
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of gold–silica core–shell nanoparticles by pulsed laser ablation in liquid and their physico-chemical properties towards photothermal cancer therapy†
René
Riedel
a,
Nora
Mahr
a,
Chenyang
Yao
b,
Aiguo
Wu
 b,
Fang
Yang
*ab and
Norbert
Hampp
*a
b,
Fang
Yang
*ab and
Norbert
Hampp
*a
aPhysical Chemistry Department of University of Marburg, Marburg, Germany. E-mail: hampp@uni-marburg.de
bCixi Institute of Biomedical Engineering, CAS Key Laboratory of Magnetic Materials and Devices & Key Laboratory of Additive Manufacturing Materials of Zhejiang Province, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, P.R. China. E-mail: yangf@nimte.ac.cn
First published on 9th January 2020
Abstract
Due to the increasing scientific and biomedical interest in various nanoparticles (NPs) with excellent properties and the onset of their commercial use, a convenient and adjustable physical method for improved efficiency needs to be used for enabling their tech-scale production. Recently, great progress has been made in the large-scale production of NPs with a simple structure by pulsed laser ablation in liquid (PLAL). In this work, we synthesized gold–silica core–shell NPs by improved PLAL and provided a guide on how to investigate their physico-chemical properties and association with biological effects towards cancer photothermal therapy (PTT). By means of this method, reproducible and scalable liquid phase NPs with less toxicity and good stability can be realized for tech-scale production based on its further adjustment and modification. Moreover, a more complete investigation of the associations between the physico-chemical properties of functional NPs with complex structure and their biological effects may enable more targeted NPs towards specific requirements of biomedical applications.
Introduction
The field of nanobiotechnology has experienced a rapid development over the past decades. The focus of NP applications in medicine, often referred to as nanomedicine,1 certainly lies on bioimaging and treatment of diseases. A huge variety of nanoparticles with different sizes, shapes, chemical compositions and functionalisations have been synthesised and investigated for their mechanical, physical and optical properties as well as for their biomedical behaviours.2 However, the conventional method of wet chemistry NP production requires the usage of additional substances such as stabilisers as well as a huge amount of work and time. Synthesis pathways are rather complicated and the outcome contains side products that are difficult to remove.2–5 Consequently, experimental results therefrom are often controversial. For example, the behaviour and toxicity of gold nanoparticles (AuNPs) tested in vivo strongly depend on the properties of stabilisers.6Hence, especially in the field of nanomedicine, the production of clean nanoparticles with long-time stability is highly desired and an indispensable requirement for biomedical applications. As an alternative to chemical pathways, physical methods of NP synthesis are the emerging strategies in nanotechnology and serve as a promising route for the large-scale production of pure NPs. Laser ablation is normally used for surface processing technology and in the interface research domain. The produced NP had been an undesired side-product for a long time. Only in 1991, Iida et al.7 investigated a typical NP side-product and hence revealed a new field of research. When executed on metal targets in a liquid environment using pulsed lasers, the NPs can be trapped by the liquid. Pulsed Laser Ablation in Liquid (PLAL) provides an easy way to produce NPs in solution, decreasing experimental complexity while simultaneously increasing the integrity and stability of the pure NPs. PLAL is a green method, which means it is in accordance with the 12 principles of sustainable green chemistry.8 Even the greenest method of wet chemistry NP synthesis requires complex purification and separation from the remaining metal salts in solution.9 In addition, stabilisers are needed to avoid aggregation and agglomeration and can influence the properties and behaviour of the NPs, especially in biological systems. PLAL, however, provides a simple one-step synthesis of NPs in a clean solution without heavy-metal salts and the addition of stabilisers. Although it still lacks in productivity as well in NP shape variety, it is one of the fastest, cheapest and cleanest NP synthesis methods among all the NP production techniques and highly suitable for biomedical applications.9
To date, PLAL has rarely been considered for nanomedical purposes. A major reason may be the fact that synthesizing NPs with desired compositions and properties is still challenging. In addition, the initial operation details of PLAL in processing the NPs at the nanoscale were rather puzzling for almost any considered NPs with complex structures. Nevertheless, the properties and behaviour of the pure, stable NPs prepared in vitro and in vivo can be analysed without the influence of stabilizers and other perturbing substances. The results will help to understand the biological effects of NPs and their interactions with biological components of cells and organisms. The effect of the mechanical properties of NPs on cellular uptake, especially adhesion and elasticity, has rarely been investigated to date. There is evidence that elasticity plays an important role in uptake mechanisms.10–13 For instance, macrophages are not able to phagocytose very soft samples. Thus, soft NPs can circulate in the blood stream for longer compared to hard NPs. The elasticity of NPs is highly related to their outside polymer shells, whereby the species, thickness, and the physicochemical properties are varied. Its effect can be theoretically described for better understanding.10 According to the model, the membrane wrapping of soft NPs is much more time- and energy consuming compared to the wrapping around stiff NPs, thus soft NPs are less prone to uptake. For example, PEGylated AuNPs have a smaller ratio compared to plain AuNPs.
Irradiation of noble metal NPs at a certain frequency leads to their surface plasmon resonance due to the electron oscillation after high energy absorption.14 Moreover, the scattering rate increases with the NP size.15 For small NPs, the absorbed energy is transferred to heat via electron lattice interaction to heat up the surrounding medium. This effect is fully exploited in photothermal therapy (PTT).16–19 Noble metal NPs, mostly AuNPs, serve as heating probes due to their high heating efficiency in addition of their inertia to improve biocompatibility which can improve their blood circulation half-life and tumour cell accumulation. After accumulation, the cells were irradiated with a continuous wave laser at a frequency close to the maximum absorption of the NPs. When the temperature increases to 45 °C, it results in higher apoptosis or necrosis.20 PTT provides a method for selectively destroying tumour cells by precisely targeting at the tumour area and minimizing the damage of surrounding healthy tissues.21 Silicates are present in the human body in significant quantities.22 In the past, the silicate surface of NPs was found to highly increase the biocompatibility of the NPs.23–25 Since, in addition, the silicate surface offers the possibility of uncomplicated surface chemistry, for example to covalently bind inhibitors or other molecules, silicate- or silicate-coated nanoparticles have become a popular object of investigation. In addition, silica NPs are prone to repulse each other because of their surface hydroxy groups which results in good stability.25–27 Thus, silica-coated core–shell NPs have recently gained importance. Although the silica shell provides biocompatibility, the core serves as a functional part and can be accessed through its magnetic or SPR properties. Mostly, gold is used as the core metal for its excellent SPR properties and its noble character. Gold is considered to be non-toxic. However, gold NPs can interact with components containing thiol or disulphide groups28 and should thus be shielded by means of silica coating. PLAL provides a simple way for a one-step synthesis of core–shell NPs as has been shown by previous work.
Hereby, we present a synthetic control strategy for the large-scale production of gold–silica core–shell NPs and provide an opportunity to synthesize other NPs with different shapes and structures by PLAL. In addition, we investigate their physicochemical properties, such as size, structure, and ζ-potential as well as their elasticity and adhesion which are related to cell viability.29,30 We also reveal their biological effect toward tumor treatment via PTT, such as photothermal performance in vitro, cell viability, accumulation and toxicity in various tissues.
Methods
Preparation of Au@SiO2 NP
Au@SiO2 NPs were prepared by PLAL. A ns-pulsed Nd:YAG laser (Quanta-Ray GCR-100, Spectra-Physics, 8 ns pulse duration, 532 nm, 10 Hz, laser fluence of 30 J cm−1) was focused onto a rotating gold target (5 g, Degussa) immersed in 30 ml distilled water containing 0.4 μM NaCl (ChemSolute, purity >99%) and 0.4 μM NaSiO2 (purified sodium silicate, Carl Roth). The gold target was rotated in order to avoid an overlay of impacts of consecutive laser pulses and hence to gain a higher ablation rate. The process was carried out for 15 minutes leading to an ablation mass of about 1.6 mg gold. The synthesized Au@SiO2 NPs were centrifuged for 10 minutes at 5000 rpm to separate them from the remaining sodium–water glass. The as-obtained NPs were either analysed or modified by Bovine Serum Albumin (BSA) coating. In our experiment, ablation resulted in spherical silica coated gold NPs with an average diameter of 9 nm and a coating thickness of 2 nm being generated. The dependency of the NP size and composition on laser settings was tested in order to optimise size and size distribution as well as shell thickness and ablation performance. The results can be found in the ESI.† Transmission Electron Microscopy (TEM) images (Fig. 2A) clearly show that the NPs exhibit spherical shapes and smooth surfaces. Fig. 2C shows a high-angle annular dark-field scanning TEM (HAADF-STEM) image of the NPs which consist of pure gold as the core and silica as the shell. Their absorption was demonstrated by UV-Vis spectroscopy. Using our synthesis method, the NPs were stable without the addition of tensides at a ζ-potential of −58.7 mV (for more details, see the ESI†).BSA coating
The capability of proteins or ligands to easily bind to the NPs was demonstrated using BSA. The BSA coating was prepared as reported previously.31–33 In order to coat Au@SiO2 with BSA, 10 μL of gold NP solution (1.77 × 10−8 mol L−1) and 10 μL BSA solution (6.45 × 10−5 mol L−1) were added to 980 μL of pH-adjusted HCl solution (pH 4.5) so as to have a high BSA surplus. The solutions were shaken for 30 minutes and subsequently centrifuged for 5 minutes at 5000 rpm. The supernatant was analysed by means of scanning electron microscopy (SEM) and atomic force microscopy (AFM). The results can be found in the ESI.†Ablation rate measurement
The concentration of fabricated gold NPs in solution is difficult to measure. Although it is possible to calculate the concentration from recorded UV-vis spectra,34 the evaluation is quite complex. In order to estimate the concentration, the gold target was weighed right before laser ablation. After the ablation process, the gold target was washed with distilled water, dried in air and weighed again. The difference in weight (before and after laser ablation) was assumed to be the ablated gold mass. To improve accuracy, laser ablation was performed for 150 minutes. The gold NP solution was replaced with new solution every 30 minutes to reduce absorption effects. After 150 minutes, 16.2 ± 0.5 mg gold was ablated and assumed to be fully converted into gold–silica NPs. Since TEM and SEM images revealed an average core diameter of 9 nm, the gold NP concentration was estimated to be 1.77 × 10−8 mol L−1.Particle characterisation
ζ-Potential measurements were performed using dynamic light scattering (Delsa™ Particle Analyzer, Beckman Coulter). UV-Vis spectra were recorded with a Lambda 35 UV-Vis spectrometer (Lambda 35, PerkinElmer) in the wavelength range from 350 to 1050 nm. Shape and particle size distribution of the NPs were both determined by means of scanning electron microscopy (SEM, Jeol JSM 7500-F). For this purpose, a 1 μL drop of the NP solution (1.77 × 10−10 mol L−1) was dried on a clean plate of stainless steel which had been rinsed with isopropanol and distilled water in an ultrasonic bath for 15 minutes. The dried drop was then analysed. The electron beam was driven at 10 keV and 10 μA.For higher resolution, TEM images were recorded (TEM, Jeol JEM-3010). To do so, 1 μL of the NP solution (1.77 × 10−10 mol L−1) was trickled onto a TEM graphite sheet. After drying, the TEM measurement was then carried out.
To study the mechanical properties of NPs which include elasticity and adhesion, AFM measurements were performed (MultiMode8, Bruker Corporation). 1 μL of the NP solution (106-fold diluted) was dropped onto a silicon wafer (<0.002 Ω cm2, SilChem) and air-dried for 2 hours. The NPs were characterized by conventional force spectroscopy. The whole process is described in Fig. 3B. First, the topography was measured. Subsequently, the tip was placed right above a NP with a vertical distance so that virtually no interaction forces existed between the tip and the NP. The tip was then extended towards the NP. Cantilever deflection was converted to interaction force between the tip and the sample and plotted versus cantilever vertical position, resulting in a force–distance curve. As the cantilever is intended into the NP, repulsive forces lead to its deflection. At a certain point, the cantilever was retracted from the NP. At the detachment point, adhesion forces may contribute to the interactions. Further retracting leads to an uncoupling resulting in a zero net force of the cantilever. Both process and data evaluation are described in detail in the ESI.†
For all measurements, a sharp silicon nitride cantilever (Bruker SNL-10, tip A, 2 nm tip radius, nominal spring constant 0.35 N m−1) was used. Thermal tune measurements were conducted before each measurement and a spring constant close to the nominal value was revealed. More than 1500 force–distance curves were recorded in conventional force curve mode with one ramp per NP. Although not all the data could be evaluated for each property and model, enough data existed for statistical calculations. For each mode, adhesion and Young's modulus were calculated. For the latter, different models are available which were evaluated by fitting the linear slope of the force–distance curves to the models. For evaluation, the Hertz model, DMT model and JKR model were used as described in Experimental section. For further details, please refer to the ESI.†
PeakForce Quantitative Nanomechanical (PF-QNM35) property measurements were performed with BSA-coated gold–silica NPs to locate the hard NPs within the soft BSA coating. The procedure was the same as that described above.
Photothermal performance
The Au@SiO2 NPs were evaluated for their photothermal performance using a digital MAG-V30 NIR photothermal imaging system (Wuhan Co, China) under a 520 nm laser irradiation.36 Specifically, 1.0 mL containing various concentrations (1.5 to 12 nM) of the Au@SiO2 NP aqueous solutions was added into different disposable cuvettes, and then the solutions were irradiated with a 520 nm laser for 15 minutes at 400 mW cm−2. Similarly, 1.0 mL of 12 nM Au@SiO2 NP aqueous solution was added into a disposable cuvette, and then the solution was irradiated with the 520 nm laser at diverse power densities (100, 200, 300 and 400 mW cm−2) for 15 minutes. The temperature changes were recorded every 5 seconds and thermal images were taken every minute using a NIR photothermal imaging system.Cell culture
Human cervical carcinoma cells (HeLa cells) were kindly provided by Greta Linden, working group Vazquez, Biochemistry Department of Philipps-University of Marburg, Germany. Briefly, a seed of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well was cultured in 200 μl Dulbecco's Modified Eagle's Medium (DMEM) containing 10% Fetal Bovine Serum (FBS) and incubated for 21 hours. The cell cultures were stored in a 50 ml Falcon™ polypropylene centrifuge tube at 37 °C in an atmosphere containing 5% CO2. The temperature was kept constant by means of a water bath. The setup was placed in a sealed polymethylmethacrylate box. The CO2 inlet was controlled using a house-made CO2 regulator and kept constant between 4.80 and 5.20%.
000 cells per well was cultured in 200 μl Dulbecco's Modified Eagle's Medium (DMEM) containing 10% Fetal Bovine Serum (FBS) and incubated for 21 hours. The cell cultures were stored in a 50 ml Falcon™ polypropylene centrifuge tube at 37 °C in an atmosphere containing 5% CO2. The temperature was kept constant by means of a water bath. The setup was placed in a sealed polymethylmethacrylate box. The CO2 inlet was controlled using a house-made CO2 regulator and kept constant between 4.80 and 5.20%.
For PPT performance tests, human breast cancer cell line (MCF-7 cells) was cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 units per mL of penicillin and 100 mg mL−1 of streptomycin in a 5% CO2, 37 °C incubator.37 All cells are from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cell cultures were stored in a 50 mL Falcon™ polypropylene centrifuge tube at 37 °C in an atmosphere containing 5% CO2. The temperature was kept constant by means of a water bath. The setup was placed in a sealed polymethylmethacrylate box. CO2 inlet was controlled using a house-made CO2 regulator and kept constant between 4.80 and 5.20%.
Cell viability assay
Cell viability assays were recorded using the Resazurin method as previously reported38 for evaluating the toxicity of NPs. For this purpose, 110 μL of the well containing 200 μL of cell solution were removed. The remaining cell solution contained ∼100000 cells. 10 μL of the gold–silica nanoparticle solution with different concentrations (1.5 to 12 nM) were added and incubated for 23 hours. After that, the cells were washed with Phosphate Buffered Saline (PBS). Subsequently, 100 μL of 10% resazurin solution in complete cell medium was added and incubated for 4 hours at 37 °C and 5% CO2. The fluorescence intensity was measured at 590 nm so as to investigate the presence of resazurin. 5 data points were taken. The fluorescence intensity was considered to be proportional to cell viability. The cells were suspended in cell medium (DMEM) and stored in a 50 mL Falcon™ polypropylene centrifuge tube. Their temperatures were kept constant at 37 °C by means of a water bath and a hot plate stirrer. This setup was placed in a polymethylmethacrylate box, where a concentration of 5% CO2 was maintained using a home-made CO2 flow controller. Before measurement, the cells were shaken by hand. A sample was then taken with a syringe.
Cell viability assays were recorded using the standard MTT method for the PTT efficiency of the NPs in vitro. For the purpose, 100 μL cells (100000 cells per mL) were incubated in a 96-well plate for 24 hours. Then, the culture medium was replaced by 100 μL DMEM containing various concentrations (1.5 to 12 nM) of Au@SiO2 NPs and incubated for another 23 hours. After that, 10 μL of MTT (5 mg mL−1 in PBS) was added into each well and incubated for 4 hours at 37 °C and 5% CO2. Then, the culture medium was removed carefully and 150 μL of dimethyl sulfoxide (DMSO) was added into each well. The optical density was measured using a microplate reader at a wavelength of 490 nm.
