
Stereoselective synthesis of hydrazinodihydrofurans via cascade Michael addition–substitution involving the reaction of curcumin and other β-dicarbonyls with α-hydrazinonitroalkenes†
Kalisankar
Bera
,
Narasimham
Ayyagari
,
Nishikant
Satam
 and
Irishi N. N.
Namboothiri
*
and
Irishi N. N.
Namboothiri
*
Department of Chemistry, Indian Institute of Technology Bombay, Mumbai 400076, India. E-mail: irishi@iitb.ac.in
First published on 26th November 2019
Abstract
Highly diastereoselective synthesis of 2-hydrazinated 2,3-dihydrofurans in good to excellent yields involving an interrupted Feist–Bénary type reaction by treating a wide variety of 1,3-dicarbonyl compounds, including curcumins, with α-hydrazinated nitroalkenes is reported here. The first ever enantioselective reaction of α-hydrazinonitroalkenes has also been carried out with two selected 1,3-dicarbonyls, dimedone and cyclohexanone by employing an L-t-leucine derived squaramide as the chiral organocatalyst to afford the enantio-enriched 2-hydrazinodihydrofurans as single diastereomers in good yields and with good enantioselectivities.
Introduction
Heterocycles are omnipresent in the molecular skeleton of biologically active molecules and natural products. Among the heterocycles, dihydrofurans receive considerable attention in medicinal chemistry due to their wide range of biological activities.1 The presence of a 2,3-dihydrofuran skeleton in natural products such as azadirachtin, clerodin, bicunningines, conocarpan etc. has attracted the attention of synthetic chemists (Fig. 1).2 Convenient conversion of dihydrofurans into highly functionalized tetrahydrofurans with high stereoselectivity has made the dihydrofurans versatile building blocks in organic synthesis.3 Thus, several synthetic methodologies based on chiron and chiral auxiliary approaches have been reported earlier in the literature for their stereoselective synthesis.4 Numerous metal-catalyzed5 and organocatalyzed reactions, including asymmetric interrupted Feist–Bénary reactions,6 and bromocyclization of olefinic dicarbonyls,7 have been reported for the stereoselective synthesis of 2,3-dihydrofurans. Rueping,8 Xie,9 Lu,10 Feng,11 Rodriguez,12 Aleman13 and Xie14 independently reported enantioselective Michael addition–substitution involving 1,3-dicarbonyls and α-halonitroalkenes where the presence of a good leaving group such as a Br or Cl α- to NO2 group facilitated the substitution to afford enantioenriched 2,3-dihydrofurans. Recently, Xu reported iodine/organocatalyzed addition of 1,3-dicarbonyls to activated olefins towards the synthesis of enantioenriched 2,3-dihydrofurans (Scheme 1a).15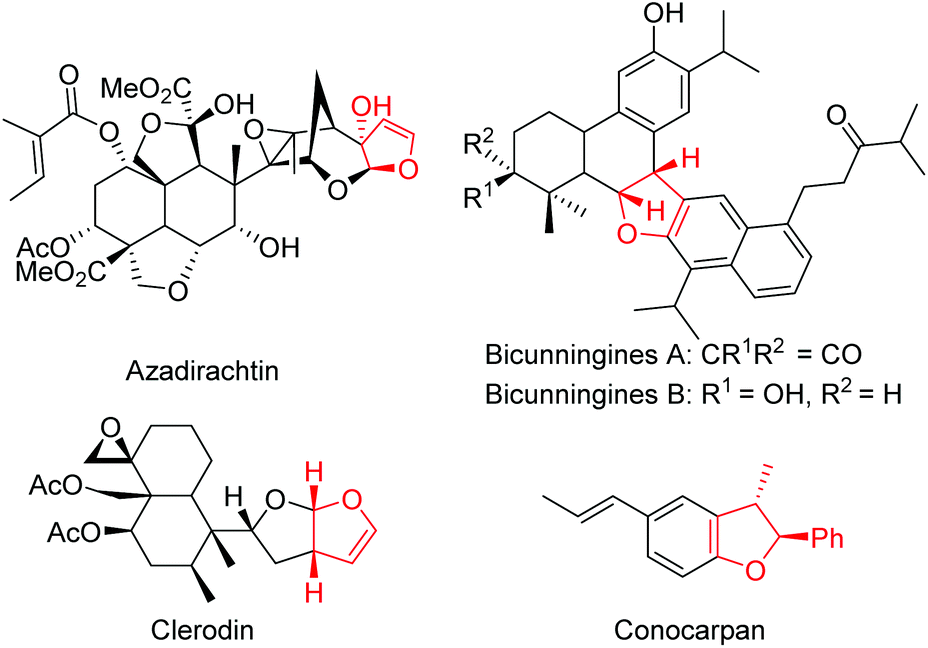 | ||
| Fig. 1 Dihydrofuran containing natural products. | ||
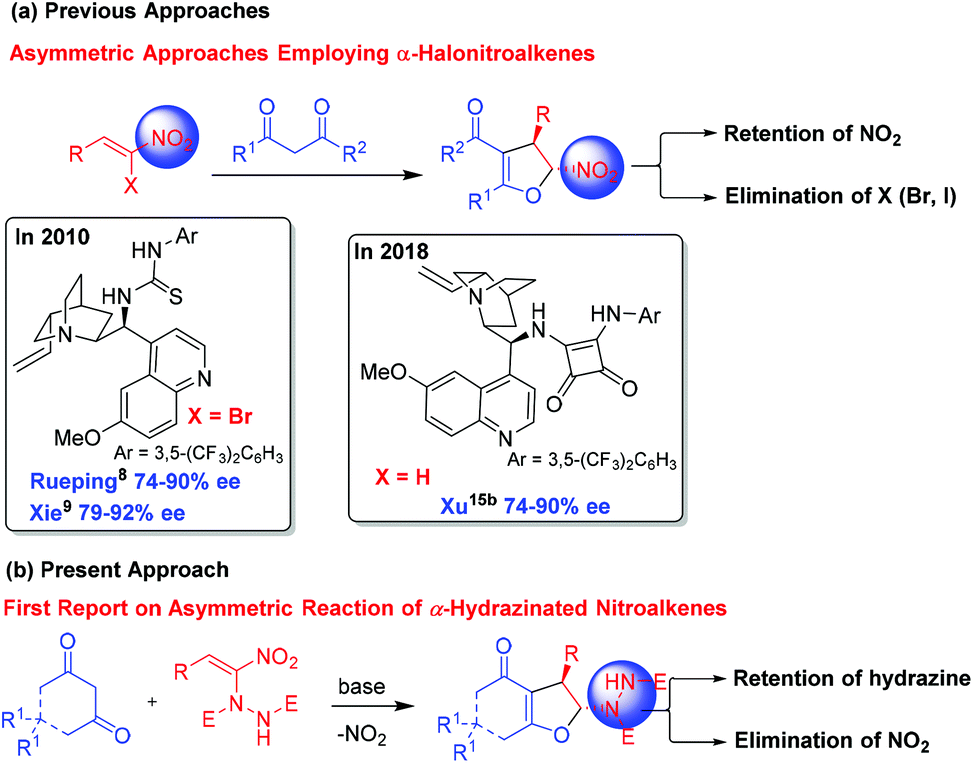 | ||
| Scheme 1 Enantioselective synthesis of dihydrofurans from nitroalkenes and 1,3-dicarbonyls. | ||
From another perspective, curcumins constitute the yellow pigment in turmeric which exhibits a wide range of activities16 such as anticancer,17 anti-oxidant,18 anti-inflammatory,19 anti-metastatic,20 anti-HIV protease,21 and anti-Alzheimer's activities,22 and many others.23 Our group for the first time employed curcumins as Michael donors in organic synthesis.24 We reported a stereoselective synthesis of functionalized cyclohexanones and dihydrofurans via cascade double Michael addition and Michael addition–cyclization involving curcumins and nitroalkenes.24 A catalytic asymmetric version of the double Michael reaction of curcumins with nitroalkenes in the presence of a cinchonine-thiourea catalyst afforded functionalized cyclohexanones in an enantio-enriched manner.25 While similar reaction of curcumins with chalcones provided the expected double Michael adducts, the reaction stopped at the single Michael stage in the case of azodicarboxylates.26 Other groups also explored the reactivity of curcumins in Michael additions. Ye's group reported a tertiary amine-thiourea catalyzed single Michael addition of curcumins to nitroalkenes.27 Yan's group disclosed the formation of spiro-oxindoles from curcumins and oxindolylidene malononitrile via a double Michael addition.28 Formation of enantio-enriched spiroindanediones from curcumins and 2-arylidene-1,3-indanediones in the presence of quinine as the chiral catalyst was reported by Zhang and co-workers.29
As part of our ongoing research on the synthesis and applications of Morita–Baylis–Hillman (MBH) and Rauhut–Currier (RC)30 adducts of nitroalkenes,31 we have reported the synthesis of α-hydrazinonitroalkenes.32 Subsequent reaction of α-hydrazinonitroalkenes with phenols/naphthols and thioindoles afforded hydrazinated arenodihydrofurans and thienoindoles, respectively.33 Herein, we report the reaction of α-hydrazinonitroalkenes with curcumins and other 1,3-dicarbonyls in the stereoselective synthesis of fused dihydrofurans bearing a key hydrazinodicarboxylate moiety (Scheme 1b).
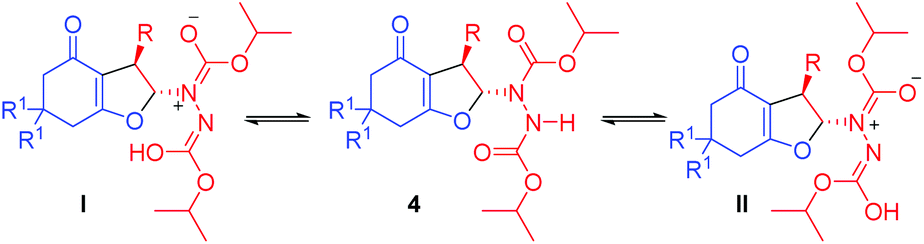
We envisioned that base mediated Michael addition of 1,3-dicarbonyls 1 to α-hydrazinated nitroalkenes 2 would initially give rise to intermediate 3 which on enolization followed by O-alkylation of the enolate of 3 by nucleophilic substitution of the NO2 group would afford 2,3-dihydrofurans 4 (Scheme 2). Unlike in the case of α-halonitroalkenes where substitution of the halide takes place during the intramolecular cyclization (O-alkylation),8–14 substitution of the nitro group is expected here affording hydrazinodihydrofurans 4. Therefore, we report herein the first diastereo- and enantioselective synthesis of fused dihydrofurans bearing a stable hydrazinodicarboxylate moiety.
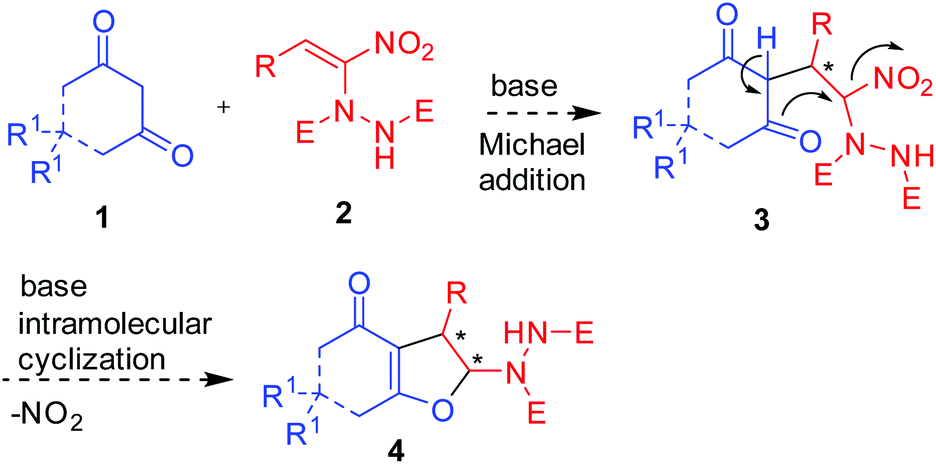 | ||
| Scheme 2 Proposed Michael addition–intramolecular cyclization sequence for the synthesis of hydrazinodihydrofurans. | ||
Results and discussion
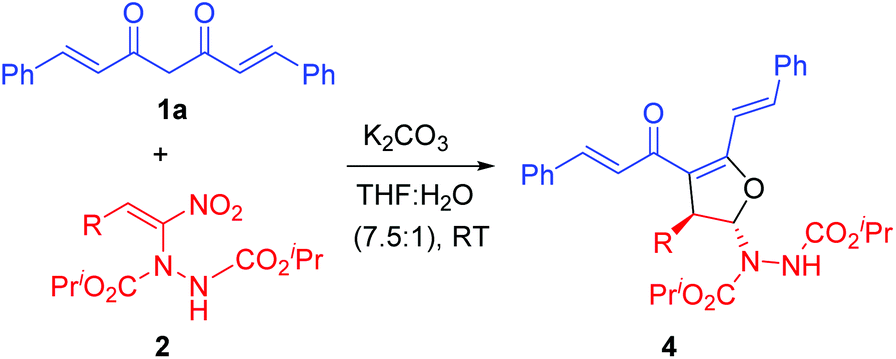
In order to study the feasibility of our hypothesis (Scheme 2), initially we reacted curcumin 1a and α-hydrazinated nitroalkene 2a under our previously optimized conditions,24 that is, in the presence of K2CO3 in THF–water (7.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Table 1, entry 1). To our delight, we isolated the 2,3-dihydrofuran 4a as a single diastereomer in excellent yield (92%, entry 1). Later, we employed selected hydrazino derivatives of nitroalkenes, namely, p-methoxynitrostyrene 2b, p-chloronitrostyrene 2c and 2-nitrovinylthiophene 2d, which also provided the corresponding dihydrofurans 4b–d in excellent yields (94–98%, entries 2–4). It is important to note that nitroalkenes 2a–d bearing diverse β-aryl groups, regardless of their electronic nature, delivered the products 4a–d in nearly quantitative yields (92–98%) in 10–18 h.
:1, Table 1, entry 1). To our delight, we isolated the 2,3-dihydrofuran 4a as a single diastereomer in excellent yield (92%, entry 1). Later, we employed selected hydrazino derivatives of nitroalkenes, namely, p-methoxynitrostyrene 2b, p-chloronitrostyrene 2c and 2-nitrovinylthiophene 2d, which also provided the corresponding dihydrofurans 4b–d in excellent yields (94–98%, entries 2–4). It is important to note that nitroalkenes 2a–d bearing diverse β-aryl groups, regardless of their electronic nature, delivered the products 4a–d in nearly quantitative yields (92–98%) in 10–18 h.
In order to explore the scope of curcumins, we treated various curcumins 1b–f with a representative α-hydrazinated nitroalkene 2a and the results are summarized in Table 2. Curcumins bearing electron rich aryl groups 1b–d and heteroaryl groups 1e–f reacted smoothly with α-hydrazinated nitroalkene 2a to provide 2,3-dihydrofurans 5a–e as single diastereoisomers in 92–98% yields (entries 1–5).
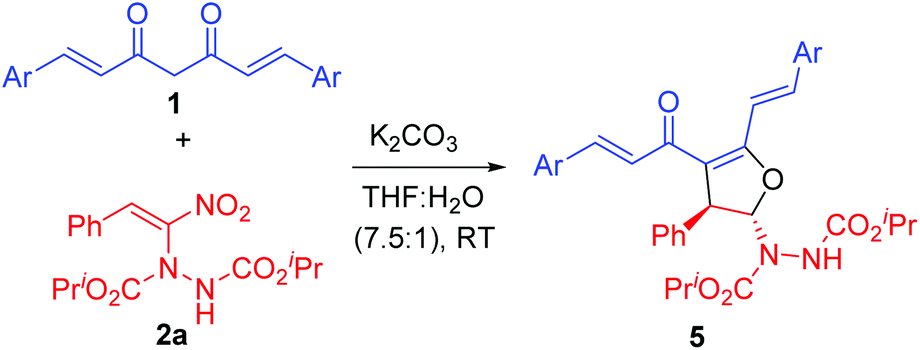
The above results confirmed that the electronic nature of the aryl groups in curcumins 1a–f also did not have any adverse effect on the outcome of the reaction as the products 4a–d and 5a–e were formed in excellent yields. While the reaction times were shorter for unsubstituted curcumin analog 1a (10–18 h, Table 1), substituted ones required more time for completion (20–28 h, Table 2).
Having established the reactivity of curcumins 1 with hydrazinonitroalkenes 2, further scope of this cascade Michael addition–substitution strategy was investigated with various 1,3-dicarbonyls 1g–l (Fig. 2) and selected nitroalkene 2a and nitrodiene 2g under the above conditions, that is, in the presence of K2CO3 in THF–water (7.5:1). It is noteworthy that, diverse 1,3-dicarbonyl compounds such as acyclic 1,3-diketones 1g-h, β-ketoesters 1i–j and cyclic 1,3-diketones 1k–l well tolerated the reaction conditions to afford the corresponding dihydrofurans 6, 7 and 8 as single diastereoisomers in good to high yields (Scheme 3). Notably, 1,3-dicarbonyls 1g–i and 1k–l were more reactive than curcumins 1a–f and the corresponding products 6a–c, 7a and 8 were formed in high yields (80–92%) in 6–8 h. Hydroxycoumarin 1j was the only Michael donor which required an extended time (12 h) and the yield was also below normal (77%).
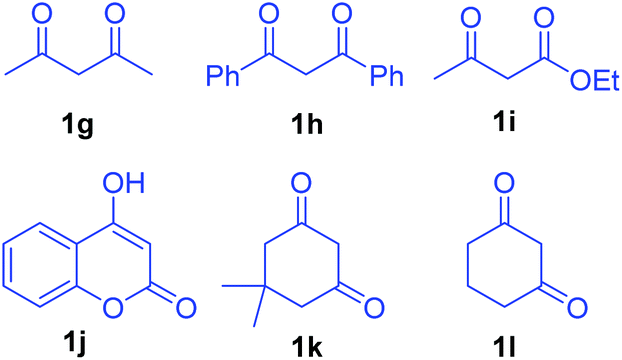 | ||
| Fig. 2 Various 1,3-dicarbonyl compounds 1g–l. | ||
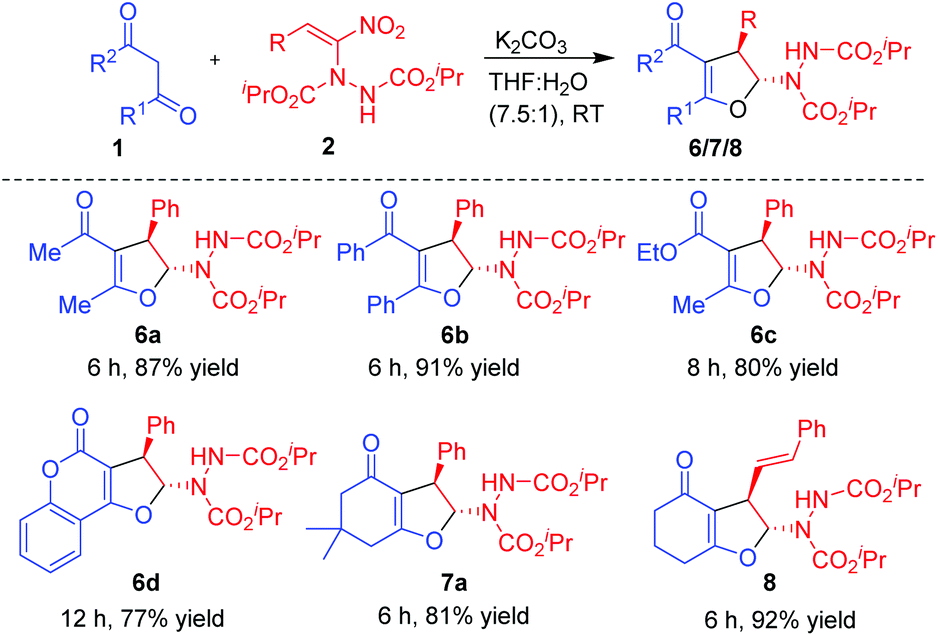 | ||
| Scheme 3 | ||
After successful diastereoselective synthesis of 2-hydrazinated 2,3-dihydrofurans in good to excellent yields from various 1,3-dicarbonyl compounds 1a–l and α-hydrazinated nitroalkenes 2a–g, we focused our attention to develop an enantioselective variant of this method and bifunctional (Brønsted base–Brønsted acid) organocatalysts C1–C12 were screened for this purpose (Fig. 3).34–36
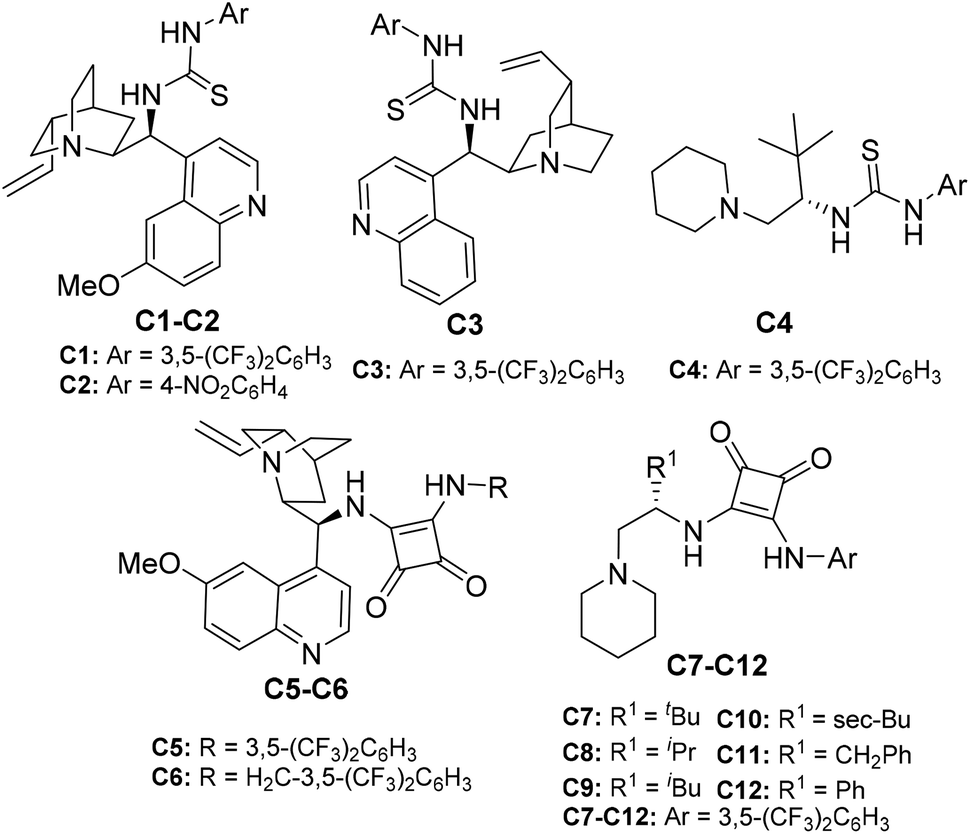 | ||
| Fig. 3 Catalysts screened. | ||
At the outset, we have treated dimedone 1k with nitroalkene 2a in the presence of 10 mol% of quinine-thiourea C1 in THF at room temperature (Table 3, entry 1). It took 3 days for complete consumption of 1k and the product was a mixture of 2,3-dihydrofuran 7a and Michael adduct 3a. But gratifyingly, the mixture on treatment with K2CO3 in THF–water (7.5:1) under microwave at 80 °C for 20 min provided the 2,3-disubstituted dihydrofuran 7a as a single diastereomer in good yield (70%) and with moderate enantioselectivity (61% ee, entry 1).37
|
|
|||||
|---|---|---|---|---|---|
| Entry | C | Solvent | Time/d | Yieldb/% | eec/% |
| a Catalyst. b After silica gel column chromatography. c Determined by chiral HPLC. d 5 mol% of the catalyst was used. e 20 mol% of the catalyst was used. f 0 °C. g 60 °C, complex mixture. h ∼20% conversion. | |||||
| 1 | C1 | THF | 3 | 70 | 61 |
| 2 | C2 | THF | 3 | 68 | 48 |
| 3 | C3 | THF | 3 | 68 | 46 |
| 4 | C4 | THF | 3 | 69 | 71 |
| 5 | C5 | THF | 3 | 74 | 66 |
| 6 | C6 | THF | 3 | 74 | 67 |
| 7 | C7 | THF | 3 | 72 | 72 |
| 8 | C8 | THF | 3 | 70 | 53 |
| 9 | C9 | THF | 3 | 65 | 19 |
| 10 | C10 | THF | 3 | 70 | 52 |
| 11 | C11 | THF | 3 | 64 | 30 |
| 12 | C12 | THF | 3 | 64 | 65 |
| 13 | C7 | Cl(CH2)2Cl | 3 | 72 | 70 |
| 14 | C7 | Mesitylene | 3 | 70 | 43 |
| 15d | C7 | THF | 4 | 72 | 73 |
| 16e | C7 | THF | 2.5 | 52 | 75 |
| 17f | C7 | THF | 7 | 60 | 75 |
| 18g | C7 | THF | 1 | — | — |
| 19 | — | THF | 2 | —h | — |
Furthermore, in order to improve the yield and enantioselectivity of dihydrofuran 7a, we screened other cinchona alkaloid and amino acid-derived thioureas C2–4 and squaramides C5–12 (entries 2–12). It was found that quinine-derived squaramides C5 and C6 provided 2,3-dihydrofuran 7a, with better enantioselectivity (66–67% ee, entries 5 and 6), compared to cinchonine derived thiourea catalysts C1, C2 and C3 (46–61% ee, entries 1–3). However, L-tert-leucine-derived thiourea C4 provided dihydrofuran 7a with slightly improved enantioselectivity (71% ee, entry 4). Further screening of amino acid-derived squaramide catalysts C7–12 (entries 7–12) revealed that L-tert-leucine-derived squaramide C7 was the best in terms of enantioselectivity (72% ee) even though the yield was slightly lower (72%, entry 7) when compared to that obtained with C5 and C6 (entries 5 and 6).
The above result prompted us to examine the role of solvent in a L-tert-leucine-squaramide C7 catalyzed cascade Michael addition–substitution sequence. Though the chlorinated solvent ethylene dichloride provided comparable yield (72%) and enantioselectivity (70% ee), the enantioselectivity dropped drastically to 43% in mesitylene (entries 13 and 14). Lowering the loading of catalyst C7 to 5 mol% still afforded the product 7a in 72% yield and with 73% enantioselectivity although an additional day was required for completion (entry 15). The reaction performed with higher quantity of the catalyst C7 caused a substantial drop in the yield (52%) though marginal improvement in the ee could be achieved (75%, entry 16). Long reaction time (7 d) and drop in the yield (60%) were encountered when the reaction was conducted at 0 °C (entry 17). Even gently heating the reaction mixture at 60 °C for a day afforded only a complex mixture (entry 18). Finally, there was marginal (∼20%) conversion of the starting materials when a control reaction was carried out in the absence of the catalyst (entry 19).38
Having optimized the enantioselective version of the sequential Michael addition–substitution for the synthesis of dihydrofurans 7, the scope of the reaction was investigated using dimedone 1k as the donor with various α-hydrazinated nitroalkenes 2a–l and the results are summarized in Table 4. It is noteworthy that nitroalkenes 2 bearing a wide variety of β-aryl groups participated in the reaction and afforded the products 7 in moderate to good yields (52–72%) and good to high ee's (69–81% ee). The yields were moderate (52–60%) only in the case of strongly electron donating aryl and heteroaryl groups as well as styrenyl substituted nitroalkenes 2c–d and 2j–k (entries 3–4 and 10–12). Other nitroalkenes bearing electroneutral 2a, weakly electron donating 2b, weakly electron withdrawing 2e–f, strongly electron withdrawing 2g and fused aryl 2h–i afforded the corresponding products in good yield (65–72%, entries 1, 2 and 5–9). As for the enantioselectivity, no appreciable substituent effect was observed and the ee's remained in a relatively narrow range of 69–81%.
|
|
||||
|---|---|---|---|---|
| Entry | 2, R | 7 | Yielda/% | eeb/% |
| a After silica gel column chromatography. b Determined by chiral HPLC. | ||||
| 1 | 2a, C6H5 | 7a | 72 | 73 |
| 2 | 2b, 4-MeC6H4 | 7b | 65 | 71 |
| 3 | 2c, 4-OMeC6H4 | 7c | 55 | 75 |
| 4 | 2d, 3,4-OCH2OC6H3 | 7d | 60 | 76 |
| 5 | 2e, 4-ClC6H4 | 7e | 67 | 69 |
| 6 | 2f, 2-BrC6H4 | 7f | 69 | 75 |
| 7 | 2g, 4-NO2C6H4 | 7g | 60 | 73 |
| 8 | 2h, 2-naphthyl | 7h | 71 | 79 |
| 9 | 2i, 1-naphthyl | 7i | 68 | 76 |
| 10 | 2j, 2-furyl | 7j | 54 | 71 |
| 11 | 2k, 2-thienyl | 7k | 52 | 74 |
| 12 | 2l, styrenyl | 7l | 60 | 81 |
The high ee observed for the nitrodiene derivative 2l (Table 4, entry 12) prompted us to treat 2l with another cyclic 1,3-diketone 1l which resulted in the formation of dihydrofuran 8 as a single diastereoisomer in moderate yield (61%), but with high enantioselectivity (81% ee, Scheme 4). Unfortunately, attempted enantioselective reactions of curcumins 1a–f and other 1,3-dicarbonyl compounds 1g–j with nitroalkenes 2 in the presence of squaramide C7 as the chiral catalyst did not provide satisfactory results under our optimized reaction conditions due to the poor enantioselectivity obtained with these 1,3-dicarbonyls.
 | ||
| Scheme 4 | ||
It is pertinent to mention that all the 2,3-dihydrofuran derivatives 4a–d, 5a–5e, 6a–d, 7a–l and 8 exhibited broadening of signals in 1H NMR at room temperature which is attributable to a dynamic phenomenon on the NMR time scale (Fig. 4).39 In order to further understand this dynamic phenomenon, variable temperature (VT) 1H NMR experiments were performed by taking dihydrofuran 7f as the model sample. At room temperature (298 K), all the protons of 7f appeared as broad signals. This signal broadening was particularly pronounced for iso-propyl methyl (δ 1.20–1.30) and methine (δ 4.90–5.05) protons. Subsequently, 1H NMR spectra of 7f were recorded at higher temperatures (313 K, 323 K and 328 K). Interestingly, gradual sharpening of the iso-propyl methine protons (δ 4.90–5.05), the NC–H proton of the dihydrofuran ring (δ 6.30) and the N–H proton (δ 6.60) was observed in the 313–323 K temperature range which culminated in the highest temperature possible in CDCl3 (328 K). Similarly, the diastereotopic methylenes (δ 2.20–2.45) in the cyclohexenone ring and the four aromatic protons (δ 7.00–7.60) became sharp and well resolved in this temperature range. The only signal that remained unchanged and unresolved was the benzylic proton (δ 4.80).
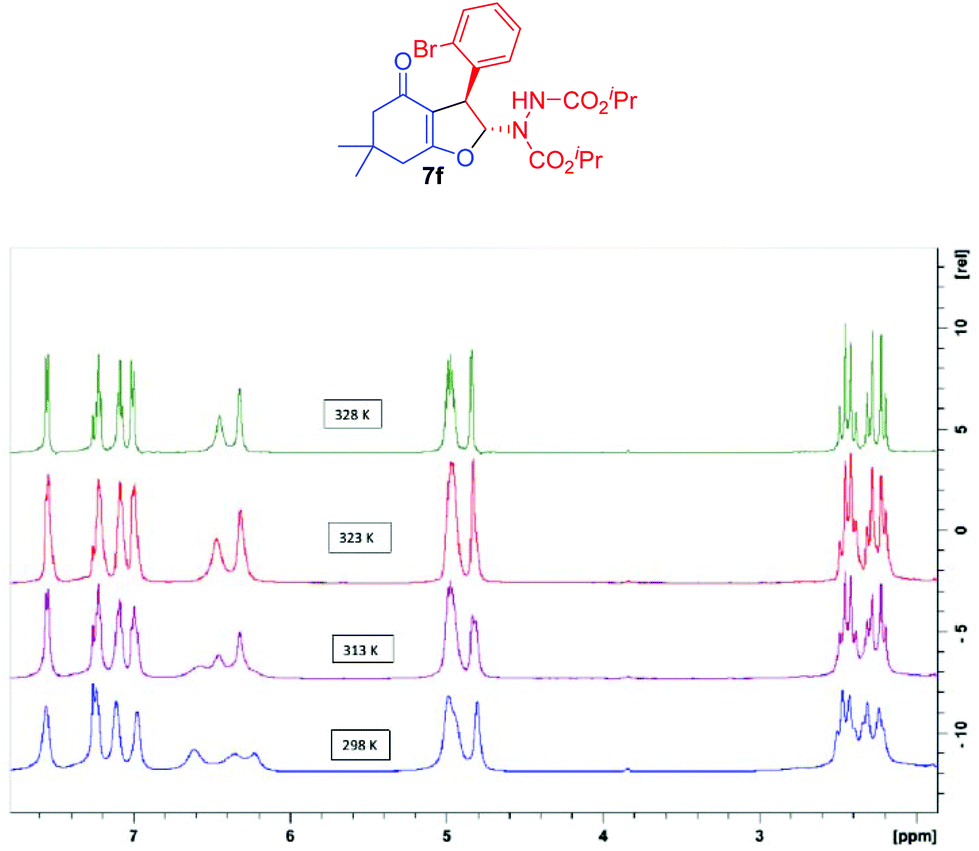 | ||
| Fig. 4 Variable temperature 1H NMR spectra of dihydrofuran 7f. | ||
It is obvious that the C–N bonds in the two carbamate moieties have a partial double bond character which restricts their rotation leading to several rotamers (Fig. 5) causing broadening of the signals at lower temperature. At higher temperature, faster rotation about the C–N bonds takes place resulting in sharp averaged signals. In order to obtain sharp signals and make unambiguous assignment, 1H and 13C NMR spectra of compounds 7g–i were also recorded at higher temperature (328 K).
 | ||
| Fig. 5 Possible isomers of compound 7. | ||
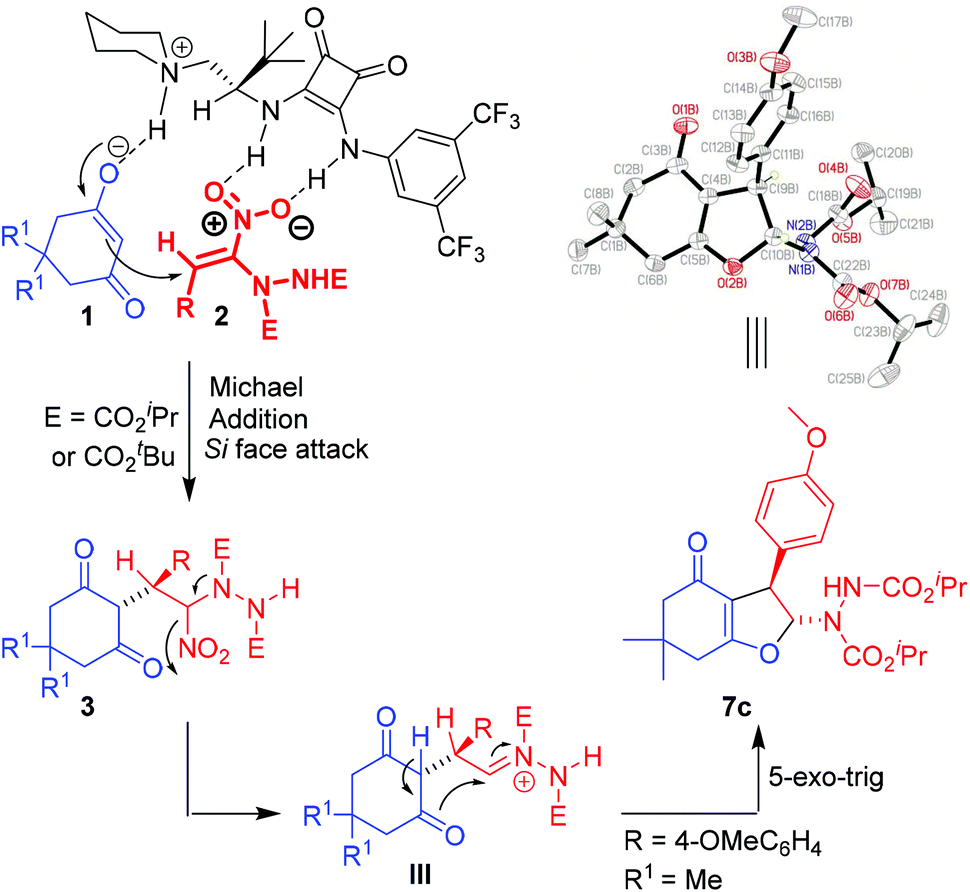
X-ray analysis of 2,3-disubstituted dihydrofuran 7c confirmed the trans stereochemistry between the hydrazinodicarboxylate moiety and the aryl group and the absolute configuration at the two adjacent chiral centers to be (R, R). The absolute configuration of the chiral centers in other 2,3-disubstituted dihydrofurans 7a–l and 8 could be assigned by analogy.40
The proposed mechanism involves deprotonation of the cyclic 1,3-dicarbonyl compound 1k/1l by the piperidine moiety of the catalyst C7 to generate the enolate (Scheme 5). The enolate then takes part in Michael addition to the α-hydrazinated nitroalkene 2 from the Si face. This face selective addition leading to the intermediate Michael adduct is facilitated by H-bonding between the nitro group of 2 and the two N–H groups of the squaramide moiety of C7. Formation of hydrazonium intermediate IIIvia elimination of the nitro group and enolization of the 1,3-dicarbonyl moiety of intermediate III with the assistance of K2CO3 followed by intramolecular 5-exo-trig cyclization result in the formation of enantioenriched 2,3-disubstituted dihydrofurans 7 and 8.41
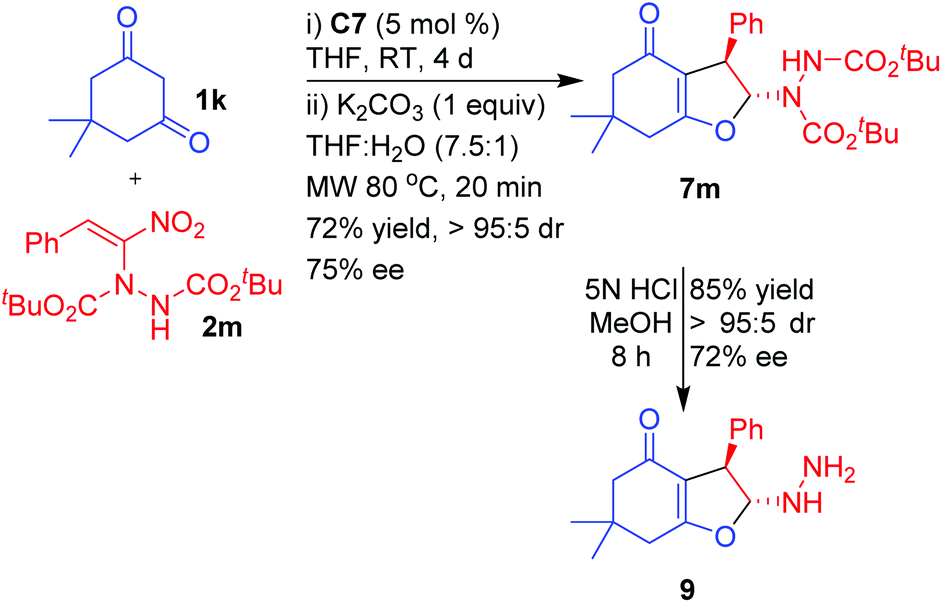
As part of demonstrating further scope and synthetic applications of the products, a di-tertiary-butyl azodicarboxylate (DBAD) adduct of nitroalkene 2m was treated with dimedone 1k under the optimized reaction conditions (Scheme 6). The corresponding dihydrofuran 7m was isolated in good yield (72%), with excellent diastereoselectivity (>95:5) and high enantioselectivity (75% ee). Compound 7m was subjected to hydrolysis under acidic conditions using methanolic HCl by stirring at room temperature for 8 h to afford 2-hydrazinyl 2,3-dihydrofuran 9 in excellent yield (85%) and without any change in diastereoselectivity (>95:5) or appreciable drop in the enantioselectivity (72%). Unlike in the case of hydrazinodicarboxylate adducts 4–8, hydrazinodihydrofuran 9 did not exhibit any appreciable line broadening in 1H and 13C NMR spectra at room temperature, thus confirming that the line broadening observed in the case of 4–8 was primarily due to the carbamate moiety and not due to reversible ring opening of the dihydrofuran moiety. The reversible ring opening in compounds 4–9 can also be ruled out based on the near retention of the stereochemical integrity of the dihydrofuran even after hydrolysis of the carbamate moieties in 7m.42
 | ||
| Scheme 6 | ||
The interrupted Feist–Bénary type reaction described here involves an unusual substitution of the nitro group that facilitates the initial Michael addition thus enabling us to introduce a hydrazinodicarboxylate moiety on the dihydrofuran ring. This reactivity is in contrast to that of α-halonitroalkenes wherein the halogen undergoes elimination affording nitrodihydrofurans.8–14
Experimental section
General experimental details
The melting points recorded are uncorrected. NMR spectra (1H, 1H decoupled 13C, 1H–1H COSY, 1H–1H NOESY, and 1H–1H HSQC) were recorded with TMS as the internal standard. The coupling constants (J values) are given in Hz. High resolution mass spectra were recorded under ESI Q-TOF conditions. Enantioselectivities were determined using a chiral HPLC system equipped with a PDA-detector. Specific rotations were measured for solutions of samples of known concentrations in CHCl3 using a polarimeter equipped with a sodium vapor lamp. X-ray data were collected on a diffractometer equipped with a graphite monochromated Co Kα radiation source. The structure was solved by direct methods shelxs97 and refined by full-matrix least squares against F2 using shelxl97 software. Catalysts C1–C12 were prepared by literature methods.36 MBH-adducts 2a–g are known in the literature and were prepared by the general procedure reported in the literature.32General procedure for the cascade Michael addition and O-alkylation of 1,3-dicarbonyl compounds 1 to α-hydrazinated nitroalkenes 2
To a solution of curcumin 1a–f (0.31 mmol, 1.1 equiv.) or other 1,3-dicarbonyl 1g–l (0.34 mmol, 1.2 equiv.) and α-hydrazinated nitroalkene 2 (0.28 mmol, 1.0 equiv.) in THF (1.5 mL) and H2O (0.2 mL) was added K2CO3 (25 mg, 0.18 mmol, 1.0 equiv.) at room temperature. The resulting reaction mixture was stirred until the reaction was completed (monitored by TLC). After concentration of the reaction mixture in vacuo, the crude residue was purified by silica gel column chromatography by eluting with ethyl acetate/petroleum ether (10–20%) to afford pure 4/5/6.Diisopropyl 1-(4-cinnamoyl-3-phenyl-5-((E)-styryl)tetrahydrofuran-2-yl)hydrazine-1,2-dicarboxylate (4a)
Yellow solid; yield 91 mg (92%); mp 98–100 °C; IR (KBr, cm−1) 3062 (w), 2984 (m), 2932 (w), 1729 (vs), 1622 (s), 1374 (m), 1235 (m), 1106 (s), 974 (s), 739 (vs); 1H NMR (400 MHz, CDCl3) δ 1.20 (d, J = 6.2 Hz, 6H), 1.26 (d, J = 6.2 Hz, 6H), 4.75–4.85 (unresolved d, 1H), 5.00 (septet, J = 6.2 Hz, 2H), 6.30 (br s, 1H), 6.49–6.51 (unresolved d, 1H), 6.68 (d, J = 15.5 Hz, 1H), 7.28–7.32 (unresolved m, 6H), 7.33–7.40 (m, 7H), 7.44 (d, J = 16.4 Hz, 1H), 7.56 (d, J = 15.5 Hz, 1H), 7.61 (d, J = 7.0 Hz, 2H), 8.01 (d, J = 16.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9, 22.1, 51.6, 70.7, 71.8, 95.2, 116.3, 116.8, 125.3, 127.9, 128.1, 128.4, 127.9, 128.9, 129.0, 129.5, 129.8, 130.2, 135.2, 136.0, 139.5, 141.4, 142.0, 154.4, 156.6, 164.1, 186.2; MS (ES + ) m/z (rel intensity) 583 ([M + 3]+, 10), 582 ([M + 2]+, 40), 581 (MH+, 100), 378 (15), 377 (40); HRMS (ES+) calcd for C35H37N2O6 (MH+) 581.2652, found 581.2649.Diisopropyl 1-(4-cinnamoyl-3-(4-methoxyphenyl)-5-((E)-styryl)tetrahydrofuran-2-yl)hydrazine-1,2-dicarboxylate (4b)
Yellow solid; yield 139 mg (94%); mp 104–106 °C; IR (KBr, cm−1) 3058 (w), 2984 (w), 1728 (s), 1622 (m), 1374 (m), 1265 (m), 1180 (m), 1106 (s), 1034 (m), 975 (m), 739 (s); 1H NMR (400 MHz, CDCl3) δ 1.20 (d, J = 6.1 Hz, 6H), 1.27 (d, J = 6.1 Hz, 6H), 3.76 (s, 3H), 4.70–4.80 (unresolved d, 1H), 4.89 (septet, J = 6.1 Hz, 2H), 6.30 (br s, 1H), 6.54–6.56 (unresolved d, 1H), 6.70 (d, J = 15.5 Hz, 1H), 6.81 (d, J = 8.5 Hz, 2H), 7.28–7.39 (m, 10H), 7.43 (d, J = 16.5 Hz, 1H), 7.57 (d, J = 15.5 Hz, 1H), 7.60 (d, J = 8.5 Hz, 2H), 8.00 (d, J = 16.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9, 22.1, 50.7, 55.4, 70.6, 71.7, 95.3, 114.8, 116.4, 116.9, 125.2, 128.1, 128.4, 128.9, 129.0, 129.8, 130.2, 133.4, 135.2, 136.0, 139.4, 141.9, 154.4, 156.6, 159.2, 164.0, 186.2; MS (ES+) m/z (rel intensity) 613 ([M + 3]+, 10), 612 ([M + 2]+, 40), 611 ([M + 1]+, 100), 407 (35); HRMS (ES+) calcd for C36H39N2O7 (MH+) 611.2757, found 611.2744.Diisopropyl 1-(3-(4-chlorophenyl)-4-cinnamoyl-5-((E)-styryl)tetrahydrofuran-2-yl)hydrazine-1,2-dicarboxylate (4c)
