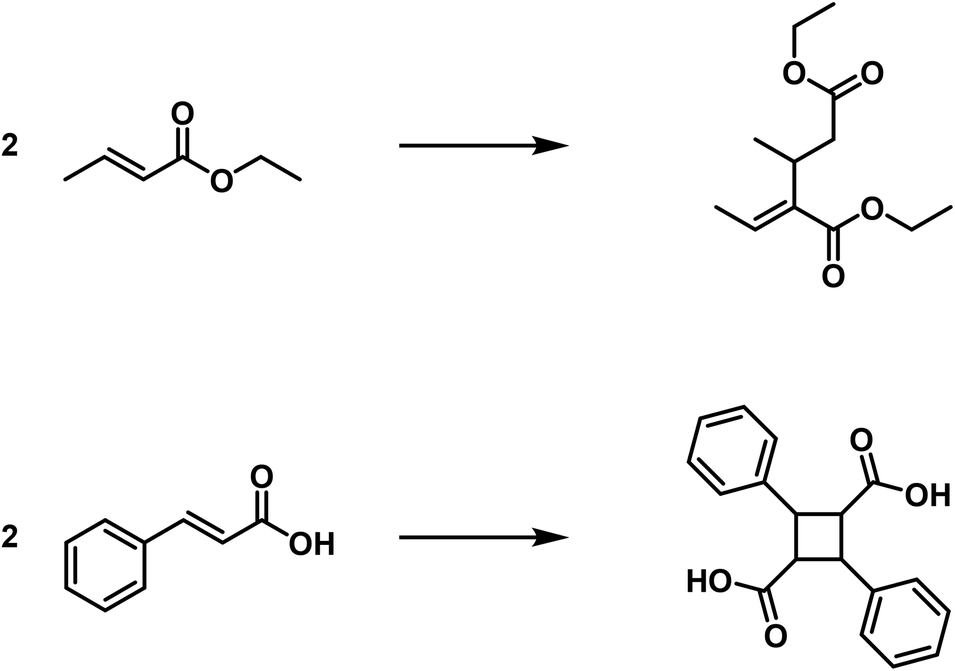
This category includes the aldol condensation, the benzoin formation, polymerisation of formaldehyde to glycolic aldehyde and sugar; furthermore the polymerisation of nitriles11 and of cyanamide to dicyanamide and many others. Further examples of this type are the formation of distyrene from styrene,12 diisobutylene from isobutylene13 as well as the dimer of acrylic acid ester from acrylic acid ester.14 These reactions should be referred to as “fake polymerisation processes” or as “condensing polymerisation processes”. They are analogous to the actual condensation processes, wherein molecules of different composition are combined in a similar manner. These processes are also often referred to as “condensation processes” (compare the benzoin formation). I would like to refer to the products of this group as “fake polymerisation products” or as “condensed polymerisation products”.
A comparison of the polymerisation of the croton ester to dicroton ester15 and the cinnamic acid to α-truxillic acid clearly shows the difference between the two types of polymerisation.
True polymerisation processes can lead to cyclic systems, namely 4-, 6- or even 8-membered rings or very high molecular weight polymerisation products are formed.
Under which conditions cycle formation occurs, and in which cases high molecular weight polymerisation products are formed, cannot be concluded on the basis of currently available results. Subtle changes of the constitution often lead to a completely different course of the polymerisation. For instance, cinnamic acid polymerizes to α-truxillic acid,16 whereas cinnamic acid esters are transformed into high molecular weight products.17 Phenyl ethylene (styrene) is transformed into high molecular weight metastyrene [polystyrene]; in contrast, the asymmetric 1,1-diphenylethylene reacts to a cyclobutane derivative. Finally, butadiene derivatives may react to dimolecular polymerisation products, for instance to cyclobutane derivatives or cyclohexene-rings, or they may be polymerised to high molecular products. For the ethylene compounds there is no correlation between constitution and inclination to polymerise.
