
Polarization of tumor-associated macrophage phenotype via porous hollow iron nanoparticles for tumor immunotherapy in vivo†
Ke
Li
 ,
Lu
Lu
,
Chencheng
Xue
,
Ju
Liu
,
Ye
He
,
Jun
Zhou
,
Zengzilu
Xia
,
Liangliang
Dai
*,
Zhong
Luo
*,
Yulan
Mao
and
Kaiyong
Cai
*
,
Lu
Lu
,
Chencheng
Xue
,
Ju
Liu
,
Ye
He
,
Jun
Zhou
,
Zengzilu
Xia
,
Liangliang
Dai
*,
Zhong
Luo
*,
Yulan
Mao
and
Kaiyong
Cai
*
Key Laboratory of Biorheological Science and Technology of Ministry of Education, College of Bioengineering, Chongqing University, Chongqing 400044, China. E-mail: dailiangliangnbe@sina.cn; luozhong918@cqu.edu.cn; kaiyong_cai@cqu.edu.cn; Fax: +86-23-65102877; Tel: +86-23-65102507
First published on 4th November 2019
Abstract
Tumor-associated macrophages (TAMs) are the most important components in the tumor immunosuppressive microenvironment, promoting tumor growth and metastasis. Although TAMs have become one of the hot topics of tumor immunotherapy, challenges still remain to achieve TAM-targeted re-polarization therapy. In this work, porous hollow iron oxide nanoparticles (PHNPs) were synthesized for loading a P13K γ small molecule inhibitor (3-methyladenine, 3-MA) and further modified by mannose to target TAMs. The delivery system named PHNPs@DPA-S-S-BSA-MA@3-MA showed good efficiency for targeting TAMs. The inflammatory factor NF-κB p65 of macrophages was activated by the combination of PHNPs and 3-MA, which synergistically switched TAMs to pro-inflammatory M1-type macrophages. As a result, it activated immune responses and inhibited tumor growth in vivo. The study provides an intracellular switch of the TAM phenotype for targeted TAM therapy.
1. Introduction
Immunotherapy is the most popular treatment for cancer.1–3 The tumor immunosuppressive microenvironment (TIME) is one of the most important characteristics that affects the therapeutic effect on solid tumors.4–6 TAMs are a vital element of the TIME,7,8 especially in triple negative breast cancer.9,10 Besides, TAMs are the key regulator of the immune system and solid tumors.11 Many studies indicated that TAMs are closely related to the occurrence,12 growth,13 metastasis and drug resistance of tumors.14–17 Therefore, the therapy that focuses on targeting TAMs has become a hot topic in immunotherapy.18–20 Currently, targeting TAM therapy includes targeted elimination of TAMs and re-polarization TAMs into pro-inflammatory M1-type macrophages.21,22 Anti-colony factor-1 receptor (anti-CSF-1R) small interfering RNA (siRNA) was loaded onto M2-like TAM dual-targeting nanoparticles (M2NPs) to develop a targeted immunotherapeutic approach that specifically blocks survival signals from TAMs and removes them from melanoma.23 Treatment with HA-coated and mannan-bound MnO2 particles (Man-HA-MnO2) polarizes TAMs to the pro-inflammatory M1-type, which could reduce tumor hypoxia and regulate chemotherapy resistance.24The rapid development of nanomaterials provides potential platforms for tumor therapy.25–28 Iron oxide nanoparticles have been widely used in clinical therapy for tumor therapy and tumor diagnosis.29,30 For example, ferumoxytol is the Food and Drug Administration (FDA)-approved iron oxide nanoparticle polarizing anti-inflammatory M2-type macrophages to pro-inflammatory M1-type macrophages to activate anti-tumor immune response in the TIME.31 Superparamagnetic iron oxide nanoparticles (SPIONs) coated with PEI were used to activate macrophages and increase several genes relevant to the M1-type.32 Moreover, iron oxide magnetic nanoparticles coated with a myeloid-derived suppressor cell membrane (MNP@MDSC) were used for photothermal therapy (PTT) to induce immunogenic cell death and switch tumor infiltrating macrophages.33 Nevertheless, the immune response induced by iron oxide nanoparticles alone was limited and could not inhibit the tumor growth significantly. On the other hand, the PTEN/PI3K γ/mTOR signalling pathway has been proved to control the polarization of macrophages.34–36 In addition, PI3-kinase γ in macrophages regulates the critical switch between the immune response and suppression during cancer and inflammation.37,38 The PI3K γ/Akt signalling pathway can inhibit NF-κB p65 activation promoting immune suppression during tumor growth. The hypoxia tumor cells secrete exosomes to activate macrophages to the M2 phenotype via the activation of the PTEN/PI3K γ/mTOR signalling pathway.39 Therefore, a small molecule inhibitor of PI3K γ selectively inhibiting the expression of PI3K γ protein could activate and prolong the expression of NF-κB p65 protein, thus promoting immune response and remodelling the tumor microenvironment through re-polarization of TAMs to pro-inflammatory M1-type macrophages. Hence, to combine iron oxide nanoparticles with the intracellular signalling pathway to switch the TAM phenotype would be a promising alternative for targeting TAM therapy.
In this work, PHNPs were synthesized to load the small molecule inhibitor of PI3K γ (3-methyladenine) and blocked with bovine serum albumin (BSA). Then, carbonylated mannose was used for surface modification to target macrophages. Considering the high redox properties of macrophages, disulfide bonds were introduced into the nanocarrier during modification to achieve GSH-responsive release of 3-methyladenine,40 resulting in the formation of a nanocarrier of PHNPs@DPA-S-S-BSA-MA@3-MA (Scheme 1a). The mannose-modified nanoparticles could effectively target TAMs and avoid uptake by other cells. PHNPs loaded with 3-MA could effectively upregulate the expression of NF-κB p65 through the cellular signal pathway of macrophages, leading to PHNPs@DPA-S-S-BSA-MA@3-MA more efficiently promoting the re-polarization of TAMs into pro-inflammatory M1-type macrophages. After re-polarization, macrophages could reshape the immunosuppressive microenvironment, increase the secretion and release of immune-promoting factors, and decrease the secretion of immunosuppressive factors (Scheme 1b). Thus, we hypothesized that the system of PHNPs@DPA-S-S-BSA-MA@3-MA would efficiently relieve the immune-suppression, activate the immune response and inhibit tumor growth via polarizing macrophage phenotypes from the M2-type to the pro-inflammatory M1-type.
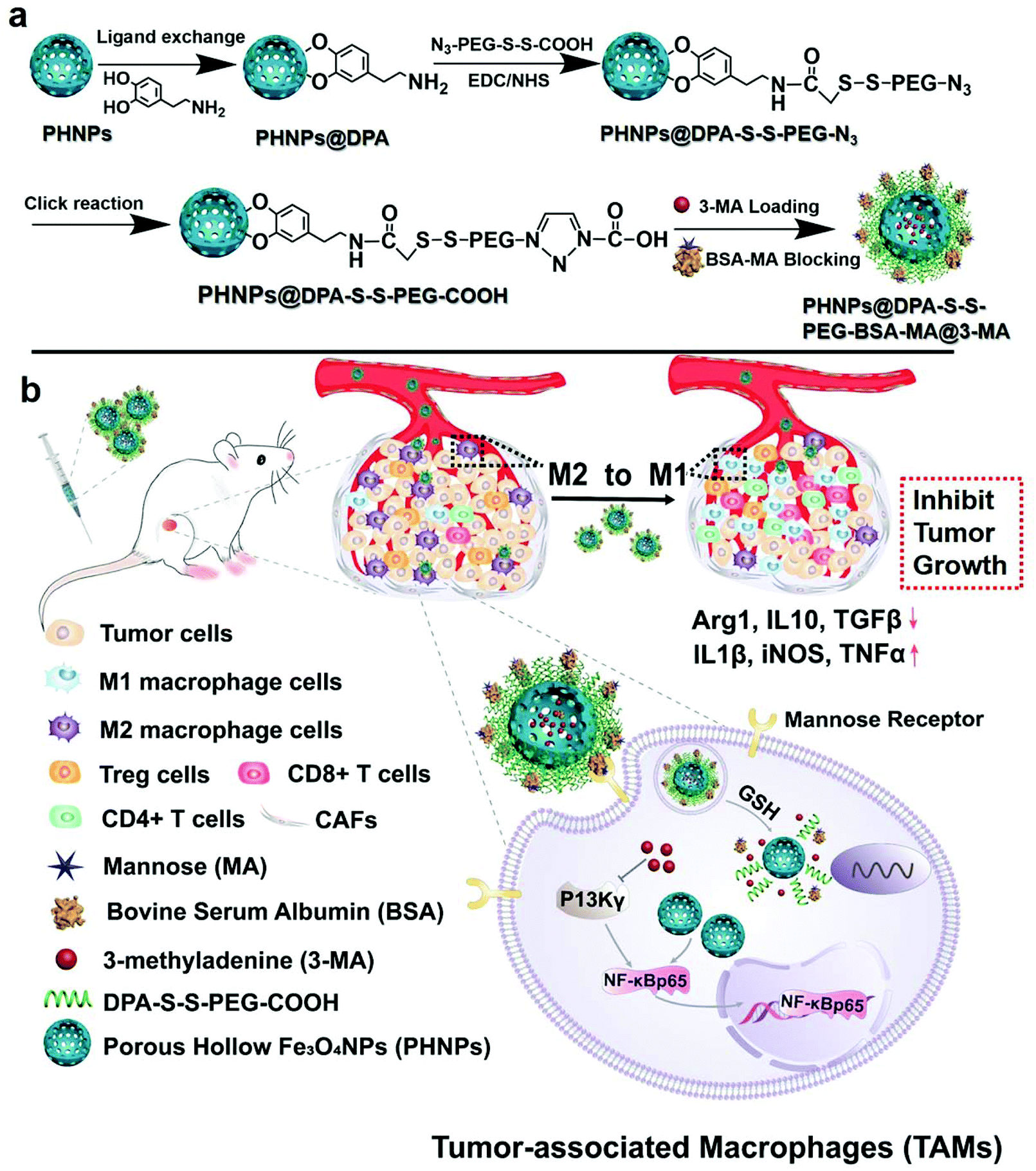 | ||
| Scheme 1 Synthesis of PHNPs@DPA-S-S-PEG-BSA-MA@3-MA (a), and (b) PHNPs and 3-MA re-polarize TAMs to the M1-type through activating the protein of NF-κB p65, then remodelling the immunosuppressive microenvironment, thus activating immune response and inhibiting tumor growth. | ||
2. Experimental section
2.1 Materials
Poly(ethylene glycol) (HO-mPEG-OH, Mn: 1000 g mol−1), N-hydroxy succinimide (NHS), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), triethylamine (TEA), dichloromethane (DCM), 4-dimethylaminopyridine (DMAP), 1-octadecene, iron pentacarbonyl, and N,N-dimethylformamide (DMF) were purchased from Sigma-Aldrich (Beijing, China). The primary antibodies including rabbit anti-PI3Kγ/anti-NF κBp65/anti-p-NF κBp65/anti-bax/anti-bcl 2/anti-IL-1β/anti-TNF-α/anti-TGF β/anti-IL-10, and mouse anti-β-actin were supplied by Cell Signalling Technology (CST, USA). Secondary antibodies of anti-rabbit IgG and anti-mouse IgG were bought from Beyotime (Jiangsu, China). Related antibodies used in immune cell detection were bought from Biolegend (California, USA) and eBioscience (SanDiego, CA).2.2 Material characterization
To determine the structures of synthetic products, 1H NMR spectra were recorded with a Bruker Avance 500 MHz. Fourier transform infrared spectroscopy (FTIR, model 6300, BioRad Co. Ltd, USA) was used to characterize the surface modification process. Transmission electron microscopy (TEM, LIBRA 200 CS, Carl Zeiss Co, Germany) was used to reveal the morphology of Fe3O4 PHNPs. The sizes and size distribution of Fe3O4 PHNPs were measured by zeta potential measurement (Nano ZS90 Zeta sizer, Malvern Instruments Co. Ltd, UK) equipped with dynamic light scattering (DLS). An ultraviolet spectrophotometer was used to determine the concentration of 3-MA.2.3 Preparation of PHNPs@DPA-S-S-BSA-MA@3-MA
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm min−1 for 8 min.