Yellow solid; yield 86 mg (98%); mp 99–101 °C; IR (KBr, cm−1) 3056 (w), 2986 (w), 1729 (m), 1623 (w), 1266 (m), 1106 (w), 740 (vs); 1H NMR (400 MHz, CDCl3) δ 1.20 (d, J = 6.2 Hz, 6H), 1.27 (d, J = 6.2 Hz, 6H), 4.75–4.85 (unresolved d, 1H), 5.01 (septet, J = 6.0 Hz, 2H), 6.43 (br s, 1H), 6.54–6.56 (unresolved d, 1H), 6.64 (d, J = 15.4 Hz, 1H), 7.31–7.40 (m, 12H), 7.44 (d, J = 16.3 Hz, 1H), 7.58 (d, J = 15.4 Hz, 1H), 7.60 (d, J = 6.4 Hz, 2H), 7.97 (d, J = 16.3 Hz, 1H); confirmed by 1H–1H COSY and NOESY experiments; 13C NMR (100 MHz, CDCl3) δ 21.8, 22.0, 50.8, 70.7, 71.8, 94.8, 115.8, 116.5, 124.9, 128.0, 128.3, 128.9, 129.0, 129.2, 129.5, 129.9, 130.3, 133.7, 134.9, 135.8, 139.7, 139.9, 142.3, 154.2, 156.6, 164.3, 185.8; confirmed by 1H–13C HSQC experiment; MS (ES+) m/z (rel intensity) 617 ([M + 3]+, 20), 616 ([M + 2]+, 20), 615 ([M + 1]+, 55), 578 (40), 557 (100); HRMS (ES+) calcd for C35H36N2O6Cl (MH+) 615.2262, found 615.2263.Diisopropyl 1-(4-cinnamoyl-3-(thienyl)-5-((E)-styryl)tetrahydrofuran-2-yl)hydrazine-1,2-dicarboxylate (4d)
Yellow solid; yield 79 mg (96%); mp 80–82 °C; IR (KBr, cm−1) 2984 (w), 1728 (vs), 1623 (s), 1375 (m), 1237 (m), 1106 (s), 975 (m), 739 (m); 1H NMR (400 MHz, CDCl3) δ 1.20 (d, J = 6.0 Hz, 6H), 1.30 (d, J = 6.0 Hz, 6H), 4.95–5.15 (m, septet and unresolved d overlapped, 3H), 6.30–6.60 (br s and unresolved d overlapped, 2H), 6.79 (d, J = 16.0 Hz, 1H), 6.99 (dd, J = 4.9, 3.6 Hz, 1H), 7.08–7.12 (unresolved m, 1H), 7.24 (d, J = 4.9 Hz, 1H), 7.29–7.47 (m, 9H), 7.61 (d, J = 6.9 Hz, 2H), 7.62 (d, J = 15.4 Hz, 1H), 7.97 (d, J = 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 22.0 (×2), 46.6, 70.7, 71.8, 94.7, 116.0, 116.6, 124.9, 125.6, 125.9, 127.7, 128.1, 128.4, 128.9, 129.0, 129.8, 130.2, 135.1, 135.8, 139.8, 142.1, 144.8, 154.2, 156.4, 164.0, 185.8; MS (ES+) m/z (rel intensity) 589 ([M + 3]+, 13), 588 ([M + 2]+, 39), 587 (MH+, 100), 383 (23); HRMS (ES+) calcd for C33H35N2O6S (MH+) 587.2216, found 587.2214.Diisopropyl 1-(4-((E)-3-(4-hydroxy-3-methoxyphenyl)acryloyl)-5-(4-hydroxy-3-methoxystyryl)-3-phenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (5a)
Orange solid; yield 191 mg (93%); mp 124–126 °C; IR (KBr, cm−1) 3335 (br m), 2985 (w), 1726 (s), 1588 (vs), 1514 (vs), 1375 (m), 1283 (vs), 975 (m); 1H NMR (400 MHz, CDCl3) δ 1.21 (d, J = 6.1 Hz, 6H), 1.26 (d, J = 6.1 Hz, 6H), 3.75 (s, 3H), 3.86 (s, 3H), 4.77–4.78 (unresolved d, 1H), 4.99 (septet, J = 6.1 Hz, 2H), 6.29 (br s, 2H), 6.35–6.45 (unresolved d, 1H), 6.49 (d, J = 15.8 Hz, 1H), 6.58–6.65 (br s, 2H), 6.82, 6.85 (ABq, J = 8.2 Hz, 2H), 6.91 (d, J = 8.1 Hz, 1H), 7.06 (d, J = 8.1 Hz, 1H), 7.12 (s, 1H), 7.23–7.28 (m, 1H), 7.31–7.42 (m, 5H), 7.48 (d, J = 15.4 Hz, 1H), 7.87 (d, J = 15.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9, 22.0, 51.5, 55.9, 56.1, 70.7, 71.7, 95.0, 109.0, 109.2, 114.3, 114.8, 114.9, 115.3, 123.3, 123.4, 123.6, 127.7 (×2), 127.9, 128.5, 129.3, 139.8, 141.8, 141.9, 146.8, 147.2, 147.8, 148.1, 154.4, 156.7, 164.7, 186.1; MS (ES+) m/z (rel intensity) 675 ([M + 3]+, 14), 674 ([M + 2]+, 43), 673 (MH+, 100), 557 (37); HRMS (ES+) calcd for C37H41N2O10 (MH+) 673.2761, found 673.2755.Diisopropyl 1-(4-((E)-3-(3,4-dimethoxyphenyl)acryloyl)-5-(3,4-dimethoxystyryl)-3-phenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (5b)
Orange solid; yield 199 mg (95%); mp 110–112 °C; IR (KBr, cm−1) 3313 (m), 2982 (m), 2934 (m), 2835 (w), 1726 (vs), 1619 (s), 1590 (vs), 1512 (vs), 1265 (vs), 1139 (m), 1106 (m), 1025 (s), 976 (vs); 1H NMR (400 MHz, CDCl3) δ 1.21 (d, J = 5.9 Hz, 6H), 1.25 (d, J = 5.9 Hz, 6H), 3.77 (s, 3H), 3.84 (s, 3H), 3.89 (s, 3H), 3.91 (s, 3H), 4.70–4.80 (unresolved d, 1H), 4.92–5.00 (m, 2H), 6.25 (br s, 1H), 6.49 (d, J = 15.8 Hz, 1H), 6.57–6.62 (unresolved d, 1H), 6.65 (s, 1H), 6.75 (d, J = 8.2 Hz, 1H), 6.84 (d, J = 8.1 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 7.10 (d, J = 8.1 Hz, 1H), 7.14 (s, 1H), 7.23–7.28 (unresolved m, 1H), 7.31–7.42 (m, 5H), 7.48 (d, J = 15.4 Hz, 1H), 7.88 (d, J = 15.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.8, 21.9, 51.4, 55.7, 55.8, 55.9 (×2), 70.3, 71.5, 94.8, 109.1, 109.2, 110.9, 111.0, 114.5, 115.2, 122.5, 123.1, 123.5, 127.6, 127.8, 128.0, 128.9, 129.2, 139.4, 141.3, 141.8, 149.0, 149.2, 150.6, 151.0, 154.3, 156.4, 164.5, 185.8; MS (ES+) m/z (rel intensity) 703 ([M + 3]+, 12), 702 ([M + 2]+, 45), 701 ([M + 1]+, 100); HRMS (ES+) calcd for C39H45N2O10 (MH+) 701.3074, found 701.3070.Diisopropyl 1-(4-((E)-3-(4-methoxyphenyl)acryloyl)-5-(4-methoxystyryl)-3-phenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (5c)
Dark yellow solid; yield 193 mg (94%); mp 93–95 °C; IR (KBr, cm−1) 3300 (br w), 2982 (m), 2935 (w), 1726 (s), 1619 (s), 1589 (vs), 1511 (m), 1298 (m), 1254 (vs), 1172 (m), 1108 (m), 1033 (m), 975 (m); 1H NMR (400 MHz, CDCl3) δ 1.20 (d, J = 6.1 Hz, 6H), 1.26 (d, J = 6.1 Hz, 6H), 3.78 (s, 3H), 3.82 (s, 3H), 4.72–4.80 (unresolved d, 1H), 4.99 (septet, J = 6.1 Hz, 2H), 6.20 (br s, 1H), 6.40–6.50 (unresolved d, 1H), 6.55 (d, J = 15.6 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H), 7.22–7.28 (m, 2H), 7.32–7.42 (m, 5H), 7.53 (d, J = 15.4 Hz, 1H), 7.52–7.54 (unresolved m, 1H), 7.90 (d, J = 15.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.8, 21.9, 51.4, 55.3, 55.4, 70.4, 71.5, 94.9, 114.2, 114.3, 114.5, 115.4, 123.1, 127.6, 127.8 (×2), 128.7, 129.2, 129.5, 129.9, 138.9, 141.4, 141.6, 154.3, 156.5, 160.9, 161.2, 164.3, 186.0; MS (ES+) m/z (rel intensity) 643 ([M + 3]+, 10), 642 ([M + 2]+, 40), 641 (MH+, 100), 578 (25), 557 (60), 437 (20); HRMS (ES+) calcd for C37H41N2O8(MH+) 641.2863, found 641.2870.Diisopropyl-1-(3-phenyl-4-((E)-3-(pyridin-3-yl)acryloyl)-5-((E)-2-(pyridin-3-yl)vinyl)-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (5d)
Light yellow solid; yield 162 mg (98%); mp 94–96 °C; IR (KBr, cm−1) 3291 (br w), 2981 (w), 2933 (w), 1725 (s), 1588 (s), 1511 (m), 1252 (s), 1172 (m), 1106 (m), 1032 (m), 973 (m); 1HNMR (400 MHz, CDCl3) δ 1.15 (d, J = 6.2 Hz, 6H), d 1.19 (d, J = 6.2 Hz, 6H), 4.70–4.80 (unresolved d, 1H), 4.90–5.00 (unresolved septet, 2H), 6.25 (br s, 1H), 6.45–6.55 (unresolved d, 1H), 6.64 (d, J = 15.5 Hz, 1H), 7.10–7.36 (m, 8H), 7.44 (br d, J = 13.3 Hz, 2H), 7.75–7.90 (m, 2H), 8.40–8.48 (m, 1H), 8.43 (s, 2H), 8.50 (d, J = 15.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9, 22.1, 51.0, 70.0, 71.4, 95.5, 116.7, 118.7, 123.7, 123.9, 126.8, 127.8, 129.4, 130.7, 131.6, 133.5, 134.3, 134.9, 138.1, 141.0, 149.6, 149.8, 150.6, 154.5, 157.2, 163.6, 185.5; MS (ES+) m/z (rel intensity) 585 ([M + 3]+, 6), 584 ([M + 2]+, 38), 583 (MH+, 100), 557 (20), 379 (20), 292 (22), 279 (25), 206 (20), 190 (15); HRMS (ES+) calcd for C33H35N4O6 (MH+) 583.2557, found 583.2550.Diisopropyl 1-(4-((E)-3-(furan-2-yl)acryloyl)-5-((E)-2-(furan-2-yl)vinyl)-3-phenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (5e)
Dark yellow solid; yield 146 mg (92%); mp 90–92 °C; IR (KBr, cm−1) 3281 (br w), 2982 (m), 2931 (w), 1726 (vs), 1617 (vs), 1600 (vs), 1533 (m), 1468 (m), 1236 (s), 1107 (s), 972 (s), 746 (m); 1H NMR (400 MHz, CDCl3) δ 1.18 (d, J = 6.0 Hz, 6H), 1.23 (d, J = 6.0 Hz, 6H), 4.70–4.80 (unresolved d, 1H), 4.97 (sept, J = 6.0 Hz, 2H), 6.20 (br s, 1H), 6.35 (d, J = 1.6 Hz, 1H), 6.42 (dd, J = 3.1, 1.6 Hz, 1H), 6.47 (d, J = 3.1 Hz, 1H), 6.54 (d, J = 2.0 Hz, 1H), 6.65 (unresolved d, 1H), 7.18 (d, J = 15.8 Hz, 1H), 7.26–7.37 (m, 8H), 7.45–7.47 (unresolved, 1H), 7.78 (d, J = 15.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.8, 21.9, 51.4, 70.5, 71.6, 94.9, 112.4 (×2), 113.4, 114.6, 115.3, 116.4, 122.7, 125.8, 127.6, 127.8, 128.2, 129.2 (×2), 141.3, 144.5, 151.8, 152.3, 154.3, 156.4, 163.4, 185.4; MS (ES+) m/z (rel intensity) 583 (MNa+, 50), 561 (MH+, 100), 357 (55); HRMS (ES+) calcd for C31H33N2O8 (MH+) 561.2237, found 561.2235.Diisopropyl 1-(4-acetyl-5-methyl-3-phenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (6a)