However, some statements can be made about the stability of polymerisation products and their tendency to depolymerise. Four-membered rings are generally less stable than six-membered rings. Especially in recent years, following the study of ketenes a great number of observations have been made about the decay of four-membered rings.18 Baeyer's theory of ring strain has found extensive confirmation. As mentioned earlier, the tendency to decay depends on the number of atoms in the ring. Heterocyclic four-membered rings, particularly in the case of several heteroatoms, are commonly not as stable as cyclobutane derivatives that possess strong carbon–carbon bonds. The stability, especially that of the cyclobutane derivatives, is strongly modified by substituents, and the ring system can be very much loosened—for example, by introducing several phenyl groups or carbonyl moieties. Thus, we have the same experience as with linear carbon chains, where it is known that the stability is reduced by phenyl rings. Furthermore, molecules are known to be unstable, in which two or more carbonyl groups are attached to a carbon atom.
In comparison to the four-membered rings, six-membered ring derivatives, particularly isocyclic six-membered rings, are stable; in heterocyclic rings, cleavages occur more frequently.
In the case of the high molecular polymerisation products the stability also changes strongly with composition and substituents. Whether smooth depolymerisation to monomolecular compounds occurs will, in all cases, not only be due to the decomposition temperature of the polymerisation product, but will also depend on the stability of the monomolecular compounds. As will be shown below, taking into account these aspects, all known stable polymerisation products may well be represented by chemical formulas with normal valences.†
Four-membered rings
Heterocyclic four-membered rings are still hardly observed as polymerisation products of unsaturated compounds. As has already been pointed out, the dimolecular nitroso compounds belong in this category, whose easy decomposition is understandable because the N–O bond shows low stability.‡ Further examples include the little-studied dimolecular condensation product of thioacetone19 and of the phenyl isocyanate,20 which is depolymerized again at high temperatures by cleavage of the four-membered ring.More frequently, however, four-membered ring formation has been observed in the polymerisation of ethylene derivatives. To mention examples, I refer to the polymerisation of cinnamic acid to truxillic acid, of 1,1-diphenylethylene to tetraphenylcyclobutane,5 and also of dibenzalacetone,21 the methylene malonic acid22 and the dicarboxyglutaconic acid esters.23 The constitution of the dimolecular polymerisation products has been proven by detailed investigations in several cases.§