000 rpm min−1 for 8 min.
The loading of 3-MA into PHNPs@DPA-S-S-PEG-COOH largely depends on the hydrophobic–lipophilic interactions. 200 mg of PHNPs@DPA-S-S-PEG-COOH and 200 mg of 3-methyladenine in an aqueous solution were used and stirred for 48 h. Then the product was washed with water to remove the unloaded 3-methyladenine. The product was obtained by freeze-drying after centrifugal precipitation. After that, 200 mg of PHNPs@DPA-S-S-PEG-COOH, EDC and NHS were dissolved in water and stirred for 4 h. Then 200 mg of BSA-MA was dropped into the above solution mixture. After stirring for 48 h, the product was washed with water. The PHNPs@DPA-S-S-PEG-BSA-MA@3-MA was finally obtained by lyophilization.
2.4 Drug release behaviour
PHNPs@DPA-S-S-PEG-BSA-MA@3-MA was dispersed in PBS. The solution mixture was placed on a shaker and stirred at 37 °C. After 2 h, reduced glutathione was dropped into the mixture and the concentration of GSH was 0, 2 and 10 mM, respectively. At the given points, centrifugation was performed and the supernatant (100 μL) was taken out and refreshed with PBS-dissolved GSH with the same volume and concentration. The release of 3-methyladenine was determined by using a UV spectrophotometer at 272.5 nm.2.5 Cell culture
HUVEC, MDA-MB-231 cells, and RAW 264.7 cells were provided by the Department of Oncology in Xinqiao Hospital of the Third Military Medical University. THP-1 cells were provided by Army Military Medical University (Chongqing). The MDA-MB-231 cells, HUVEC, and RAW 264.7 cells were cultured with high-glucose DMEM supplemented with 10% FBS (Gibco), penicillin (100 U mL−1) and streptomycin (100 μg mL−1) at 37 °C under a 5% CO2 atmosphere. The THP-1 cells were cultured in RPMI-1640 with 10% FBS (Gibco) and 1% antibiotic–antimycotic solution (Gibco, Thermo Fisher Scientific) at 37 °C under a 5% CO2 atmosphere. For differentiation of THP-1 to macrophages, THP-1 cells were treated with 50 nM PMA for 24 h to make the cells differentiate into THP-1 macrophages. The PMA-containing medium was removed after 24 h and the cells were washed three times with PBS to remove the PMA.43,442.6 Cell cytotoxicity of MDA-MB-231 and RAW 264.7 cells
The MDA-MB-231 and RAW 264.7 cells were treated with PHNPs, 3-methyladenine and PHNPs@DPA-S-S-PEG-BSA-MA@3-MA. After the administration of different concentrations of PHNPs, the cell samples were incubated for 24 h. Then, the mixture of MTT solution and non-serum-containing medium was added. The cell samples were further incubated for 4 h before adding DMSO. The results were recorded using ELISA (Bio-Rad 680, USA) at 490 nm. The experimental control was the non-treated cells. For the cytotoxicity evaluation of 3-methyladenine (0–100 μM) and PHNPs (0–200 μg mL−1), the same experimental steps were as the above experiment.2.7 Cellular uptake
The endocytosis of nanoparticles in RAW 264.7 cells was quantitatively analysed by FCM and visualized by CLSM, respectively. In addition, the endocytosis of NPs in MDA-MB-231 cells and HUVEC was also quantitatively detected by FCM (BD, Biosciences). Firstly, RAW 264.7 cells, HUVEC, and MDA-MB-231 cells were cultured with DMEM which contained PHNPs@DPA-S-S-BSA@FITC (50 μg mL−1) and PHNPs@DPA-S-S-BSA-MA@FITC (50 μg mL−1) for 2 and 4 h, respectively. Then the cells were detected by FCM. Secondly, RAW 264.7 cells were seeded on a confocal microscopy observation dish and cultured in DMEM containing PHNPs@DPA-S-S-BSA-MA@FITC and PHNPs@DPA-S-S-BSA@FITC for 12 and 24 h. Then the samples were stained with Hoechst 33258 for 10 min and observed by CLSM (TCS SP5, Leica, Germany).262.8 Western blotting analysis of RAW 264.7 cells and MDA-MB-231 cells in vitro
Specifically, when 70% confluence of RAW 264.7 and MDA-MB-231 cells was reached, they were divided into 2 batches and treated separately. In the first batch, RAW 264.7 cells and MDA-MB-231 cells were treated with different concentrations of 3-MA for 24 h, respectively. For the second batch, RAW 264.7 cells and MDA-MB-231 cells were treated with a non-drug-containing medium, 3-MA (66.9 μM), 200 μg mL−1 of PHNPs@DPA-S-S-BSA-MA, and 200 μg mL−1 of PHNPs@DPA-S-S-BSA-MA@3-MA (containing 66.9 μM of 3-MA). These treated cells were cleaned with PBS, digested with trypsin, centrifuged, lysed, and centrifuged to collect total proteins. The BCA kit was used to measure the concentrations of various proteins. Specifically, the same amount of proteins was suspended into the buffer solution. Quantity One software was used to measure the greyscale intensity of various proteins.372.9 Quantitative RT-PCR (qRT-PCR) analysis of the repolarization of RAW 264.7 cells and the co-culture of MDA-MB-231 cells and THP-1 macrophages in vitro
Specifically, THP-1 macrophages were cultured in a transwell chamber and the MDA-MB-231 cells were cultured in an upper chamber. When 70% confluence of the cells was reached, a non-drug-containing medium, 3-MA (66.9 μM), 200 μg mL−1 of PHNPs@DPA-S-S-BSA-MA, and 200 μg mL−1 of PHNPs@DPA-S-S-BSA-MA@3-MA (containing 66.9 μM of 3-MA) were added into the transwell. For comparison, RAW 264.7 and THP-1 macrophages were not co-cultured with MDA-MB-231 cells. Non-drug containing DMEM, 3-MA (66.9 μM), PHNPs (200 μg mL−1), and PHNP@3-MA (200 μg mL−1) (containing 66.9 μM of 3-MA) were also added. After co-culture for 24 h, the cells were digested with trypsin, centrifuged and collected. The total RNA of these samples was ascertained by using the total RNA kit (Omega, USA). Subsequently, the next experiment was performed according to the protocol and the instructions of the PrimeScript RT reagent kit (Takara, Japan). The synthesized primer (Sangon, China) sequences are listed in Tables S1 and S2.†2.10 FCM analysis of the repolarization of RAW 264.7 cells and the co-culture of MDA-MB-231 and THP-1 macrophages in vitro
THP-1 macrophages (co-cultured with MDA-MB-231 cells) and RAW 264.7 cells hatched with PHNPs@DPA-S-S-BSA-MA, 3-MA, and PHNPs@DPA-S-S-BSA-MA@3-MA for 24 h. The acquired cells were stained with anti-CD86-PE and anti-CD206-APC for 0.5 h and the phenotype of macrophages was detected by FCM.2.11 Development of the tumor-bearing mouse model
All animal experiments were conducted under the guidelines established by the Chinese Institutional Animal Care and Use Committee with the approval of the Animal Ethics Committee in the Third Military Medical University (SYXK-PLA-20120031). The Balb/c female mice (average weight around 18 g) were provided by Xinqiao Hospital of the Third Military Medical University. Each mouse was injected with 100 μL of saline buffer containing 2 × 106 units of MDA-MB-231 cells on the right side of thigh. The mice were used for the subsequent experiments until the volume of the tumor reached 60 mm3.462.12 Evaluation of the immunotherapy effect in vivo