Yellow solid; yield 98 mg (87%); IR (neat, cm−1) 3281 (m), 2982 (m), 2942 (w), 1727 (vs), 1674 (m), 1600 (s), 1376 (s), 1294 (m), 1221 (s), 1182 (m), 1107 (s), 930 (s); 1H NMR (500 MHz, CDCl3) δ 1.19 (d, J = 6.0 Hz, 6H), 1.23–1.30 (unresolved d, 6H), 1.85 (s, 3H), 2.33 (s, 3H), 4.40–4.45 (unresolved d, 1H), 4.85–5.14 (m, 2H), 6.15 (br s, 1H), 6.53–6.82 (unresolved d, 1H), 7.13–7.25 (m, 3H), 7.27–7.34 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 14.8, 21.9, 22.0, 29.7, 51.3, 70.5, 71.6, 95.4, 115.0, 127.6 (×2), 129.1, 141.3, 154.2, 156.5, 167.7, 195.3; MS (ES+) m/z (rel intensity) 427 ([MNa]+, 100), 414 (9), 385 (8); HRMS (ES+) calcd for C21H28N2NaO6 (MNa+) 427.1840, found 427.1836.Diisopropyl 1-(-4-benzoyl-3,5-diphenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (6b)
Colorless solid; yield 135 mg (91%); mp 73–75 °C; IR (film, cm−1) 3293 (br m), 2982 (s), 1727 (vs), 1616 (s), 1596 (s), 1574 (m), 1492 (m), 1373 (s), 1295 (s), 1237 (s), 1105 (s), 697 (m); 1H NMR (500 MHz, CDCl3) δ 1.26 (d, J = 6.0 Hz, 6H), 1.30 (d, J = 6.0 Hz, 6H), 4.80–5.20 (br m, 3H), 6.45 (br s, 1H), 6.60–6.70 (unresolved d, 1H), 7.02–7.14 (m, 4H), 7.16–7.25 (m, 3H), 7.27–7.33 (m, 2H), 7.33–7.42 (m, 4H), 7.44–7.52 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 21.9, 22.1, 54.2, 70.5, 71.7, 95.1, 115.5, 127.4, 127.9 (×2), 128.2, 129.0, 129.1, 129.4, 129.5, 130.5, 131.7, 138.8, 140.7, 154.4, 156.4, 162.9, 192.6; MS (ES+) m/z (rel intensity) 567 (MK+, 50), 552 ([MNa + 1]+, 35), 551 (MNa+, 100), 529 (35); HRMS (ES+) calcd for C31H32N2O6Na (MNa+) 551.2153, found 551.2156.Diisopropyl 1-(4-(ethoxycarbonyl)-5-methyl-3-phenyl-2,3-dihydrofuran-2-yl)hydrazine-1,2-dicarboxylate (6c)
Colorless sticky oil; yield 98 mg (80%); IR (neat, cm−1) 3292 (br m), 2982 (s), 2938 (m), 1727 (vs), 1700 (vs), 1651 (s), 1379 (s), 1316 (m), 1297 (m), 1239 (m), 1214 (m), 1106 (s), 960 (m), 933 (m), 760 (m), 701; 1H NMR (500 MHz, CDCl3) δ 1.00–1.04 (unresolved t, 3H), 1.22 (d, J = 6.2 Hz, 6H), 1.28 (d, J = 6.2 Hz, 6H), 2.32 (s, 3H), 3.91–4.06 (m, 2H), 4.35–4.40 (unresolved d, 1H), 4.89–5.13 (m, 2H), 6.35 (br s, 1H), 6.40–6.50 (unresolved d, 1H), 7.11–7.25 (m, 3H), 7.26–7.34 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 14.2 (×2), 21.9, 22.0, 50.7, 59.6, 70.5, 71.6, 96.3, 106.6, 127.2, 127.7, 128.7, 141.8, 154.4, 156.3, 165.4, 167.4; MS (ES+) m/z (rel intensity) 473 (MK+, 10), 458 ([MNa + 1]+, 21), 457 (MNa+, 100), 444 (8), 431 (18); HRMS (ES+) calcd for C22H30N2O7Na (MNa+) 457.1945, found 451.1954.Diisopropyl 1-(4-oxo-3-phenyl-3,4-dihydro-2H-furo[3,2-c]chromen-2-yl)hydrazine-1,2-dicarboxylate (6d)
Colorless solid; yield 101 mg (77%); mp 83–85 °C; IR (film, cm−1) 3280 (w), 2980 (w), 1726 (vs), 1645 (m), 1386 (m), 1292 (m), 1270 (m), 1240 (m), 1103 (m), 1036 (m); 1H NMR (500 MHz, CDCl3) δ 1.24 (d, J = 6.0 Hz, 6H), 1.27 (d, J = 6.0 Hz, 6H), 4.74–4.80 (unresolved d, 1H), 5.00 (septet, J = 6.0 Hz, 2H), 6.45 (br s, 1H), 6.75–6.85 (unresolved d, 1H), 7.26–7.37 (m, 6H), 7.39 (d, J = 8.4 Hz, 1H), 7.59 (t, J = 8.4 Hz, 1H), 7.71 (d, 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9 (×2), 22.0, 22.1, 48.3, 71.0, 72.3, 99.7, 104.7, 112.2, 117.3, 123.2, 124.2, 127.7, 128.0, 129.3, 133.0, 138.7, 154.2, 155.5, 156.3, 159.5, 165.9; MS (ES+) m/z (rel intensity) 505 (MK+, 60), 490 ([MNa + 1]+, 29), 489 (MNa+, 100), 468 (15), 467 (47), 440 (11); HRMS (ES+) calcd for C25H26N2O7Na (MNa+) 489.1632, found 489.1631.General procedure for the enantio- and diastereoselective cascade Michael addition-O-alkylation of 1,3-dicarbonyls 1k–l to α-hydrazinated nitroalkenes 2
A solution of 1,3-dicarbonyl 1k/1l (0.5 mmol, 1.0 equiv.), α-hydrazinated nitroalkenes 2 (0.75 mmol, 1.5 equiv.), and catalyst C7 (12.3 mg, 0.025 mmol, 5 mol%) in THF (1 mL) was stirred at room temperature until 1,3-dicarbonyl 1k/1l was completely consumed (4 d). The solvent was evaporated under reduced pressure and the excess α-hydrazinated nitroalkene 2 was removed by flash column chromatography using EtOAc–petroleum ether (5–50%, gradient elution) as the eluent to obtain a mixture of 7 or 8 and the Michael adduct. To this mixture in THF (1.5 mL) and H2O (0.2 mL) was added K2CO3 (1 equiv., 69.1 mg) and the reaction mixture was heated at 80 °C under microwave for 20 min. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography by eluting with EtOAc–petroleum ether (10–50%).Diisopropyl 1-((2R,3R)-6,6-dimethyl-4-oxo-3-phenyl-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7a)
White solid; yield 160 mg, 72%; mp 112–114 °C; IR (film, cm−1) 3412 (br), 3295 (br), 2982 (w), 1727 (s), 1632 (s), 1455 (w), 1395 (m), 1287 (m), 1215 (m), 1144 (w), 1107 (m), 1041 (w), 919 (m), 851 (w), 738 (m); 1H NMR (400 MHz, CDCl3) δ 1.12 (s, 3H), 1.17 (s, 3H), 1.25 (d, J = 6.1 Hz, 6H), 1.26 (d, J = 6.1 Hz, 6H), 2.20, 2.30 (ABq, J = 5.6 Hz, 2H), 2.35, 2.45 (ABq, J = 11.0 Hz, 2H), 4.40–4.50 (br unresolved, 1H), 4.94–5.00 (m, 2H), 6.17–6.45 (br unresolved, 2H), 7.21–7.24 (m, 3H), 7.28–7.32 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 22.0 (×2), 28.9 (×2), 34.3, 37.7, 47.4, 51.2, 70.5, 71.9, 98.6, 114.5, 127.3, 127.4, 128.9, 140.3, 154.2, 156.2, 175.0, 193.8; MS (ES+) m/z (rel intensity) 447 ([M + 3]+, 6), 446 ([M + 2]+, 28), 445 ([M + 1]+, 100), 241 (15); HRMS (ES+) calcd for C24H33N2O6 (MH+) 445.2339, found 445.2333; [α]25D −43.67° (c 1.0, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 11.4 min, tR (minor) = 13.7 min; 73% ee.Diisopropyl 1-((2R,3R)-6,6-dimethyl-4-oxo-3-p-tolyl-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7b)
White solid; yield 149 mg, 65%; mp 59–61 °C; IR (film, cm−1) 3284 (br m), 2979 (m), 1730 (vs), 1633 (s), 1467 (w), 1390 (m), 1284 (m), 1238 (m), 1219 (m), 1145 (w), 1109 (m), 1042 (w), 920 (w), 765 (m), 746 (m); 1H NMR (500 MHz, CDCl3) δ 1.11 (s, 3H), 1.16 (s, 3H), 1.25 (d, J = 6.2 Hz, 6H), 1.27 (d, J = 6.2 Hz, 6H), 2.20, 2.28 (ABq, J = 8.8 Hz, 2H), 2.35, 2.45 (ABq, J = 17.6 Hz, 2H), 2.30 (s, 3H), 4.35–4.45 (br unresolved, 1H), 4.93–5.09 (m, 2H), 6.11–6.55 (br unresolved, 2H), 7.05–7.15 (br unresolved, 4H); 13C NMR (125 MHz, CDCl3) δ 21.2, 22.0 (×2), 28.9, 29.1, 34.3, 37.8, 46.9, 51.3, 70.6, 71.9, 98.5, 114.8, 127.3, 129.7, 136.9, 137.3, 154.3, 156.2, 174.8, 193.8; MS (ES+) m/z (rel intensity) 461 ([M + 3]+, 4), 460 ([M + 2]+, 22), 459 ([M + 1]+, 100), 452 (3); HRMS (ESI) calcd for C25H35N2O6 (MH+) 459.2490, found 459.2491; [α]25D −54.05° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 10.8 min, tR (minor) = 13.6 min; 71% ee.Diisopropyl 1-((2R,3R)-3-(4-methoxyphenyl)-6,6-dimethyl-4-oxo-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7c)
White solid; yield 130 mg, 55%; mp 143–145 °C; IR (film, cm−1) 3286 (w), 2981 (m), 1729 (vs), 1630 (vs), 1513 (m), 1467 (w), 1390 (s), 1287 (m), 1248 (s), 1212 (m), 1179 (w), 1145 (w), 1108 (m), 1036 (m), 918 (m), 857 (w); 1H NMR (400 MHz, CDCl3) δ 1.11 (s, 3H), 1.16 (s, 3H), 1.24 (d, J = 5.7 Hz, 6H), 1.26 (d, J = 5.7 Hz, 6H), 2.20–2.30 (unresolved m, 2H), 2.35, 2.45 (ABq, J = 11.8 Hz, 2H), 3.76 (s, 3H), 4.35–4.45 (br unresolved, 1H), 4.90–5.05 (m, 2H), 6.10–6.60 (br unresolved, 2H), 6.83 (d, J = 7.6 Hz, 2H), 7.05–7.15 (br unresolved, 2H); 13C NMR (100 MHz, CDCl3) δ 22.0 (×2), 28.9 (×2), 34.3, 37.8, 46.6, 51.3, 55.4, 70.6, 71.9, 98.6, 114.4, 114.7, 128.5, 132.4, 154.3, 156.2, 158.9, 175.2, 193.8; MS (ES+) m/z (rel intensity) 477 ([M + 3]+, 6), 476 ([M + 2]+, 30), 475 ([M + 1]+, 100), 271 (60); HRMS (ES+) calcd for C25H35N2O7 (MH+) 475.2444, found 475.2458; [α]25D −117.81° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 14.7 min, tR (minor) = 21.1 min; 75% ee. Selected X-ray data (CCDC 1947973†): C50H67N3O15, M = 950.06, triclinic, space group P1, a = 10.4124(3) Å, b = 11.3067(3) Å, c = 11.3573(3) Å, α = 91.46°, β = 98.011°, γ = 104.143°, V = 1(6) Å3, DX = 1.231 Mg m−3, Z = 1, F(000) = 508.0, λ = 1.54184 Å, μ = 0.75 cm−1, total/unique = 43857/7928 [R(int) = 0.113], T = 293(2) K, θ range = 3.9 to 65.0°, final R[I > 2σ(I)]: R1 = 0.063, wR2 = 0.163, R(all data): 0.0721, wR2 = 0.179. Flack parameter = 0.0(2).
Diisopropyl 1-((2R,3R)-3-(benzo[d][1,3]dioxol-5-yl)-6,6-dimethyl-4-oxo-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7d)
White solid; yield 146 mg, 60%; mp 158–160 °C; IR (film, cm−1) 3199 (w), 2957 (w), 1722 (vs), 1602 (vs), 1386 (w), 1369 (m), 1282 (m), 1246 (m), 1125 (w), 1104 (w), 1023 (w), 915 (vw), 888 (vw), 748 (w); 1H NMR (400 MHz, CDCl3) δ 1.09 (s, 3H), 1.15 (s, 3H), 1.24–1.27 (br unresolved, 12H), 2.22, 2.30 (ABq, J = 7.2 Hz, 2H), 2.38, 2.46 (ABq, J = 14.8 Hz, 2H), 4.30–4.40 (br unresolved, 1H), 4.95–4.96 (br m, 2H), 5.90 (d, J = 4.3 Hz, 2H), 6.20–6.45 (br m, 2H), 6.60–6.75 (br m, 3H); 13C NMR (100 MHz, CDCl3) δ 22.0 (×2), 28.7, 28.9, 34.2, 37.7, 47.1, 51.2, 70.6, 71.9, 98.6, 101.1, 107.7, 108.6, 114.5, 120.8, 134.1, 146.9, 148.1, 154.2, 156.2, 175.1, 193.8; MS (ES+) m/z (rel intensity) 490 ([M + 2]+, 10), 489 ([M + 1]+, 34), 271 (100); HRMS (ES+) calcd for C25H33N2O8 (MH+) 489.2237, found 489.2237; [α]25D −120.46° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 14.3 min, tR (minor) = 19.6 min; 76% ee.Diisopropyl 1-((2R,3R)-3-(4-chlorophenyl)-6,6-dimethyl-4-oxo-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7e)
White solid; yield 160 mg, 67%; mp 106–108 °C; IR (film, cm−1) 3399 (br, s), 2978 (m), 2923 (m), 1731 (s), 1632 (s), 1492 (w), 1389 (m), 1265 (m), 1108 (m), 741 (s); 1H NMR (400 MHz, CDCl3) δ 1.12 (s, 3H), 1.17 (s, 3H), 1.25 (d, J = 6.1 Hz, 6H), 1.28 (d, J = 6.1 Hz, 6H), 2.20, 2.30 (ABq, J = 4.0 Hz, 2H), 2.35, 2.50 (ABq, J = 10.8 Hz, 2H), 4.38–4.48 (br unresolved, 1H), 4.96–4.99 (m, 2H), 6.12–6.41 (br unresolved, 2H), 7.16 (d, J = 6.0 Hz, 2H), 7.28 (br unresolved, 2H); 13C NMR (100 MHz, CDCl3) δ 22.0, 22.1, 28.9, 34.4, 37.8, 46.9, 51.3, 70.9, 72.1, 98.6, 114.3, 128.9, 129.2, 133.3, 138.8, 154.2, 156.3, 175.7, 193.7; MS (ES+) m/z (rel intensity) 482 ([M + 4]+, 10), 481 ([M + 3]+, 40), 480 ([M + 2]+, 29), 479 ([M + 1]+, 100); HRMS (ES+) calcd for C24H32N2O6Cl (MH+) 479.1949, found 479.1951; [α]25D −96.16° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 11.1 min, tR (minor) = 15.6 min; 69% ee.Diisopropyl 1-((2R,3S)-3-(2-bromophenyl)-6,6-dimethyl-4-oxo-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7f)
White solid; yield 180 mg, 69%; mp 78–80 °C; IR (film, cm−1) 3283 (m), 2982 (s), 1735 (vs), 1637 (vs), 1467 (m), 1440 (w), 1396 (s), 1288 (m), 1236 (m), 1219 (m), 1184 (w), 1145 (w), 1108 (s), 1040 (m), 938 (w), 919 (m), 848 (w), 757 (s); 1H NMR (300 MHz, CDCl3 + 55 °C) δ 1.12 (s, 3H), 1.20 (s, 3H), 1.21–1.30 (m, 12H), 2.21, 2.30 (ABq, J = 16.0 Hz, 2H), 2.39, 2.49 (ABq, J = 18.0 Hz, 2H), 4.80–4.85 (m, 1H), 4.87–5.06 (m, 2H), 6.25–6.35 (br unresolved, 1H), 6.40–6.50 (br unresolved, 1H), 6.99 (dd, J = 7.7, 1.6 Hz, 1H), 7.09 (td, J = 7.7, 1.6 Hz, 1H), 7.22 (td, J = 7.7, 1.3 Hz, 1H), 7.55 (dd, J = 7.7, 1.3 Hz, 1H); 13C NMR (125 MHz, CDCl3, +55 °C) δ 22.0 (×2), 22.1 (×2), 28.6, 29.3, 34.2, 38.1, 47.9, 51.5, 70.6, 71.9, 97.9, 113.6, 124.3, 128.0, 129.1, 129.2, 133.6, 139.3, 154.1, 156.1, 176.5, 193.0; MS (ES+) m/z (rel intensity) 564 ([MK + 3]+, 21), 563 ([MK + 2]+, 100), 562 ([MK + 1]+, 20), 561 ([MK]+, 91), 548 (11), 547 (51), 546 (12), 545 (55); HRMS (ES+) calcd for C24H31KN2O6 (MK+) 561.0997, found 561.0996; [α]25D −34.476° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 12.0 min, tR (minor) = 15.6 min; 75% ee.Diisopropyl 1-((2R,3R)-6,6-dimethyl-3-(4-nitrophenyl)-4-oxo-2,3,4,5,6,7-hexahydro-benzofuran-2-yl)hydrazine-1,2-dicarboxylate (7g)
White solid; yield 147 mg, 60%; mp 88–90 °C; IR (film, cm−1) 3295 (br w), 2983 (m), 2934 (m), 1729 (vs), 1632 (vs), 1522 (m), 1468 (w), 1391 (m), 1348 (m), 1288 (m), 1218 (m), 1246 (w), 1107 (s), 1042 (w), 918 (m), 739 (m); 1H NMR (400 MHz, CDCl3) δ 1.13 (s, 3H), 1.18 (s, 3H), 1.24–1.30 (m, 12H), 2.22, 2.23 (ABq, J = 3.3 Hz, 2H), 2.40, 2.50 (ABq, J = 10.6 Hz, 2H), 4.50–4.60 (br unresolved, 1H), 4.95–5.00 (m, 2H), 6.20–6.55 (br unresolved, 2H), 7.42 (br d, J = 8.4 Hz, 2H), 8.17 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.8 (×2), 21.9, 22.0, 28.8, 34.3, 37.6, 47.3, 51.0, 70.8, 71.9, 97.4, 113.5, 124.1, 128.6, 147.2, 147.7, 154.0, 156.4, 176.1, 193.7; MS (ES+) m/z (rel intensity) 492 ([M + 3]+, 26), 491 ([M + 2]+, 30), 490 ([M + 1]+, 100), 404 (6), 271 (8); HRMS (ES+) calcd for C24H32N3O8 (MH+) 490.2189, found 490.2184; [α]25D −120.38° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 1.0 mL min−1, λ = 250 nm), tR (major) = 9.6 min, tR (minor) = 15.4 min; 73% ee.Diisopropyl 1-((2R,3R)-6,6-dimethyl-3-(naphthalen-2-yl)-4-oxo-2,3,4,5,6,7-hexahydro-benzofuran-2-yl)hydrazine-1,2-dicarboxylate (7h)
Light yellow solid; yield 175 mg 71%; mp 83–85 °C; IR (film, cm−1) 3284 (w), 2982 (m), 1729 (vs), 1634 (vs), 1508 (w), 1467 (w), 1397 (s), 1375 (m), 1287 (m), 1245 (m), 1217 (m), 1182 (w), 1144 (w), 1107 (s), 1042 (w), 920 (w), 752 (s); 1H NMR (300 MHz, CDCl3, +55 °C) δ 1.14 (s, 3H), 1.20 (s, 3H), 1.20–1.27 (m, 6H), 1.30 (d, J = 6.2 Hz, 6H), 2.22, 2.30 (ABq, J = 16.5 Hz, 2H), 2.40, 2.50 (ABq, J = 18.0 Hz, 2H), 4.63 (d, J = 3.5 Hz, 1H), 4.98 (sept, J = 6.2 Hz, 1H), 5.00–5.11 (m, overlaps with sept, 1H), 6.42–6.55 (br, overlaps with d, 1H), 6.49 (d, J = 3.5 Hz, 1H), 7.34 (dd, J = 8.4, 1.5 Hz, 1H), 7.37–7.47 (m, 2H), 7.66 (br s, 1H), 7.74–7.85 (m, 3H); 13C NMR (75 MHz, CDCl3, +55 °C) δ 21.8, 21.9, 22.0 (×2), 28.6, 28.8, 34.2, 37.8, 47.9, 51.3, 70.6, 71.8, 98.6, 114.7, 125.4, 125.7, 126.1, 126.3, 127.6, 128.0, 128.7, 132.9, 133.7, 137.8, 154.2, 156.1, 175.2, 193.3; MS (ES+) m/z (rel intensity) 518 ([MNa + 1]+, 26), 517 ([MNa]+, 100); HRMS (ESI) calcd for C28H34N2NaO6 (MNa+) 517.2309, found 517.2306; [α]25D −150.58° (c 0.75, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 12.6 min, tR (minor) = 18.4 min; 79% ee.Diisopropyl 1-((2R,3R)-6,6-dimethyl-3-(naphthalen-1-yl)-4-oxo-2,3,4,5,6,7-hexahydro-benzofuran-2-yl)hydrazine-1,2-dicarboxylate (7i)
White solid; yield 168 mg, 68%; mp 205–207 °C; IR (film, cm−1) 3238 (w), 2980 (m), 1729 (vs), 1633 (vs), 1467 (w), 1398 (m), 1251 (m), 1210 (m), 1182 (w), 1146 (w), 1106 (m), 1043 (w), 920 (w), 764 (m); 1H NMR (300 MHz, CDCl3, +55 °C) δ 1.08–1.42 (br m, 12H), 1.17 (s, 3H), 1.25 (s, 3H), 2.30, 2.42 (ABq, J = 16.5 Hz, 2H), 2.45, 2.53 (ABq, J = 18.0 Hz, 2H), 4.96 (sept, J = 6.2 Hz, 1H), 5.05–5.15 (br unresolved, 1H), 5.15–5.25 (br unresolved, 1H), 6.30–6.40 (br unresolved, 1H), 6.45–6.65 (br unresolved, 1H), 7.10 (d, J = 7.6, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.43–7.52 (m, 2H), 7.75 (d, J = 7.6 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 8.25–8.40 (br unresolved, 1H); 13C NMR (125 MHz, CDCl3, +55 °C) δ 21.9, 22.1, 22.2, 28.5, 29.3, 34.3, 38.0, 44.7, 51.5, 70.7, 72.0, 98.1, 114.1, 124.2, 125.4, 125.8, 126.5, 128.2, 128.9, 132.1, 134.6, 136.0, 154.1, 156.4, 175.9, 193.6; MS (ES+) m/z (rel intensity) 518 ([MNa + 1]+, 28), 517 ([MNa]+, 100); HRMS (ES+) calcd for C28H34N2NaO6 (MNa+) 517.2309, found 517.2306; [α]25D 4.44° (c 0.5, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 90/10, flow rate = 0.5 mL min−1, λ = 268 nm), tR (major) = 27.2 min, tR (minor) = 16.0 min; 76% ee.Diisopropyl 1-((2R,3R)-3-(furan-2-yl)-6,6-dimethyl-4-oxo-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7j)
Light yellow solid; yield 117 mg, 54%; mp 61–63 °C; IR (film, cm−1) 3305 (br s), 2981 (s), 1738 (vs), 1635 (s), 1386 (w), 1241 (w), 1145 (vw), 1105 (w), 1042 (w), 916 (m), 764 (m), 737 (m); 1H NMR (400 MHz, CDCl3) δ 1.05 (s, 3H), 1.09 (s, 3H), 1.20–1.35 (br m, 12H), 2.20, 2.30 (ABq, J = 10.0 Hz, 2H), 2.35, 2.45 (ABq, J = 18.0 Hz, 2H), 4.48–4.55 (br unresolved, 1H), 4.90–5.05 (m, 2H), 6.10–6.20 (br unresolved, 1H), 6.25–6.30 (br unresolved, 1H), 6.40–6.70 (br unresolved, 2H), 7.30–7.35 (br unresolved, 1H); 13C NMR (100 MHz, CDCl3) δ 22.0 (×2), 28.6 (×2), 34.3, 37.7, 41.5, 51.2, 70.5, 71.9, 95.2, 107.1, 110.6, 112.3, 142.2, 152.0, 154.1, 156.1, 175.3, 193.6; MS (ES+) m/z (rel intensity) 437 ([M + 3]+, 4), 436 ([M + 2]+, 26), 435 ([M + 1]+, 100), 231 (60); HRMS (ES+) calcd for C22H31N2O7 (MH+) 435.2131, found 435.2145; [α]25D −101.75° (c 0.5, CHCl3); HPLC: Chiralpack IA (petroleum ether/i-PrOH = 70/30, flow rate = 1.0 mL min−1, λ = 250 nm), tR (major) = 6.1 min, tR (minor) = 8.7 min; 71% ee.Diisopropyl 1-((2R,3R)-6,6-dimethyl-4-oxo-3-(thiophen-2-yl)-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7k)
White solid; yield 117 mg, 52%; mp 73–75 °C; IR (film, cm−1) 3289 (m), 2981 (s), 2879 (m), 1728 (vs), 1633 (vs), 1516 (w), 1469 (w), 1393 (s), 1286 (m), 1243 (m), 1218 (m), 1181 (w), 1144 (w), 1106 (s), 1038 (s), 917 (s), 855 (w), 825 (w), 781 (m), 736 (m), 700 (m); 1H NMR (400 MHz, CDCl3) δ 1.10 (s, 3H), 1.16 (s, 3H), 1.25–1.35 (m, 12H), 2.25, 2.30 (ABq, J = 8.4 Hz, 2H), 2.38, 2.45 (ABq, J = 6.4 Hz, 2H), 4.68–4.78 (br unresolved, 1H), 4.95–5.05 (br unresolved, 2H), 6.30–6.60 (br unresolved, 2H), 6.85–7.00 (br unresolved, 2H), 7.15–7.20 (br unresolved, 1H); 13C NMR (100 MHz, CDCl3) δ 22.0 (×2), 28.5 (×2), 34.2, 37.9, 43.0, 51.3, 70.7, 72.1, 98.3, 114.5, 124.6, 125.3, 127.3, 143.8, 154.2, 156.2, 175.3, 193.7; MS (ES+) m/z (rel intensity) 453 ([M + 3]+, 9), 452 ([M + 2]+, 26), 451 ([M + 1]+, 100), 247 (27); HRMS (ES+) calcd for C22H31N2O6S (MH+) 451.1903, found 451.1922; [α]25D −81.31° (c 0.5, CHCl3); HPLC: Chiralpack IA (petroleum ether/i-PrOH = 70/30, flow rate = 1.0 mL min−1, λ = 250 nm), tR (major) = 6.0 min, tR (minor) = 15.2 min; 74% ee.(E)-Diisopropyl 1-((2R,3R)-6,6-dimethyl-4-oxo-3-styryl-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7l)
Light yellow sticky liquid; yield 141 mg, 60%; IR (neat, cm−1) 3297 (m), 2983 (s), 2939 (m), 2881 (w), 1727 (vs), 1631 (s), 1497 (w), 1468 (m), 1389 (m), 1376 (m), 1285 (m), 1240 (s), 1182 (w), 1145 (w), 1108 (s), 1042 (m), 963 (w), 918 (m), 826 (w), 759 (m); 1H NMR (500 MHz, CDCl3) δ 1.11 (s, 3H), 1.16 (s, 3H), 1.23–136 (m, 12H), 2.23, 2.31 (ABq, J = 16.3 Hz, 2H), 2.34, 2.41 (ABq, J = 17.7 Hz, 2H), 4.05–4.12 (br unresolved, 1H), 4.92–5.09 (m, 2H), 6.22 (dd, J = 15.5, 7.0 Hz, 1H), 6.30–6.60 (br m, 2H), 7.22 (t, J = 7.2 Hz, 1H), 7.29 (t, J = 7.2 Hz, 2H), 7.36 (d, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 22.0, 22.1 (×3), 28.5, 29.0, 34.3, 37.8, 44.9, 51.4, 70.2, 70.5, 71.9, 96.4, 113.8, 126.6, 127.3, 127.6, 128.5, 132.3, 136.9, 154.3, 156.1, 175.1, 194.0; MS (ES+) m/z (rel intensity) 494 ([MNa + 1]+, 20), 493 ([MNa]+, 92), 313 (100); HRMS (ES+) calcd for C26H34N2NaO6 (MNa+) 493.2309, found 493.2306; [α]25D −99.39° (c 0.5, CHCl3); HPLC: Chiralpack IC (petroleum ether/i-PrOH = 97/3, flow rate = 0.5 mL min−1, λ = 244 nm), tR (major) = 12.4 min, tR (minor) = 16.7 min; 81% ee.(E)-Diisopropyl 1-((2R,3R)-4-oxo-3-styryl-2,3,4,5,6,7-hexahydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (8)
Light yellow sticky liquid; yield 135 mg, 61%; IR (neat, cm−1) 3284 (w), 2981 (m), 2939 (w), 1728 (vs), 1627 (s), 1467 (w), 1453 (w), 1391 (m), 1375 (m), 1297 (m), 1243 (m), 1227 (m), 1181 (m), 1107 (m), 1042 (w), 963 (w), 916 (w), 758 (w); 1H NMR (500 MHz, CDCl3) δ 1.18–1.35 (br m, 12H), 1.98–2.08 (br m, 2H), 2.25–2.41 (m, 2H), 2.46–2.52 (br unresolved, 2H), 4.02–4.10 (br unresolved, 1H), 4.90–5.05 (m, 2H), 6.18 (dd, J = 15.8, 8.5 Hz, 1H), 6.42–6.61 (br m, 2H), 7.21 (t, J = 7.5 Hz, 1H), 7.28 (t, J = 7.5 Hz, 2H), 7.35 (d, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 21.6, 22.0, 22.0, 23.9, 36.9, 44.5, 70.4, 71.8, 96.0, 115.1, 126.6, 127.3, 127.6, 128.5, 132.2, 136.8, 154.3, 156.0, 175.8, 194.7; MS (ES+) m/z (rel intensity) 482 ([MK + 1]+, 25), 481 ([MK]+, 100), 479 (17); HRMS (ES+) calcd for C24H30N2KO6 (MK+) 481.1735, found 481.1734; [α]25D −52.54° (c 1.0, CHCl3); HPLC: Chiralcel OD-H (petroleum ether/i-PrOH = 95/5, flow rate = 0.5 mL min−1, λ = 250 nm), tR (major) = 33.0 min, tR (minor) = 45.3 min; 81% ee.Di-tert-butyl 1-((2R,3R)-6,6-dimethyl-4-oxo-3-phenyl-2,3,4,5,6,7-hexa-hydrobenzofuran-2-yl)hydrazine-1,2-dicarboxylate (7m)
A solution of dimedone (71 mg, 0.5 mmol, 1.0 equiv.), α-hydrazinated nitroalkene 2m (284 mg, 0.75 mmol, 1.5 equiv.) and catalyst C7 (12.3 mg, 0.025 mmol, 5 mol%) in THF (1 mL) was stirred at room temperature until dimedone was completely consumed (4 d). The solvent was evaporated under reduced pressure and the excess α-hydrazinated nitroalkene 2m was removed by flash column chromatography using EtOAc–petroleum ether (5–50%, gradient elution) as the eluent to obtain a mixture of 7m and the Michael adduct. To this mixture in THF (1.5 mL) and H2O (0.2 mL) was added K2CO3 (69.1 mg, 1 equiv.) and the reaction mixture was heated at 80 °C under microwave for 20 min. The solvent was evaporated under reduced pressure and the residue was purified by silica gel column chromatography by eluting with EtOAc–petroleum ether (30–50%) to afford pure 7m. White solid; yield 170 mg, 72%; mp 163–165 °C; IR (film, cm−1) 3306 (br s), 2978 (vs), 1732 (vs), 1633 (vs), 1493 (s), 1445 (s), 1391 (s), 1368 (s), 1308 (m), 1144 (s), 1049 (s), 915 (s), 847 (s), 737 (vs), 700 (s); 1H NMR (400 MHz, CDCl3, 323 K) δ 1.11 (s, 3H), 1.15 (s, 3H), 1.45 (s, 9H), 1.48 (s, 9H), 2.20, 2.24 (ABq, J = 16.1 Hz, 2H), 2.38, 2.43 (ABq, J = 17.6 Hz, 2H), 4.42 (s, 1H), 6.38 (s, 1H), 7.18–7.21 (m, 2H), 7.25–7.30 (m, 3H); 13C NMR (100 MHz, CDCl3, 323 K) δ 28.1, 28.3, 28.8, 28.9, 34.1, 37.9, 47.7, 51.4, 81.9, 83.3, 98.7, 114.7, 127.2, 127.4, 128.8, 140.7, 153.4, 155.4, 175.2, 193.4; HRMS (ES+) calcd for C26H37N2O6 (MH+) 473.2646, found 473.2641; [α]25D −51.72° (c 0.5, CHCl3); HPLC: Chiralcel AD-H (petroleum ether/i-PrOH = 90/10, flow rate = 1.0 mL min−1, λ = 268 nm), tR (major) = 23.7 min, tR (minor) = 25.5 min; 75% ee.(2R,3R)-2-Hydrazinyl-6,6-dimethyl-3-phenyl-3,5,6,7-tetrahydrobenzo-furan-4(2H)-one (9)
To a solution of dihydrofuran 7m (136 mg, 0.5 mmol, 1.0 equiv.) in MeOH (5.0 mL) was added 5 N HCl (3.0 mL) at room temperature. The resulting reaction mixture was stirred until the reaction was complete (8 h, monitored by TLC). The crude product was purified by silica gel column chromatography by eluting with ethyl acetate/petroleum ether (50–60%). Yellow liquid; yield 116 mg, 85%; IR (film, cm−1) 3316 (br m), 3029 (w), 2959 (m), 1618 (vs), 1453 (m), 1424 (m), 1402 (m), 1207 (s), 1144 (m), 1119 (m), 1045 (m), 913 (m), 891 (m), 700 (m); 1H NMR (400 MHz, CDCl3) δ 1.14 (s, 6H), 2.21, 2.25 (ABq, J = 16.4 Hz, 2H), 2.43 (ABq collapsed to s, 2H), 4.08 (s, 1H), 5.69 (s, 1H), 7.11 (d, J = 7.3 Hz, 2H), 7.18–7.23 (m, 1H), 7.26 (t, J = 7.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 28.4, 29.5, 34.3, 38.1, 51.1, 52.9, 110.4, 110.5, 114.8, 127.3, 128.9, 139.6, 175.8, 194.9; HRMS (ES+) calcd for C16H20N2O2Na (MNa+) 295.1417, found 295.1415; [α]25D −21.80° (c 0.5, CHCl3); HPLC: Chiralcel AD-H (petroleum ether/i-PrOH = 80/20, flow rate = 1.0 mL min−1, λ = 240 nm), tR (major) = 29.6 min, tR (minor) = 34.6 min; 72% ee.Conclusions
2-Hydrazinated 2,3-dihydrofurans have been synthesized in a highly diastereoselective fashion in good to excellent yields by treating a wide variety of 1,3-dicarbonyl compounds, including curcumins, with α-hydrazinated nitroalkenes in the presence of K2CO3 in aqueous THF at room temperature. The enantioselective version of the above interrupted Feist–Bénary type reaction has been successfully developed by treating selected 1,3-dicarbonyls such as dimedone and cyclohexanone with α-hydrazinonitroalkenes in the presence of an L-t-leucine derived squaramide as the chiral catalyst. The enantio-enriched 2-hydrazinodihydrofurans fused to a cyclohexane ring were formed in moderate to good yields, with absolute diastereoselectivities and good to very good enantioselectivities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
INNN thanks SERB-DST for financial assistance. KSB thanks the CSIR, NA and NS thank the UGC for a senior research fellowship. The authors thank Prof R. J. Butcher, Howard University, USA, for help with X-ray data.References
- (a) J. P. Michael, Nat. Prod. Rep., 2000, 17, 603–620 RSC; (b) J. P. Michael, Nat. Prod. Rep., 1997, 14, 605–618 RSC; (c) P. A. Jacobi and H. G. Selnick, J. Org. Chem., 1990, 55, 202–209 CrossRef CAS; (d) B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef CAS.
- Books: (a) F. M. Dean and M. V. Sargent, in Comprehensive Heterocyclic Chemistry, ed. C. W. Bird and G. W. H. Cheeseman, Pergamon, New York, 1984, vol. 4, Part 3, p. 531 Search PubMed; (b) G. Vernin, The Chemistry of Heterocyclic Flavoring and Aroma Compounds, Ellis Horwood, Chichester, U.K., 1982 Search PubMed. Reviews: (c) H. Maruoka, F. Okabe and K. Yamagata, Adv. Chem. Res., 2012, 15, 79–102 CAS; (d) N. Oskar and B. Harald, ACS Symp. Ser., 2003, 847, 213–218 CrossRef; (e) B. M. Fraga, Nat. Prod. Rep., 1992, 9, 217–241 RSC; (f) A. T. Merrit and S. V. Ley, Nat. Prod. Rep., 1992, 9, 243–287 RSC; (g) T. G. Kilroy, T. P. O'Sullivan and P. J. Guiry, Eur. J. Org. Chem., 2005, 4929–4949 CrossRef CAS; (h) R. Jacques, R. Pal, N. A. Parker, C. E. Sear, P. W. Smith, A. Ribaucourt and D. M. Hodgson, Org. Biomol. Chem., 2016, 14, 5875–5893 RSC.
- Reviews: (a) X.-L. Hou, Z. Yang, K.-S. Yeung and H. N. C. Wong, Prog. Heterocycl. Chem., 2005, 17, 142–171 CAS; (b) M. C. Elliott, J. Chem. Soc., Perkin Trans. 1, 2002, 2301–2323 RSC; (c) M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407–2474 CrossRef CAS PubMed. Selected articles: (d) P. Mueller, G. Bernardinelli, Y. F. Allenbach, M. Ferri and S. Grass, Synlett, 2005, 1397–1400 CrossRef CAS; (e) H. Ishitani and K. Achiwa, Heterocycles, 1997, 46, 153–156 CrossRef CAS.
- Selected articles: (a) J.-C. Zheng, C.-Y. Zhu, X.-L. Sun, Y. Tang and L.-X. Dai, J. Org. Chem., 2008, 73, 6909–6912 CrossRef CAS PubMed; (b) R. K. Bowman and J. S. Johnson, Org. Lett., 2006, 8, 573–576 CrossRef CAS PubMed; (c) W. Xia, C. Yang, B. O. Patrick, J. R. Scheffer and C. Scott, J. Am. Chem. Soc., 2005, 127, 2725–2730 CrossRef CAS PubMed; (d) H. J. Gais, L. R. Reddy, G. S. Babu and G. Raabe, J. Am. Chem. Soc., 2004, 126, 4859–4864 CrossRef CAS PubMed; (e) F. Garzino, A. Méou and P. Brun, Eur. J. Org. Chem., 2003, 1410–1414 CrossRef CAS; (f) X. Cheng, C. D. Quintanilla and L. Zhang, J. Org. Chem., 2019, 84, 11054–11060 CrossRef CAS PubMed.
- [4 + 1] Cycloaddition: (a) S. Son and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 1046–1047 CrossRef CAS PubMed; (b) J.-L. Zhou, L.-J. Wang, H. Xu, X.-L. Sun and Y. Tang, ACS Catal., 2013, 3, 685–688 CrossRef CAS [3 + 2] Cycloaddition: (c) D. A. Evans, Z. K. Sweeney, T. Rovis and J. S. Tedrow, J. Am. Chem. Soc., 2001, 123, 12095–12096 CrossRef CAS PubMed. Heck-type reaction: (d) F. Ozawa, A. Kubo and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 1417–1419 CrossRef CAS; (e) F. Ozawa, A. Kubo, Y. Matsumoto and T. Hayashi, Organometallics, 1993, 12, 4188–4196 CrossRef CAS; (f) T. Tu, W.-P. Deng, X.-L. Hou, L.-X. Dai and X.-C. Dong, Chem. – Eur. J., 2003, 9, 3073–3081 CrossRef CAS; (g) P. Kaukoranta, K. Källström and P. G. Andersson, Adv. Synth. Catal., 2007, 349, 2595–2602 CrossRef CAS; (h) S. T. Henriksen, P.-O. Norrby, P. Kaukoranta and P. G. Andersson, J. Am. Chem. Soc., 2008, 130, 10414–10421 CrossRef CAS PubMed; (i) L. Penn, A. Shpruhman and D. Gelman, J. Org. Chem., 2007, 72, 3875–3879 CrossRef CAS PubMed; (j) C. Wu and J. Zhou, J. Am. Chem. Soc., 2014, 136, 650–652 CrossRef CAS PubMed; (k) Y.-H. Miao, Y.-Z. Hua and M.-C. Wang, Org. Biomol. Chem., 2019, 17, 7172–7181 RSC; (l) A. G. Mundsson, K. P. J. Gustafson, B. K. Mai, B. Yang, F. Himo and J.-E. Backvall, ACS Catal., 2018, 8, 12–16 CrossRef; (m) O. El-Sepelgy, A. Brzozowska, J. Sklyaruk, Y. K. Jang, V. Zubar and M. Rueping, Org. Lett., 2018, 20, 696–699 CrossRef CAS PubMed; (n) M. L. Piotrowski and M. A. Kerr, Org. Lett., 2018, 20, 7624–7627 CrossRef CAS PubMed; (o) I. Geibel, M. Schmidtmann and J. Christoffers, Org. Biomol. Chem., 2017, 15, 7824–7829 RSC; (p) K. Li, F.-L. Zhu, Z.-T. Liu, J. Tong and X.-P. Hu, Org. Biomol. Chem., 2018, 16, 742–749 RSC; (q) T. Shi, S. Teng, Y. Wei, X. Guo and W. Hu, Green Chem., 2019, 21, 4936–4940 RSC; (r) J. A. Carmona, V. Hornillos, P. Ramírez-Lopez, A. Ros, J. Iglesias-Sigüenza, E. Gomez-Bengoa, R. Fernandez and J. M. Lassaletta, J. Am. Chem. Soc., 2018, 140, 11067–11075 CrossRef CAS PubMed; (s) X. Cheng, Z. Wang, C. D. Quintanilla and L. Zhang, J. Am. Chem. Soc., 2019, 141, 3787–3791 CrossRef CAS PubMed; (t) Y. Zhu, I. Colomer, A. L. Thompson and T. J. Donohoe, J. Am. Chem. Soc., 2019, 141, 6489–6493 CrossRef CAS PubMed; (u) Y.-C. Zhang, B.-W. Zhang, R.-L. Geng and J. Song, Org. Lett., 2018, 20, 7907–7911 CrossRef CAS PubMed.
- Cycloadditions: (a) S. Bisht, R. Rani and R. K. Peddinti, J. Org. Chem., 2018, 83, 75–84 CrossRef CAS PubMed; (b) J.-P. Croisetiere and C. Spino, J. Org. Chem., 2018, 83, 5609–5618 CrossRef CAS PubMed. Application of MBH adducts: (c) Y. Cheng, Y. Han and P. Li, Org. Lett., 2017, 19, 4774–4777 CrossRef CAS PubMed; (d) S.-T. Du, Z. Sun, W. Liu and W.-W. Liao, Org. Lett., 2017, 19, 6598–6601 CrossRef CAS PubMed; (e) P. Zhang, X. Guo, C. Liu, W. Li and P. Li, Org. Lett., 2019, 21, 152–155 CrossRef CAS PubMed. Cascade reactions: (f) S. C. Sahoo, R. Maity and S. C. Pan, ACS Omega, 2019, 4, 2792–2803 CrossRef CAS PubMed; (g) Y. Tangella, K. L. Manasa, V. L. Nayak, M. Sathish, B. Sridhar, A. Alarifi, N. Nagesh and A. Kamal, Org. Biomol. Chem., 2017, 15, 6837–6853 RSC; (h) M. Balha, B. Mondal and S. C. Pan, Org. Biomol. Chem., 2019, 17, 6557–6561 RSC; (i) Y. Ye, L. Wang and R. Fan, J. Org. Chem., 2010, 75, 1760–1763 CrossRef CAS PubMed; (j) M. A. Calter, R. M. Phillips and C. Flaschenriem, J. Am. Chem. Soc., 2005, 127, 14566–14567 CrossRef CAS PubMed; (k) M. A. Calter and A. Korotkov, Org. Lett., 2011, 13, 6328–6330 CrossRef CAS PubMed.
- Y. Zhao, X. Jiang and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2013, 52, 8597–8601 CrossRef CAS PubMed.
- M. Rueping, A. Parra, U. Uria, F. Besseliévre and E. Merino, Org. Lett., 2010, 12, 5680–5683 CrossRef CAS PubMed.
- L.-P. Fan, P. Li, X.-S. Li, D.-C. Xu, M.-M. Ge, W.-D. Zhu and J.-W. Xie, J. Org. Chem., 2010, 75, 8717–8722 CrossRef PubMed.
- X. Dou, F. Zhong and Y. Lu, Chem. – Eur. J., 2012, 18, 13945–13948 CrossRef CAS PubMed.
- (a) J. Feng, L. Lin, K. Yu, X. Liu and X. Feng, Adv. Synth. Catal., 2015, 357, 1305–1310 CrossRef CAS; (b) J. Feng, X. Yuan, W. Luo, L. Lin, X. Liu and X. Feng, Chem. – Eur. J., 2016, 22, 15650–15653 CrossRef CAS PubMed.
- V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140–2143 CrossRef CAS PubMed.
- C. Jarava-Barrera, F. Esteban, C. Navarro-Ranninger, A. Parra and J. Aleman, Chem. Commun., 2013, 49, 2001–2003 RSC.
- J.-W. Zhang, L.-S.-H. Yu, J.-L. Dong, Q.-C. Sun and J.-W. Xie, Synlett, 2018, 603–608 CAS.
- (a) X.-L. Zhang, C.-K. Tang, A.-B. Xia, K.-X. Feng, X.-H. Du and D.-Q. Xu, Eur. J. Org. Chem., 2017, 3152–3160 CrossRef CAS; (b) X.-L. Zhang, K.-X. Feng, A.-B. Xia, Y.-Y. Zheng, C. Li, X.-H. Du and D.-Q. Xu, Eur. J. Org. Chem., 2018, 2918–2925 CrossRef CAS.
- Reviews: (a) I. Chattopadhyay, K. Biswas, U. Bandopadhyay and R. K. Banerjee, Curr. Sci., 2004, 87, 44–53 CAS; (b) A. Goel, B. K. Kunnumakkara and B. B. Aggarwal, Biochem. Pharmacol., 2008, 75, 787–809 CrossRef CAS PubMed.
- (a) P. Anand, C. Sundaram, S. Jhurani, A. B. Kunnumakkara and B. B. Aggarwal, Cancer Lett., 2008, 267, 133–164 CrossRef CAS PubMed; (b) P. V. Leyon and G. Kuttan, J. Exp. Clin. Cancer Res., 2003, 22, 77–83 CAS; (c) B. B. Aggarwal, A. Kumar and A. C. Bharti, Anticancer Res., 2003, 23, 363–398 CAS.
- (a) B. B. Aggarwal and S. Shishodia, Biochem. Pharmacol., 2006, 71, 1397–1421 CrossRef CAS PubMed; (b) O. P. Sharma, Biochem. Pharmacol., 1976, 25, 1811–1812 CrossRef CAS PubMed.
- (a) A. N. Nurfina, M. S. Reksohadiprodjo, H. Timmerman, U. A. Jenie, D. Sugiyanto and H. van der Goot, Eur. J. Med. Chem., 1997, 32, 321–328 CrossRef CAS; (b) Ref. 18b .
- F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS PubMed.
- A. Mazumder, N. Neamati, S. Sunder, J. Schulz, H. Pertz, E. Eich and Y. Pommier, J. Med. Chem., 1997, 40, 3057–3063 CrossRef CAS PubMed.
- L. G. Menon, R. Kuttan and G. Kuttan, Cancer Lett., 1999, 141, 159–165 CrossRef CAS.
- For various biological activities of curcumin–metal complexes: Y. M. Song, J. P. Xu, L. Ding, Q. Hou, J. W. Liu and Z. L. Zhu, J. Inorg. Biochem., 2009, 103, 396–400 CrossRef CAS PubMed.
- N. Ayyagari, D. Jose, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2011, 52, 258–262 CrossRef CAS.
- N. Ayyagari and I. N. N. Namboothiri, Tetrahedron: Asymmetry, 2012, 23, 605–610 CrossRef CAS.
- N. Ayyagari, A. Mehta, E. Gopi, I. Deb, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron, 2013, 69, 5973–5980 CrossRef CAS.
- W. Li, W. Wu, F. Yu, H. Huang, X. Liang and J. Ye, Org. Biomol. Chem., 2011, 9, 2505–2511 RSC.
- X.-G. Yin, X.-Y. Liu, Z.-P. Hu and M. Yan, Org. Biomol. Chem., 2012, 10, 1506–1509 RSC.
- N.-H. Luo, X. Sun, W.-T. Wei, X.-J. Zhang and M. Yan, Tetrahedron: Asymmetry, 2013, 24, 402–408 CrossRef CAS.
- Selected books/reviews on MBH reaction: (a) D. Basavaiah and R. T. Naganaboina, New J. Chem., 2018, 42, 14036–14066 RSC; (b) S. Bhowmik and S. Batra, Curr. Org. Chem., 2014, 18, 3078–3119 CrossRef CAS; (c) M.-X. Zhao, Y. Wei and M. Shi, The chemistry of the Morita-Baylis-Hillman reaction, in RSC Catal. Ser, ed. M. Shi, F.-J. Wang, M.-X. Zhao and Y. Wei, Royal Society of Chemistry, Cambridge, 2011, vol. 8, pp. 1–561 Search PubMed; (d) E. Ciganek, Org. React., 1997, 51, 201–350 CAS; (e) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS PubMed; (f) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614–4628 CrossRef CAS. RC reaction: (g) P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 6213–6226 CrossRef CAS; (h) C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 CrossRef CAS; (i) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS.
- Reviews/accounts: (a) K. Kaur and I. N. N. Namboothiri, Chimia, 2012, 66, 913–920 CrossRef CAS PubMed; (b) D. K. Nair, T. Kumar and I. N. N. Namboothiri, Synlett, 2016, 2425–2442 CAS; (c) W.-Y. Huang, S. Anwar and K. Chen, Chem. Rec., 2017, 17, 363–381 CrossRef CAS PubMed.
- M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Biomol. Chem., 2006, 4, 2525–2528 RSC.
- (a) V. Mane, J. Pandey, N. Ayyagari, R. Kale and I. N. N. Namboothiri, Org. Biomol. Chem., 2016, 14, 2427–2438 RSC; (b) V. Mane, T. V. Baiju and I. N. N. Namboothiri, ACS Omega, 2018, 3, 17617–17628 CrossRef CAS.
- Organocatalysis, selected books/reviews: (a) A. Berkessel and H. Groeger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, Germany, 2005 CrossRef; (b) Organocatalysis in Organic Synthesis, in Tetrahedron, ed. P. Kocovsky and A. V. Malkov, Elsevier, Amsterdam, Netherlands, 2005, vol. 62, p. 260 Search PubMed; (c) H. Pellissier, Recent Developments in Asymmetric Organocatalysis, Royal Society of Chemistry, Cambridge, UK, 2010 Search PubMed; (d) Science of Synthesis: Asymmetric Organocatalysis 2, Bronsted Base and Acid Catalysts, and Additional Topics, ed. K. Maruoka, Georg Thieme Verlag, Stuttgart, Germany, 2012, p. 974 Search PubMed; (e) M. Waser, Asymmetric Organocatalysis in Natural Product Syntheses, Springer-Verlag, Wien, 2012, p. 197 CrossRef; (f) Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions and Applications, Privileged Catalysts, ed. P. I. Dalko, Wiley-VCH, Weinheim, Germany, 2013, vol. 1–3 Search PubMed; (g) S. J. Connon, Chem. Commun., 2008, 2499–2510 RSC; (h) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed; (i) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785–795 CrossRef CAS; (j) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701–1716 CrossRef CAS; (k) B. List, Top. Curr. Chem., 2010, 291, 395–456 Search PubMed; (l) E. M. Carreira, (Ed.), Synthesis, 2011, issue 12 (special issue on organocatalysis).
- Chiral squaramides, selected reviews: (a) J. Aleman, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 6890–6899 CrossRef CAS PubMed; (b) S. Karahan and C. Tanyeli, Tetrahedron Lett., 2018, 59, 3725–3737 CrossRef CAS; (c) P. Li, X. Hu, X.-Q. Dong and X. Zhang, Molecules, 2016, 21, 1–14 Search PubMed; (d) J. V. Alegre-Requena, E. Marques-Lopez, R. P. Herrera and D. D. Diaz, CrystEngComm, 2016, 18, 3985–3995 RSC; (e) F. E. Held and S. B. Tsogoeva, Catal. Sci. Technol., 2016, 6, 645–667 RSC; (f) M. Zabka and R. Sebesta, Molecules, 2015, 20, 15500–15524 CrossRef CAS PubMed; (g) F. Vetica, R. M. de Figueiredo, M. Orsini, D. Tofani and T. Gasperi, Synthesis, 2015, 2139–2184 CAS; (h) P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253–281 CrossRef CAS; (i) M. Tsakos and C. G. Kokotos, Tetrahedron, 2013, 69, 10199–10222 CrossRef CAS; (j) B.-L. Zhao, J.-H. Li and D.-M. Du, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS PubMed; (k) X. Han, H.-B. Zhou and C. Dong, Chem. Rec., 2016, 16, 897–906 CrossRef CAS PubMed. The first article: (l) J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed.
- (a) C1: B. Vakulya, S. Varga, A. Csampai and T. Soos, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS PubMed; (b) C2: J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC; (c) C3: K. Bera and I. N. N. Namboothiri, Org. Lett., 2012, 14, 980–983 CrossRef CAS PubMed; (d) C4: Y. Gao, Q. Ren, L. Wang and J. Wang, Chem. – Eur. J., 2010, 16, 13068–13071 CrossRef CAS PubMed; (e) C5: W. Yang and D.-M. Du, Org. Lett., 2010, 12, 5450–5453 CrossRef CAS PubMed; (f) C6: H. Jiang, M. W. Paixão, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775–2783 CrossRef CAS PubMed; (g) C7–C9: K. Bera and I. N. N. Namboothiri, Org. Biomol. Chem., 2014, 12, 6425–6431 RSC; (h) C10–C12: K. Bera, N. S. Satam and I. N. N. Namboothiri, J. Org. Chem., 2016, 81, 5670–5680 CrossRef CAS PubMed.
- The achiral base, K2CO3, was not added in the first step to ensure that the possible background reaction does not affect the enantioselectivity of the product.
- These optimization studies suggested that the background reaction in the first step and the harsh conditions required in the second step may be responsible for the moderate enantioselectivity.
- similar phenomenon was observed in the case of hydrazinodicarboxylates of nitroalkenes (ref. 32), triazoles and arenodihydrofurans (ref. 33a) as well as thienoindoles (ref. 33b).
- The hydrazinodihydrofurans reported here are sufficiently stable for isolation and characterization. There was no evidence of any isomerization or reversible ring opening of these aminals. Possible aromatization via elimination of the hydrazinodicarboxylate moiety or air oxidation was also not observed.
- Selected recent reports on organocatalytic reactions involving a nitro group as a leaving group: (a) M. S. Manna and S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 130–133 CrossRef CAS PubMed; (b) A. N. Dinh, R. R. Noorbehesht, S. T. Toenjes, A. C. Jackson, M. A. Saputra, S. M. Maddox and J. L. Gustafson, Synlett, 2018, 2155–2160 CAS.
- However, reversible ring opening as a marginal secondary phenomenon causing minor stereochemical scrambling in compounds 4–9 cannot be completely ruled out. This is because small multiple signals are observed on either side of the methyl signal in the 1H NMR spectrum of compound 9 and the ee dropped marginally from 75% in 7m to 72% in 9. This requires further investigation and the results will be reported in due course.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all the new compounds, HPLC profiles for all the new enantio-enriched compounds and X-ray data for compound 7c. CCDC 1947973. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ob01974j |
| This journal is © The Royal Society of Chemistry 2020 |