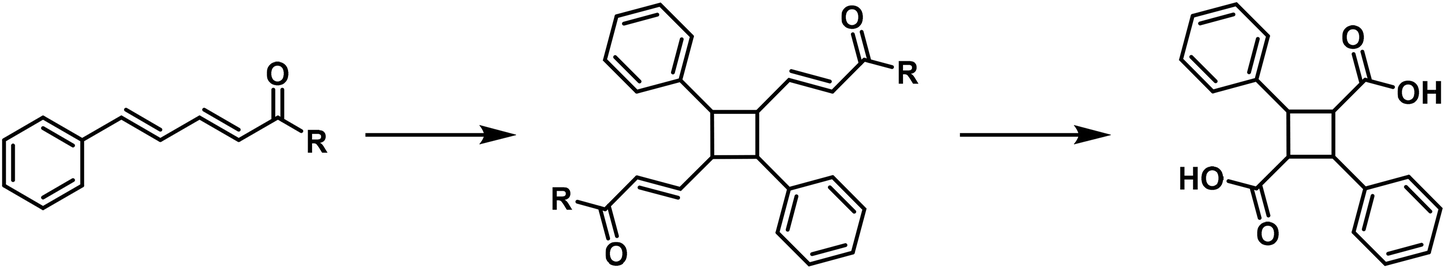
Numerous butadiene derivatives are also converted into cyclobutane derivatives by polymerisation, for example cinnamylidene malonic acid,25 cinnamylidene benzyl cyanide,26 cinnamylidene acetophenone,27 cinnamylidene acetylacetone28 and α-methyl cinnamylidene acetic acid.29
The constitution of several of these polymerisation products has been proven by oxidation, resulting in α-truxillic acid. The polymerisation of butadiene derivatives occurs according to the following general reaction:
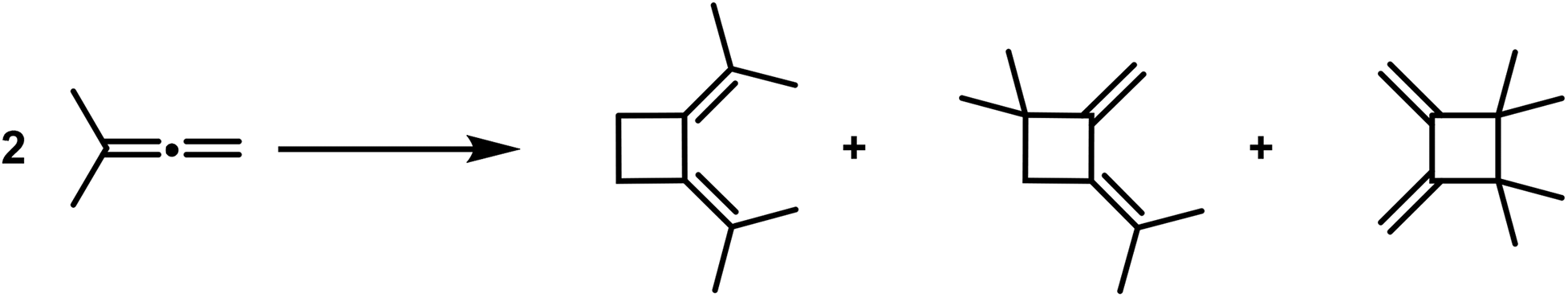
Cyclobutane derivatives are also obtained from the polymerisation of the allenes and ketenes, both classes of compounds possessing twin double bonds [cumulated double bonds]. Regarding the polymerisation of allenes, in recent years well-executed studies have been published by Lebedew32,33 that show that e.g. the dimethyl-allene affords the following polymerisation products:
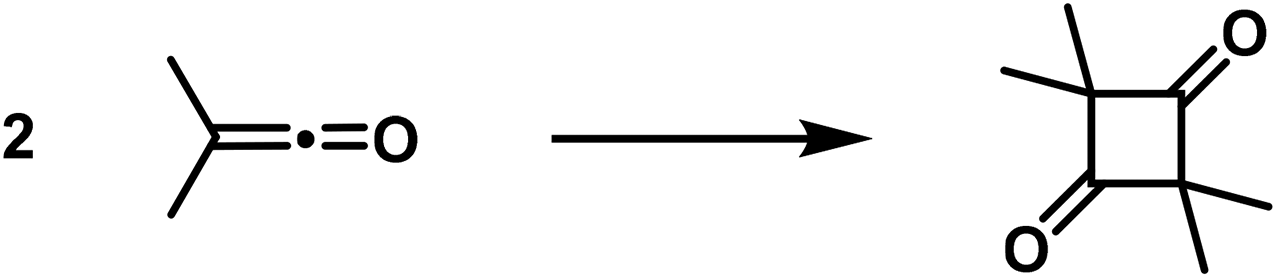
Six- and eight-membered rings
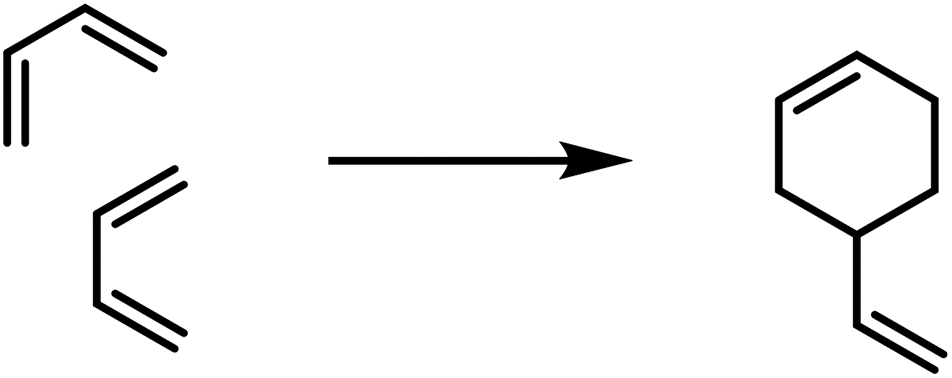
Six-membered rings as polymerisation products were obtained for aldehydes (paraldehyde), in the case of Schiff bases (formaldehyde-anilin), for thio-aldehydes and thioketones, and finally for cyan acid derivatives (cyanuric acid). These polymerisation products can be partially re-transformed into the monomolecular compounds by heating.In the case of ethylene derivatives the combination of three equivalents to a cyclohexane ring has not been observed. In contrast, butadiene derivatives may afford cyclohexene derivatives by reaction of two equivalents.32 In this case one equivalent reacts in a 1,2-orientation with another molecule in 1,4-orientation. In the case of phenylbutadiene¶ this kind of polymerisation is the main reaction, for butadiene and isoprene35 only to a lesser extent:
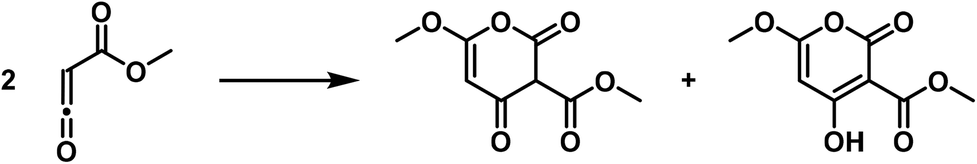
Finally, carbonyl-substituted ketenes polymerize to six-membered rings, pyronone derivatives, albeit these compounds show subsequent rearrangement.37
It has been claimed that eight-membered rings can be formed according to the assumptions made by Harries38via polymerisation of butadienes. In this context also the formation of dianthracene from anthracene should be considered, provided one accepts the chemical formula put forward in the textbook by Meyer–Jacobson.39
Finally one may include in this group of polymerisations the formation of a series of six- and eight-membered rings that may be viewed as dimolecular products of unstable three- or four-membered rings, for instance the disalicylide.40||
