
From simple quinoxalines to potent oxazolo[5,4-f]quinoxaline inhibitors of glycogen-synthase kinase 3 (GSK3)†
Frédéric
Lassagne
 *a,
Camille
Duguépéroux
a,
Carlos
Roca
b,
Concepcion
Perez
c,
Ana
Martinez
bd,
Blandine
Baratte
ef,
Thomas
Robert
ef,
Sandrine
Ruchaud
ef,
Stéphane
Bach
*ef,
William
Erb
a,
Thierry
Roisnel
a and
Florence
Mongin
*a
*a,
Camille
Duguépéroux
a,
Carlos
Roca
b,
Concepcion
Perez
c,
Ana
Martinez
bd,
Blandine
Baratte
ef,
Thomas
Robert
ef,
Sandrine
Ruchaud
ef,
Stéphane
Bach
*ef,
William
Erb
a,
Thierry
Roisnel
a and
Florence
Mongin
*a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: florence.mongin@univ-rennes1.fr
bCentro de Investigaciones Biologicas-CSIC, Ramiro de Maeztu 9, 28040 Madrid, Spain. E-mail: ana.martinez@csic.es
cInstituto de Química Médica-CSIC, Juan de la Cierva 3, 28006 Madrid, Spain
dCentro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Instituto Carlos III, 28031 Madrid, Spain
eSorbonne Université, CNRS, FR2424, Plateforme de criblage KISSf (Kinase Inhibitor Specialized Screening facility), Station Biologique de Roscoff, CS 90074, 29682 Roscoff Cedex, France. E-mail: bach@sb-roscoff.fr
fSorbonne Université, CNRS, UMR 8227, Integrative Biology of Marine Models, Station Biologique de Roscoff, CS 90074, 29688 Roscoff Cedex, France
First published on 27th November 2019
Abstract
2,7-Disubstituted oxazolo[5,4-f]quinoxalines were synthesized from 6-amino-2-chloroquinoxaline in four steps (iodination at C5, substitution of the chloro group, amidation and copper-catalysed cyclization) affording 28 to 44% overall yields. 2,8-Disubstituted oxazolo[5,4-f]quinoxaline was similarly obtained from 6-amino-3-chloroquinoxaline (39% overall yield). For the synthesis of other oxazolo[5,4-f]quinoxalines, amidation was rather performed before substitution; moreover, time-consuming purification steps were avoided between the amines and the final products (38 to 54% overall yields). Finally, a more efficient method involving merging of the last two steps in a sequential process was developed to access more derivatives (37 to 65% overall yields). Most of the oxazolo[5,4-f]quinoxalines were evaluated for their activity on a panel of protein kinases, and a few 2,8-disubstituted derivatives proved to inhibit GSK3 kinase. While experiments showed an ATP-competitive inhibition on GSK3β, structure–activity relationships allowed us to identify 2-(3-pyridyl)-8-(thiomorpholino)oxazolo[5,4-f]quinoxaline as the most potent inhibitor with an IC50 value of about 5 nM on GSK3α.
Introduction
Oxazolo[5,4-f]quinoxalines, to our knowledge, have never been synthesized before. Their structures can be seen either as a pyrazine fused to a benzoxazole or as an oxazole fused to a quinoxaline1 (Fig. 1, top left). Oxazolo[4,5-h]quinolines, which are analogues of oxazolo[5,4-f]quinoxalines, lacking one nitrogen (Fig. 1, top right), have shown anti-allergic activities.2 Thus, it is of interest to access oxazolo[5,4-f]quinoxalines and study their biological behaviour in order to find original properties.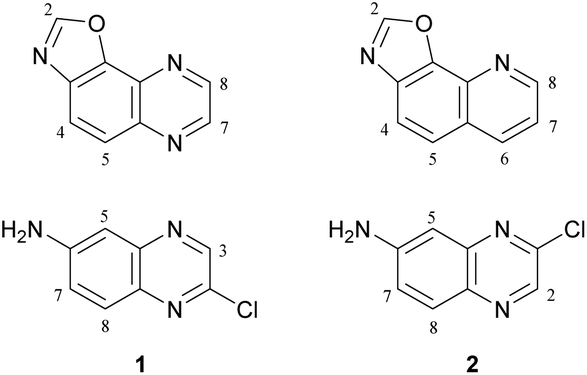 | ||
| Fig. 1 Top: Structures of oxazolo[5,4-f]quinoxaline (left) and oxazolo[4,5-h]quinoline (right). Bottom: Planned quinoxaline substrates. | ||
It has been shown in 2011 that 2-quinolinol can be functionalized either at its 6- or at its 7-position by nitration at room temperature using potassium nitrate in sulfuric acid or nitric acid in acetic acid, respectively.3 Subsequent conversion to the corresponding chlorides (reflux with phosphorus oxychloride and phosphorus pentachloride) and nitro reduction3 can easily furnish appropriate substrates (Fig. 1, bottom) for the synthesis of various 2,7- and 2,8-disubstituted oxazolo[5,4-f]quinoxalines.
Recently, our random screening of original polyaromatic compounds revealed inhibitors of protein kinases.4 Following these studies, we report in the present paper the synthesis of original oxazolo[5,4-f]quinoxalines and evaluation of their activity on a panel of protein kinases. Docking studies are carried out, leading to a potent inhibitor of GSK3.
Results and discussion
6-Amino-2-chloroquinoxaline (1) was converted into 2,7-disubstituted oxazolo[5,4-f]quinoxalines in four steps (Scheme 1). An approach to obtain benzoxazole is by constructing a ring from 2-haloanilide.5 Therefore, inspired by the reported bromination of 6-aminoquinoxaline at C5,6 we treated a solution of 1 in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dioxane–water with an excess of iodine and sodium bicarbonate at room temperature for 4 h. Under these conditions, the 5-iodo derivative 3, formed regioselectively, was isolated in 88% yield.
:1 dioxane–water with an excess of iodine and sodium bicarbonate at room temperature for 4 h. Under these conditions, the 5-iodo derivative 3, formed regioselectively, was isolated in 88% yield.
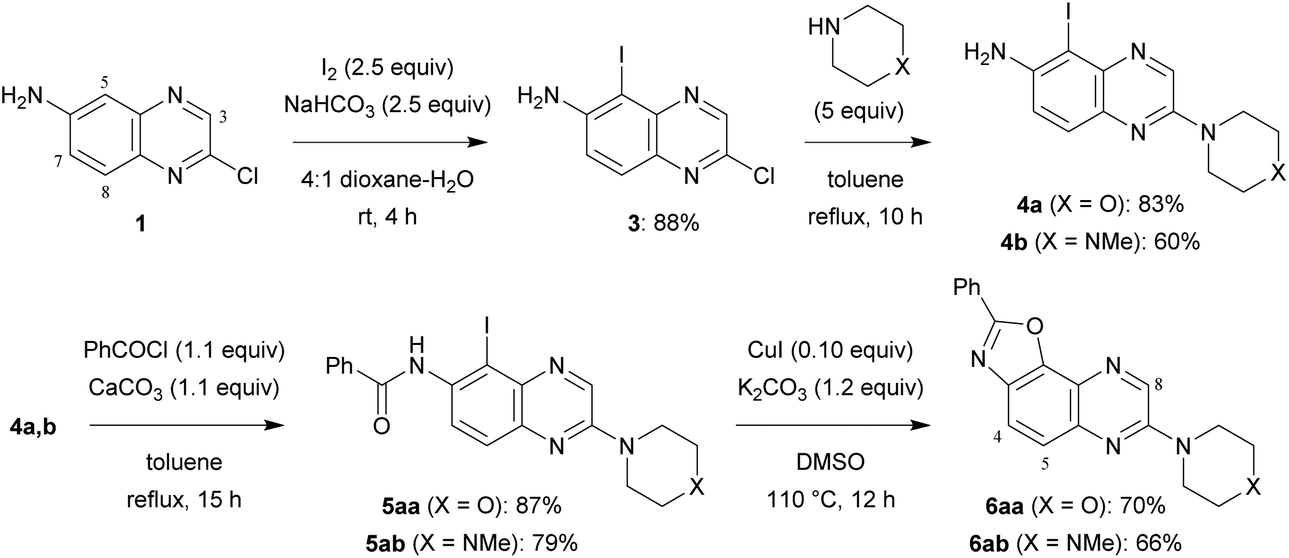 | ||
| Scheme 1 Synthesis of 2,7-disubstituted oxazolo[5,4-f]quinoxalines 6aa and 6ab. | ||
In order to progress towards compounds of biological interest, the chloro group was successfully replaced by morpholino and 4-methylpiperazino at the reflux temperature of toluene, affording 4a,b. Their reaction with benzoyl chloride in the presence of calcium carbonate under similar conditions led to benzamides 5aa and 5ab.
Next, we sought simple and efficient conditions under which we could attempt the synthesis of benzoxazole.5,7 Convinced with the interest in copper-catalysed reactions, we decided to use catalytic copper(I) iodide in the presence of potassium carbonate, and selected dimethylsulfoxide as the solvent in order to avoid an additional ligand. As expected, after 12 h at 110 °C, 2,7-disubstituted oxazolo[5,4-f]quinoxalines 6aa and 6ab were isolated in good yields.
A similar four-step synthesis, employing isonicotinoyl chloride instead of benzoyl chloride, was used to obtain 2,8-disubstituted oxazolo[5,4-f]quinoxaline 10a (Scheme 2).
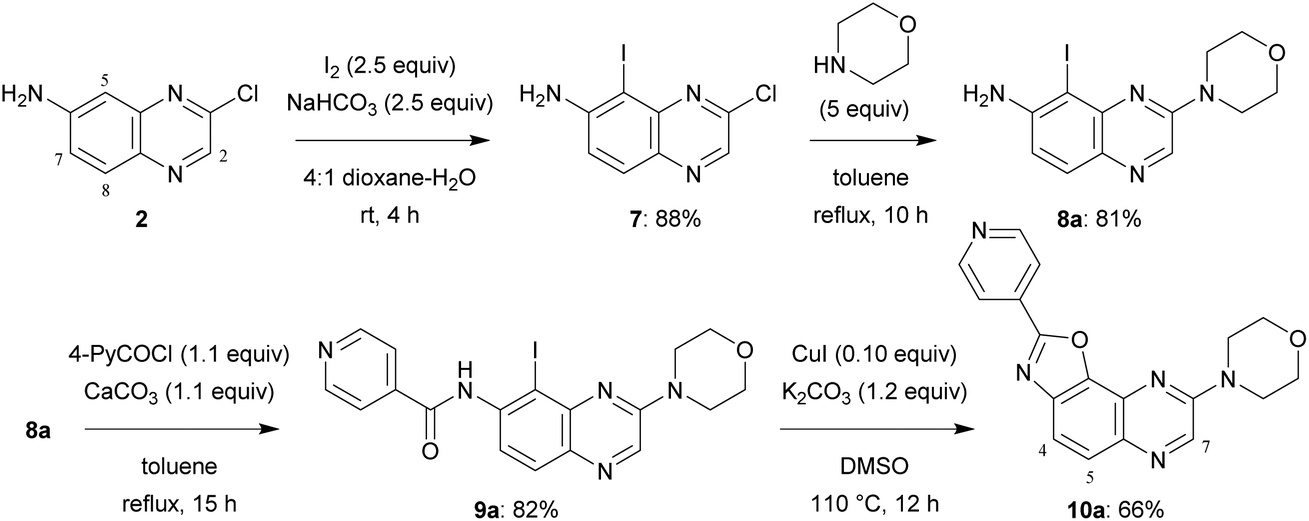 | ||
| Scheme 2 Synthesis of 2,8-disubstituted oxazolo[5,4-f]quinoxaline 10a. | ||
For the purpose of moving towards a more convergent strategy, we reversed the order of the steps and performed amidation before substitution of the chloro group (Scheme 3). A further benefit of this approach would be the possibility to carry out substitution under milder conditions; indeed, the reaction should be favoured with a less electron-donating group such as aroylamino at C6 than the previous amino group. In a preliminary attempt, iodide 3 was treated with nicotinoyl chloride; to solve the solubility issue sometimes encountered with toluene, acetonitrile containing 5 equivalents of pyridine was rather employed. Next, chlorocarboxamide 11b was reacted with morpholine in dimethylsulfoxide, a solvent also suitable for the formation of benzoxazole, and an excess of potassium carbonate was introduced right from the beginning of the reaction. After the required time to achieve nucleophilic substitution at room temperature, catalytic copper(I) iodide was introduced and the mixture containing 5ba was heated to 110 °C in order to allow cyclization to take place. Under these conditions, 2,7-disubstituted oxazolo[5,4-f]quinoxaline 6ba was isolated after purification with an overall yield of 51% in this three-step process (Scheme 3, top).