When the tumor volume grew to around 60 mm3, all tumor-bearing mice were randomly divided into 4 groups (12 mice in each group). All samples (saline, PHNPs-S-S-BSA-MA, 3-MA, and PHNPs-S-S-BSA-MA@3-MA) were injected through the caudal vein. The injected dose was 20 mg kg−1 d−1 of PHNPs-S-S-BSA-MA, 1.45 mg kg−1 d−1 of 3-MA, and 20 mg kg−1 d−1 of PHNPs-S-S-BSA-MA@3-MA. Each sample was injected every 3 days. During the whole experiment, the body weight and tumor volume of the mice were recorded periodically. The size of the tumor was calculated using the following formula: Vt = ab2/2 (where a is the maximum diameter of the tumor and b is the minimum diameter of the tumor). The tail vein administration lasted for about 21 days and the surviving mice continued to be cared for until death. The survival curves were plotted for all the mice over the course of treatment based on the recorded observations. At 0 day, 7 day, 14 day and 21 day, mice were anesthetized with 4% chloral hydrate (100 g/800 μM) and the tumor-bearing mice were imaged.2.13 Macrophage re-polarization and T cell proliferation
At the end of the treatment, the mice were euthanized to collect the tumor tissues (n = 6). After that, the tumor tissue of 1 g was weighed using a digital display balance and cut into pieces. 500 μL of serum-free DMEM was used to transfer the tumor tissue into a 15 mL centrifuge tube, and 3 mL of the tumor tissue digestive fluid (tumor tissue dissociation kit, Miltenyi, Germany) was added to digest the tumor tissue at 37 °C for 0.5 h. Then DMEM containing serum was added to the upper centrifuge tube to terminate digestion. After that, the cells were collected by centrifuging and re-suspended in PBS. The following experiments were performed according to the instructions for antibody staining. The acquired cells were stained with L/D dye (BV510), anti-CD45-AF700, anti-CD3-APC-CY7, anti-CD4-FITC, anti-CD8-PerCP-CY5.5, anti-CD45R-PE and anti-CD49b-PE-CY7 and detected by FCM (Gallios, Beckman Coulter, USA) for measuring the CD4+ T cells, CD8+ T cells, B cells and NK cells. For Treg cells, the acquired cells were stained with L/D dye (BV510), anti-CD45-AF700, anti-CD4-FITC and anti-CD25 (BV421), and then fixed and permeabilized (Bioscience). Then the cells were stained with anti-Foxp3-Alexa Fluor 647. These samples were detected by FCM separately. For the re-polarization of the TAMs, the acquired cells were stained with L/D dye (BV510), anti-CD45-AF700, anti-CD11b-FITC, anti-F4/80-Brilliant Violet 421, anti-CD86-PE and anti-CD206-APC and detected by FCM to analyze the M1-type macrophages and M2-type macrophages.2.14 Histological analysis of the biosafety of various samples and cancer cell apoptosis
After the mice were euthanized, tumors and important organs (heart, liver, spleen, lungs and kidneys) were harvested and fixed in formalin solution separately. According to the instructions of making paraffin sections and the H&E staining procedure, these tissue samples were dehydrated, dewaxed and stained to make the H&E sections. Finally, these tissues were observed with an optical microscope separately. TUNEL staining was further used to characterize the treatment-induced apoptosis of tumor cells. The tumor tissues were made into paraffin sections and dewaxed. These processed sections were stained according to the TUNEL staining procedure. Finally, these tissues were observed with a CLSM (Leica TCS SP8, Germany).2.15 Immunofluorescence (IFC) and western blotting assays of the tumor tissues
To further characterize the TAM re-polarization (anti-CD 11b, CD 86 and CD 206) and the expression of inflammatory associated proteins (pro-inflammatory associated proteins: TNF-α, IL-1β, iNOS; anti-inflammatory associated proteins: IL-10, TGF-β, arginase I) in vivo, immunofluorescence assays were performed.33 To protect the antigens in tissues, these tissues were made into frozen sections. Briefly, the frozen sections were taken out from a −20 °C refrigerator and blocked with an immunofluorescent blocking solution. After the addition of a diluted primary antibody, the samples were incubated at 4 °C overnight and added with a proportionally diluted secondary antibody with a fluorescent label at 37 °C for 1 h in the dark. Hoechst 33258 was used to stain the nuclei of cells. Finally, an anti-fluorescence quencher was added for sealing. Subsequently, these sections were observed with the CLSM (Leica TCS SP8, Germany).2.16 Quantitative qRT-PCR analysis of the tumor tissue
The expression of the inflammatory-associated genes was measured by quantitative RT-PCR. Briefly, 0.5 g of the tumor tissues were cut into pieces, added liquid nitrogen to freeze the tissues, and then ground using a mortar. 1.5 mL of RZ lysis buffer were added into an EP tube (4 mL). The tissues were transferred into the tubes separately and the samples were disrupted by the cell ultrasonic fragmentation apparatus placed in an ice bath separately. The supernatant contained various RNA. The total RNA of these samples was ascertained using the total RNA kit (Omega, USA). Subsequently, the next experiment was performed according to the protocol and the instructions of the PrimeScript RT reagent kit (Takara, Japan).3. Results and discussion
3.1 Preparation and characterization of mannose-modified BSA to block 3-MA-loaded PHNPs
To synthesize mannose-modified BSA to block 3-methyladenine (3-MA)-loaded PHNPs (PHNPs@DPA-S-S-BSA-MA@3-MA), the hollow mesoporous Fe3O4 nanoparticles (PHNPs) were synthesized according to previous studies.41,45–47 Furthermore, the molecular weights and structures of N3-PEG-S-S-COOH and MA-COOH42 were determined by Mass Spectrometry (MS) (Fig. S4a†) and 1H NMR (Fig. S1–S4†). According to the images (Fig. 1a and b; a1 and b1) of the transmission electron microscopy (TEM), the average diameter of the present PHNPs and PHNPs@DPA-S-S-BSA-MA@3-MA was around 20 nm, the results of which are consistent with those of the dynamic light scattering (DLS) (Fig. 1d). X-ray diffraction (XRD) results (Fig. 1c) showed that the surface modification did not change the crystalline feature of PHNPs. The zeta potential results (Fig. S7†) indicated that the surface charges were −14.8 mV and −19.7 mV for PHNPs and PHNPs@DPA-S-S-BSA-MA in HEPES solution (pH value 7.4), respectively. Fourier transform infrared spectroscopy (FTIR) was used to characterize the surface modification process of PHNPs. The results show that PHNPs were successfully synthesized and surface functionalized (Fig. S5†).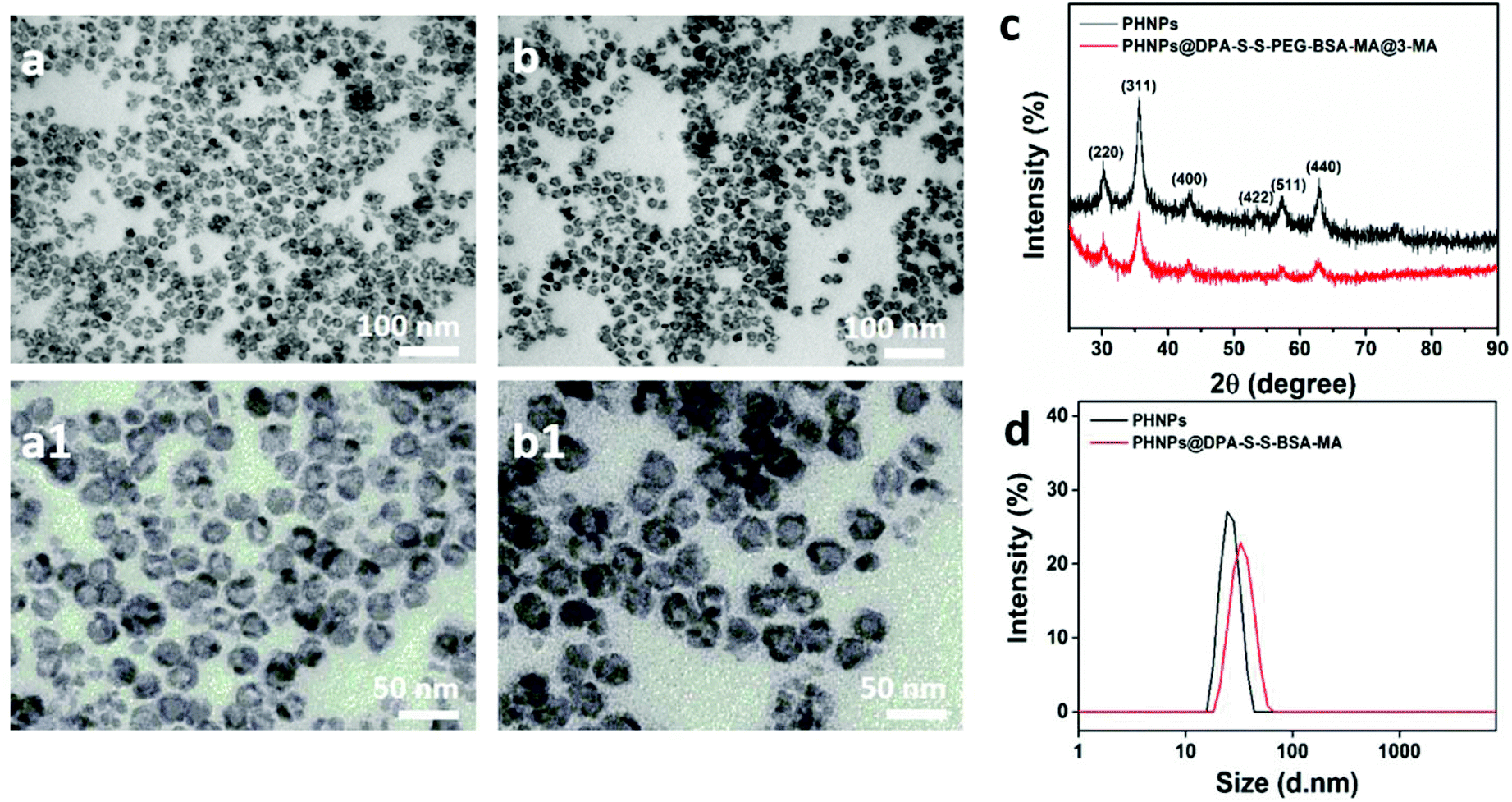 | ||
| Fig. 1 TEM images of PHNPs (a, a1) and PHNPs@DPA-S-S-BSA-MA (b, b1); (c) XRD patterns of PHNPs and PHNP@DPA-S-S-BSA-MA powder; (d) the size distribution of PHNPs and PHNPs@DPA-S-S-BSA-MA measured by DLS. | ||
To control the release of 3-MA, PHNPs were subsequently blocked with mannose modified BSA, which could target the TAMs with a high expression of mannose receptors. The synthesis process of PHNPs@DPA-S-S-BSA-MA@3-MA is shown in Scheme 1. A disulfide bond was introduced by an amidation reaction, followed by a carboxyl group on the surface of PHNPs by a click reaction. Afterwards, 3-MA was loaded into the cavity of PHNPs@DPA-S-S-PEG-COOH, followed by the coating of BSA which was modified with mannose via the amidation reaction between the carboxyl and amino groups. The system was defined as PHNPs@DPA-S-S-BSA-MA@3-MA as shown in Scheme 1a. According to the full absorption spectrum of 3-MA, it was found that there was a characteristic absorption peak at 272.5 nm. The loading efficiency of 3-MA was determined as 4.99%.
3.2 Glutathione-responsive 3-MA release
The release of 3-MA from PHNPs@DPA-S-S-BSA-MA@3-MA at different concentrations of GSH was measured (Fig. 2a). PHNP@DPA-S-S-BSA-MA@3-MA solutions were dispersed in tris buffer that contained 0, 2 and 10 mM of GSH, for 48 h. The cumulative release amount of 3-MA was measured by using a UV-vis spectrophotometer. When there was no GSH in the buffer, only around 20% of the total 3-MA was released from PHNPs@DPA-S-S-BSA-MA@3-MA within 48 h, indicating the good 3-MA retention capability of PHNPs@DPA-S-S-BSA-MA@3-MA. For comparison, the cumulative release amount of 3-MA from PHNPs@DPA-S-S-BSA-MA@3-MA increased to 44% and 83.5% after 48 h at 2 and 10 mM concentrations of GSH tris buffer. The above results showed that the 3-MA release was started by the GSH-mediated removal of BSA via breaking the disulphide bond and the amount released was positively correlated with the concentration of glutathione.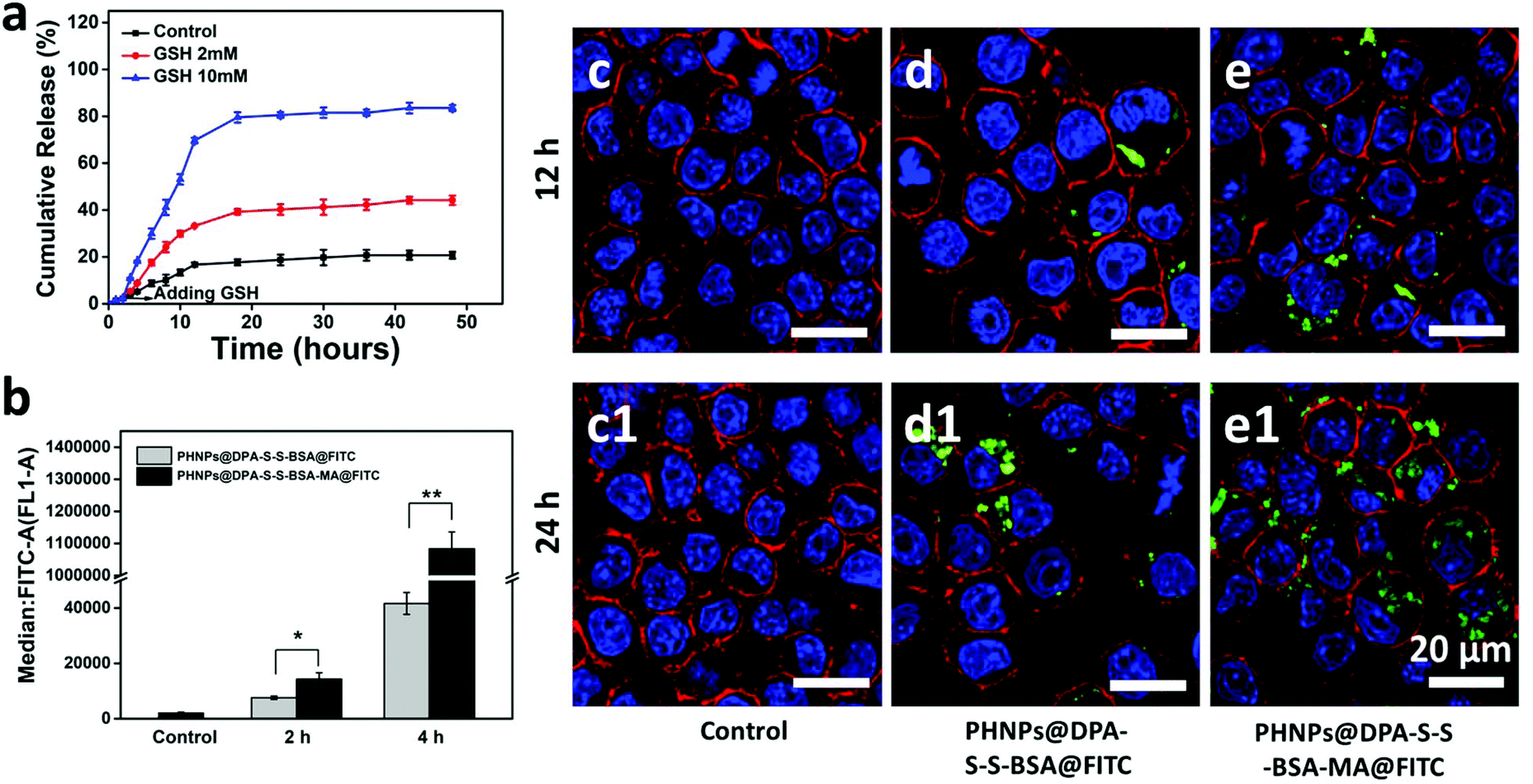 | ||
| Fig. 2 (a) Redox-responsive cumulative release amount of 3-MA from PHNPs@DPA-S-S-BSA-MA at different concentrations of GSH; (b) the median of RAW 264.7 cells for phagocytosis of PHNPs@DPA-S-S-BSA@FITC and PHNPs@DPA-S-S-BSA-MA@FITC after culturing with 2 and 4 h, detected by FCM. Characterization of phagocytosis of PHNPs@DPA-S-S-BSA@FITC and PHNPs@DPA-S-S-BSA-MA@FITC by macrophages using CLSM after 12 (c–e) and 24 h (c1–e1) of incubation. | ||
3.3 Cytotoxicity and cellular uptake of PHNPs@DPA-S-S-BSA-MA@3-MA
The cytotoxicity of PHNPs@DPA-S-S-BSA-MA was investigated via the CCK-8 assay,48 and PHNPs@DPA-S-S-BSA-MA@3-MA showed good cytotoxicity to both MDA-MB-231 cells and RAW 264.7 cells (Fig. S9c†). 3-MA and PHNPs@DPA-S-S-BSA-MA also had good cytotoxicity to both MDA-MB-231 cells and RAW 264.7 cells (Fig. S9a–d†). The results suggest that PHNPs@DPA-S-S-BSA-MA@3-MA had good cytotoxicity in vitro.To demonstrate the macrophage targeting of PHNPs@DPA-S-S-BSA-MA in vitro, FITC-modified PHNPs@DPA-S-S-BSA-MA (PHNPs@DPA-S-S-BSA-MA@FITC) and PHNPs@DPA-S-S-BSA (PHNPs@DPA-S-S-BSA@FITC) were synthesized in advance. As shown in Fig. 2b, PHNPs@DPA-S-S-BSA-MA@FITC has higher cellular uptake than PHNPs@DPA-S-S-BSA@FITC of the RAW 264.7 cells. Furthermore, the cellular uptake of MDA-MB-231 cells and HUVEC were also investigated (Fig. S8†). The above results of cellular uptake confirm that mannose modified PHNPs@DPA-S-S-BSA (PHNPs@DPA-S-S-BSA-MA) had higher endocytosis for macrophages with mannose-receptor expression, but not for MDA-MB-231 cells and HUVEC, and the cellular endocytosis was positively correlated with the incubation time. Meanwhile, to visually characterize the phagocytosis of nanoparticles, the phagocytosis of macrophages was characterized by CLSM after incubation for 12 and 24 h. The mannose modified PHNPs@DPA-S-S-BSA (PHNPs@DPA-S-S-BSA-MA) displayed relatively high uptake efficiency by macrophages (Fig. 2c–e and c1–e1).
3.4 PHNP@DPA-S-S-BSA-MA@3-MA induced polarization of macrophages in vitro
According to a previous study,31 iron oxide nanoparticles suppress tumor growth though inducing TAM re-polarization. In addition, PI3K γ inhibitors could convert TAMs into pro-inflammatory M1 macrophages that promote the immune response and inhibit tumor growth by inhibiting PI3K γ and promoting NF-κB p65 expression. 3-Methyladenine is an effective autophagy inhibitor, and studies have found that it could also inhibit PI3K γ expression. Thus, the PHNPs@DPA-S-S-BSA-MA@3-MA would have a good effect on switching the TAMs into the pro-inflammatory M1-type.To prove the effect above, THP-1 macrophages (co-cultured with MDA-MB-231 cells) and RAW 264.7 cells (cultured alone) were hatched with PHNPs@DPA-S-S-BSA-MA, 3-MA, and PHNPs@DPA-S-S-BSA-MA@3-MA. Flow cytometry was used to detect the macrophage phenotype. PHNPs@DPA-S-S-BSA-MA@3-MA had better effects on decreasing the expression of CD 206 (Fig. 3a & Fig. S12a†) and increasing the expression of CD 86 (Fig. 3b & Fig. S12b†) than the groups of PHNPs@DPA-S-S-BSA-MA and 3-MA. The qRT-PCR results showed that PHNPs@DPA-S-S-BSA-MA, 3-MA, and PHNPs@DPA-S-S-BSA-MA@3-MA inhibited the expression of anti-inflammatory genes (Fig. 3c & Fig. S13a†) and promoted the expression of pro-inflammatory genes (Fig. 3d & Fig. S13b†), while the group co-cultured with the tumor cells increased the expression of anti-inflammatory genes and pro-inflammatory genes compared with the untreated group. Consistent with the flow cytometry results, PHNPs@DPA-S-S-BSA-MA@3-MA had a better effect on the expression of pro-inflammatory genes and inhibited the expression of anti-inflammatory genes than other groups.
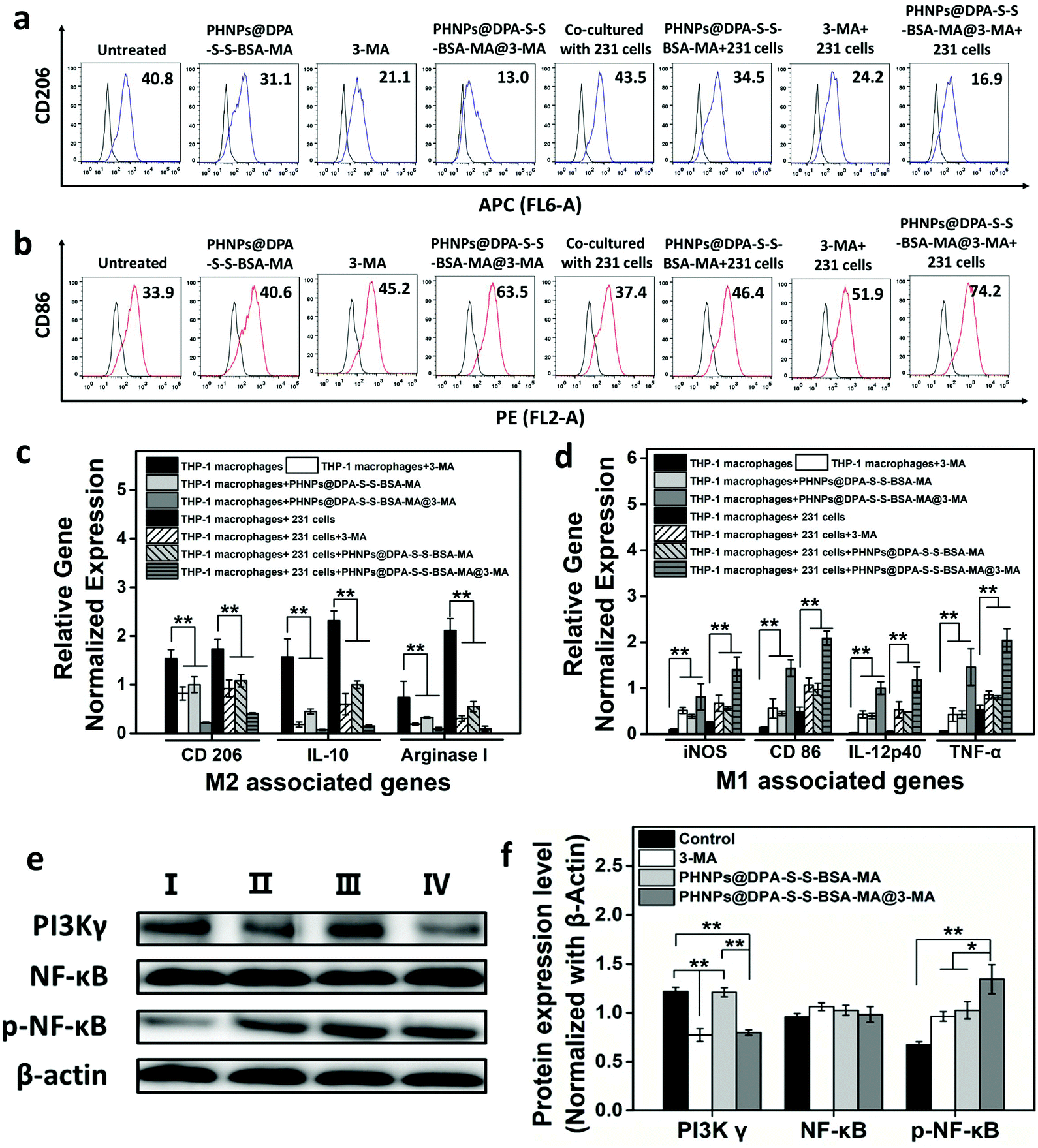 | ||
| Fig. 3 Flow cytometry analysis of the expression of CD 206 (a) and CD 86 (b) in THP-1 macrophages. qRT-PCR analysed the gene expression of the THP-1 macrophages. (c) M2 associated genes and (d) M1 associated genes (n = 3). (e) Western blotting of RAW 264.7 cells after administration of the control (I), 66.9 μM of 3-MA (II). 200 μg mL−1 of PHNPs@DPA-S-S-BSA-MA (III) and 200 μg mL−1 of PHNPs@DPA-S-S-BSA-MA@3-MA (IV). (f) Quantitative analysis of PI3K γ, NF-κBp65 and p-NF-κBp65 expressions in the RAW 264.7 cells. Error bars are standard error of the mean (n = 3), *p < 0.05, **p < 0.01. | ||
To further verify how PHNPs@DPA-S-S-BSA-MA@3-MA regulates the expression of macrophages at the molecular level, the expression of key proteins was evaluated with western blotting. The expression of major proteins in macrophages cultured at different concentrations of 3-MA for 24 h was investigated. The results and gray scale statistics (Fig. S11a and b†) showed that 3-MA effectively suppressed the PI3K γ protein expression and activated the phosphorylation of NF-κB p65. It was positively correlated with the effect and concentration. In MDA-MB-231 cells, 3-MA had a similar trend effect (Fig. S10a and b†). The results (Fig. 3e) show that PHNPs@DPA-S-S-BSA-MA promoted the expression of NF-κB p65 protein, and 3-MA suppressed the PI3K γ protein expression and enhanced the NF-κB p65 protein expression. Interestingly, PHNPs@DPA-S-S-BSA-MA@3-MA inhibited the PI3K γ protein expression and promoted the NF-κB p65 protein expression more effectively. According to the results of gray scale statistics (Fig. 3f), the promotion effect of the PHNP@DPA-S-S-BSA-MA@3-MA group on the NF-κB p65 protein significantly differed from the other groups. In addition, a similar trend was found in MDA-MB-231 cells as measured in Fig. S10c & d.†
3.5 In vivo activation of immune response and macrophage re-polarization of PHNPs@DPA-S-S-BSA-MA@3-MA
To investigate the macrophage re-polarization and activation of immune response with PHNPs@DPA-S-S-BSA-MA@3-MA in vivo, tumor-infiltrating immune cells were quantitatively measured by FCM. It is well-known that TAM re-polarization could activate the immune response and suppress the proportion of Treg cells. Firstly, we characterized the amount of TAMs and associated immune cells in tumor-bearing mice after 21 days of different treatments. According to quantitative results (Fig. 4a and Fig. S16 ESI†), the groups of PHNPs@DPA-S-S-BSA-MA, 3-MA and PHNPs@DPA-S-S-BSA-MA@3-MA showed low levels of the M2-type and high levels of the M1-type compared with the saline group (control). The PHNP@DPA-S-S-BSA-MA@3-MA group (the proportion of M1-type macrophages was 18.7% and that of the M2-type macrophages was 4.11%) showed better effects on TAM re-polarization compared with the PHNP@DPA-S-S-BSA-MA group (the proportion of M1-type macrophages was 9.31% and that of the M2-type macrophages was 9.53%) and 3-MA (the proportion of M1-type macrophages was 11.4% and that of the M2-type macrophages was 8.58%). Secondly, as for the immune-related immune cells, the groups of PHNPs@DPA-S-S-BSA-MA, 3-MA and PHNPs@DPA-S-S-BSA-MA@3-MA displayed high levels of CD8+ T cells, CD4+ T cells, NK cells and B cells and low levels of Treg cells compared with saline (Fig. 4b and Fig. S15 ESI†).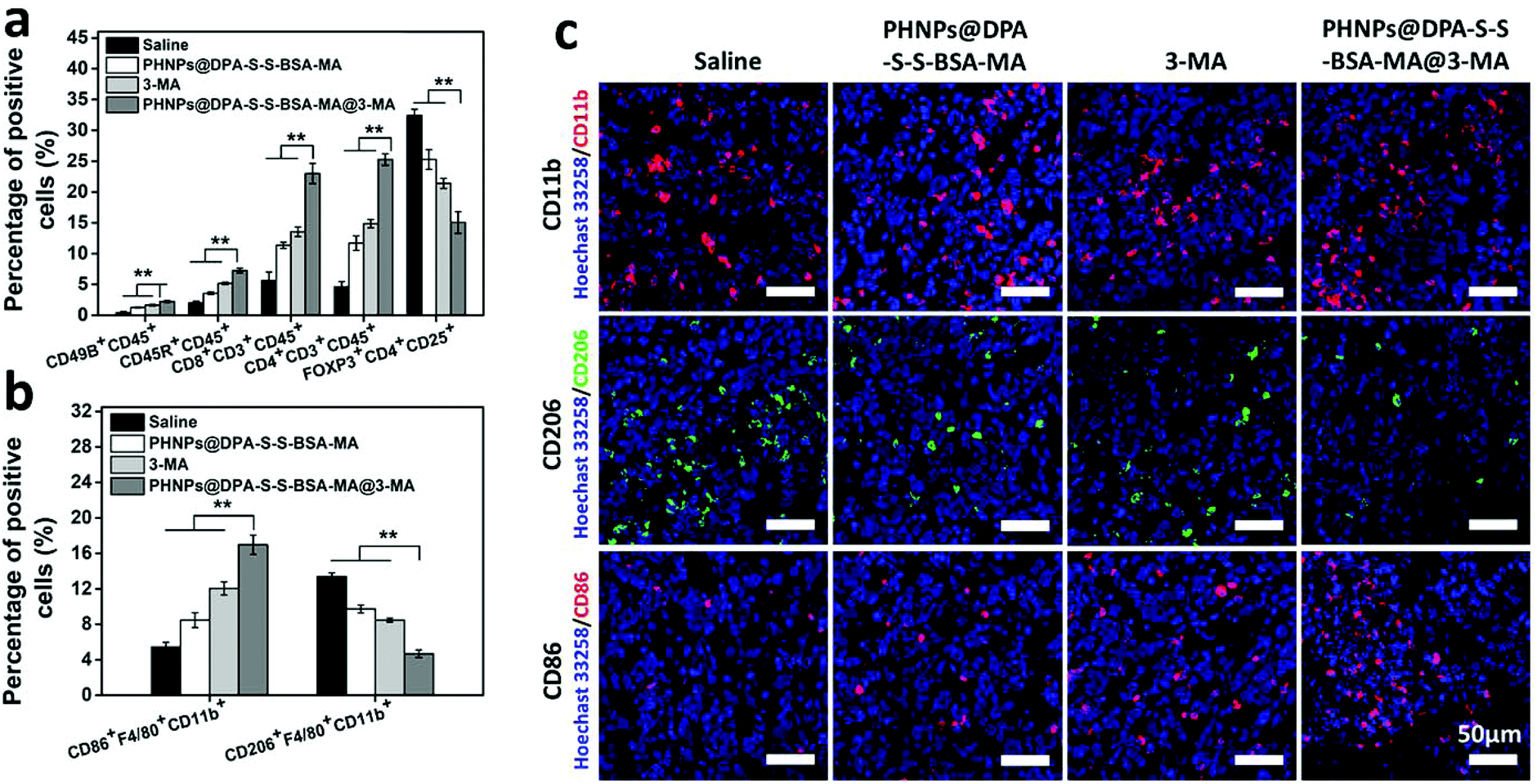 | ||
| Fig. 4 PHNPs@DPA-S-S-BSA-MA@3-MA switches the TAMs’ M2-type toward the M1-type and activates the immune system: (a) FCM detects the macrophage phenotype in tumors after PHNP@DPA-S-S-BSA-MA@3-MA treatment (n = 6); (b) FCM detects immune cells in tumors after various treatments (n = 6). (c) Immunofluorescence image of tumor sections stained with a macrophage marker of CD11b (red), the M2 macrophage marker of CD206 (green) and the M1 macrophage marker of CD86 (red) after the administration of PHNPs@DPA-S-S-BSA-MA@3-MA. Scale bar 50 μm. | ||
The proportion of CD8+ T cells with the PHNP@DPA-S-S-BSA-MA@3-MA group was 25.8%, showing higher levels than the other groups (the proportion with the saline group was 4.89%, that with the PHNP@DPA-S-S-BSA-MA group was 11.6% and that with the 3-MA group was 14.4%). As for the proportion of CD4+ T cells, with the PHNP@DPA-S-S-BSA-MA@3-MA group it was 23.8%, showing higher levels than the other groups (with the saline group it was 2.43%, with the PHNP@DPA-S-S-BSA-MA group it was 9.49% and with the 3-MA group it was 12.4%). At the same time, the proportion of B cells and NK cells with the PHNP@DPA-S-S-BSA-MA@3-MA group showed higher levels than other groups (B cells: with the saline group it was 1.58%, with the PHNP@DPA-S-S-BSA-MA group it was 3.55%, with the 3-MA group it was 5.09% and with the PHNP@DPA-S-S-BSA-MA@3-MA group it was 7.53%; NK cells: with the saline group it was 0.52%, with the PHNP@DPA-S-S-BSA-MA group it was 1.40%, with the 3-MA group it was 1.55% and with the PHNP@DPA-S-S-BSA-MA@3-MA group it was 2.50%). It should be mentioned that the proportion of Treg cells in the tumor with the PHNP@DPA-S-S-BSA-MA@3-MA group was 12.6%, showing lower levels than the other groups (with the saline group it was 34.3%, with the PHNP@DPA-S-S-BSA-MA group it was 29.5% and with the 3-MA group it was 22.7%). The results were consistent with other studies.31
The immunofluorescence of tumor tissues was characterized by CLSM to further characterize TAM polarization. According to the results shown in Fig. 4c, the total amount of macrophages in the tumor tissue was approximately the same. However, the M2-type macrophages in the tumor tissue with the PHNP@DPA-S-S-BSA-MA group presented a significantly decreasing trend compared with other groups. Meanwhile, the M1-type macrophages showed the opposite trend, and the PHNP@DPA-S-S-BSA-MA@3-MA group showed a higher proportion than other groups. It was consistent with the data in ref. 33 and 40.
3.6 PHNPs@DPA-S-S-BSA-MA@3-MA regulating the secretion of immune-related factors
According to the above results, PHNPs@DPA-S-S-BSA-MA@3-MA could effectively improve the tumor immunosuppressive microenvironment, activate immune cells and inhibit Treg cells. The immune factors involved in the remodelling of the immune microenvironment were further investigated. According to the immunofluorescence results of the tumor tissue (Fig. 5a), the immunity promotion factors such as IL-1β, iNOS and TNF-α, PHNPs@DPA-S-S-BSA-MA, 3-MA and PHNPs@DPA-S-S-BSA-MA@3-MA presented a significantly increasing trend compared with the saline group. While the immunosuppressive factors such as IL-10, TGF-β and arginase I showed the opposite trend (Fig. 5b). qRT-PCR was performed to reveal the gene expression levels of the tumor tissues. The PHNP@DPA-S-S-BSA-MA@3-MA group increased the expression of M1-type macrophage related genes (Fig. 5c) and decreased the expression of M2-type macrophage related genes (Fig. 5d) compared with the other groups. As revealed by the western blot results (Fig. 5e and Fig. S14, ESI†), the proteins of IL-1β and TNF-α of PHNP@DPA-S-S-BSA-MA, 3-MA and PHNP@DPA-S-S-BSA-MA@3-MA groups presented a significantly increasing trend compared with the saline group, while the proteins of IL-10 and TGF-β showed the opposite trend.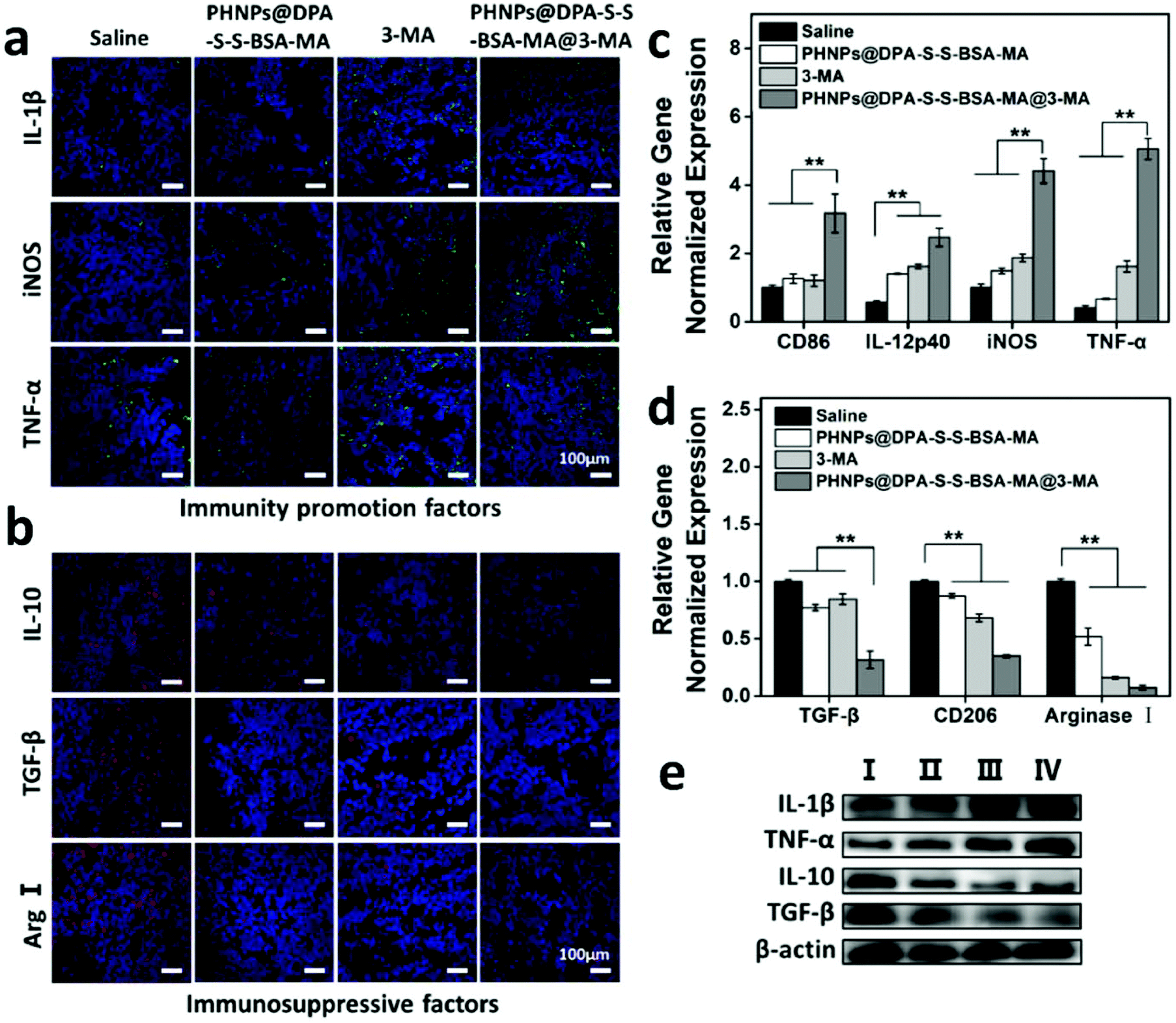 | ||
| Fig. 5 PHNPs@DPA-S-S-BSA-MA@3-MA regulated the immunity promotion factors and immunosuppressive factors: (a) representative immune-fluorescence images of tumor sections stained with the immunity promotion factors. (b) Representative immunofluorescence images of tumor sections stained with the immunosuppressive factors. Magnification 400×; scale bar 100 μm. (c, d) qRT-PCR analysed the expression of immunity promotion associated genes and immunosuppressive associated genes of the tumor issues. Error bars are standard error of the mean, **p < 0.01. (e) Western blotting was measured to analyse the expression of immunity promotion protein and immunosuppressive protein after the administration of saline (I), PHNPs@DPA-S-S-BSA-MA (II), 3-MA (III) and PHNPs@DPA-S-S-BSA-MA@3-MA (IV), respectively. | ||
3.7 The antitumor effect of PHNPs@DPA-S-S-BSA-MA@3-MA in vivo
To investigate the antitumor efficacy of PHNPs@DPA-S-S-BSA-MA@3-MA in vivo, MDA-MB-231 tumor-bearing mice were used. The images of tumor-bearing Balb/c mice (Fig. 6a) show that PHNPs@DPA-S-S-BSA@3-MA showed the best therapeutic effect among other groups. The relative tumor volume V/V0 (Fig. 6c) showed a similar trend. Fig. 6b & d show the tumor photographs and tumors weight in each group. The survival curve of mice also displayed a similar trend (Fig. S18, ESI†). Considering all the above results, the PHNP@DPA-S-S-BSA-MA@3-MA group showed the best anti-tumor efficacy in vivo when comparing with other groups. It significantly suppressed tumor growth and prolonged the mice survival time.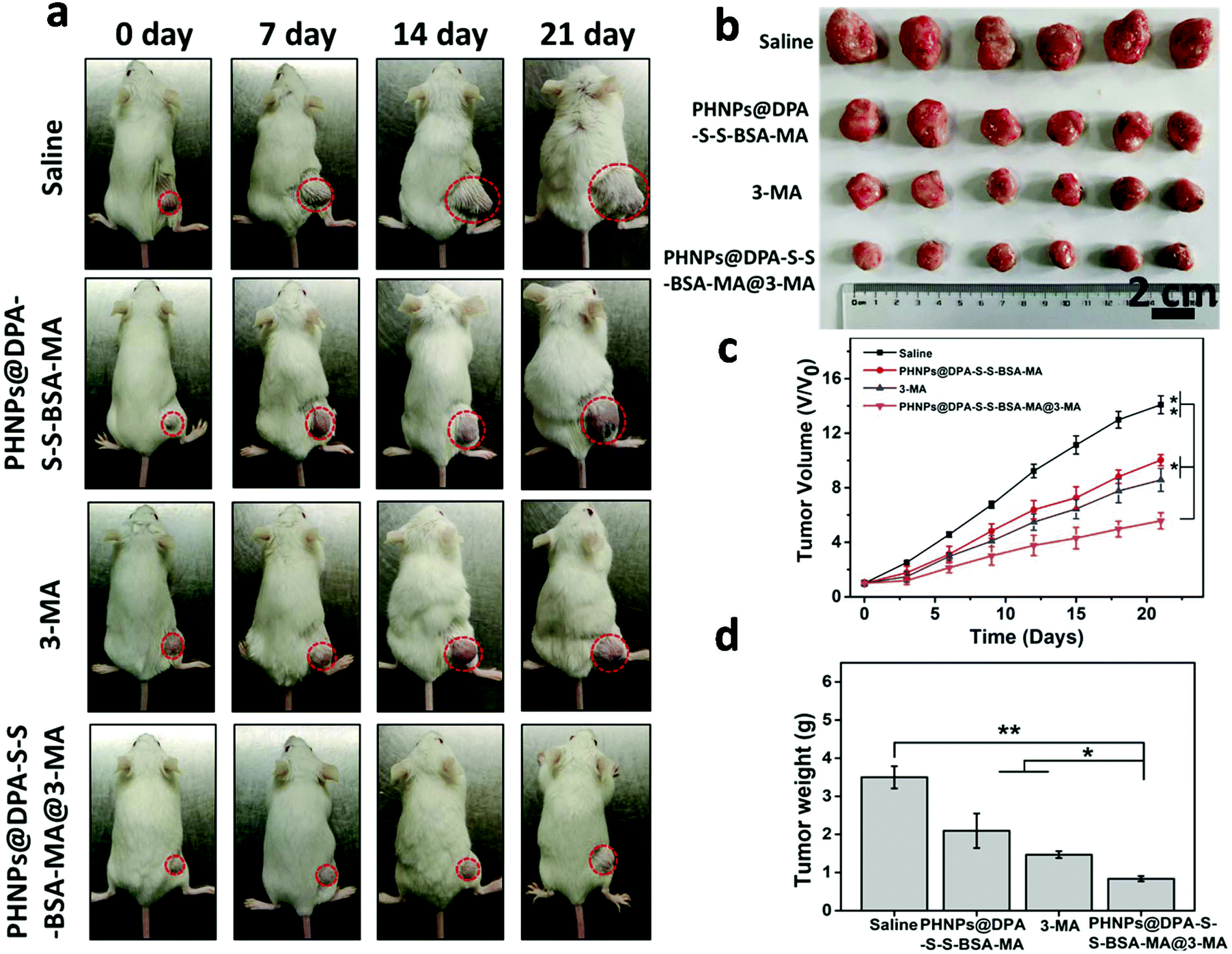 | ||
| Fig. 6 (a) The images of tumor-bearing Balb/c mice. (b) The photographs of the tumours after different treatments for 21 days. (c) Tumor volumes (V/V0) and (d) tumor weight (n = 6). Error bars are standard error of the mean, *p < 0.05, **p < 0.01. | ||
H&E images of the main organs of the tumor-bearing mice were characterized to evaluate the biocompatibility of PHNPs@DPA-S-S-BSA-MA@3-MA in vivo. According to Fig. S19† and mouse body weight (Fig. S17 ESI†), PHNPs@DPA-S-S-BSA-MA@3-MA presented no obvious toxic and side effects on the main organs and tissues of mice, indicating that PHNPs@DPA-S-S-BSA-MA@3-MA also had good biocompatibility in vivo. Subsequently, TUNEL and H&E staining was performed to demonstrate the antitumor effect of PHNPs@DPA-S-S-BSA-MA@3-MA in vivo. As shown in Fig. 7a & b, the PHNP@DPA-S-S-BSA-MA@3-MA group induced the most severe tumor cell apoptosis, as shown by the condensation of chromatin and cell shrinkage in H&E images. At the same time, in TUNEL images, a large amount of FITC dots co-located with cell nuclei is shown.
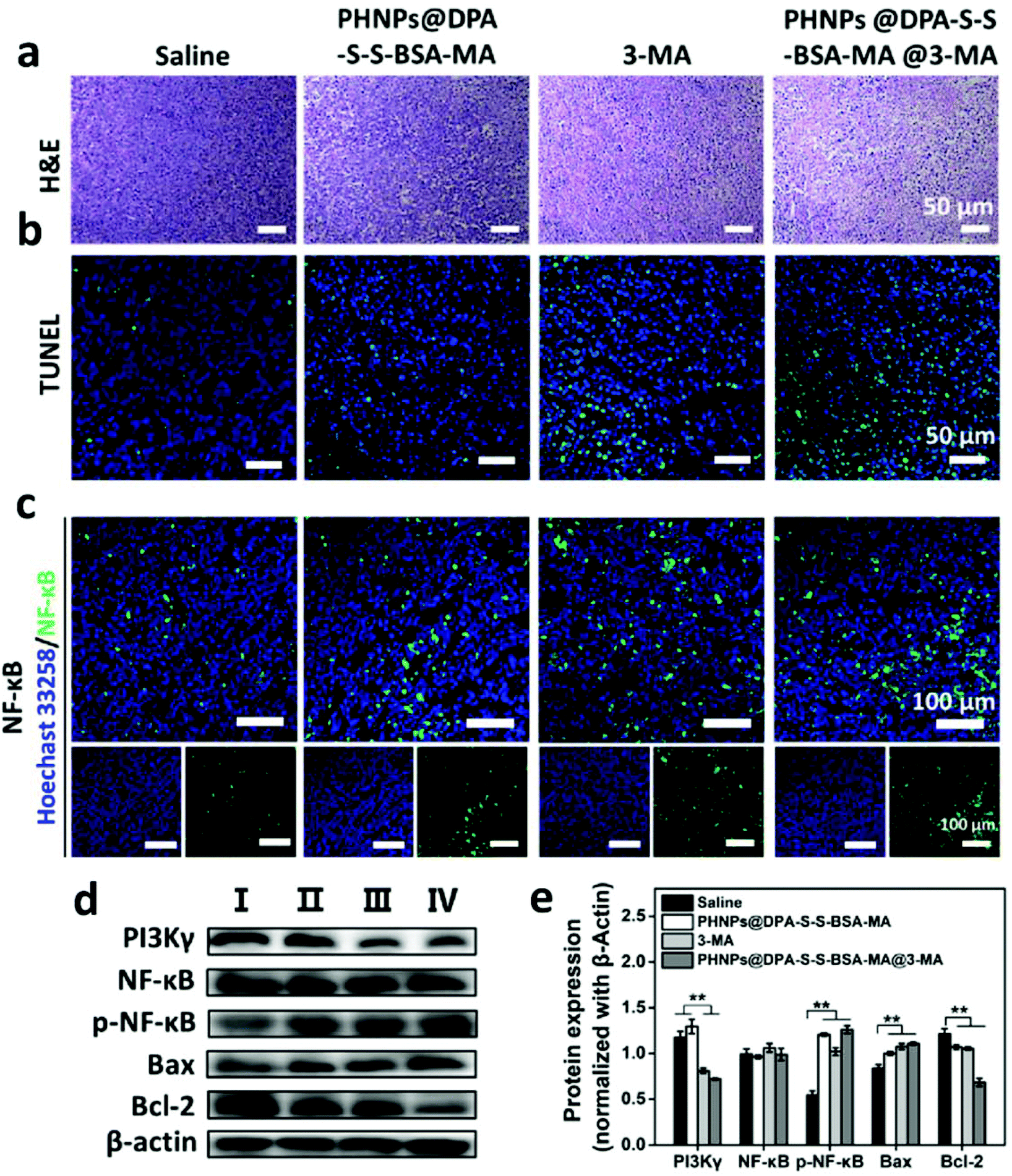 | ||
| Fig. 7 Histological characterization. (a, b) Images of H&E and TUNEL staining of tumor issues. (c) IFC images of tumor tissues stained with anti-NF-κBp65. (d) Western blotting investigated the expressions of PI3K γ, NF-κBp65, p-NF-κBp65, Bax and Bcl-2 in tumor sections under different treatments, saline (I), PHNPs@DPA-S-S-BSA-MA (II), 3-MA (III) and PHNPs@DPA-S-S-BSA-MA @3-MA (IV). (e) Quantitative analysis of the expressions of PI3K γ, NF-κB p65, p-NF-κB p65, Bax and Bcl-2 in the tumor sections under different treatments. Error bars are standard error of the mean (n = 3), **p < 0.01. | ||
To further reveal how PHNPs@DPA-S-S-BSA-MA@3-MA regulates the re-polarization of TAMs to pro-inflammatory M1 type macrophages in vivo, the expression levels of PI3K γ, NF-κB p65, p-NF-κB p65, Bax and Bcl-2 proteins in tumor tissues were detected. The western blotting assay (Fig. 7d) and grayscale statistics (Fig. 7e) results showed that 3-MA and PHNPs@DPA-S-S-BSA-MA@3-MA effectively inhibited the expression of PI3K γ protein compared with the saline and PHNP@DPA-S-S-BSA-MA groups. They effectively activated the expression of p-NF-κB p65 compared with the saline group. Moreover, PHNP@DPA-S-S-BSA-MA, 3-MA and PHNP@DPA-S-S-BSA-MA groups effectively activated the expression of Bax and inhibited protein Bcl-2. The IFC results of NF-κB p65 protein in tumor tissues further displayed that PHNP@DPA-S-S-BSA-MA, 3-MA and PHNP@DPA-S-S-BSA-MA@3-MA groups activated the expression of NF-κB p65 in contrast to the saline group in vivo. The results were similar to those of in vitro. Moreover, in vivo, 3-MA activated the expression of NF-κB p65 protein by inhibiting the expression of PI3K γ protein. PHNPs@DPA-S-S-BSA-MA also activated NF-κB p65 protein expression through other signalling pathways. This indicated that PHNPs@DPA-S-S-BSA-MA @3-MA effectively inhibited the expression of PI3K γ protein, and more effectively activated the expression of NF-κB p65 compared with PHNPs@DPA-S-S-BSA-MA and 3-MA. Previous studies showed that the activation of NF-κB p65 could effectively promote the re-polarization of TAMs to pro-inflammatory M1-type macrophages. Hence, PHNPs@DPA-S-S-BSA-MA@3-MA activated NF-κB p65 protein by inhibiting the expression of PI3K γ protein, thus promoting the re-polarization of TAMs. Taken together, we confirmed our hypothesis that the PHNP@DPA-S-S-BSA-MA@3-MA system could efficiently relieve the immunosuppression, activate the immune response and inhibit tumor growth via switching macrophage phenotypes from M2-type to pro-inflammatory M1-type.
4. Conclusions
In summary, we designed and developed a novel PHNP@DPA-S-S-BSA-MA@3-MA system for efficiently targeting TAMs in this study. The system significantly switched macrophages into the pro-inflammatory M1-type, improved the pro-inflammatory genes and inhibited the anti-inflammatory genes. In addition, this system effectively inhibited PI3K γ protein expression in macrophages and tumor cells, and upregulated NF-κB p65 protein expression. Similar results were obtained in anti-tumor tests in vivo. This system was proved to activate NF-κB p65 protein expression by inhibiting the expression of PI3K γ protein and switch the TAM phenotype into the pro-inflammatory M1-type. After re-polarization, macrophages could re-model the immunosuppressive microenvironment via increasing CD8+ T cells, CD4+ T cells, B cells and NK cells, as well as the release of immune-promoting factors; on the other hand, via reducing the proportion of Treg cells, it decreases the secretion of immunosuppressive factors and finally leads to the inhibition of tumor growth with high efficiency.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51825302, 21734002, 31700827 & 51673032), the State Key Project of Research and Development (Grant No. 2016YFC1100300 & 2017YFB0702603), the Natural Science Foundation of Chongqing Municipal Government (CXTDX201601002), the China Postdoctoral Science Foundation funded project (2017M622971 & 2018T110946), the Innovation Team in University of Chongqing Municipal Government (CXTDX201601002), and the Fundamental Research Funds for the Central Universities (10611CDJXZ238826).References
- M. Vanneman and G. Dranoff, Nat. Rev. Cancer, 2012, 12, 237–251 CrossRef CAS PubMed.
- L. Jeanbart and M. A. Swartz, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14467–14472 CrossRef CAS.
- P. Kvistborg and J. W. Yewdell, Science, 2018, 359, 516–517 CrossRef CAS.
- H. Tsukamoto, K. Fujieda, A. Miyashita, S. Fukushima, T. Ikeda, Y. Kubo, S. Senju, H. Ihn, Y. Nishimura and H. Oshiumi, Cancer Res., 2018, 78, 5011–5022 CrossRef CAS PubMed.
- Y. Huang, J. Yuan, E. Righi, W. S. Kamoun, M. Ancukiewicz, J. Nezivar, M. Santosuosso, J. D. Martin, M. R. Martin, F. Vianello, P. Leblanc, L. L. Munn, P. Huang, D. G. Duda, D. Fukumura, R. K. Jain and M. C. Poznansky, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17561–17566 CrossRef CAS.
- M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, L. M. Coussens, D. I. Gabrilovich, S. Ostrand-Rosenberg, C. C. Hedrick, R. H. Vonderheide, M. J. Pittet, R. K. Jain, W. Zou, T. K. Howcroft, E. C. Woodhouse, R. A. Weinberg and M. F. Krummel, Nat. Med., 2018, 24, 541–550 CrossRef CAS PubMed.
- R. Ostuni, F. Kratochvill, P. J. Murray and G. Natoli, Trends Immunol., 2015, 36, 229–239 CrossRef CAS PubMed.
- J. W. Pollard, Nat. Rev. Cancer, 2004, 4, 71–78 CrossRef CAS PubMed.
- R. A. Franklin, W. Liao, A. Sarkar, M. V. Kim, M. R. Bivona, K. Liu, E. G. Pamer and M. O. Li, Science, 2014, 344, 921–925 CrossRef CAS PubMed.
- M. Niu, S. Valdes, Y. W. Naguib, S. D. Hursting and Z. Cui, Mol. Pharm., 2016, 13, 1833–1842 CrossRef CAS PubMed.
- D. G. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed.
- Y. Shi, Y. F. Ping, W. Zhou, Z. C. He, C. Chen, B. S. J. Bian, L. Zhang, L. Chen, X. Lan, X. C. Zhang, K. Zhou, Q. Liu, H. Long, T. W. Fu, X. N. Zhang, M. F. Cao, Z. Huang, X. Fang, X. Wang, H. Feng, X. H. Yao, S. C. Yu, Y. H. Cui, X. Zhang, J. N. Rich, S. Bao and X. W. Bian, Nat. Commun., 2017, 8, 15080 CrossRef CAS.
- J. Hao, F. Yan, Y. Zhang, A. Triplett, Y. Zhang, D. A. Schultz, Y. Sun, J. Zeng, K. A. T. Silverstein, Q. Zheng, D. A. Bernlohr, M. P. Cleary, N. K. Egilmez, E. Sauter, S. Liu, J. Suttles and B. Li, Cancer Res., 2018, 78, 2343–2355 CrossRef CAS.
- B. Z. Qian and J. W. Pollard, Cell, 2010, 41, 39–51 CrossRef PubMed.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Cell, 2017, 168, 707–723 CrossRef CAS PubMed.
- M. De Palma and C. E. Lewis, Cancer Cell, 2013, 23, 277–286 CrossRef CAS PubMed.
- B. Ruffell and L. M. Coussens, Cancer Cell, 2015, 27, 462–472 CrossRef CAS PubMed.
- R. Noy and J. W. Pollard, Immunity, 2014, 41, 49–61 CrossRef CAS PubMed.
- J. M. Brown, L. Recht and S. Strober, Clin. Cancer Res., 2017, 23, 3241–3250 CrossRef PubMed.
- J. B. Mitchem, D. J. Brennan, B. L. Knolhoff, B. A. Belt, Y. Zhu, D. E. Sanford, L. Belaygorod, D. Carpenter, L. Collins, D. Piwnica-Worms, S. Hewitt, G. M. Udupi, W. M. Gallagher, C. Wegner, B. L. West, A. Wang-Gillam, P. Goedegebuure, D. C. Linehan and D. G. DeNardo, Cancer Res., 2013, 73, 1128–1141 CrossRef CAS PubMed.
- A. Patel and S. Sant, Biotechnol. Adv., 2016, 34, 803–812 CrossRef CAS PubMed.
- M. Ovais, M. Guo and C. Chen, Adv. Mater., 2019, 31, 1808303 CrossRef PubMed.
- Y. Qian, S. Qiao, Y. Dai, G. Xu, B. Dai, L. Lu, X. Yu, Q. Luo and Z. Zhang, ACS Nano, 2017, 11, 9536–9549 CrossRef CAS PubMed.
- M. Song, T. Liu, C. Shi, X. Zhang and X. Chen, ACS Nano, 2016, 10, 633–647 CrossRef CAS PubMed.
- J. Liu, H. Liang, M. Li, Z. Luo, J. Zhang, X. Guo and K. Cai, Biomaterials, 2018, 157, 107–124 CrossRef CAS PubMed.
- L. Dai, K. Li, M. Li, X. Zhao, Z. Luo, L. Lu, Y. Luo and K. Cai, Adv. Funct. Mater., 2018, 28, 1707249 CrossRef.
- J. Conde, C. Bao, Y. Tan, D. Cui, E. R. Edelman, H. S. Azevedo, H. J. Byrne, N. Artzi and F. Tian, Adv. Funct. Mater., 2015, 25, 4183–4194 CrossRef CAS PubMed.
- L. Dai, X. Li, X. Duan, M. Li, P. Niu, H. Xu, K. Cai and H. Yang, Adv. Sci., 2019, 6, 1801807 CrossRef PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- C. Li, Nat. Mater., 2014, 13, 110–115 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- V. Mulens-Arias, J. M. Rojas, S. Perez-Yague, M. P. Morales and D. F. Barber, Biomaterials, 2015, 52, 494–506 CrossRef CAS PubMed.
- G.-T. Yu, L. Rao, H. Wu, L.-L. Yang, L.-L. Bu, W.-W. Deng, L. Wu, X. Nan, W.-F. Zhang, X.-Z. Zhao, W. Liu and Z.-J. Sun, Adv. Funct. Mater., 2018, 28, 1801389 CrossRef.
- S. Joshi, A. R. Singh, M. Zulcic and D. L. Durden, Mol. Cancer Res., 2014, 12, 1520–1531 CrossRef CAS PubMed.
- L. Zhang, C. Huang, Y. Guo, X. Gou, M. Hinsdale, P. Lloyd and L. Liu, J. Immunol., 2015, 195, 5404–5414 CrossRef CAS PubMed.
- E. Sahin, S. Haubenwallner, M. Kuttke, I. Kollmann, A. Halfmann, A. M. Dohnal, L. Chen, P. Cheng, B. Hoesel, E. Einwallner, J. Brunner, J. B. Kral, W. C. Schrottmaier, K. Thell, V. Saferding, S. Bluml and G. Schabbauer, J. Immunol., 2014, 193, 1717–1727 CrossRef CAS PubMed.
- M. M. Kaneda, K. S. Messer, N. Ralainirina, H. Li, C. J. Leem, S. Gorjestani, G. Woo, A. V. Nguyen, C. C. Figueiredo, P. Foubert, M. C. Schmid, M. Pink, D. G. Winkler, M. Rausch, V. J. Palombella, J. Kutok, K. McGovern, K. A. Frazer, X. Wu, M. Karin, R. Sasik, E. E. Cohen and J. A. Varner, Nature, 2016, 539, 437–442 CrossRef CAS PubMed.
- O. De Henau, M. Rausch, D. Winkler, L. F. Campesato, C. Liu, D. H. Cymerman, S. Budhu, A. Ghosh, M. Pink, J. Tchaicha, M. Douglas, T. Tibbitts, S. Sharma, J. Proctor, N. Kosmider, K. White, H. Stern, J. Soglia, J. Adams, V. J. Palombella, K. McGovern, J. L. Kutok, J. D. Wolchok and T. Merghoub, Nature, 2016, 539, 443–447 CrossRef CAS PubMed.
- L. Liu, H. Yi, H. He, H. Pan, L. Cai and Y. Ma, Biomaterials, 2017, 134, 166–179 CrossRef CAS PubMed.
- X. Wang, G. Luo, K. Zhang, J. Cao, C. Huang, T. Jiang, B. Liu, L. Su and Z. Qiu, Cancer Res., 2018, 78, 4586–4598 CrossRef CAS PubMed.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- W. Yu, C. Liu, Y. Liu, N. Zhang and W. Xu, Pharm. Res., 2010, 27, 1584–1596 CrossRef CAS PubMed.
- Z. Niu, Q. Shi, W. Zhang, Y. Shu, N. Yang and B. Chen, Nat. Commun., 2017, 8, 766 CrossRef PubMed.
- A. R. D. Reeves, K. L. Spiller, D. O. Freytes, G. V. Novakovic and D. L. Kaplan, Biomaterials, 2015, 73, 272–283 CrossRef CAS PubMed.
- K. Y. Cai, J. Li, Z. Luo, Y. Hu, Y. Hou and X. Ding, Chem. Commun., 2011, 47, 7719–7721 RSC.
- G. Yang, L. Xu, Y. Chao, J. Xu, X. Sun, Y. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef PubMed.
- J. Zhou, J. H. Li, X. W. Ding, J. Liu, Z. Luo, Y. Liu, Q. Ran and K. Cai, Nanotechnology, 2015, 26, 425101 CrossRef PubMed.
- J. Zhou, M. H. Li, Y. Hou, