
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Time-controllable roll-up onset of polythiophene sheets into nanotubes that exhibit circularly polarized luminescence†
N.
Kameta
 * and
T.
Shimizu
* and
T.
Shimizu
Nanomaterials Research Institute, Department of Materials and Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: n-kameta@aist.go.jp; Fax: +81-29-861-4545; Tel: +81-29-861-4478
First published on 8th January 2020
Abstract
Self-assembly of a polythiophene-conjugated glycolipid exclusively produced square sheets a few micrometers on each side. Seventeen hours after the sheets were dispersed in ethanol at 25 °C, they suddenly started to roll up, and eventually they were completely transformed into nanotubes. The onset timing of the roll-up was temperature-dependent. The roll-up involved rearrangement of the molecular packing within the bilayer membranes, which was accompanied by strengthening of the intermolecular hydrogen bonds, alteration of the polythiophene aggregation mode and enhancement of supramolecular chirality due to chiral packing. The nanotubes exhibited not only strong fluorescence derived from J-type aggregation of the polythiophene aromatic moiety but also circularly polarized luminescence (CPL) originating from the left-handed helicity of the polythiophene main chain backbone. Because the CPL onset was concurrent with the sheet roll-up, the CPL onset was also able to be controlled by varying the temperature. Such delayed CPL onset has never been reported in chiral supramolecular structures, in which CPL onset and helicity inversion usually begin immediately upon application of a stimulus and then progress either quickly or gradually. Our findings can be expected to facilitate the development of new stimulus-responsive supramolecular structures that can be used for delayed-action capsules or optical switching devices.
Introduction
Introduction of stimulus-responsiveness into supramolecular nanostructures permits drastic changes in their morphologies and their chemical, physical, and mechanical properties. It has therefore attracted considerable attention for the development of functional soft materials for biological, medical, informational, environmental, and energy-related applications.1–13 In general, stimulus-responsive supramolecular systems begin to respond as soon as the stimulus is applied, although the response can then progress quickly or gradually, depending on the stability of the supramolecular structure and the strength of the stimulation.14–16 In contrast, delayed responses to stimuli have rarely been reported.15,16Most supramolecular nanostructures are formed directly by self-assembly of rationally designed low-molecular-weight, oligomeric, or polymeric amphiphiles, although seed nanostructures can be used to grow into nanofibers with controlled lengths by means of living supramolecular polymerization.17–21 Supramolecular nanotubes22–26 are often formed by transformations of precursors such as ribbon-, toroid-, ring-, sheet-, or disk-like nanostructures. Specifically, twisted and/or helically coiled ribbon-like nanostructures transform into nanotubes through a “closing pitch” mechanism or a “growing width” mechanism.27–32 In the former, the ribbon width remains constant while the pitch gradually shortens; whereas in the latter, the pitch remains constant while the width gradually increases. Toroid- and ring-like nanostructures assemble into nanotubes by stacking on top of one another,33–37 and sheet-like nano- and microstructures roll up into nanotubes that have whirling patterns.38–43 Zipping of the both face to face edgings of the rolled-up sheet results in perfectly tubular shapes. Disk-like nanostructures grow into fragments of the membrane walls of nanotubes.44,45 Securement of lifetimes of such precursors has a potential to be useful for time-controllable onset of the transformation into nanotubes. However, the precursors have a wide range of lifetimes. In fact, most precursors are often observable with the nanotubes. In addition, the selective isolation of the precursors at the metastable state has not been achieved yet.
Herein we report time-controllable roll-up onset of polythiophene sheets into nanotubes. The onset timing of the roll-up strongly depended on the ambient temperature. The nanotubes emitted not only strong fluorescence derived from J-type aggregation of the polythiophene aromatic moiety but also circularly polarized luminescence (CPL) originating from the left-handed helicity of the polythiophene main chain backbone; in contrast the precursor sheets emitted neither fluorescence nor CPL. In other words, we have developed a system that exhibits delayed-onset fluorescence and CPL.
Results and discussion
Selective construction of nanotubes and sheets
As previously reported by our group, the self-assembly of compounds formed by condensation reactions between synthetic glycolipids and aromatic boronic acids independently produces nanoparticles, nanofibers, or nanotubes, depending on the chemical structures of the compounds.46–48 In this study, we investigated the self-assemblies of two poly(thiopheneboronic acid) derivatives conjugated with different glycolipids, PTB-GlcOle and PTB-GlcSte (Scheme 1). They were synthesized by polymerization of the corresponding monomers that were in turn obtained by condensation reactions between thiopheneboronic acid and N-(9-cis-octadecenoyl)-β-D-glucopyranosylamine49 and N-stearoyl-β-D-glucopyranosylamine.50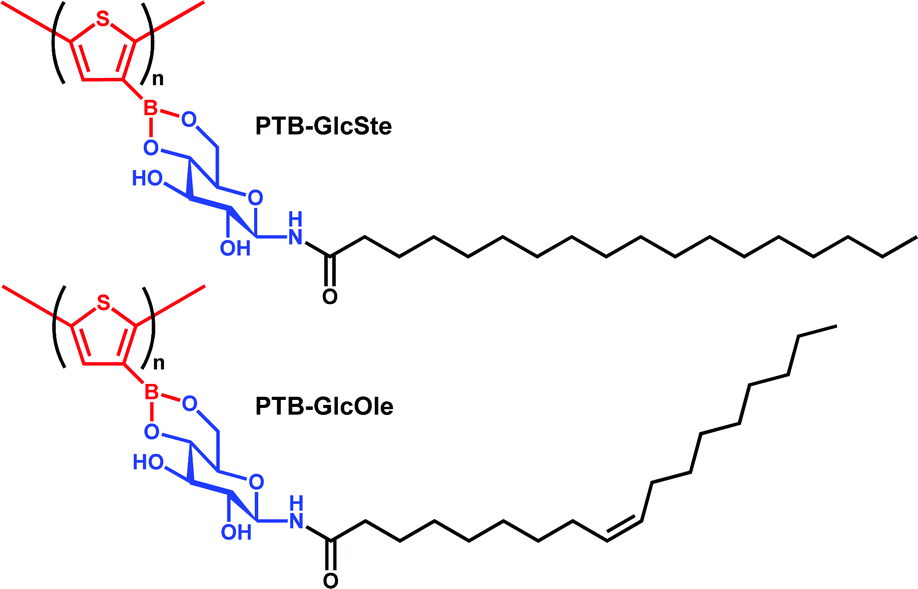 | ||
| Scheme 1 Chemical structures of PTB-GlcSte and PTB-GlcOle polymers. | ||
The two polymers were self-assembled as follows. Films of PTB-GlcOle and PTB-GlcSte were prepared by solvent evaporation of the solutions of PTB-GlcOle (1 mg) and PTB-GlcSte (1 mg) in toluene (1 mL). Each film was dispersed in dehydrated ethanol (1 mL) at 25 °C. Scanning electron microscopy revealed that in ethanol solution, PTB-GlcOle self-assembles into nanotubes with diameters of 250–350 nm and lengths of a few micrometers (Fig. 1a), and scanning transmission electron microscopy indicated that the nanochannels were 40–50 nm in diameter (Fig. S1†). In contrast, PTB-GlcSte self-assembled into square sheets a few micrometers on each side (Fig. 1b). Tapping mode atomic force microscopy indicated that the thickness of the sheets were about 50–65 nm (Fig. S2†). Although the hydrocarbon chains of both glycolipid moieties had the same number of carbons, we were able to control the morphology of the self-assembled structure by choosing the saturated or unsaturated lipid.
 | ||
| Fig. 1 Scanning electron microscopy images of (a) PTB-GlcOle-nanotubes, (b) PTB-GlcSte-sheets, (c) intermediates of the transformation from the PTB-GlcSte-sheets to PTB-GlcSte-nanotubes and (d) PTB-GlcSte-nanotubes. | ||
Powder X-ray diffraction (XRD) analysis and infrared (IR) spectroscopy provided information about the molecular packing of the nanotubes (hereafter referred to as the PTB-GlcOle-nanotubes) and the sheets (hereafter referred to as the PTB-GlcSte-sheets). The XRD patterns showed a single diffraction peak in the small-angle region (Fig. S3†), and the membrane-stacking periodicities (d) of the PTB-GlcOle-nanotubes and PTB-GlcSte-sheets were 4.44 and 4.89 nm, respectively. These findings indicate that both structures consist of stacked bilayer membranes (Fig. 2a, S4 and S5†). The thickness (50–65 nm) of the PTB-GlcSte-sheets, estimated from atomic force microscopy, was approximately 10–13 times the d value, the indication being that 10–13 bilayers stacked to form the sheets. Because the wide-angle regions in the XRD patterns of both structures had peaks at 0.41 and 0.38 nm (Fig. S3†), we assigned the lateral packing of the hydrocarbon chains as an orthorhombic perpendicular type51,52 (Fig. S4 and S5†). The subcell type suggested that PTB-GlcOle and PTB-GlcSte packed in an interdigitated fashion39 within the bilayers. The IR spectra of the PTB-GlcOle-nanotubes and PTB-GlcSte-sheets showed the presence of amide hydrogen bonds (Fig. S3†). The wavenumbers of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration band (amide-I) and the N–H deformation vibration band (amide-II) indicated that intermolecular hydrogen bonds stabilize the bilayers of both structures (Fig. S4 and S5†).
O stretching vibration band (amide-I) and the N–H deformation vibration band (amide-II) indicated that intermolecular hydrogen bonds stabilize the bilayers of both structures (Fig. S4 and S5†).
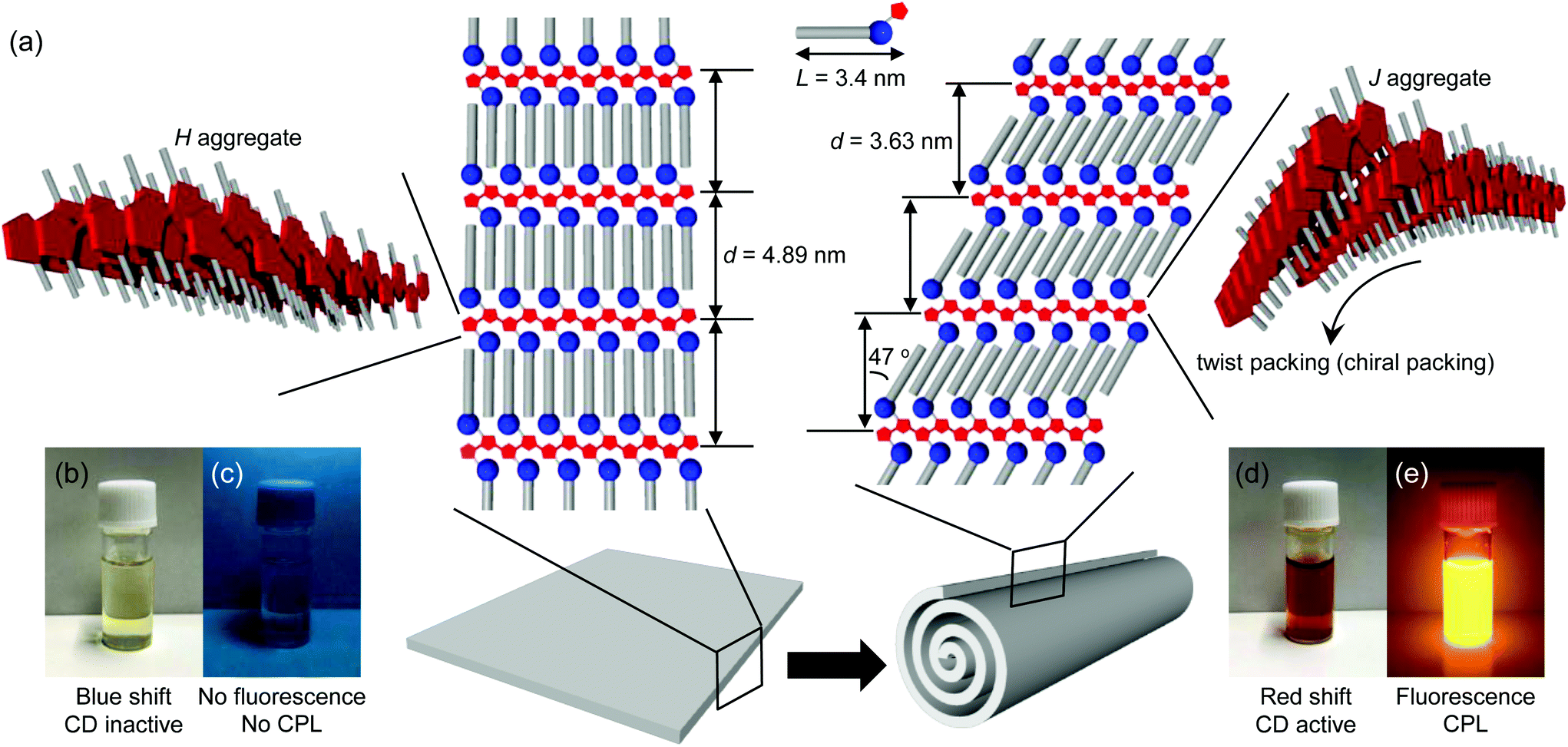 | ||
| Fig. 2 (a) Schematic representation of the molecular packing of a PTB-GlcSte-sheet and a PTB-GlcSte-nanotube. (b) Photograph of an ethanol dispersion of the PTB-GlcSte-sheets. (c) Photograph of an ethanol dispersion of the PTB-GlcSte-sheets under UV light irradiation. (d) Photograph of an ethanol dispersion of the PTB-GlcSte-nanotubes. (e) Photograph of an ethanol dispersion of the PTB-GlcSte-nanotubes under UV light irradiation. | ||
UV–vis and fluorescence spectroscopy yielded information about the aggregation modes of the polythiophene aromatic moieties within the bilayers. The absorption band (λmax = 450 nm) of the polythiophene aromatic moiety in the PTB-GlcOle-nanotubes dispersed in ethanol at 25 °C was red-shifted compared with that (λmax = 390 nm) of the polythiophene aromatic moiety of free PTB-GlcOle molecules dissolved in ethanol at 70 °C (Fig. S7†). In contrast, the absorption band (λmax = 332 nm) of the polythiophene aromatic moiety in the PTB-GlcSte-sheets dispersed in ethanol at 25 °C was blue-shifted compared with that (λmax = 380 nm) of the polythiophene aromatic moiety of free PTB-GlcSte molecules dissolved in ethanol at 70 °C (Fig. S7†). These shifts indicate that the polythiophene aromatic moiety formed a J-type aggregate in the PTB-GlcOle-nanotubes and an H-type aggregate in the PTB-GlcSte-sheets (Fig. 2a).53 The fluorescence properties of the polythiophene aromatic moieties strongly depended on the aggregation mode. The H-type aggregate in the PTB-GlcSte-sheets showed no fluorescence, whereas the J-type in the PTB-GlcOle-nanotubes showed a fluorescence band at around 560 nm (Fig. S7†).
Time control of roll-up onset of sheets into nanotubes
On completion of the self-assembly, the PTB-GlcOle-nanotubes and PTB-GlcSte-sheets were collected on membrane filters (pore size, 200 nm) and then re-dispersed in ethanol at a certain temperature. Aliquots were taken up from the dispersions every 15–60 minutes and examined by scanning electron microscopy. The PEG-GlcOle-nanotubes retained their tubular morphology for over a month at 25 °C. In contrast, 17 hours after re-dispersion at 25 °C, the PTB-GlcSte-sheets suddenly started to roll up (Fig. 1c), and within 1 hour, the sheets were completely transformed into nanotubes (Fig. 1d). The nanotubes (hereafter referred to as PTB-GlcSte-nanotubes) showed vestiges of the roll-up on their surface, which was remarkably different from the smooth surface of the PTB-GlcOle-nanotubes that formed by self-assembly (Fig. 1a and d).The roll-up event involved a molecular packing rearrangement while retaining the bilayer structures. The d spacing of the PTB-GlcSte-nanotubes was shorter (3.63 nm) than that of the PTB-GlcSte-sheets (4.89 nm), which are referred to hereafter as precursor sheets; however, the PTB-GlcSte-nanotubes had a wide-angle XRD pattern similar to that of the precursor sheets (Fig. S3†). These findings imply that the PTB-GlcSte monomers were tilted by 47° and were packed in an interdigitated manner within the bilayers of the PTB-GlcSte-nanotubes (Fig. 2a and S6†). The amide-I band was observed at a lower frequency in the PTB-GlcSte-nanotubes than that in the precursor sheets (Fig. S3†), indicating that the molecular tilting strengthened the intermolecular hydrogen bonding within the bilayers of the nanotubes. This result suggests that stabilization of the bilayers could be a driving force for the roll-up. The roll-up event never occurred in methanol, propanol, other alcohols, aliphatic and aromatic solvents. Ethanol solvation to the sheets and incorporation of ethanol molecules into the molecular packing within the bilayers will be a trigger of the roll-up event accompanied with the molecular packing rearrangement.
In the roll-up process, the aggregation mode of the polythiophene aromatic moiety also changed from H-type to J-type (Fig. 2a and S8†), and the absorption band of the PTB-GlcSte-nanotubes was red-shifted (λmax = 430 nm, Fig. 3a and S7†) compared with that of free PTB-GlcSte molecules in ethanol (λmax = 380 nm; Fig. S7†), whereas the absorption band of the precursor sheets was blue-shifted (λmax = 332 nm, Fig. 3a and S7†). A similar J-type aggregation mode was observed for the polythiophene moiety in the PTB-GlcOle-nanotubes (Fig. S7†). The timing of the disappearance of the blue-shifted absorption band of the precursor sheets and the appearance of the red-shifted absorption band of the PTB-GlcSte-nanotubes was consistent with the starting and ending times of the roll-up of the sheets into the nanotubes (Fig. 3b), as estimated by scanning electron microscopy. The change in the color of the dispersed solutions corresponding to the change in the absorption spectra was visible to the naked eye (Fig. 2b and d).
 | ||
| Fig. 3 (a) Absorption spectra of the H-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-sheets (blue) and the J-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-nanotubes (red) dispersed in ethanol: [PTB-GlcSte-sheet] = [PTB-GlcSte-nanotube] = 1.0 × 10−5 M. (b) Time course of the transformation from the H-type aggregate to the J-type aggregate, as indicated by the change in the absorbance at 450 nm. (c) Circular dichroism spectra of the H-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-sheets (blue) and the J-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-nanotubes (red) dispersed in ethanol: [PTB-GlcSte-sheet] = [PTB-GlcSte-nanotube] = 1.0 × 10−5 M. (d) Time course of the transformation of the nonchiral H-type aggregate to the chiral J-type aggregate, as indicated by the change in the circular dichroism intensities at 404 and 466 nm. | ||
The circular dichroism (CD) spectra of the precursor sheets and PTB-GlcSte-nanotubes had Cotton bands at the similar absorption wavelength as that of the polythiophene aromatic moiety bonded to a D-glucose moiety acting as the source of chirality in PTB-GlcSte (Fig. 3c). The Cotton bands were assignable to supramolecular CD54 because free PTB-GlcSte molecules were CD-inactive in the same wavelength region (Fig. S9†). The CD intensity of the PTB-GlcSte-nanotubes was greater than that of the precursor sheets. This finding is ascribable to amplification due to chiral (twist) packing, which was induced when the molecules tilted within the bilayers upon formation of the nanotubes (Fig. 2a).27–32 The split-type CD spectrum (the shape and the negative and positive signs) was consistent with the left-handed helicity of the polythiophene main chain backbone.55–57 The helical direction in the PTB-GlcSte-nanotubes was similar to that in the PTB-GlcOle-nanotubes, which had the same chiral source, the D-glucose moiety (Fig. S9†). The change in the CD spectrum due to the roll-up of the precursor sheets into the PTB-GlcSte-nanotubes exhibited a sigmoid time dependence (Fig. 3d), which was compatible with the time dependence of the change in the UV–vis spectra (Fig. 3b).
Delayed induction of CPL
CPL has attracted much attention because of its applications for optical displays, photoelectric devices, information storage arrays, and chiroptical materials.58–61 Self-assembly of chiral fluorescent molecules into supramolecular helical structures is an important method for inducing CPL and enhancing its intensity.62 Introducing stimulus-responsiveness into a helical structure would permit CPL to be turned on and off and helicity to be inverted. In general, these events begin to occur immediately upon application of the stimulus, and then proceed quickly or gradually.62 However, the delayed roll-up of the precursor sheets to form the PTB-GlcSte-nanotubes observed in this study has the potential to be useful for novel time controllable induction of CPL.When the precursor sheets rolled up to form the PTB-GlcSte-nanotubes 17 hours after the re-dispersion in ethanol at 25 °C, visible fluorescence derived from the π–π* transition of the polythiophene moiety appeared at 563 nm (Fig. 2e, 4a, b and S8†). Furthermore, the delayed appearance of the fluorescence band was accompanied by a positive CPL band (Fig. 4c and d). The sigmoid time dependence of the CPL band was coincident with that of the supramolecular CD resulting from the roll-up of the precursor sheets into the PTB-GlcSte-nanotubes (Fig. 3c and d), indicating that the CPL originated from the left-handed helicity of the polythiophene main chain backbone in the nanotube membrane walls.63–65 The CPL intensity can be evaluated in terms of the luminescence dissymmetry factor (glum), which is defined as 2(IL − IR)/(IL + IR), where IL and IR represent the intensities of left- and right-handed CPL, respectively.66 The value of glum ranges from +2 for an ideal left-handed CPL to −2 for an ideal right-handed CPL, and a glum value of 0 indicates no CPL. The glum value of the PTB-GlcSte-nanotubes was calculated to be 3.1 × 10−3, which is comparable to the values for previously reported supramolecular structures62 and the value for the PTB-GlcOle-nanotubes (4.4 × 10−3, Fig. S10†).
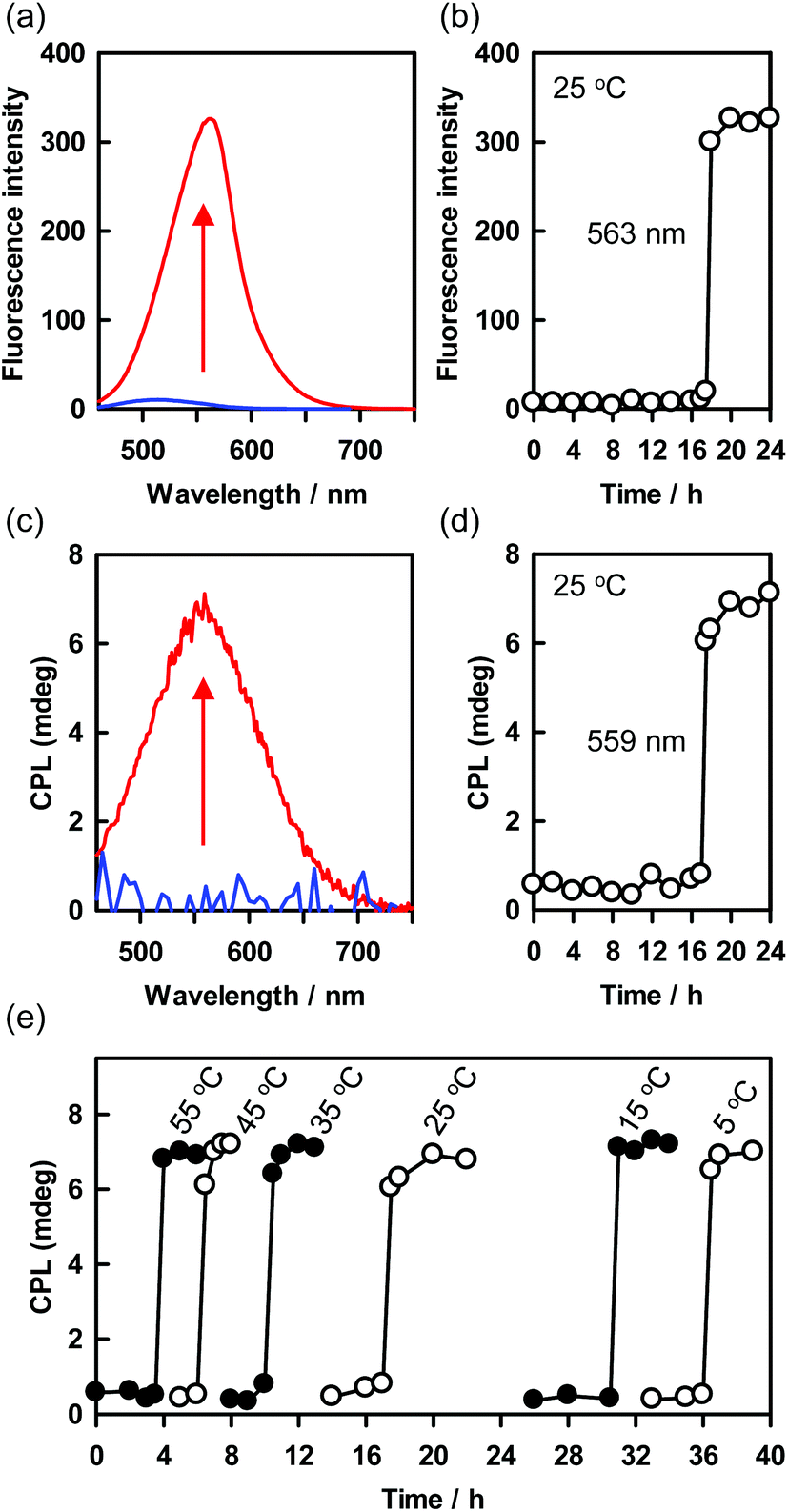 | ||
| Fig. 4 (a) Fluorescence spectra of the H-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-sheets (blue) and the J-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-nanotubes (red) dispersed in ethanol: [PTB-GlcSte-sheet] = [PTB-GlcSte-nanotube] = 1.0 × 10−6 M. The excitation wavelengths were 332 nm for the sheets and 430 nm for the nanotubes. (b) Time course of the transformation from the H-type aggregate to the J-type aggregate, as indicated by the change in the fluorescence intensity at 563 nm. (c) Circularly polarized luminescence spectra of the H-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-sheets (blue) and the J-type aggregate of the polythiophene aromatic moiety in the PTB-GlcSte-nanotubes (red) dispersed in ethanol: [PTB-GlcSte-sheet] = [PTB-GlcSte-nanotube] = 1.0 × 10−6 M. The excitation wavelengths were 332 nm for the sheets and 430 nm for the nanotubes. (d) Time course of the transformation of the nonchiral H-type aggregate to the chiral J-type aggregate, as indicated by the change in the circularly polarized luminescence intensities at 559 nm. (e) Temperature dependence of the time for the CPL onset accompanied with the roll-up onset of the PTB-GlcSte-sheets into the PTB-GlcSte-nanotubes. | ||
Notably, the onset timing of the CPL appearance, that is, the onset of the roll-up of the precursor sheets, depended on the temperature of the ethanol dispersion of the sheets (Fig. 4e). For example, the onset of CPL after the re-dispersion of the precursor sheets began at 3.5 hours at 55 °C and at 36 hours at 5 °C. However, the duration of the roll-up process (that is, the interval between the CPL appearance and the time at which it reached its maximum value) was approximately 1 hour at all the tested temperatures (Fig. 4e). This finding suggests that the rate of transformation of the precursor sheets to the PTB-GlcSte-nanotubes was insensitive to temperature. The self-assembly process reached the same chiral configuration every time. Since the Cotton effects, CPL and helical conformation should be affected by external chiral sources, the present system will be useful for chiral-sensing and -catalyst.67–70
It should be noted that although the system described herein provides a method to achieve delayed-onset CPL, there is no method for turning off the CPL, because the PTB-GlcSte-nanotubes never unrolled to re-form the precursor sheets.
Conclusions
We prepared the polythiophene-conjugated glycolipid that self-assembled in ethanol into square sheets a few micrometers on each side. The sheets dispersed in ethanol remained stable for hours but then suddenly rolled up to form nanotubes. The onset timing of the roll-up process depended on ambient temperature. The roll-up involved the molecular rearrangement of the PTB-GlcSte packing within the bilayers, which was associated with strengthening of the intermolecular hydrogen bonds, alteration of the aggregation mode of the polythiophene aromatic moiety, and enhancement of supramolecular chirality due to chiral packing. The nanotubes exhibited not only strong fluorescence derived from J-type aggregation of the polythiophene aromatic moiety but also CPL originating from the left-handed helicity of the polythiophene main chain backbone; in contrast, the precursor sheets exhibited no fluorescence or CPL. Because the CPL onset was coincident with roll-up of the sheets into nanotubes, the timing of the CPL onset were able to be controlled by varying the temperature. Delayed-onset CPL has never been reported for supramolecular structures; in these structures, the turning-on and -off of the CPL and the helicity inversion usually occur as soon as the stimulus is applied to the system, or they start immediately and then quickly or gradually proceed. Our findings should facilitate the development of new stimulus-responsive supramolecular structures that can be used for delayed-action capsules or optical switching devices.Experimental
Synthesis of monomers, TB-GlcSte and TB-GlcOle
3-Thiopheneboronic acid (TB) was purchased from Tokyo Chemical Co. and used without purification. N-Stearoyl-β-D-glucopyranosylamine (GlcSte) and N-(9-cis-octadecenoyl)-β-D-glucopyranosylamine (GlcOle) were synthesized as described previously.49,50 Dehydration reactions between 3-thiopheneboronic acid (0.13 g, 1 mmol) and the glycolipids (0.45 g, 1 mmol) in refluxing toluene were carried out in a reaction vessel equipped with a Dean–Stark trap. After 3 hours, the solvent was completely removed by a rotary evaporator. The obtained residues were washed several times with THF and were stored in a vacuum desiccator. The yields of the condensation compounds, TB-GlcSte and TB-GlcOle, were 93% and 91%, respectively.CH–), 5.13 (d, J = 5.5 Hz, 1H; OH-2), 4.10 (m, 1H; H-6), 3.79 (t, J = 9.0 Hz, 1H; H-1), 3.57 (m, 1H; H-6), 3.45–3.38 (m, 3H; H-3, H-4, H-5), 3.22 (m, 1H; H-2), 2.08 (m, 2H; –COCH2–), 1.98 (m, 2H; –CH2CHCH–), 1.47 (m, 2H; –COCH2CH2–), 1.24 (m, 22H; –CH2–), 0.86 (t, J = 7.0 Hz, 3H; –CH3). Elemental analysis calcd (%) for C28H46BNO6S: C 62.56, H 9.00, N 2.61; found C 62.88, H 8.72, N 2.69.
Synthesis of polymers, PTB-GlcSte and PTB-GlcOle
TB-GlcSte (64 mg, 0.12 mmol) and TB-GlcOle (64 mg, 0.12 mmol) were separately polymerized in toluene (1 mL) in the presence of FeCl3 (73 mg, 0.44 mmol) for 24 hours at room temperature under an atmosphere of nitrogen. After evaporation of the solvent, methanol was added to the residue, and the methanolic supernatant was decanted from any insoluble residue. The yields of PTB-GlcSte and PTB-GlcOle crystalized from the supernatant were 73% and 79%, respectively.Characterization of PTB-GlcSte and PTB-GlcOle: Mn (number-average molecular weight) and polydispersity index (PDI = Mw (weight-average molecular weight)/Mn) were determined by means of size-exclusion chromatography (Shimadzu LC-20AD, SPD-M20A UV detector, Shodex KF-803 column, THF eluent, 1.0 mL min−1 flow rate) with a polystyrene standard (Fig. S13†). The values were as follows: Mn = 1.32 × 104 and PDI = 1.037 for PTB-GlcSte; Mn = 1.22 × 104 and PDI = 1.048 for PTB-GlcOle. The IR spectra of the polymers exhibited an out-of-plane vibration band at 788 cm−1 (Fig. S14†), indicating that the polymerization occurred at the 2- and 5-positions of the thiophene moieties. The absorption maxima of PTB-GlcSte and PTB-GlcOle in ethanol solution at 60 °C were observed at 387 and 372 nm, respectively, whereas those of the monomers were at less than 250 nm (Fig. S15†). Fluorescence bands of PTB-GlcSte and PTB-GlcOle in ethanol solution at 70 °C appeared at 509 and 507 nm, respectively, whereas the monomers showed no fluorescence (Fig. S15†). All the spectroscopy results were consistent with the polymers having extensive conjugation in the polythiophene backbone.
Morphological observation
Ethanol dispersions of the PTB-GlcSte and PTB-GlcOle self-assemblies were dropped onto an amorphous carbon grid, and excess ethanol was allowed to evaporate at room temperature. Images of the residues were obtained with a scanning electron microscope (Hitachi S-4800) operated at 10 keV. The PTB-GlcSte-sheets were observed with an atomic force microscope (Nanoscope IIIA, Digital Instruments) operating in tapping mode under ambient conditions. Microfabricated silicone cantilever tips (NCH-10 V) with a resonance frequency of ≈320 kHz and a spring constant of about 42 N m−1 were used. The scan rate was varied from 0.5 to 1.5 Hz.Molecular packing analysis
XRD patterns of lyophilized self-assemblies of PTB-GlcSte and PTB-GlcOle were measured with a Rigaku diffractometer (type 4037) using graded d-space elliptical side-by-side multilayer optics, monochromated Cu Kα radiation (40 kV, 30 mA), and an imaging plate (R-Axis IV). The exposure time was 5 min with a 150 mm camera length. The structures were also analyzed by means of Fourier transform IR spectroscopy on an instrument (FT-620, JASCO) operated at 4 cm−1 resolution and equipped with an unpolarized beam, an attenuated total reflection accessory system (Diamond MIRacle, horizontal attenuated total reflection accessory with a diamond crystal prism, PIKE Technologies), and a mercury cadmium telluride detector.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Iketani Science and Technology Foundation (grant no. 0311037-A).References
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- B. Rybtchinski, ACS Nano, 2011, 5, 6791 CrossRef CAS PubMed.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashizume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. E. A. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2013–2078 CrossRef PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- Intelligent Stimuli-Responsive Materials: From Well-Defined Nanostructures to Applications, ed. Q. Li, Wiley, 2013 Search PubMed.
- S. Ogi, T. Fukui, M. Lue, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2014, 53, 14363–14367 CrossRef CAS PubMed.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- A. Aliprandi, M. Mauno and L. D. Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- T. Komatsu, Nanoscale, 2012, 4, 1910–1918 RSC.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- J. Jiang, G. Ouyang, L. Zhang and M. Liu, Chem. – Eur. J., 2017, 23, 9439–9450 CrossRef CAS PubMed.
- T. Shimizu, Bull. Chem. Soc. Jpn., 2018, 91, 623–668 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- A. Brizard, R. Oda and I. Huc, Top. Curr. Chem., 2005, 256, 167 CrossRef CAS PubMed.
- C. Valery, F. Artzner and M. Paternostre, Soft Matter, 2011, 7, 9583–9594 RSC.
- I. W. Hamley, Angew. Chem., Int. Ed., 2014, 53, 6866–6881 CrossRef CAS PubMed.
- T. G. Barclay, K. Constantopoulos and J. Matisons, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- S. Tu, S. H. Kim, J. Joseph, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2011, 133, 19125–19130 CrossRef CAS PubMed.
- S. Yagai, M. Yamauchi, A. Kobayashi, T. Karatsu, A. Kitamura, T. Ohba and Y. Kikkawa, J. Am. Chem. Soc., 2012, 134, 18205–18208 CrossRef CAS PubMed.
- Y. Kim, T. Kim and M. Lee, Polym. Chem., 2013, 4, 1300–1308 RSC.
- T. Fukino, H. Joo, Y. Hisada, M. Obana, H. Yamagishi, T. Hikima, M. Takata, N. Fujita and T. Aida, Science, 2014, 344, 499–504 CrossRef CAS PubMed.
- N. Kameta, M. Masuda and T. Shimizu, Chem. – Eur. J., 2015, 21, 8832–8839 CrossRef CAS PubMed.
- K. Yoshida, H. Minamikawa, S. Kamiya, T. Shimizu and S. Isoda, J. Nanosci. Nanotechnol., 2007, 7, 960–964 CrossRef CAS PubMed.
- H. Yui, H. Minamikawa, R. Danev, K. Nagayama, S. Kamiya and T. Shimizu, Langmuir, 2008, 24, 709–713 CrossRef CAS PubMed.
- X. Zhang, T. Bera, W. Liang and J. Fang, J. Phys. Chem. B, 2011, 115, 14445–14449 CrossRef CAS PubMed.
- A. Uesaka, M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Langmuir, 2012, 28, 6006–6012 CrossRef CAS PubMed.
- M. C. di Gregorio, M. Varenik, M. Gubitosi, L. Travaglini, N. V. Pavel, A. Jover, F. Meijide, O. Regev and L. Galantini, RSC Adv., 2015, 5, 37800–37806 RSC.
- B. Shen, Y. He, Y. Kim, Y. Wang and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 2382–2386 CrossRef CAS PubMed.
- T. Shimizu, N. Kameta, W. Ding and T. Shimizu, Langmuir, 2016, 32, 12242–12264 CrossRef CAS PubMed.
- R. Takahashi, H. Kawauchi, N. Kameta, J. H. Lee, S. Fujii, T. Shimizu and K. Sakurai, J. Phys. Chem. B, 2019, 123, 4340–4345 CrossRef CAS PubMed.
- N. Kameta, K. Ishikawa, M. Masuda, M. Asakawa and T. Shimizu, Chem. Mater., 2012, 24, 209–214 CrossRef CAS.
- N. Kameta, M. Masuda and T. Shimizu, Chem. Commun., 2015, 51, 11104–11107 RSC.
- N. Kameta, M. Aoyagi and M. Asakawa, Chem. Commun., 2017, 53, 10116–10119 RSC.
- S. Kamiya, H. Minamikawa, J. H. Jung, B. Yang, M. Masuda and T. Shimizu, Langmuir, 2005, 21, 743–750 CrossRef CAS PubMed.
- K. Ishikawa, N. Kameta, M. Aoyagi, M. Asakawa and T. Shimizu, Adv. Funct. Mater., 2013, 23, 1677–1683 CrossRef CAS.
- N. Garti and K. Sato, Crystallization and Polymorphism of Fats and Fatty Acids, Marcel Dekker, New York, 1988, pp. 139–187 Search PubMed.
- N. Yamada, K. Okuyama, T. Serizawa, M. Kawasaki and S. Oshima, J. Chem. Soc., Perkin Trans. 2, 1996, 12, 2707–2713 RSC.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- A. R. A. Palmans, E. W. Meijer and J. R. Reynolds, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS PubMed.
- B. M. W. Langeveld-Voss, R. A. J. Janssen and E. W. Meijer, J. Mol. Struct., 2000, 521, 285–301 CrossRef CAS.
- K. P. R. Nilsson, J. D. M. Olsson, P. Konradsson and O. Inganas, Macromolecules, 2004, 37, 6316–6321 CrossRef CAS.
- C. R. G. Grenier, S. J. George, T. J. Joncheray, E. W. Meijer and J. R. Reynolds, J. Am. Chem. Soc., 2007, 129, 10694–10699 CrossRef CAS PubMed.
- J. R. Brandt, F. Salerno and M. J. Matthew, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- T. Harada, Polym. J., 2018, 50, 679–687 CrossRef CAS.
- W. Ma, C. Hao, M. Sun, L. Xu, C. Xu and H. Kuang, Mater. Horiz., 2018, 5, 141–161 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011–1024 CrossRef CAS.
- G. Fukuhara, M. Imai, D. Fuentealba, Y. Ishida, H. Kurohara, C. Yang, T. Mori, H. Uyama, C. Bohne and Y. Inoue, Org. Biomol. Chem., 2016, 14, 9741–9750 RSC.
- T. Ikai, K. Takayama, Y. Wada, S. Minami, C. Apiboon and K. Shinohara, Chem. Sci., 2019, 10, 4890–4895 RSC.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS.
- W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 2689 CrossRef PubMed.
- W. Ma, H. Kuang, L. Wang, L. Xu, W.-S. Chang, H. Zhang, M. Sun, Y. Zhu, Y. Zhao, L. Liu, C. Xu, S. Link and N. A. Kotov, Sci. Rep., 2013, 3, 1934 CrossRef PubMed.
- M. Sun, L. Xu, A. Qu, P. Zhao, T. Hao, W. Ma, C. Hao, X. Wen, F. M. Colombari, A. F. de Moura, N. A. Kotov, C. Xu and H. Kuang, Nat. Chem., 2018, 10, 821–830 CrossRef CAS PubMed.
- X. Zhao, L. Xu, M. Sun, W. Ma, X. Wu, C. Xu and H. Kuang, Nat. Commun., 2017, 8, 2007 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of monomers and polymers, the characterization, morphological observation, molecular packing analysis and spectroscopic measurements. See DOI: 10.1039/c9nr08032e |
| This journal is © The Royal Society of Chemistry 2020 |