Real-time monitoring of cell viability
Generally, adhesive cells can lead to cell death when the loss of cell attachment to the extracellular matrix occurs. Previous research showed that a mechanical mass sensor could be employed for label-free, real-time monitoring of intoxication in terms of the loss in cell adhesion by using the oscillating system because the cell adhesion properties may be related to signs of cell death.29 With the fast-screening-technique, cell viability can be continuously assessed. When a cell dies, its adhesion forces decrease quickly. The oscillating system experiences a change in amplitude at a given frequency as its mass changes. When a cell attached on the oscillating system dies, it will be shrunk and detached. The oscillation presents an exponential attenuation which can be described by a damping process. Thus, a damping coefficient, here referred to as a B-value is an indication for amplitude damping rate in order to quantitatively describe cell viability. In the method, which means the real-time, label-free monitoring system of cell viability mentioned above,39 HeLa cells were allowed to attach onto an oscillating AFM cantilever (SNL-10, k = 0.12 N m−1, f0 = 23 kHz, Bruker Corporation). For that purpose, a drop of 100 μL of HeLa cell solution surrounded the installed cantilever, while the AFM was driven in Intermediate Mode at a given frequency of 22 kHz, so that the cell sediment eventually attaches to the surface of the cantilever. After 30 minutes, the cell solution drop was removed and replaced with a mixture of cell medium and NP solution with varying concentrations. The amplitude of the cantilever was recorded and plotted against time. The B value was calculated from the exponential attenuation by decreasing the amplitude. More technical details can be found in the ESI.†In vitro PPT
MCF-7 cells were cultured in a 96-well plate at 37 °C and 5% CO2 for 24 hours.40 100 μL culture medium was replaced with fresh medium at 12 nM of Au@SiO2. The NP was added to MCF-7 cells incubated for 12 hours. After that, cells were washed with PBS for several times and 100 μL fresh medium was added. Thereafter, the cells with NPs and control groups were both irradiated (520 nm laser, 30 minutes) using different power densities (100 and 400 mW cm−2). Then, MCF-7 cells were cultured at 37 °C and 5% CO2 for another 12 hours. By MTT assay, the cell viability was evaluated.Animal models and in vivo toxicity
Female BALB/c mice (4–6 weeks old) were purchased from Nanjing Cavins Biotechnology Co., Ltd (Nanjing, China) and used under protocols approved by the Regional Ethics Committee for Animal Experiments at Ningbo University, China (Permit No. SYXK (Zhe) 2019-0005). For in vivo toxicity, healthy female BALB/c mice were intravenously injected with 100 μL of PBS solution (control group) and 100 μL Au@SiO2 NPs (12 nM, in PBS solution). After 14 days, the major organs (heart, liver, spleen, lungs and kidneys) from two groups of mice were subjected to the Haematoxylin and Eosin (H&E) staining (Fig. 7).41In vivo biodistribution
Healthy female BALB/c mice were intravenously injected with 100 μL Au@SiO2 NPs (12 nM, in PBS solution).42 At different times after injection (12, 24 and 72 h), the obtained-main organs (heart, liver, spleen, lungs, kidneys and brain) were put into 50 mL centrifuge tubes, and then 2 mL of HNO3 and 6 mL of HCl were mixed with the above tubes in the water bath (100 °C) for 12 hours. When the temperature of the solution was cooled to about 25 °C, it was then filtered with a 0.22 μm membrane, and then the volume of solution was made up to 10 mL by adding pure water. The content of Au from various organs at different times post-injection was measured using an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES, PE Optima 2100DV, PerkinElmer, USA).Results and discussion
Nanoparticle characterisation
As expected, due to the ablation mechanism of PLAL, the spherical NPs were successfully synthesized. The compositions of the synthesised NPs were investigated by means of TEM and HAADF-STEM, from which their morphology and size distribution were more evident relative to the TEM images. The measurements clearly revealed a core–shell structure of the NPs wherein the core consists of pure gold and the coating is silica (Fig. 2D). The core–shell structure was also visualised using SEM. Particle size was determined by evaluating more than 3000 NPs on SEM images and had an average size of 13.0 nm (shell thickness 2 nm) with a narrow size distribution (see in Fig. S3†). It is liable that the silica coating leads to a confinement of the gold core growth resulting in a narrow particle size distribution which was not only proven by SEM but also by the narrow SPR peak width in the UV-vis spectra (Fig. 1B). The SPR peak wavelength appeared at 522 nm which is a typical value for Au@SiO2 NPs of this size. The NP size was dependent on laser settings and was optimized for a fluence of 30 J s−1 leading to a NP size of 13 nm. This size promises a long blood circulation time, since nanoparticles with a diameter below 10 nm are excreted by the kidneys and nanoparticles with a diameter above 100 nm are excreted by the immune system.43,44 For nanoparticles with a diameter between 10 nm and 100 nm, the blood circulation time decreases with the size of the nanoparticle according to current knowledge. At 13 nm, we are at the lower limit of this range. However, NP ζ-potential did hardly vary upon laser settings but only upon the coating surface, in this situation, which means BSA coating. The synthesised NPs had a ζ-potential of −58.7 mV (±13.9 mV, 208 measurements) and are hence negatively charged. The strong repulsion between the NPs causes long-time stability. The results show that UV/VIS spectra and ζ-potential measurements provide a good indication of the stability of the NPs. Comparison between UV-vis spectra taken right after the synthesis and 21 days later presents nearly no difference in absorption which is proof for non-agglomerating particles (Fig. 2). Hence, we are convinced that the presented NPs having long-time stabilization without the addition of stabilisers will be used broadly in further biomedical applications.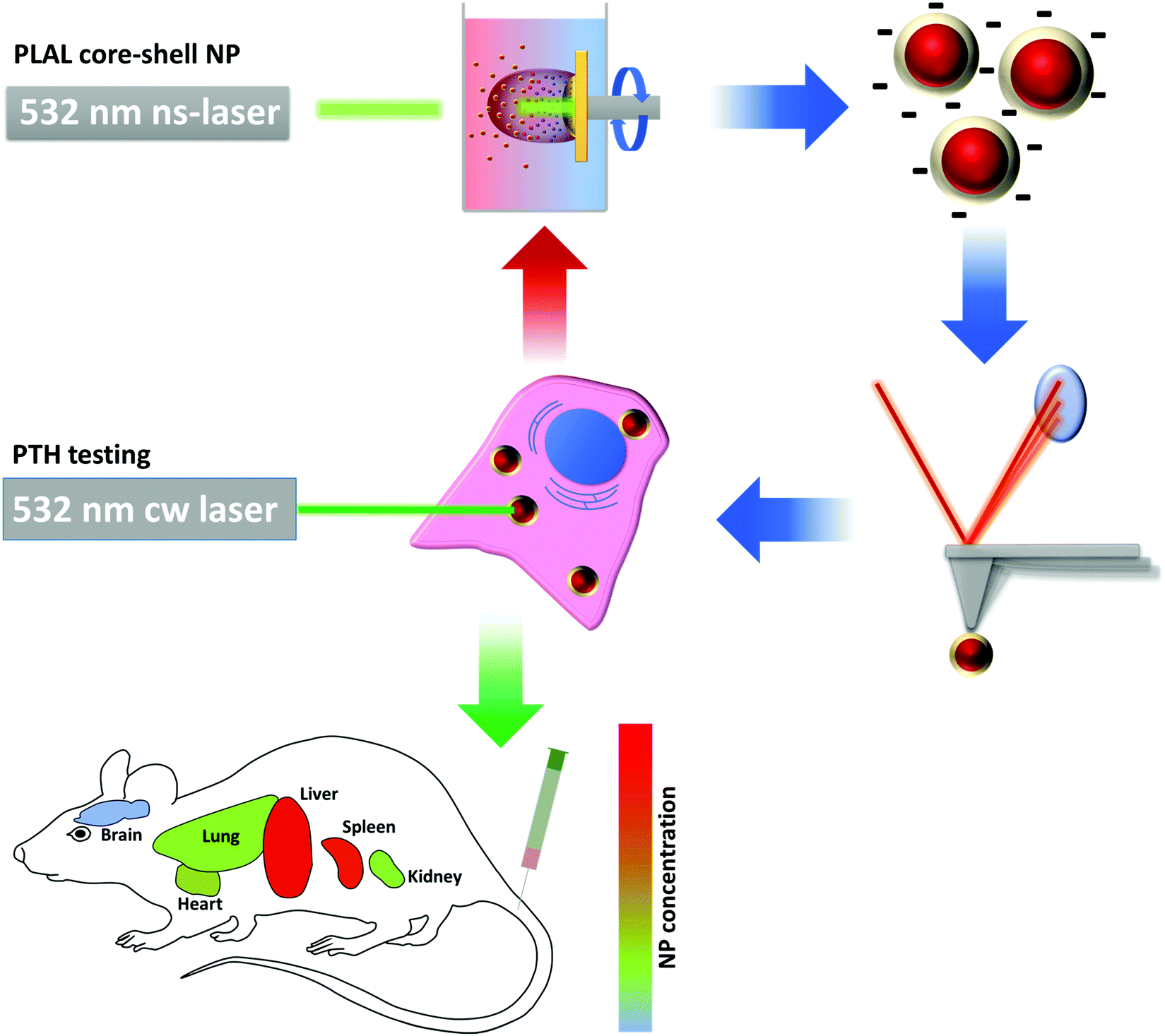 | ||
| Fig. 1 Schematic illustration of the process cycle of nanoparticle (NP) synthesis. Nanoparticles are produced using Pulsed Laser Ablation in Liquid (PLAL). After synthesis, the nanoparticles were characterized in view of their physico-chemical properties. Cell viability and photothermal efficiency (PTH) tests were done in vitro, biodistribution and cell toxicity were measured in vivo. Optimization cycles were applied to achieve the desired NP properties. | ||
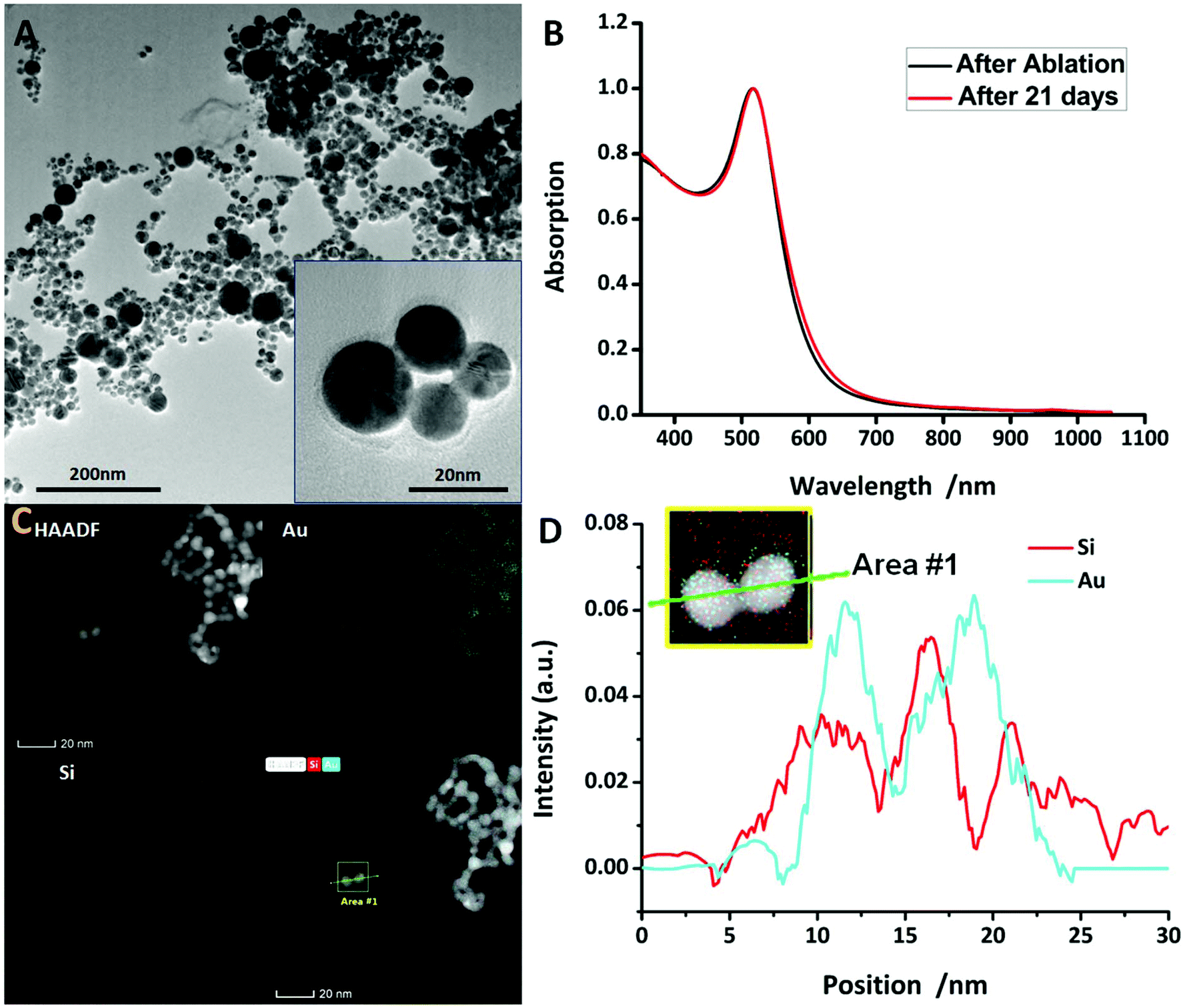 | ||
| Fig. 2 (A) TEM images of the Au@SiO2 NPs. (B) UV-vis absorbance spectra of the Au@SiO2 NPs after ablation and 21 days later. (C) HAADF-STEM image and the corresponding EDX elemental mapping of the Au@SiO2 NPs. The scale bar corresponds to 20 nm. (D) The distribution of Si and Au elements along the line in area #1, where two gold–silica core–shell NPs are shown. | ||
Mechanical properties
The mechanical properties, which in this situation mean elasticity and adhesion, were extracted from force–distance curves measured using Force Spectroscopy mode in AFM. Young's modulus was derived from the slope of the retract part in force–distance curves. Fig. 3A shows an example of a measured force–distance curve after baseline correction. The blue curve shows the extension line, whereas the red line depicts the retraction line. Adhesion effects can be clearly seen since the minimum point of the retraction line is much lower than the minimum point of the extension line. There are several models that describe the behaviour of a body that is intended into another body or plane. The two most appropriate models, proposed by Derjaguin, Muller and Toporov (DMT)45 and Johnson, Kendall and Roberts (JKR)46 were considered for data evaluation, since they consider adhesion effects between a NP and indentation (more details can be found in the ESI†). For comparison, the Hertz model,47 which assumes no adhesion, was also evaluated. Adhesion force could simply be read out from the absolute value of the lowest point of the retraction line of the force–distance curve. Table 1 provides an overview of the mechanical properties. Evaluating the force–distance curves of the Au@SiO2 NPs gave an average value of 496.5 ± 184.1 MPa (JKR) and 733.9 ± 409.6 MPa (DMT). Although applying these two methods led to different values, the true value is expected to be in-between, but closer to JKR due to distinct adhesion forces (again, more details can be found in the ESI†). Although bulk gold has a very high elasticity modulus of 78 GPa,48 Ramos et al. measured an elasticity modulus of 100 GPa (ref. 49) for six-fold icosahedral Au@SiO2 NP 22 nm in size. PEGylation of pure Au NPs, however, seem to strongly decrease the elasticity modulus. Del Pino et al. found the average elasticity modulus of PEGylated Au NPs with a size comparable to the NP synthesised in this work to be at around 20 MPa.13 Hydrogel NPs exhibit a very low elasticity of less than 2 MPa.50 The high elasticity modulus of the hard gold core can be lowered by coating with a polymer or any other softer shell. In this case, SiO2 lowers the elasticity modulus to a value much smaller than that of pure Au NPs or bulk gold. An elasticity modulus of around 500 MPa is still high and it can be expected to be internalised into cells rather than into softer particles. On the other hand, according to previous studies12,51 and theoretical calculations,10 NPs with higher elasticity are more prone to macrophage endocytosis compared to NPs with low elasticity. According to these findings, silica coated NPs are expected to have a longer blood circulation time compared to pure gold NPs. However, there are other aspects that determine the blood circulation time of NPs, too.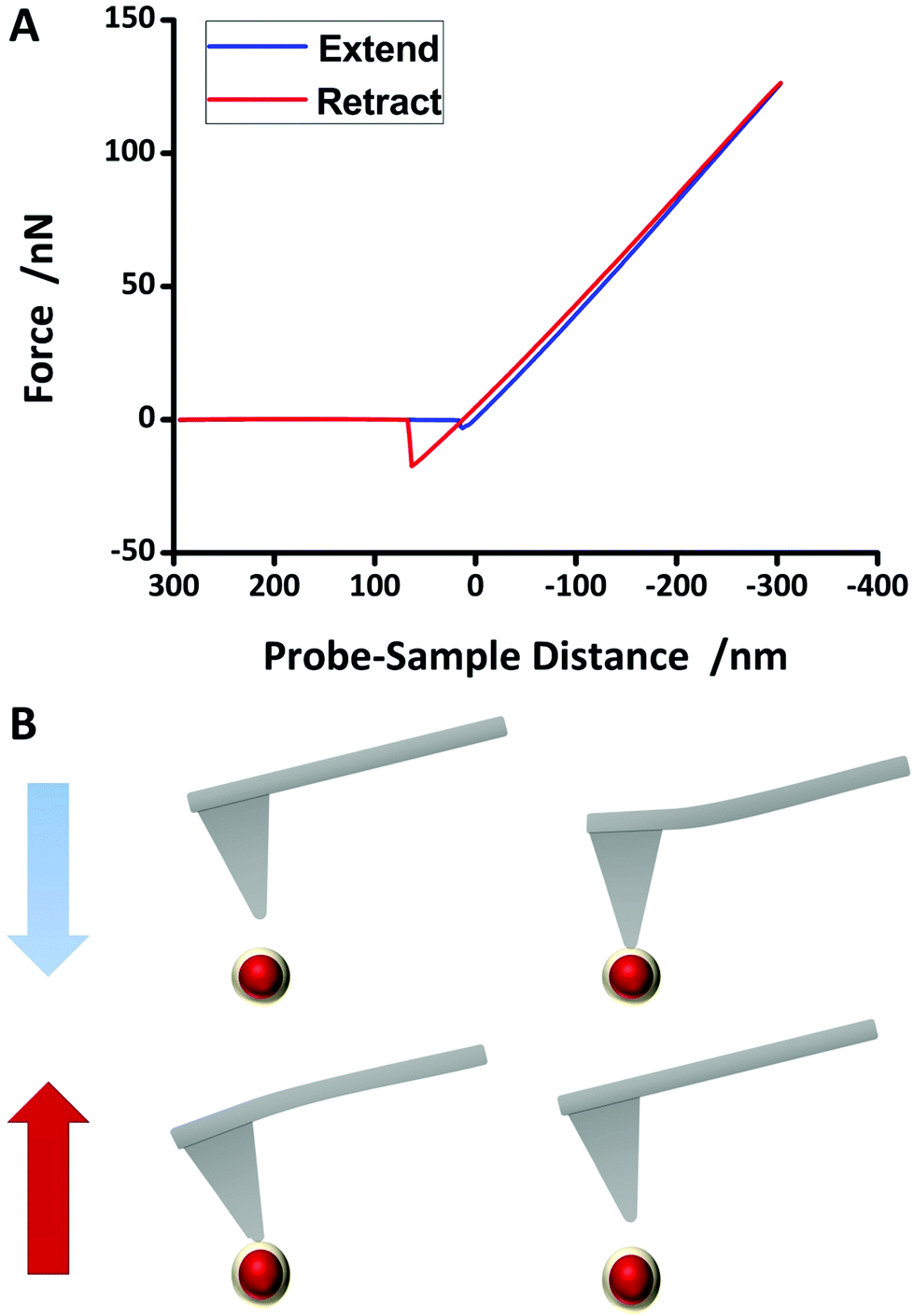 | ||
| Fig. 3 Force spectroscopy process using atomic force microscopy. A tip fixed at the end of a cantilever is located right above a nanoparticle so that the interaction force between the tip and NP is approximately zero. The tip is then extended towards the NP (blue line in (A)). Attractive forces increase slowly, but as the tip approaches further, repulsive forces dominate and the cantilever is deflected. The tip is intended into the NP up to a certain indentation depth, then it is retracted from the NP (red line in (A)). Due to possible adhesion forces, the detachment of the tip from the NP may be shifted. Adhesion force was read from the minimum of retract line. Hertz, DMT and JKR models were fit to the slope of the retraction line. More details can be found in the ESI† of this paper. | ||
| SPR peak | 522 nm | Adhesion | 13.6 nN (± 0.5 nN) |
|---|---|---|---|
| Size | 13.0 nm (±6.0 nm) | Elasticity (DMT) | 733.9 MPa (±409.6 MPa) |
| Hydrodynamic size | 28.3 nm (±9.5 nm) | Elasticity (JKR) | 496.5 MPa (±184.2 MPa) |
| ζ-Potential | −58.7 mV (±13.9 mV) | Elasticity (Hertz) | 978.2 MPa (±353.7 MPa) |
Nanoparticle heating capability
Hyperthermia is well known to induce cell death and is widely used for thermal therapy in clinical trials due to the low thermotolerance of tumours.17 In order to investigate the ability of Au@SiO2 to be applied for PTT, heating experiments were conducted using a continuous laser with a wavelength of 520 nm close to the SPR wavelength. Different NP concentrations in PBS buffer were used and revealed that at the highest concentration that was investigated in this study (12 nM), the solution can be heated after 15 minutes of irradiation and during this time the temperature increased gradually. For inducing the death of tumour cells, a temperature of at least 43 °C is required.52 Hence, the optimal variations of NPs must show a good heating behaviour which is suitable for PTT by heating experiment. Fig. 4A and B show the dependency of the temperature on irradiation power (400 mW) with different concentrations of the NPs and revealed that only 12 nM NPs can reach 43 °C. Therefore, 12 nM NPs will be applied to increase the temperature to a value above 43 °C after 15 min. However, the optimal dosage of the NPs should allow precisely controllable PTT by adjustable external laser irradiation power. Fig. 4C and D show the dependence of the temperature on 12 nM with different irradiation power. Only 400 mW (power on the surface of solution) could suffice at 43 °C with short irradiation time, because the damage to the surrounding healthy tissues would be minimized.53 In order to verify the efficiency of PTT related to the NPs, 100 mW power laser irradiation needs to be utilized to improve PTT efficiency in vitro to avoid unnecessary damage caused by high power laser irradiation.54 The silica coating of the NP is not expected to affect the heating capability.55,56 Instead, the silica coating has a shape-conserving effect on the NP and prevents the shape deforming due to the melting of the gold core after laser irradiation.57,58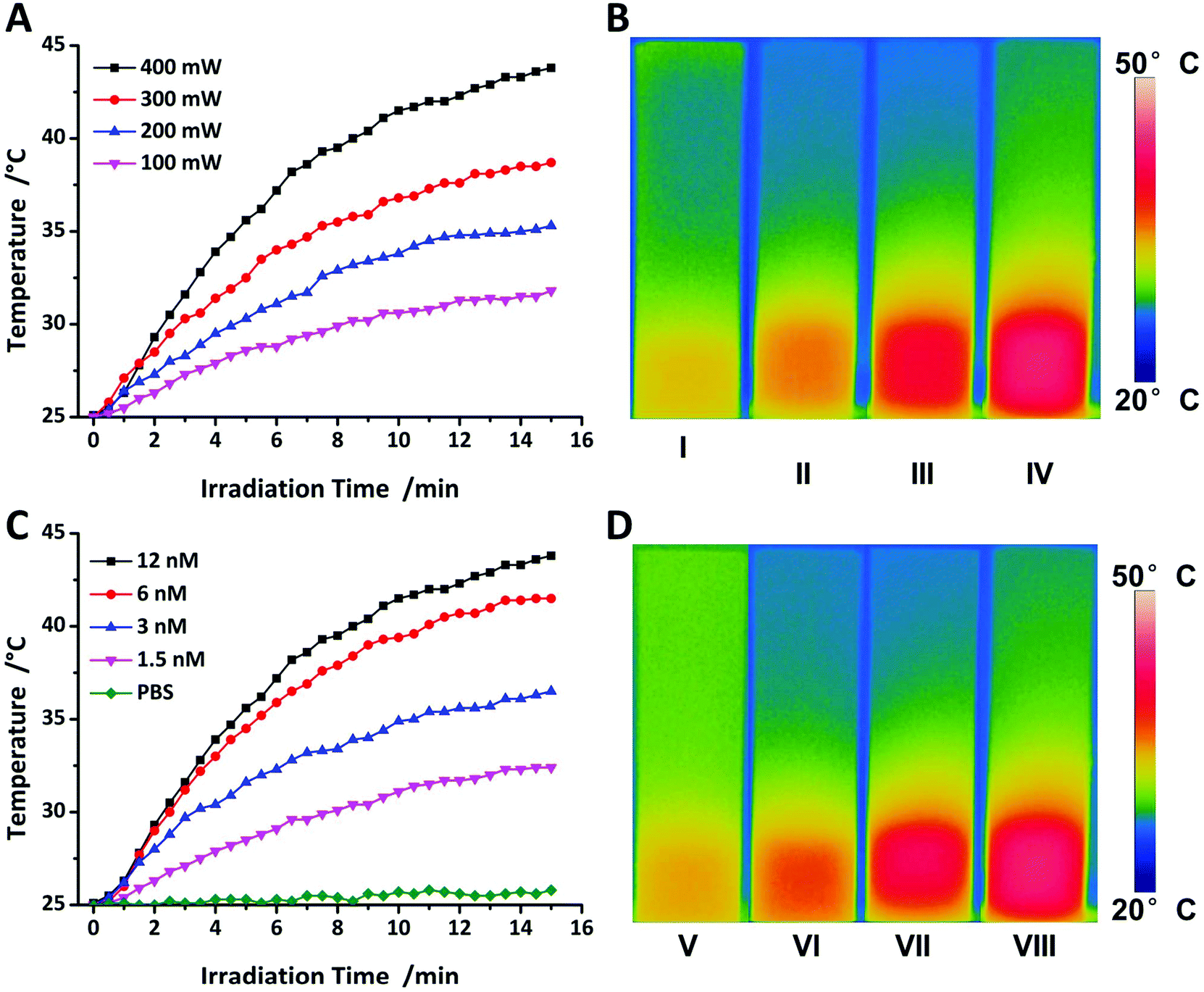 | ||
| Fig. 4 Temperature evaluation of the Au@SiO2 NPs by exposure to a 520 nm laser (A) 12 nM with different power densities, (C) 400 mW cm−2 with various concentrations. NIR photothermal images of the Au@SiO2 NP aqueous solution under different irradiation conditions for 15 minutes, (B) 12 nM with different power densities, (D) 400 mW cm−2 with various concentrations. | ||
Cell viability
We evaluated the cell viability of HeLa cells with varying concentrations 1.5, 3, 6 and 12 nM by resazurin tests and using a real-time monitoring system (Table 2). Although the cell viability decreases slightly with increasing NP concentration, it generally remains at a high level. Resazurin tests revealed a viability of 87.83% after 12 nM NP exposure to HeLa cells for 24 hours. In real-time monitoring of the cell viability method, the B-value extracted from the amplitude decreases by detachment of cells. The low B-value results reveal a low amplitude damping rate when the NPs are incubated with the cells. It proved that the NPs have good biocompatibility. Vácha et al. demonstrated that the NPs are expected to be ingested by cells via receptor-mediated endocytosis.59 The results of the B-values obtained from the real-time cell viability measurements are between 3 × 10−5 s−1 and 7 × 10−5 s−1, indicating high cell biocompatibility compared with the results of Yang et al.39 The B-value findings are in the range of PEGylated Au NPs of the same size which are known to be biocompatible. Both test findings, B-value and resazurin test, are complementary. Although the B-value can be regarded as an indicator of acute toxicity within the early stage of toxicity of NP exposure to the cells, the resazurin test clarifies the toxicity after 24 hours.| NP concentration | 12 nM | 6 nM | 3 nM | 1.5 nM |
|---|---|---|---|---|
| B-Value after real-time monitoring test | −6.945 × 10−5 s−1 | −4.988 × 10−5 s−1 | −4.001 × 10−5 s−1 | −3.423 × 10−5 s−1 |
| Cell viability according to resazurin test | 87.83% | 90.96% | 93.26% | 94.81% |
PTT efficiency in vitro
In order to assess the PTT efficiency of the NPs, MTT assay was utilized to evaluate the toxicity by continuous laser irradiation (520 nm). The results shown in Fig. 5A undoubtedly confirm the cell viability of MCF-7 cells incubated without/with the NPs under 100 mW laser irradiation. The cells without NPs are dying during photothermal treatment due to hyperthermia and the cells with the NPs are dying by apoptosis during photothermal treatment due to the PTT effect of the NPs. In Fig. 5C, the black dots represent the relative ratio increasing constantly at 100 mW, thus it can be seen that the PTT efficiency perfectly correlates with the materials, in our case, which means the AuNP@SiO2. Fig. 5B emphasizes the cell viability of MCF-7 cells incubated without/with the NPs under 400 mW laser irradiation. It can be obviously seen that hyperthermia can lead to the death of many cells even without the NPs. In Fig. 5C, the red dots present cell deaths which are related to the PTT efficiency of the NPs in the early stage of the treatment (<15 minutes). As a result, the PTT effect can surely be enhanced with the NPs under low power of laser irradiation to avoid damaging healthy cells or even tissues by hyperthermia.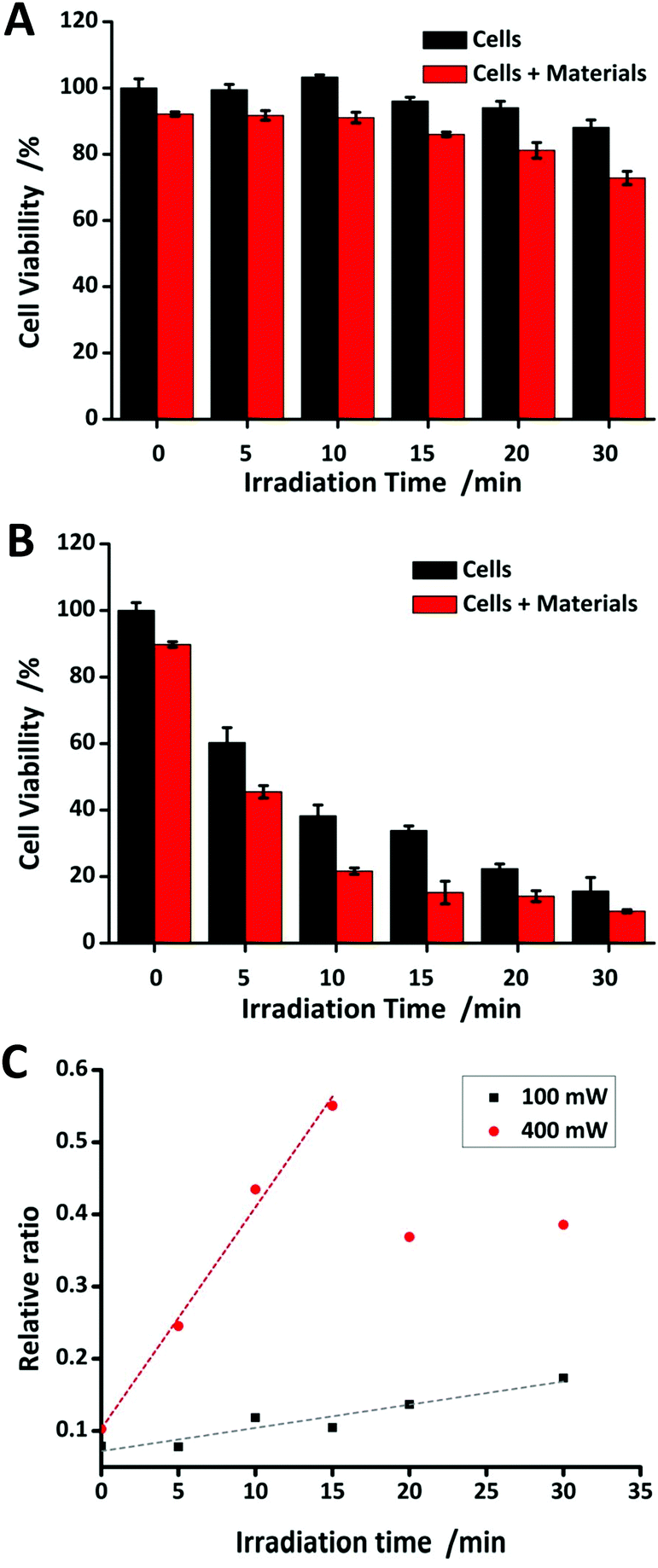 | ||
| Fig. 5 Cell viabilities of MCF-7 cells incubated with 12 nM Au@SiO2 NPs in (A) 100 mW cm−2 and (B) 400 mW cm−2, after being irradiated with a 520 nm laser for different times. (C) The relationship between cell deaths and irradiation time in 100 and 400 mW cm−2. | ||
In vivo toxicity study
In general, NP behaviour in living mechanisms is not well understood, although a lot of investigations have been done so far.44,60–63 The main reason is the strongly varying conditions of the experiments, especially synthesis routes, stabilisers and by-products of the synthesised NPs.44 To confirm the role of toxicity of the NPs in vivo, good biodistribution is a crucial indication for further PTT therapy in vivo and other biomedical applications, since NP behaviour in vivo cannot be exactly predicted from in vitro tests. With our NPs, a clear result can be obtained that is not affected by stabilisers or other molecules. We intravenously injected 100 μL NP solution into the mouse tail. In order to quantitatively assess toxicity of the NPs in vivo, ICP-MS was utilized for measuring the NP contents in the main organs which include heart, liver, spleen, lungs, kidneys and brain after 24 hours. In Fig. 6, the results showed that most of the NPs were found in liver and spleen. This might be due to the high accumulation of the NPs in the liver and spleen by the induced immune system. However, after 24 hours, the NPs were also detected in other organs, indicating a long blood circulation time and good distribution of the NPs. Interestingly, no NPs were found in the brain and this might be due to the blood brain barrier. The amount of accumulated NPs in all organs decreased with time, which is an indication of good biodegradability. Hence, the NPs can be widely used in biomedical applications.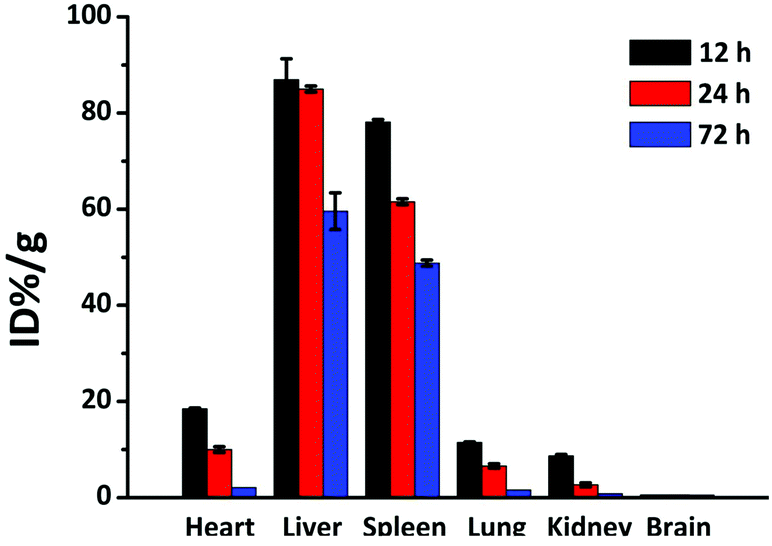 | ||
| Fig. 6 Biodistribution of gold in major mice organs at 12, 24 and 72 h post-injection intravenously. | ||
Furthermore, to further investigate the toxicity to the main organs, the histological assessment of tissues was performed to determine whether different concentrations of Au@SiO2 NPs could induce tissue damage, inflammation, or lesions at 14 days after the injection. The pathological observation of the major organs in vivo with varying concentrations of the NPs (1.5, 3, 6 and 12 nM) showed that there was no damage in the heart, liver, spleen, lungs and kidneys (see Fig. 7). The pathological analysis implies that the Au@SiO2 NPs synthesized by PLAL have excellent biocompatibility and great potential in further biomedical applications. No morphological changes of the mice were observed after NP injection, leading to the conclusion that the NP concentration was not too high for an appropriate evaluation of the results.64–66
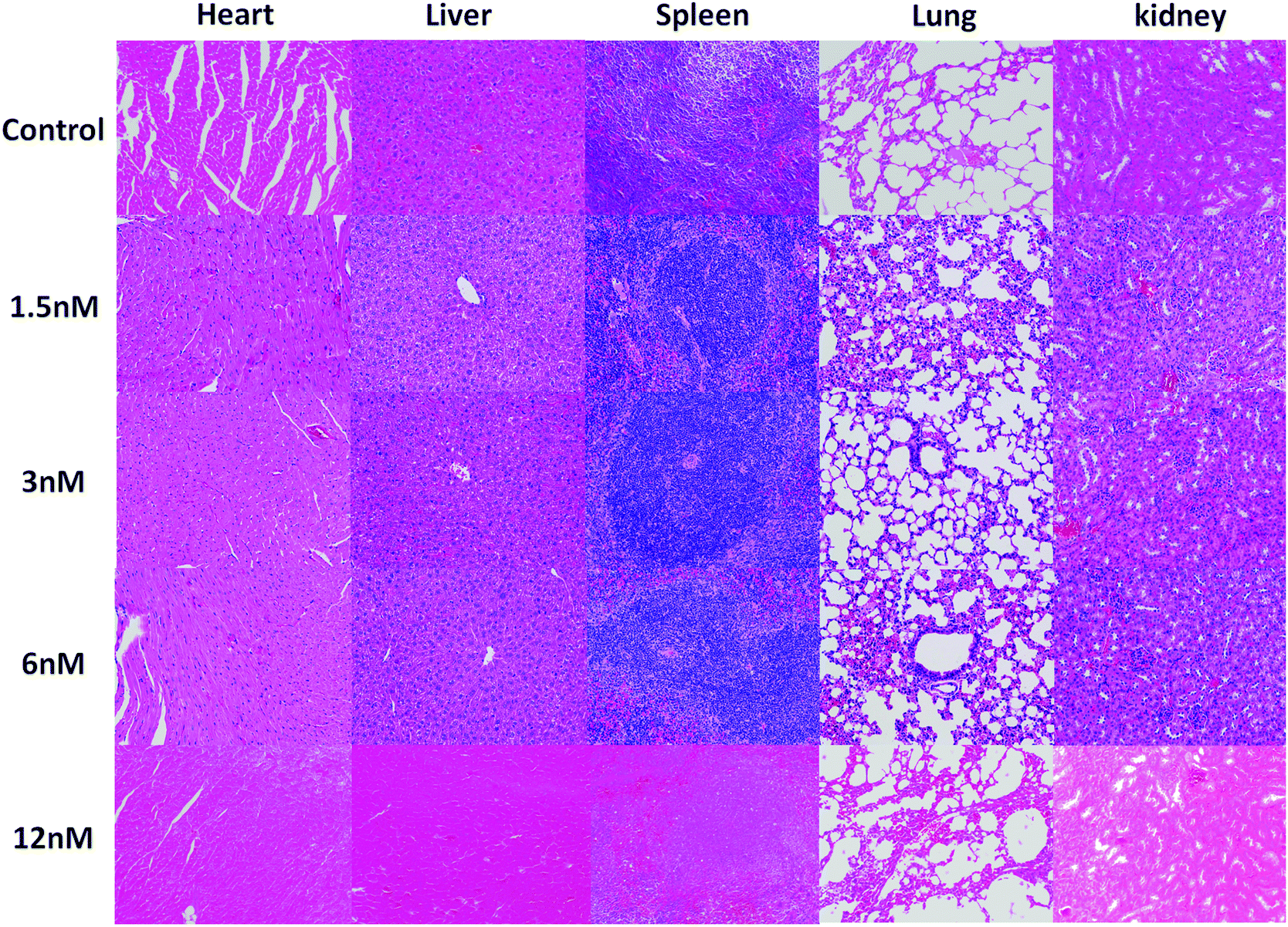 | ||
| Fig. 7 H&E staining images of the major organs collected from mice after different treatments on the 14th day (scale bar: 100 μm). | ||
Conclusions
Pulsed laser ablation of a gold target in distilled water containing small quantities of sodium chloride and sodium waterglass was employed to synthesise Au@SiO2 core–shell nanoparticles in an easy and inexpensive way. Electron microscopy images revealed an average NP diameter of 13.0 nm (shell thickness 2 nm). The NPs were clean and stable over at least 21 days without the addition of stabilisers. Biocompatibility could be proven in vitro by means of resazurin viability test and real-time viability monitoring. The NP elasticity modulus was at 0.5 GPa and hence about 200 times lower than the elasticity modulus of pure gold NPs. They are expected to be more resistant to macrophage endocytosis compared to pure gold NPs and more prone to cell incubation compared to softer NPs. The NPs could easily be coated with bovine serum albumin. The coating was observed by electron microscopy and atomic force microscopy. In vitro and in vivo tests showed low toxicity of the NPs on HeLa cells and MCF-7 cells as well as on BALB/c mice. Investigation of the biodistribution of our NPs in mice exhibited NPs in heart, liver, spleen, lungs and kidneys, with the largest proportion found in spleen and liver. Long-time observation revealed the clearance of the NPs from the organs. The NPs could not be observed in the brain, which suggests that the brain–blood-barrier was not passed by the NPs. Photothermal efficiency could be proved by laser irradiation, although irradiation at 532 nm also damaged biological tissue. However, the gold silica, non-toxic and bioavailable NPs fabricated in this work provide the basic for future research. The synthesised NPs can be further modified to adopt the properties as desired, e.g. by surface silanisation with cancer cell ligands.Over the last decade, remarkable progress has been made in scaling up the synthesis of liquid phase NPs by PLAL. This physical method of the synthesis of NPs has been, in general, much improved, but the adaptation of traditional synthesis is not sufficient. Therefore, in this work, it is now modified, and this has enabled the tech-scale production of the NPs with complex structure. We have demonstrated an easy and effective way to synthesize gold–silica core–shell NPs by optimizing PLAL and provided a guide on how to investigate their physico-chemical properties underlying their biological effects of application towards cancer PTT. Using this method, we were able to investigate the nanomechanical properties and biomedical behaviour as well as the PTT efficiency independent of the influence of any stabilisers or other molecules. Previous work on the fabrication of silicate coated noble metal NPs via PLAL used noble metal salts dissolved in the liquid and silica as the target.67,68 Trace amounts of these salts may have toxic effects. In our work, we used sodium silicate which could be well separated and is non-toxic. In line with this approach, the reproducible and scalable liquid phase NPs with less toxicity and good stability can be realized for tech-scale production by further adjustment and modification. Moreover, a more complete study of associations between the physico-chemical properties of the NPs and their biological effects may enable more useful and advanced NP synthesis towards specific requirements of biomedical applications. For instance, in order to satisfy the requirements of advanced diagnosis or therapy based on functional NPs in vivo, well-designed NPs with excellent specific properties can be synthesized by modified PLAL. In essence, this is the final purpose. Together, these efforts on improving the conditions of PLAL could enable the controllable tech-scale production of various functional NPs for the increasing industrial interest and the onset of commercialization in a wide range of biomedical studies and clinical applications in the future. This, in turn, would greatly promote a safe, sustainable use and high-precision system based on PLAL as well as effectively boost PLAL technique innovation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51803228 and U1432114), the National Key R&D Program of China (2018YFC0910601) and the Zhejiang Provincial Natural Science Foundation of China (LGF18H180017). The authors are very thankful to the fine mechanics workshop of Philipps-University of Marburg, Germany, for constructing the rotating target setup. Also, we are thankful to Greta Linden, working group Vazquez, Biochemistry Department of Philipps-University of Marburg, Germany, for performing the resazurin cell viability tests and providing HeLa cells for real-time monitoring tests.References
- R. A. Freitas, Nanomedicine, 2005, 1, 2–9 CrossRef CAS PubMed.
- D. L. Fedlheim and C. A. Foss, Metal Nanoparticles: Synthesis, Characterization, and Applications, Taylor & Francis, 2001 Search PubMed.
- L. Wei, J. Lu, H. Xu, A. Patel, Z.-S. Chen and G. Chen, Drug Discovery Today, 2015, 20, 595–601 CrossRef CAS PubMed.
- H. Daraee, A. Eatemadi, E. Abbasi, S. Fekri Aval, M. Kouhi and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 410–422 CrossRef CAS PubMed.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, Nano Res., 2015, 8, 1771–1799 CrossRef CAS.
- Y. Iida, A. Tsuge, Y. Uwamino, H. Morikawa and T. Ishizuka, J. Anal. At. Spectrom., 1991, 6, 541–544 RSC.
- S. L. Y. Tang, R. L. Smith and M. Poliakoff, Green Chem., 2005, 7, 761–762 RSC.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- X. Yi, X. Shi and H. Gao, Phys. Rev. Lett., 2011, 107, 98101 CrossRef PubMed.
- L. Sun, R. Riedel, S. G. Stanciu, F. Yang, N. Hampp, L. Xu and A. Wu, J. Mater. Chem. B, 2018, 6, 2960–2971 RSC.
- S. L. Tao and T. A. Desai, J. Controlled Release, 2005, 109, 127–138 CrossRef CAS PubMed.
- P. Del Pino, F. Yang, B. Pelaz, Q. Zhang, K. Kantner, R. Hartmann, N. Martinez de Baroja, M. Gallego, M. Möller and B. B. Manshian, et al. , Angew. Chem., Int. Ed., 2016, 55, 5483–5487 CrossRef CAS PubMed.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- S. Eustis, M. A. El-sayed and M. Kasha, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- L. C. Kennedy, L. R. Bickford, N. A. Lewinski, A. J. Coughlin, Y. Hu, E. S. Day, J. L. West and R. A. Drezek, Small, 2011, 7, 169–183 CrossRef CAS PubMed.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed.
- P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
- G. A. Sotiriou, F. Starsich, A. Dasargyri, M. C. Wurnig, F. Krumeich, A. Boss, J.-C. Leroux and S. E. Pratsinis, Adv. Funct. Mater., 2014, 24, 2818–2827 CrossRef CAS.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- S. Gai, G. Yang, P. Yang, F. He, J. Lin, D. Jin and B. Xing, Nano Today, 2018, 19, 146–187 CrossRef CAS.
- E. J. King and M. McGeorge, Biochem. J., 1938, 32, 417 CrossRef CAS PubMed.
- G. A. Sotiriou, T. Sannomiya, A. Teleki, F. Krumeich, J. Vörös and S. E. Pratsinis, Adv. Funct. Mater., 2010, 20, 4250–4257 CrossRef CAS PubMed.
- Q. Zeng, Y. Zhang, W. Ji, W. Ye, Y. Jiang and J. Song, ACS Appl. Mater. Interfaces, 2014, 6, 19327–19335 CrossRef CAS PubMed.
- C. Hanske, M. N. Sanz-Ortiz and L. M. Liz-Marzán, Adv. Mater., 2018, 30, 1707003 CrossRef PubMed.
- J. L. Montaño-Priede, J. P. Coelho, A. Guerrero-Martínez, O. Peña-Rodríguez and U. Pal, J. Phys. Chem. C, 2017, 121, 9543–9551 CrossRef.
- M. P. Brandon, D. M. Ledwith and J. M. Kelly, J. Colloid Interface Sci., 2014, 415, 77–84 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- J. A. Varner and D. A. Cheresh, Curr. Opin. Cell Biol., 1996, 8, 724–730 CrossRef CAS PubMed.
- L. Zhang, Z. Cao, Y. Li, J.-R. Ella-Menye, T. Bai and S. Jiang, ACS Nano, 2012, 6, 6681–6686 CrossRef CAS PubMed.
- J. Li, R. Cai, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2015, 3, 5806–5814 RSC.
- C. Fu, H. Yang, M. Wang, H. Xiong and S. Yu, Chem. Commun., 2015, 51, 3634–3636 RSC.
- S. Ge, K. Kojio, A. Takahara and T. Kajiyama, J. Biomater. Sci., Polym. Ed., 1998, 9, 131–150 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- B. Pittenger, in Nanomechanical Anal. High Perform. Mater, ed. A. Tiwari, Springer, Netherlands, 2014, pp. 31–51 Search PubMed.
- S. Wang, X. Li, Y. Chen, X. Cai, H. Yao, W. Gao, Y. Zheng, X. An, J. Shi and H. Chen, Adv. Mater., 2015, 27, 2775–2782 CrossRef CAS PubMed.
- M. Zheng, C. Yue, Y. Ma, P. Gong, P. Zhao, C. Zheng, Z. Sheng, P. Zhang, Z. Wang and L. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity, 2000 Search PubMed.
- F. Yang, R. Riedel, P. del Pino, B. Pelaz, A. H. Said, M. Soliman, S. R. Pinnapireddy, N. Feliu, W. J. Parak and U. Bakowsky, et al. , J. Nanobiotechnol., 2017, 15, 23 CrossRef PubMed.
- B. Jang, J.-Y. Park, C.-H. Tung, I.-H. Kim and Y. Choi, ACS Nano, 2011, 5, 1086–1094 CrossRef CAS PubMed.
- T.-J. Wu, Y.-K. Tzeng, W.-W. Chang, C.-A. Cheng, Y. Kuo, C.-H. Chien, H.-C. Chang and J. Yu, Nat. Nanotechnol., 2013, 8, 682 CrossRef CAS PubMed.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Chen, Q. Yang, D. W. Felsher and H. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668–7672 CrossRef CAS PubMed.
- J. F. Hillyer and R. M. Albrecht, Microsc. Microanal., 1998, 4, 481–490 CrossRef CAS PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- B. V. Derjaguin, V. M. Muller and Y. Toporov, J. Colloid Interface Sci., 1975, 53, 314–326 CrossRef CAS.
- K. L. Johnson, K. Kendall and A. D. Roberts, Proc. R. Soc. London, 1971, 324, 301–313 CrossRef CAS.
- H. Hertz, J. Reine Angew. Math., 1881, 92, 156–171 Search PubMed.
- Q. F. Gu, G. Krauss, W. Steurer, F. Gramm and A. Cervellino, Phys. Rev. Lett., 2008, 100, 45502 CrossRef CAS PubMed.
- M. Ramos, L. Ortiz-Jordan, A. Hurtado-Macias, S. Flores, T. J. Elizalde-Galindo, C. Rocha, B. Torres, M. Zarei-Chaleshtori and R. R. Chianelli, Materials, 2013, 6, 198–205 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Adv. Drug Delivery Rev., 2017, 108, 51–67 CrossRef CAS PubMed.
- A. C. Anselmo, M. Zhang, S. Kumar, D. R. Vogus, S. Menegatti, M. E. Helgeson and S. Mitragotri, ACS Nano, 2015, 9, 3169–3177 CrossRef CAS PubMed.
- R. K. Gilchrist, R. Medal, W. D. Shorey, R. C. Hanselman, J. C. Parrott and C. B. Taylor, Ann. Surg., 1957, 146, 596 CrossRef CAS PubMed.
- I. H. El-Sayed, X. Huang and M. A. El-Sayed, Cancer Lett., 2006, 239, 129–135 CrossRef CAS PubMed.
- J. R. Melamed, R. S. Edelstein and E. S. Day, ACS Nano, 2015, 9, 6–11 CrossRef CAS PubMed.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2011, 112, 2373–2433 CrossRef PubMed.
- J.-T. Song, X.-Q. Yang, X.-S. Zhang, D.-M. Yan, Z.-Y. Wang and Y.-D. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 17287–17297 CrossRef CAS PubMed.
- M. Kanehara, Y. Watanabe and T. Teranishi, J. Nanosci. Nanotechnol., 2009, 9, 673–675 CrossRef CAS PubMed.
- J. Chen, R. Zhang, L. Han, B. Tu and D. Zhao, Nano Res., 2013, 6, 871–879 CrossRef CAS.
- R. Vácha, F. J. Martinez-Veracoechea and D. Frenkel, Nano Lett., 2011, 11, 5391–5395 CrossRef PubMed.
- V. J. Mohanraj and Y. Chen, Trop. J. Pharm. Res., 2006, 5, 561–573 Search PubMed.
- T. K. Indira and P. K. Lakshmi, Int. J. Pharm. Sci. Nanotechnol., 2010, 3, 1035–1042 CAS.
- Z. Magdolenova, A. Collins, A. Kumar, A. Dhawan, V. Stone and M. Dusinska, Nanotoxicology, 2014, 8, 233–278 CrossRef CAS PubMed.
- M. Ajdary, M. A. Moosavi, M. Rahmati, M. Falahati, M. Mahboubi, A. Mandegary, S. Jangjoo, R. Mohammadinejad and R. S. Varma, Nanomaterials, 2018, 8, 634 CrossRef PubMed.
- Y.-S. Chen, Y.-C. Hung, I. Liau and G. S. Huang, Nanoscale Res. Lett., 2009, 4, 858 CrossRef CAS PubMed.
- O. Bar-Ilan, R. M. Albrecht, V. E. Fako and D. Y. Furgeson, Small, 2009, 5, 1897–1910 CrossRef CAS PubMed.
- C. Lasagna-Reeves, D. Gonzalez-Romero, M. A. Barria, I. Olmedo, A. Clos, V. M. S. Ramanujam, A. Urayama, L. Vergara, M. J. Kogan and C. Soto, Biochem. Biophys. Res. Commun., 2010, 393, 649–655 CrossRef CAS PubMed.
- J. R. González-Castillo, E. Rodriguez, E. Jimenez-Villar, D. Rodríguez, I. Salomon-García, G. F. de Sá, T. García-Fernández, D. B. Almeida, C. L. Cesar and R. Johnes, et al. , Nanoscale Res. Lett., 2015, 10, 399 CrossRef PubMed.
- E. Jiménez, K. Abderrafi, R. Abargues, J. L. Valdés and J. P. Martínez-Pastor, Langmuir, 2010, 26, 7458–7463 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07129f |
| This journal is © The Royal Society of Chemistry 2020 |