High molecular polymerisation products
In polymerisation processes very often high molecular [weight] products are formed, whose constitution is little known to date. These products are amorphous, colloidal compounds. They may often be insoluble in the respective monomolecular compound and precipitate immediately during polymerisation. This is observed for the formation of paraformaldehyde from liquid formaldehyde, and the polymerisation of vinyl bromide also belongs in this category. This is a group of polymerisation processes that are viewed as “euthymorphic polymerisation” by Kronstein.44**On the other hand, as is very often the case, high molecular weight polymerisation products can be colloidally dissolved in the monomolecular compound. The colloid is only separated upon the addition of other solvents or removal by distillation of the monomolecular compound. The gel obtained in this manner can become soluble upon repeated addition of the monomolecular compound, i.e. it represents a reversible gel. According to the investigation of Stobbe46 metastyrene [polystyrene] should be placed in this context. However, the gel may be insoluble once the monomolecular compound has been removed, as it appears to be the case for some types of natural rubber.
For all these colloids one can obviously not obtain a molecular weight, and it is of little use to attempt molecular weight determination with such substances. One may observe a certain depression of the melting point or increase of the boiling point, if the polymeric compound still contains a fraction of the monomer dispersion agent, which is commonly very difficult to remove. Molecular weight data determined in this manner show extremely strong scattering. If one carries out molecular weight determination with pure substances liberated from the monomolecular compounds, no or very little variation of melting or boiling points of the solvents is observed, as has been proven by B. Stobbe and Posnjak46 for the pure styrene and by Staudinger and Ott47 for the malonic acid anhydride.
If one wants to form an idea about the formation and constitution of such high molecular compounds, one can assume that primarily a union of unsaturated molecules has occurred. This is similar to the formation of four- and six-membered rings, but for some, possibly steric reason, the ring closure does not take place and now many, possibly hundreds, of molecules combine. This happens until a state of equilibrium has established itself between the individual large molecules that may depend on temperature, concentration and solvent.
In the case of inorganic colloids, for instance for colloidal gold, one observes that, depending on the mode of formation, the particle size varies greatly. In the same way, the organic colloids are strongly influenced; the different “formation mode”48 can be explained by the combination of numerous, simple molecules. For instance, the paraformaldehyde molecule may be represented by the following formula:

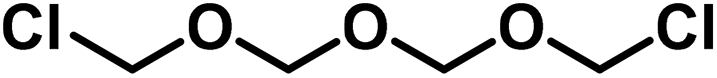
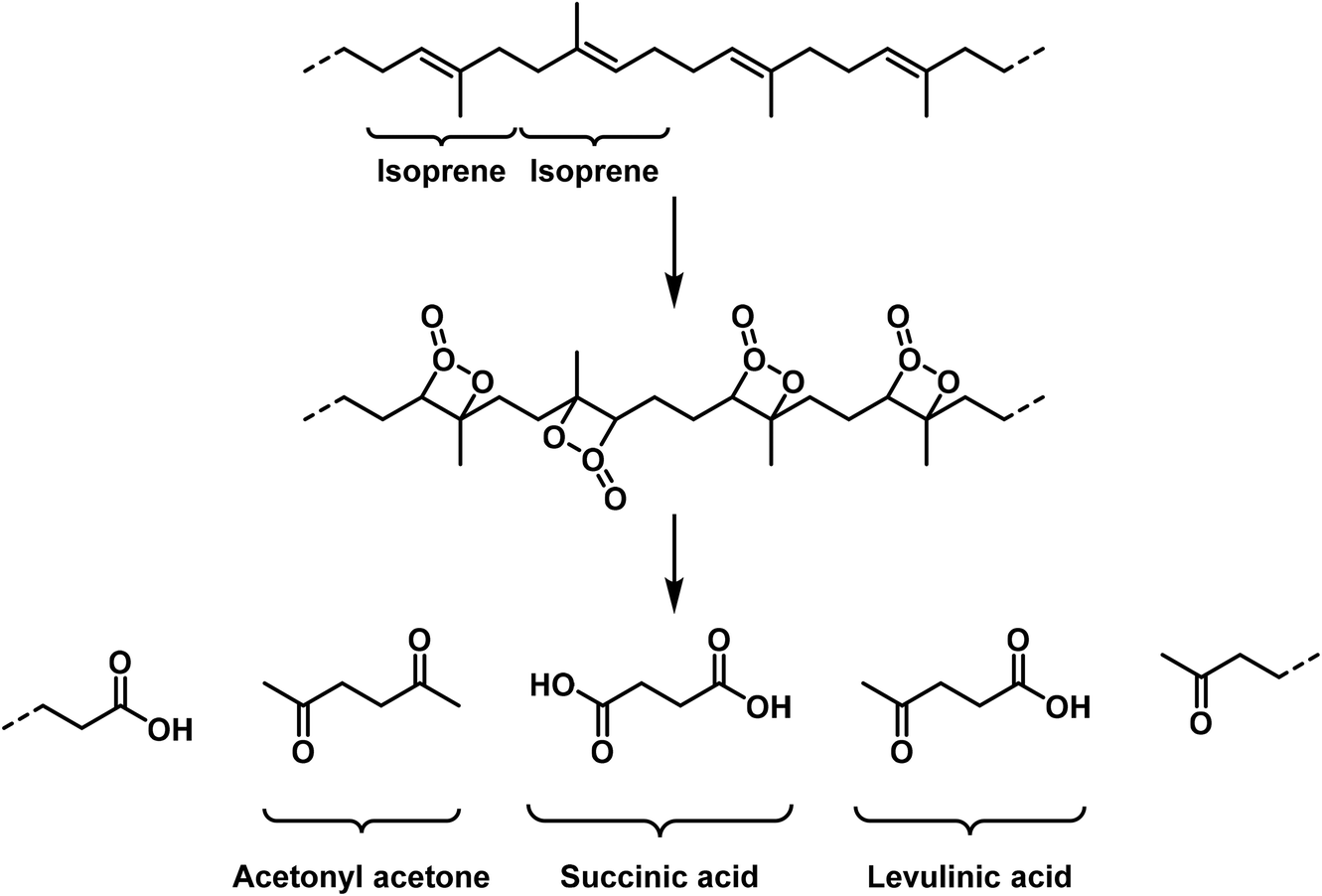
If we finally consider which unsaturated compounds afford high molecular polymerisation products, we find these for aldehydes (formaldehyde, acrolein, chloral, glyoxal and succinic aldehyde), for cyano compounds (paracyan [paracyanogen]), for ethylene derivatives (vinyl bromide, styrene, acrylic acid esters, cinnamic acid esters, methylene malonic ester,22,53 dibromo-cyclobutane,31 pyrrole and cyclopentadiene), acetylene derivatives (polymeric bromo-acetylene54) and finally also the ketenes (polymerisation products of halide- and oxygen-substituted ketenes55,56).
These high molecular weight polymerisation products may in part depolymerize upon heating. This reaction occurs in a very well-defined manner for [the polymer of] formaldehyde as well as for polycinnamic acid ethyl ester.17 However, for natural rubber isoprene is only obtained in low yields, since the degradation temperature is rather high and the monomolecular compound is quite sensitive. The polymerized vinyl bromide shows full decomposition upon heating, and depolymerisation cannot be observed.
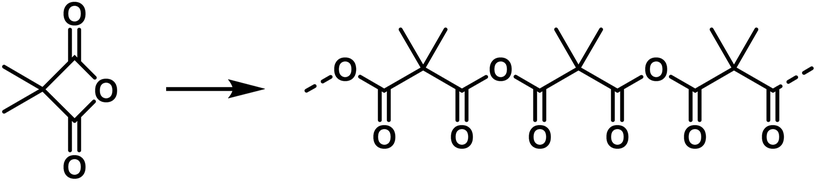
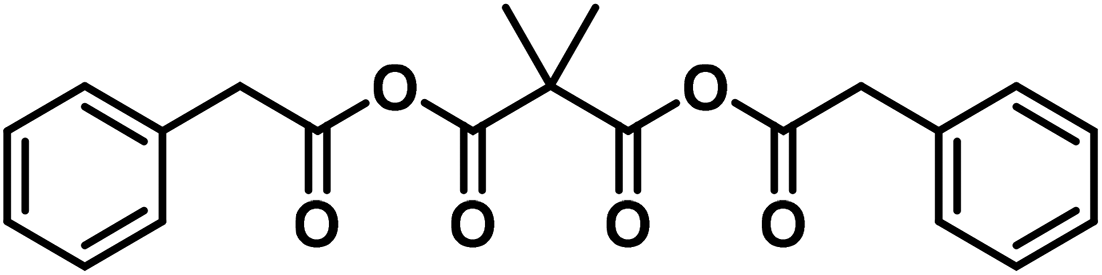
High molecular weight compounds can finally be formed from ring systems that are rather unstable due to their ring strain. In this case the monomolecular compound is usually not known, as also observed in other cases.§§ As an example, the high molecular malonic acid anhydrides belong to this group and ought to be shown in the following manner:47,58
Zürich, July 1919.
References
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073–1085 CrossRef.
- H. Staudinger and J. Fritschi, Helv. Chim. Acta, 1922, 5, 785–806 CrossRef CAS.
- G. Schroeter, Ber. Dtsch. Chem. Ges., 1916, 49, 2697–2745 CrossRef CAS.
- H. Staudinger, Die Ketene, F. Enke, Stuttgart, 1912, p. 46 Search PubMed.
- H. Hildebrand, Über die Polymerisation des asym. Diphenyläthylens, Dissertation, PhD thesis, University of Strasbourg, Straßburg, 1909 Search PubMed.
- (a) P. Pfeiffer, Justus Liebigs Ann. Chem., 1914, 404, 1–20 CrossRef CAS; (b) P. Pfeiffer, Justus Liebigs Ann. Chem., 1917, 412, 253–335 CrossRef.
- L. Lautenschläger, Autoxydation und Polymerisation ungesättigter Kohlenwasserstoffe, Dissertation, Karlsruhe, 1913 Search PubMed.
- E. Suter, Über den Einfluß von Substituenten auf die Kohlenstoffdoppelbindung, Dissertation, Zürich, 1920 Search PubMed.
- T. Weyl, Die Methoden der organischen Chemie ein Handbuch für die Arbeiten im Laboratorium. Bd. 2 Besonderer Teil Abt. 2, Thieme, Leipzig, 1911, p. 268 Search PubMed.
- A. F. Holleman, Lehrbuch der organischen Chemie, Von Veit &Co, Berlin, 13th edn, p. 115 Search PubMed.
- E. von Meyer, J. Prakt. Chem., 1888, 38, 336–343 CrossRef.
- H. Stobbe and G. Posnjak, Justus Liebigs Ann. Chem., 1909, 371, 287–302 CrossRef.
- I. Kondakow, J. Prakt. Chem., 1896, 54, 442–454 CrossRef.
- H. V. Pechmann and O. Röhm, Ber. Dtsch. Chem. Ges., 1901, 34, 427–429 CrossRef.
- H. V. Pechmann, Ber. Dtsch. Chem. Ges., 1900, 33, 3323–3341 CrossRef.
- (a) C. N. Riiber, Ber. Dtsch. Chem. Ges., 1902, 35, 2908–2909 CrossRef CAS; (b) H. Stobbe, Ber. Dtsch. Chem. Ges., 1919, 52, 666–672 CrossRef; (c) H. Stobbe, Ber. Dtsch. Chem. Ges., 1919, 52, 1021–1028 CrossRef; (d) R. Stoermer and E. Emmel, Ber. Dtsch. Chem. Ges., 1920, 53, 497–508 CrossRef; (e) R. Stoermer and G. Foerster, Ber. Dtsch. Chem. Ges., 1919, 52, 1255–1272 CrossRef.
- C. Liebermann and M. Zsuffa, Ber. Dtsch. Chem. Ges., 1911, 44, 841–849 CrossRef CAS.
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1911, 44, 521–533 CrossRef CAS.
- (a) J. Wislicenus, Z. Chem., 1869, 12, 324 Search PubMed; (b) E. Fromm and E. Baumann, Ber. Dtsch. Chem. Ges., 1889, 22, 1035–1045 CrossRef.
- A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1871, 4, 246–251 CrossRef.
- P. Praetorius and F. Korn, Ber. Dtsch. Chem. Ges., 1910, 43, 2744–2746 CrossRef.
- J. F. Bottomley and W. H. Perkin, J. Chem. Soc., Trans., 1900, 77, 294–309 RSC.
- M. Guthzeit, A. Weiss and W. Schaefer, J. Prakt. Chem., 1909, 80, 393–449 CrossRef.
- H. Wieland, Ber. Dtsch. Chem. Ges., 1904, 37, 1142–1148 CrossRef.
- (a) C. Liebermann, Ber. Dtsch. Chem. Ges., 1895, 28, 1438–1443 CrossRef; (b) C. N. Riiber, Ber. Dtsch. Chem. Ges., 1902, 35, 2411–2415 CrossRef CAS.
- H. Stobbe, Ber. Dtsch. Chem. Ges., 1912, 45, 3396–3408 CrossRef.
- H. Stobbe and C. Rücker, Ber. Dtsch. Chem. Ges., 1911, 44, 869–872 CrossRef CAS.
- (a) S. Ruhemann, J. Chem. Soc., Trans., 1904, 85, 1451–1459 RSC; (b) S. Ruhemann, Chem. Zentralbl., 1905, I, 171 Search PubMed.
- A. L. Macleod, Chem. Zentralbl., 1910, II, 1706 Search PubMed.
- H. G. Colman and W. H. Perkin, J. Chem. Soc., Trans., 1887, 51, 228–239 RSC.
- R. Willstätter and J. Bruce, Ber. Dtsch. Chem. Ges., 1907, 40, 3979–3999 CrossRef.
- S. Lebedew, Chem. Zentralbl., 1914, I, 1402 Search PubMed.
- (a) S. Lebedew, Chem. Zentralbl., 1911, II, 1915 Search PubMed; (b) S. Lebedew, Chem. Zentralbl., 1912, I, 1695 Search PubMed.
- C. N. Riiber, Ber. Dtsch. Chem. Ges., 1904, 37, 2272–2276 CrossRef CAS.
- (a) S. Lebedew, Chem. Zentralbl., 1918, I, 1002 Search PubMed; (b) S. Lebedew, Chem. Zentralbl., 1910, II, 1744 Search PubMed.
- (a) H. Staudinger and H. W. Klever, Ber. Dtsch. Chem. Ges., 1911, 44, 2212–2215 CrossRef CAS; (b) C. Harries and K. Gottlob, Justus Liebigs Ann. Chem., 1911, 383, 228–229 CrossRef CAS.
- H. Staudinger and H. Becker, Ber. Dtsch. Chem. Ges., 1917, 50, 1016–1024 CrossRef CAS.
- (a) C. Harries, Ber. Dtsch. Chem. Ges., 1905, 38, 1195–1203 CrossRef CAS; (b) S. Lebedew and N. Skawronskaja, Chem. Zentralbl., 1912, I, 1440 Search PubMed.
- V. Meyer and P. Jacobson, Lehrbuch der organischen Chemie, Leipzig, 1913, vol. II, p. 515 Search PubMed.
- A. Einhorn and H. Pfeiffer, Ber. Dtsch. Chem. Ges., 1901, 34, 2951–2953 CrossRef CAS.
- G. Schroeter and O. Eisleb, Justus Liebigs Ann. Chem., 1909, 367, 101–167 CrossRef CAS.
- G. Schroeter, Ber. Dtsch. Chem. Ges., 1919, 52, 2224–2237 CrossRef.
- R. Anschütz, Ber. Dtsch. Chem. Ges., 1919, 52, 1875–1895 CrossRef.
- A. Kronstein, Ber. Dtsch. Chem. Ges., 1902, 35, 4150–4153 CrossRef CAS.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1905, 38, 1013–1021 CrossRef CAS.
- H. Stobbe and G. Posnjak, Justus Liebigs Ann. Chem., 1909, 371, 259–286 CrossRef.
- H. Staudinger and E. Ott, Ber. Dtsch. Chem. Ges., 1908, 41, 2208–2217 CrossRef CAS.
- (a) V. Kohlschütter, Die Erscheinungsformen der Materie; Vorlesungen über Kolloidchemie, G. B. Teubner, Leipzig, 1917 Search PubMed; (b) V. Kohlschütter, Justus Liebigs Ann. Chem., 1912, 387, 86–88 CrossRef.
- F. M. Litterscheid and K. Thimme, Justus Liebigs Ann. Chem., 1904, 334, 1–49 CrossRef CAS.
- S. S. Pickles, J. Chem. Soc., Trans., 1910, 97, 1085–1090 RSC.
- (a) G. Steimmig, Ber. Dtsch. Chem. Ges., 1914, 47, 852–853 CrossRef CAS; (b) G. Steimmig, Ber. Dtsch. Chem. Ges., 1914, 47, 350–354 CrossRef CAS.
- (a) C. Harries, Ber. Dtsch. Chem. Ges., 1914, 47, 573–577 CrossRef CAS; (b) C. Harries, Ber. Dtsch. Chem. Ges., 1915, 48, 863–868 CrossRef CAS.
- E. Haworth and W. H. Perkin, J. Chem. Soc., Trans., 1898, 73, 330–345 RSC.
- F. K. Beilstein, Handbuch der Organischen Chemie. Acyclische Kohlenwasserstoffe, Oxy- und Oxo-Verbindungen, Julius Springer, Berlin, 1918, p. 245 Search PubMed.
- H. Staudinger, E. Anthes and H. Schneider, Ber. Dtsch. Chem. Ges., 1913, 46, 3539–3551 CrossRef.
- E. Ott, Justus Liebigs Ann. Chem., 1913, 401, 159–177 CrossRef.
- E. Bamberger and F. Tschirner, Ber. Dtsch. Chem. Ges., 1900, 33, 955–959 CrossRef CAS.
- A. Einhorn, Justus Liebigs Ann. Chem., 1908, 359, 145–187 CrossRef CAS.
- R. Anschütz, Justus Liebigs Ann. Chem., 1893, 273, 73–93 CrossRef.
- K. Dyckerhoff, Beiträge zur Autoxydation organischer Stoffe, Dissertation, Karlsruhe, 1910 Search PubMed.
- V. Meyer and P. Jacobson, Lehrbuch der organischen Chemie, Leipzig, 1913, vol. I, p. 301 Search PubMed.
Footnotes |
| † For the explanation of association products, for example of acetic acid, secondary valences need to be assumed in all cases. |
‡ The N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N azo group does not show a tendency to undergo polymerisation, since hypothetic binding of three nitrogen atoms would occur, which is highly unstable. N azo group does not show a tendency to undergo polymerisation, since hypothetic binding of three nitrogen atoms would occur, which is highly unstable. |
| § The dimolecular polymerisation product of benzylidene acetophenone, described by Wieland,24 needs to be investigated further. In this case a condensed polymerisation product could be present. |
| ¶ See also the work of Lebedew.32 The same acid that Lebedew described as an oxidation product of the cyclohexene derivative was also prepared by Riiber,34 who furthermore investigated the constitution of dimolecular phenylbutadiene. |
| || See the work of G. Schröter41 for further information regarding the dimolecular anhydrides of anthranilic acid. The interesting newer works of G. Schröter42 and R. Anschütz43 are not considered in this work. |
| ** The formation of cyamelide from cyanuric acid probably does not belong in this class, because cyamelide is a trimolecular compound, as reported by Hantzsch.45 |
| †† These primary products most probably possess the following constitution: OH–CH2–O–CH2–O⋯CH2–O–CH2–Cl. |
| ‡‡ See also the deviating results of Harries.52 |
| §§ For example methylene appears to polymerise to a high molecular weight (CH2)x.57 |
| This journal is © The Royal Society of Chemistry 2020 |