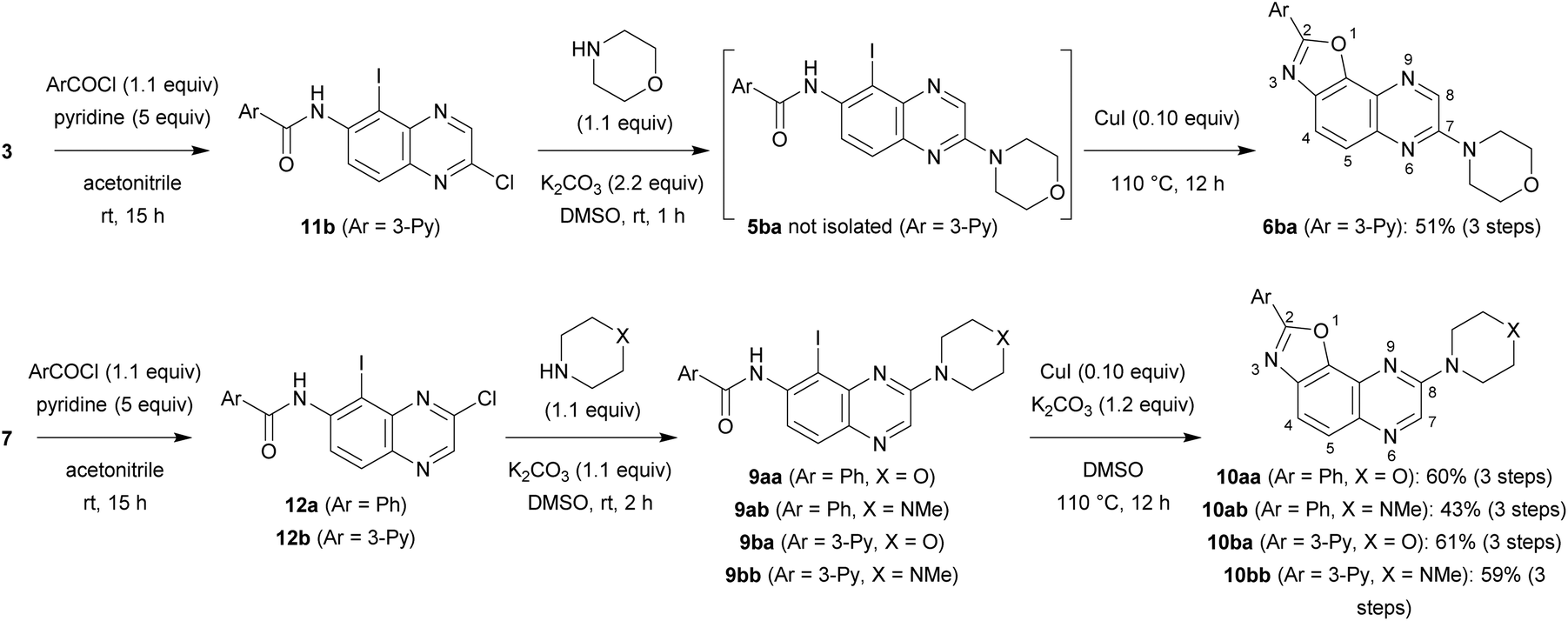 | ||
| Scheme 3 Synthesis of 2,7-disubstituted oxazolo[5,4-f]quinoxalines 6ba and 2,8-disubstituted oxazolo[5,4-f]quinoxalines 10aa, 10ab, 10ba and 10bb. | ||
In a similar way, iodide 7 was readily converted into carboxamide 12a,b using either benzoyl chloride or nicotinoyl chloride. Further replacement of the chloro group by morpholino or 4-methylpiperazino benefited from the higher reactivity of 12a,b towards nucleophiles when compared with 11b. From the compounds 9aa, 9ab, 9ba and 9bb, here only isolated from their reaction mixture by a simple work-up to ascertain their formation by NMR, cyclization was performed, leading to the new 2,8-disubstituted oxazolo[5,4-f]quinoxalines 10aa, 10ab, 10ba (unambiguously identified by X-ray diffraction; the corresponding ORTEP diagram is given in Fig. 2) and 10bb (Scheme 3, bottom). It is worth noting that there is no need to purify the intermediates of this three-step synthesis process from 7 as, after a suitable work-up, they can be directly involved in the substitution reaction and benzoxazole formation.
 | ||
| Fig. 2 ORTEP diagrams (30% probability) of 10ba (top) and 10ci (bottom). | ||
Protein kinases can be deregulated in diseases such as cancers and neurodegenerative disorders.8 Since the end of the nineties, this class of signalling biomolecules has become indispensable drug targets. As a result, many candidates are at present undergoing clinical evaluation and 50 FDA-approved kinase inhibitors are already in the market.9
In continuation of our ongoing research on novel protein kinase inhibitors,4 most of the synthesized oxazolo[5,4-f]quinoxalines as well as a few precursors were tested against a short panel of disease-related protein kinases: cyclin-dependent kinases 2 (CDK2/CyclinA), 5 (CDK5/p25) and 9 (CDK9/CyclinT), proto-oncogene kinase PIM1, CDC2-like kinase 1 (CLK1), dual specificity tyrosine phosphorylation regulated kinase 1A (DYRK1A), glycogen synthase kinase 3 (GSK3; isoform β, and possibly α or/and α/β), casein kinase 1 (CK1; isoform ε, and possibly δ/ε) and mitotic kinase Haspin (Table 1).
| Compd. | CDK2/CyclinA | CDK5/p25 | CDK9/CyclinT | PIM1 | MmCLK1 | RnDYRK1A | GSK3α | GSK3β | SscGSK3α/β | CK1ε | SscCK1δ/ε | Haspin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5aa | — | ≥100 (99) | 90 (80) | 100 (91) | 91 (95) | 100 (97) | — | 89 (92) | — | 92 (90) | — | 77 (87) |
| 5ab | — | ≥100 (100) | 84 (79) | ≥100 (≥100) | 59 (90) | 89 (96) | — | 76 (92) | — | 73 (82) | — | 68 (72) |
| 6aa | — | 100 (≥100) | 93 (81) | ≥100 (≥100) | 59 (99) | 84 (93) | — | 96 (86) | — | 83 (92) | — | 57 (60) |
| 6ab | — | ≥100 (98) | 72 (79) | ≥100 (≥100) | 44 (95) | 73 (99) | — | 89 (82) | — | 86 (78) | — | 64 (82) |
| 6ba | — | ≥100 (≥100) | ≥100 (≥100) | 99 (≥100) | — | 70 (92) | ≥100 (≥100) | 90 (≥100) | — | 81 (≥100) | — | 87 (≥100) |
| 9a | — | ≥100 (≥100) | 69 (73) | ≥100 (≥100) | — | 50 (72) | 41 (72) | 54 (64) | — | 76 (88) | — | 79 (≥100) |
| 10a | — | ≥100 (99) | 52 (79) | 78 (90) | — | 42 (74) | 24 (55) | 51 (60) | — | 72 (≥100) | — | 59 (64) |
| 9aa | 48 (87) | 96 (91) | 25 (80) | 84 (96) | — | — | 33 (82) | 37 (82) | 43 (92) | 78 (98) | — | — |
| 10aa | 70 (99) | ≥100 (98) | 40 (65) | 97 (94) | — | — | 13 (52) | 33 (59) | 29 (56) | 85 (≥100) | — | — |
| 10ab | 52 (98) | 85 (81) | 4 (61) | 75 (89) | — | — | ≤0 (54) | 12 (58) | 14 (69) | 40 (94) | — | — |
| 10ba | — | 66 (98) | 27 (45) | 23 (43) | — | 3 (11) | 9 (9) | 2 (2) | — | 65 (75) | — | 12 (23) |
| 10bb | ≥100 (≥100) | 93 (≥100) | 7 (35) | 20 (57) | — | — | 2 (4) | 6 (6) | 16 (16) | 53 (≥100) | 72 (88) | 24 (29) |
Interestingly, some of the oxazolo[5,4-f]quinoxalines and, to a lesser extent, their non-cyclized precursors proved to significantly inhibit CDK9/CyclinT, DYRK1A and GSK3 at 10 μM. In addition, even at a lower concentration of 1 μM, compounds 10ba and 10bb bearing a 3-pyridyl ring at C2 still exhibit good activity against GSK3 protein kinase. IC50 values were calculated for the most promising compounds; while micromolar values for GSK3 inhibition were found in the case of 10aa and 10ab, encouraging submicromolar values were recorded for 10ba and 10bb (Table 2).
| Entry | Product | CDK9/CyclinT | GSK3α | GSK3β | SscGSK3α/β |
|---|---|---|---|---|---|
| 1 | 9aa | 2500 | 2200 | 6800 | 8800 |
| 2 | 10aa | >10000 | 610 | 980 | 1100 |
| 3 | 10ab | 3100 | 1600 | 3100 | 1900 |
| 4 | 10ba | — | 15 | 25 | — |
| 5 | 10bb | — | 24 | 55 | 30 |
Because of its association with key biological processes (metabolism, cell signalling, cellular transport, apoptosis, proliferation, intracellular communication, etc.), the serine-threonine protein kinase GSK3 is implicated in various diseases (type 2 diabetes, bipolar disorder, schizophrenia, Alzheimer's disease, Parkinson's disease, developmental disorders and cancer), and has become an important target for drug development.10 Thus, our preliminary results prompted us to identify both the binding site and the binding mode of our best inhibitors 10ba and 10bb through (i) ATP-competition experiments and (ii) docking studies in order to optimize their structure.
(i) The kinetic mechanism of GSK3β inhibition was experimentally investigated for one of the most potent derivatives synthesized, quinoxaline 10bb, by varying the concentrations of both ATP and the tested inhibitor in the kinase reaction. The Lineweaver–Burk or double-reciprocal plots of 1/v versus 1/[ATP] at fixed concentrations of the new inhibitor 10bb showed an intersecting pattern consistent with competitive inhibition as all lines converge at or near the y-axis (Fig. 3). These results clearly show that this new family of quinoxaline derivatives and specifically compound 10bb are ATP-competitive GSK3β inhibitors because an increase in the ATP concentration (from 1 to 100 μM) does not interfere with enzyme inhibition.
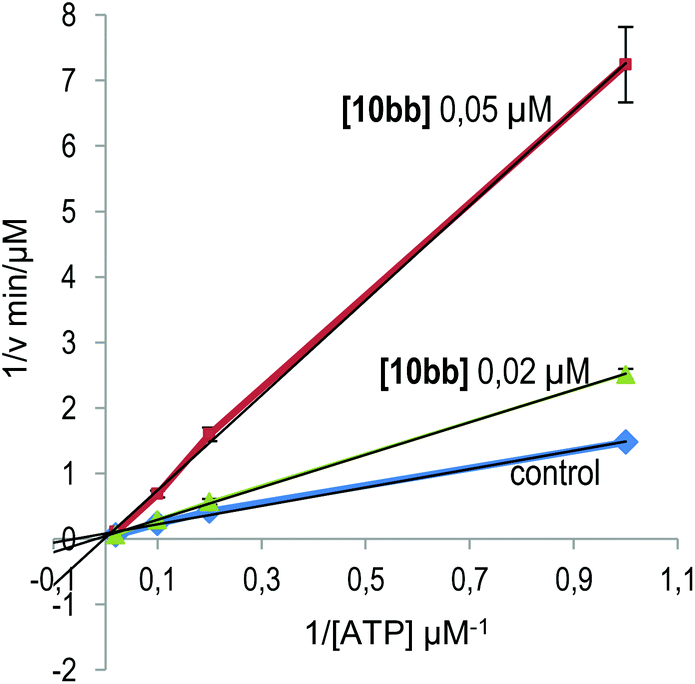 | ||
| Fig. 3 Kinetic data determined for derivative 10bb. ATP concentrations in the reaction mixture varied from 1 to 100 μM. Compound concentrations used are depicted in the plot, and the concentration of the substrate was kept constant at 12.5 μM. Each point is the mean of two different experiments, each one analysed in duplicate. | ||
(ii) In order to develop structure–activity relationships and propose novel substituents able to increase kinase inhibition potency, we performed docking studies in the ATP-binding cavity of GSK3β for compounds 10ab and 10bb. Both compounds are able to bind to the cavity through an H-bond interaction between the N6 atom of the oxazolo quinoxaline ring (see Scheme 3 for numbering) and the backbone amine group of Val135 (Fig. 4), orienting the substituent at C2 to the catalytic residue Lys85 and the substituent at C8 to the solvent. The main difference between both compounds is the substituent at C2, a phenyl ring (compound 10ab) and a pyridine ring (compound 10bb). The pyridine ring is able to interact through an H-bond interaction with Lys85, increasing the affinity and improving the inhibition of the target. Thus, in order to access more potent inhibitors, we could maintain the pyridine ring or replace it with a five-membered heterocycle that allows similar interaction. The substituent at C8 in both cases is a piperazine ring that is oriented to the solvent-exposed region. This orientation allows the introduction of different substituents in order to grow the ligand in this region. Moreover, substituents at this position with aromatic rings could be interesting to allow interaction with Arg141 through a π–cation interaction. Furthermore, H-bond acceptor groups at this position would also be able to interact with Arg141.
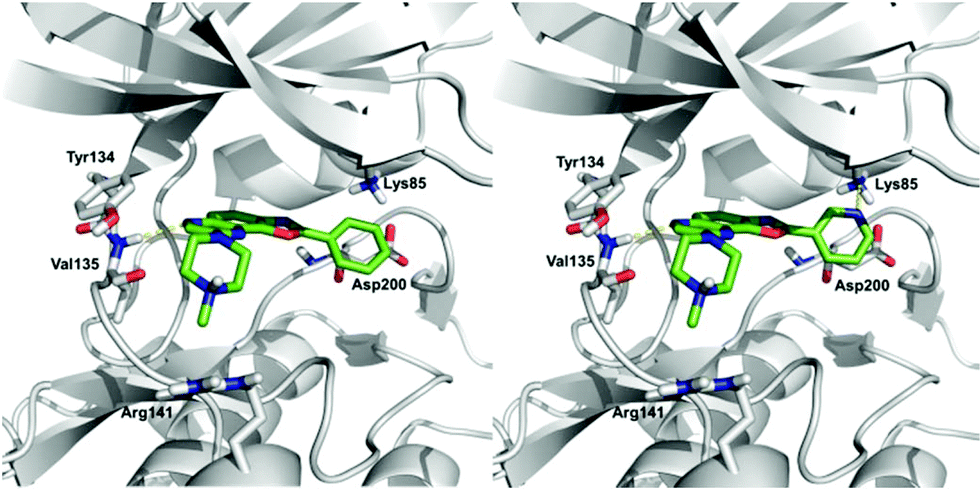 | ||
| Fig. 4 Proposed binding mode for compounds 10ab (left) and 10bb (right) in the ATP binding pocket of GSK3β. | ||
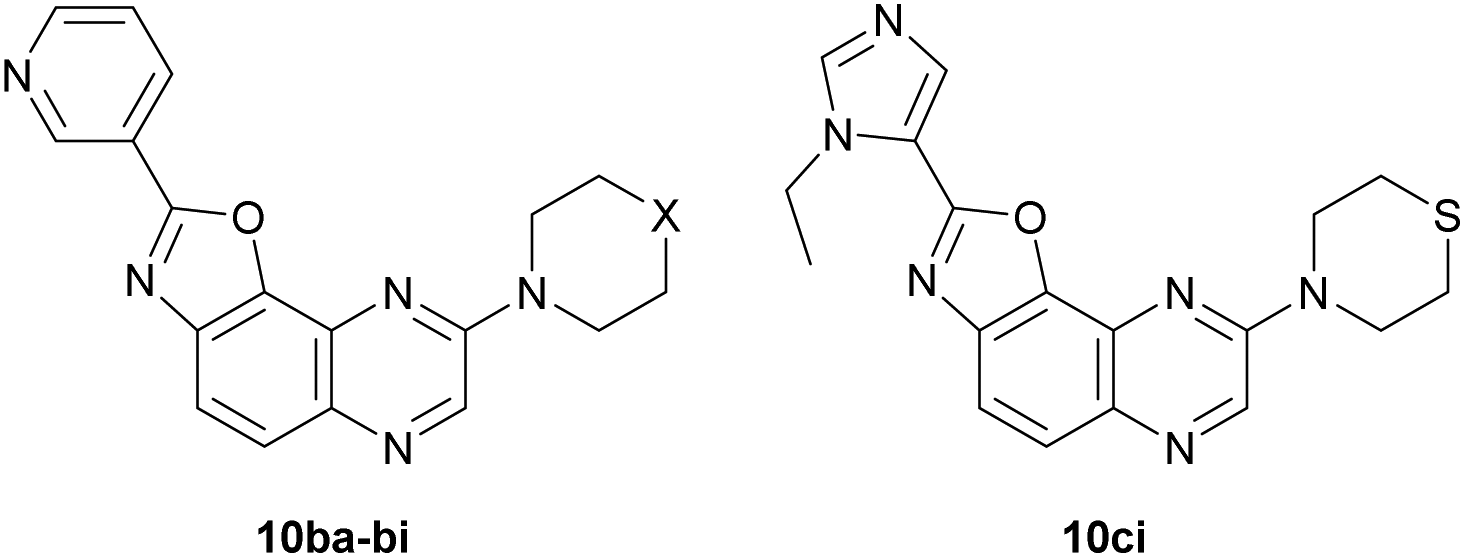
We first decided to keep the pyridine ring at C2 and replace the morpholine or 4-methylpiperazine present at C8 with various cyclic amines (Table 3, top scheme). To access the planned structures, we applied the procedure involving the merging of substitution and cyclization to obtain benzoxazole in a sequential process (Scheme 3, top). Indeed, we observed in the course of synthesis of 6ba that there was no need to isolate intermediate 5ba and to purify precursor 11b. Thus, under these conditions, 2,8-disubstituted oxazolo[5,4-f]quinoxalines 10bc–bi were prepared from the dihalogenated carboxamide 12b and isolated after purification in yields ranging from 44 to 86% (Table 3, entries 1–7).
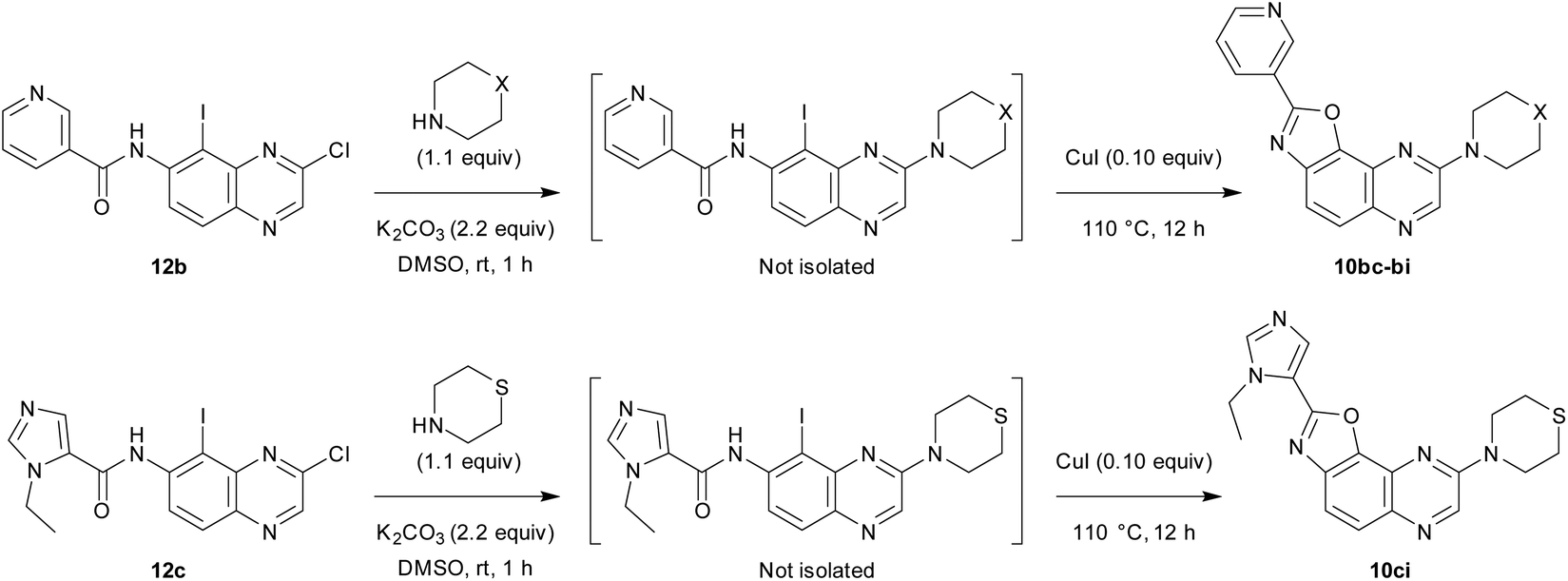

Then, we prepared 10ci, an analogue of 10bi in which the 3-pyridyl group at C2 is replaced by 1-ethyl-5-imidazolyl (Table 3, bottom scheme). The idea was to verify whether the interaction between Lys85 and the nitrogen of the ring at C2 could be improved. Compound 10ci was prepared similarly (Table 3, entry 8), and its structure was identified unambiguously by X-ray diffraction (see the ORTEP diagram in Fig. 2).
The inhibitory activities of these new compounds were evaluated for different kinases as mentioned previously and compared with those of 10ba and 10bb (Table 4). In addition, the dose-dependent effects of the most promising compounds were tested on GSK3β and GSK3α; the corresponding IC50 values are given in Table 5. Most of the derivatives lost their activities against GSK3, except compounds 10bg and 10bi which behaved similar to 10ba against GSK3β, but showed potency towards GSK3α. It is worth noting that in spite of similar ring sizes, increasing activities are noticed from 10ba to 10bg and 10bi, suggesting that either conformational changes or additional interactions might occur with these latter compounds. Finally, the dose-dependent effect of 10ba, 10bg, 10bi and 10ci was determined on DYRK1A, which showed a rather good preference of these inhibitors for GSK3 when compared with DYRK1A.
| Compd. | CDK2/CyclinA | CDK5/p25 | CDK9/CyclinT | PIM1 | RnDYRK1A | GSK3α | GSK3β | SscGSK3α/β | CK1ε | SscCK1δ/ε | Haspin |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10ba | — | 66 (98) | 27 (45) | 23 (43) | 3 (11) | 9 (9) | 2 (2) | — | 65 (75) | — | 12 (23) |
| 10bb | ≥100 (≥100) | 93 (≥100) | 7 (35) | 20 (57) | — | 2 (4) | 6 (6) | 16 (16) | 53 (≥100) | 72 (88) | 24 (29) |
| 10bc | — | 77 (97) | 31 (82) | 86 (79) | 3 (41) | 11 (19) | ≤0 (15) | — | 69 (≥100) | — | 30 (77) |
| 10bd | — | 67 (69) | 29 (69) | 29 (80) | 14 (25) | ≤0 (≤0) | 10 (16) | — | 51 (84) | — | 26 (64) |
| 10be | — | 76 (≥100) | 30 (69) | ≥100 (≥100) | 8 (51) | 9 (9) | 3 (13) | — | 74 (96) | — | 43 (54) |
| 10bf | — | 63 (84) | 1 (4) | 46 (≥100) | 16 (28) | 4 (11) | 2 (9) | — | 11 (46) | — | 7 (16) |
| 10bg | — | 58 (97) | 11 (40) | 13 (≥100) | 4 (21) | 11 (13) | 1 (5) | — | 83 (93) | — | 21 (58) |
| 10bh | — | 98 (87) | 10 (49) | 87 (≥100) | 2 (30) | 1 (8) | 5 (20) | — | 56 (85) | — | 10 (44) |
| 10bi | — | 53 (99) | 4 (69) | 18 (≥100) | 5 (19) | ≤0 (≤0) | 21 (14) | — | 44 (86) | — | 8 (63) |
| 10ci | — | 47 (95) | 9 (26) | 12 (40) | 4 (24) | 2 (5) | 8 (9) | — | 78 (≥100) | — | 16 (48) |
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | X | GSK3α | GSK3β | RnDYRK1A | |
| a Three known inhibitors (indirubine-3′-monoxime, hymenialdisine and alsterpaullone) were also measured as controls. Values obtained from the literature for known potent or selective inhibitors were also added for comparison. b IC50(GSK3β)/IC50(GSK3α) as an evaluation of the isoform selectivity. c Values obtained from the literature (radioactive method using 15 μM ATP). | ||||||
| 1 | 10ba | O | 15 | 25 | 1.7 | 200 |
| 2 | 10bb | N-Me | 24 | 55 | 2.3 | — |
| 3 | 10bc | N-Benzyl | 40 | 102 | 2.55 | — |
| 4 | 10bd | N-CH2-5-(1,3-benzodioxolyl) | 88 | 127 | 1.4 | — |
| 5 | 10be | N-Boc | 57 | 97 | 1.7 | — |
| 6 | 10bf | N–H | 24 | 68 | 2.8 | — |
| 7 | 10bg | CH2 | 11 | 33 | 3.0 | 390 |
| 8 | 10bh | CH-morpholino | 22 | 35 | 1.6 | — |
| 9 | 10bi | S | 4.8 | 22 | 4.6 | 130 |
| 10 | 10ci | — | 31 | 50 | 1.6 | 447 |
| 11 | Indirubine-3′-monoxime | 72 | 55 (22)c,11 | 0.76 | — | |
| 12 | Hymenialdisine | 3.6 | 10 (10)c,12 | 2.8 | — | |
| 13 | Alsterpaullone | 30 (4)c,13 | 22 (4)c,13 | 0.73 | — | |
| 1414 | CHIR 98014 | 7.0 | 0.6 | 0.09 | — | |
| 1515 | LY2090314 | 1.5 | 0.9 | 0.60 | — | |
| 1616 | BRD0705 | 66 | 515 | 7.8 | — | |
| 1717 | G28_14 | 33 | 218 | 6.6 | — | |
Conclusions
These results allowed us to identify the oxazolo[5,4-f]quinoxalines 10ba, 10bg and 10bi as strong ATP-competitive inhibitors of GSK3. The most potent GSK3 inhibitors reported in the literature are pyrimidine CHIR 9801414 and bis(aryl)maleimide LY2090314,15 which have subnanomolar or nanomolar IC50 values (Fig. 5, left; Table 5, entries 14 and 15); however, while CHIR 98014 selectively inhibits GSK3β, LY2090314 binds almost equally to GSK3α and GSK3β. In contrast, while inhibitors with high isoform-selectivity for GSK3α exist (e.g.BRD070516 and G28_1417 with IC50(GSK3β)/IC50(GSK3α) ratios of 7.8 and 6.6, respectively), they are less potent (Fig. 5, right; Table 5, entries 16 and 17). Our best inhibitor, the sulphur-containing molecule 10bi is potent and it binds GSK3α more easily than GSK3β (ratio of 4.6). Thus, it will be a subject of further studies aimed at identifying potent and selective inhibitors of GSK3α. It is indeed of interest to identify isoform-selective kinase inhibitors for the development of new therapies.17 Efforts devoted to cellular testing in order to identify and develop applications for this new family of heterocycles are also ongoing in the lab.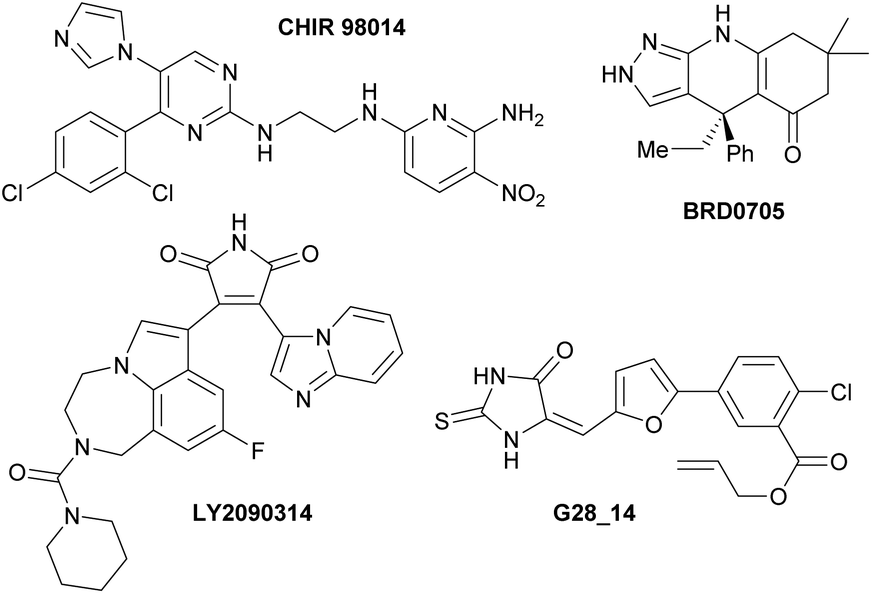 | ||
| Fig. 5 Left: Potent inhibitors of GSK3α/β CHIR 9801414 and LY2090314.15 Right: Selective inhibitors of GSK3α vs. GSK3β BRD070516 and G28_14.17 | ||
Experimental section
Procedure used to convert 12b into 10bi
To a suspension of iodinated amide 12b (0.41 g, 1.0 mmol) in dry DMSO (4 mL) were added thiomorpholine (0.11 mL, 1.1 mmol) and potassium carbonate (0.29 g, 2.2 mmol). After stirring at room temperature for 1 h, copper(I) iodide (19 mg, 0.10 mmol) was added. The tube was sealed, heated at 110 °C for 12 h and then cooled. The resulting dark mixture was poured into cold water (20 mL); the precipitate was filtered, washed with cold water (5 mL) and dissolved in chloroform. The organic layer was dried over sodium sulphate and the solvent was removed under reduced pressure. The product was purified by chromatography over silica gel (eluent: CHCl3–MeOH 97:3).
Kinetic studies
Kinetic experiments were performed by varying the concentrations of ATP (from 1 to 100 μM) and inhibitor 10bb (0.05 and 0.02 μM), while the concentration of the substrate GS2 was kept constant (12.5 μM). The ADP-Glo kinase assay18 was used. Double-reciprocal plotting of the data is depicted in Fig. 3.Docking studies
Before the docking studies, ligands and targets need to be preprocessed. Ligands were prepared for docking using the LigPrep tool,19 using the OPLS2005 force field, and by calculating the protonation state at pH = 7.2 ± 0.2. For target preprocessing, the Maestro Protein Preparation Wizard20 was used. The crystallographic structure of GSK3β in a complex with a known inhibitor (PDBID 4ACD)21 was preprocessed by adding hydrogen atoms, removing the counterions and water molecules, and protonating the residues at pH = 7.2. To develop the docking studies, we used Glide22 to perform the docking calculations using the Extra Precision Glide mode. The innerbox size of the grid was 16 × 16 × 16 Å and the outerbox size was 33.5 × 33.5 × 33.5 Å. The grid box was centred on coordinates 0.432, 9.255, 31.434, that correlate with the centre of the ATP-binding site. 10 poses per ligand were calculated and classified by the binding energy. To validate the docking protocol, we employed some GSK3β inhibitors with a known binding mode to the ATP-binding site of the enzyme.16,22,23Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Université de Rennes 1 and the Centre National de la Recherche Scientifique for supporting this research. We acknowledge the support from the Fonds Européen de Développement Régional (FEDER; D8 VENTURE Bruker AXS diffractometer). A. M. acknowledges financial support from Spanish Ministry of Innovation, Science and Universities (MICU, grant no. SAF2016-76693-R) and ISCiii (CIBERNED, grant no. CB18/05/00040). S. B. and S. R. also thank the Cancéropôle Grand Ouest (axis: Natural Sea Products in Cancer Treatment), IBiSA (French Infrastructures en sciences du vivant: biologie, santé et agronomie) and Biogenouest (Western France Life Science and Environment Core Facility Network) for supporting KISSf screening facility. We also thank Aurélie Assicot for her contribution to the study and Ludovic Paquin for the generous gift of secondary amines.Notes and references
- T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles: Structure, Reactions Synthesis and Applications, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed.
- J. H. Musser, H. Jones, S. Sciortino, K. Bailey, S. M. Coutts, A. Khandwala, P. Sonnino-Goldman, M. Leibowitz, P. Wolf and E. S. Neiss, J. Med. Chem., 1985, 28, 1255 CrossRef CAS.
- J. Deng, E. Feng, S. Ma, Y. Zhang, X. Liu, H. Li, H. Huang, J. Zhu, W. Zhu, X. Shen, L. Miao, H. Liu, H. Jiang and J. Li, J. Med. Chem., 2011, 54, 4508 CrossRef CAS PubMed.
- (a) N. Mokhtari Brikci-Nigassa, G. Bentabed-Ababsa, W. Erb, F. Chevallier, L. Picot, L. Vitek, A. Fleury, V. Thiery, M. Souab, T. Robert, S. Ruchaud, S. Bach, T. Roisnel and F. Mongin, Tetrahedron, 2018, 74, 1785 CrossRef; (b) N. Mokhtari Brikci-Nigassa, L. Nauton, P. Moreau, O. Mongin, R. E. Duval, L. Picot, V. Thiéry, M. Souab, S. Ruchaud, S. Bach, R. Le Guevel, G. Bentabed-Ababsa, W. Erb, T. Roisnel, V. Dorcet and F. Mongin, Bioorg. Chem., 2019, 82 DOI:10.1016/j.bioorg.2019.103347.
- G. Evindar and R. A. Batey, J. Org. Chem., 2006, 71, 1802 CrossRef CAS PubMed.
- X. Hui, J. Desrivot, C. Bories, P. M. Loiseau, X. Franck, R. Hocquemiller and B. Figadère, Bioorg. Med. Chem. Lett., 2006, 16, 815 CrossRef CAS PubMed.
- (a) W. R. Bowman, H. Heaney and P. H. G. Smith, Tetrahedron Lett., 1982, 23, 5093 CrossRef CAS; (b) T. Minami, T. Isonaka, Y. Okada and J. Ichikawa, J. Org. Chem., 1993, 58, 7009 CrossRef CAS; (c) B. A. Lanman, V. J. Cee, S. R. Cheruku, M. Frohn, J. Golden, J. Lin, M. Lobera, Y. Marantz, K. M. Muller, S. C. Neira, A. J. Pickrell, D. Rivenzon-Segal, N. Schutz, A. Sharadendu, X. Yu, Z.-D. Zhang, J. Buys, M. Fiorino, A. Gore, M. Horner, A. Itano, M. McElvain, S. Middleton, M. Schrag, H. M. Vargas, H. Xu, Y. Xu, X.-X. Zhang, J. Siu and R. W. Burli, ACS Med. Chem. Lett., 2011, 2, 102 CrossRef CAS PubMed; (d) J. Peng, C. Zong, M. Ye, T. Chen, D. Gao, Y. Wang and C. Chen, Org. Biomol. Chem., 2011, 9, 1225 RSC; (e) J.-H. Jia, C.-L. Jiang, X.-J. Zhang, Y.-W. Jiang and D.-W. Ma, Tetrahedron Lett., 2011, 52, 5593 CrossRef CAS; (f) V. Kavala, D. Janreddy, M. J. Raihan, C.-W. Kuo, C. Ramesh and C.-F. Yao, Adv. Synth. Catal., 2012, 354, 2229 CrossRef CAS; (g) P. J. Tambade, Y. P. Patil, Z. S. Qureshi, K. P. Dhake and B. M. Bhanage, Synth. Commun., 2012, 42, 176 CrossRef CAS; (h) A. Ahmed, R. Singha and J. K. Ray, Tetrahedron Lett., 2015, 56, 2167 CrossRef CAS; (i) T. Venu Saranya, P. Rajan Sruthi and S. Anas, Synth. Commun., 2019, 49, 297 CrossRef CAS.
- S. Klaeger, S. Heinzlmeir, M. Wilhelm, H. Polzer, B. Vick, P.-A. Koenig, M. Reinecke, B. Ruprecht, S. Petzoldt, C. Meng, J. Zecha, K. Reiter, H. Qiao, D. Helm, H. Koch, M. Schoof, G. Canevari, E. Casale, S. R. Depaolini, A. Feuchtinger, Z. Wu, T. Schmidt, L. Rueckert, W. Becker, J. Huenges, A.-K. Garz, B.-O. Gohlke, D. P. Zolg, G. Kayser, T. Vooder, R. Preissner, H. Hahne, N. Tonisson, K. Kramer, K. Goetze, F. Bassermann, J. Schlegl, H.-C. Ehrlich, S. Aiche, A. Walch, P. A. Greif, S. Schneider, E. R. Felder, J. Ruland, G. Medard, I. Jeremias, K. Spiekermann and B. Kuster, Science, 2017, 358, 1148 CrossRef CAS PubMed.
- (a) R. Roskoski Jr., Pharmacol. Res., 2019, 144, 19 CrossRef PubMed; (b) H. L. Lightfoot, F. W. Goldberg and J. Sedelmeier, ACS Med. Chem. Lett., 2019, 10, 153 CrossRef CAS PubMed.
- (a) M. K. Pandey and T. R. DeGrado, Theranostics, 2016, 6, 571 CrossRef CAS PubMed; (b) A. P. Saraswati, S. M. Ali Hussaini, N. H. Krishna, B. N. Babu and A. Kamal, Eur. J. Med. Chem., 2018, 144, 843 CrossRef CAS PubMed.
- S. Leclerc, M. Garnier, R. Hoessel, D. Marko, J. A. Bibb, G. L. Snyder, P. Greengard, J. Biernat, Y.-Z. Wu, E.-M. Mandelkow, G. Eisenbrand and L. Meijer, J. Biol. Chem., 2001, 276, 251 CrossRef CAS PubMed.
- L. Meijer, A. M. W. H. Thunnissen, A. W. White, M. Garnier, M. Nikolic, L. H. Tsai, J. Walter, K. E. Cleverley, P. C. Salinas, Y. Z. Wu, J. Biernat, E. M. Mandelkow, S. H. Kim and G. R. Pettit, Chem. Biol., 2000, 7, 51 CrossRef CAS PubMed.
- M. Leost, C. Schultz, A. Link, Y.-Z. Wu, J. Biernat, E.-M. Mandelkow, J. A. Bibb, G. L. Snyder, P. Greengard, D. W. Zaharevitz, R. Gussio, A. M. Senderowicz, E. A. Sausville, C. Kunick and L. Meijer, Eur. J. Biochem., 2000, 267, 5983 CrossRef CAS.
- A. S. Wagman, R. S. Boyce, S. P. Brown, E. Fang, D. Goff, J. M. Jansen, V. P. Le, B. H. Levine, S. C. Ng, Z.-J. Ni, J. M. Nuss, K. B. Pfister, S. Ramurthy, P. A. Renhowe, D. B. Ring, W. Shu, S. Subramanian, X. A. Zhou, C. M. Shafer, S. D. Harrison, K. W. Johnson and D. E. Bussiere, J. Med. Chem., 2017, 60, 8482 CrossRef CAS PubMed.
- J. M. Atkinson, K. B. Rank, Y. Zeng, A. Capen, V. Yadav, J. Manro, T. A. Engler and M. Chedid, PLoS One, 2015, 10, e0125028/1 CAS.
- F. F. Wagner, L. Benajiba, A. J. Campbell, M. Weiewer, J. R. Sacher, J. P. Gale, L. Ross, A. Puissant, G. Alexe, A. Conway, M. Back, Y. Pikman, I. Galinsky, D. J. Deangelo, R. M. Stone, T. Kaya, X. Shi, M. B. Robers, T. Machleidt, J. Wilkinson, O. Hermine, A. Kung, A. J. Stein, D. Lakshminarasimhan, M. T. Hemann, E. Scolnick, Y.-L. Zhang, J. Q. Pan, K. Stegmaier and E. B. Holson, Sci. Transl. Med., 2018, 10, eaam8460/1 CrossRef CAS PubMed.
- Y. Wang, X. Dou, L. Jiang, H. Jin, L. Zhang, L. Zhang and Z. Liu, Eur. J. Med. Chem., 2019, 171, 221 CrossRef CAS.
- ADP-Glo™ Kinase Assay Technical Manual: https://www.promega.es/resources/protocols/technical-manuals/0/adp-glo-kinase-assay-protocol/(July 30, 2019).
- Schrödinger Release 2016-4: LigPrep, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- M. G. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput. Aided Mol. Des., 2013, 27, 221 CrossRef PubMed.
- S. Berg, M. Bergh, S. Hellberg, K. Högdin, Y. Lo-Alfredsson, P. Söderman, S. von Berg, T. Weigelt, M. Ormö, Y. Xue, J. Tucker, J. Neelissen, E. Jerning, Y. Nilsson and R. Bhat, J. Med. Chem., 2012, 55, 9107 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177 CrossRef CAS PubMed.
- (a) S. H. Liang, J. M. Chen, M. D. Normandin, J. S. Chang, G. C. Chang, C. K. Taylor, P. Trapa, M. S. Plummer, K. S. Para, E. L. Conn, L. Lopresti-Morrow, L. F. Lanyon, J. M. Cook, K. E. G. Richter, C. E. Nolan, J. B. Schachter, F. Janat, Y. Che, V. Shanmugasundaram, B. A. Lefker, B. E. Enerson, E. Livni, L. Wang, N. J. Guehl, D. Patnaik, F. F. Wagner, R. Perlis, E. B. Holson, S. J. Haggarty, G. El Fakhri, R. G. Kurumbail and N. Vasdev, Angew. Chem., Int. Ed., 2016, 55, 9601 CrossRef CAS; (b) F. F. Wagner, L. Benajiba, A. J. Campbell, M. Weiewer, J. R. Sacher, J. P. Gale, L. Ross, A. Puissant, G. Alexe, A. Conway, M. Back, Y. Pikman, I. Galinsky, D. J. Deangelo, R. M. Stone, T. Kaya, X. Shi, M. B. Robers, T. Machleidt, J. Wilkinson, O. Hermine, A. Kung, A. J. Stein, D. Lakshminarasimhan, M. T. Hemann, E. Scolnick, Y.-L. Zhang, J. Q. Pan, K. Stegmaier and E. B. Holson, Sci. Transl. Med., 2018, 10, eaam8460/1 CAS; (c) F. X. Tavares, J. A. Boucheron, S. H. Dickerson, R. J. Griffin, F. Preugschat, S. A. Thomson, T. Y. Wang and H.-Q. Zhou, J. Med. Chem., 2004, 47, 4716 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental procedures and analyses of the compounds and NMR spectra. CCDC 1947178 (10ba) and 1964834 (10ci). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob02002k |
| This journal is © The Royal Society of Chemistry 2020 |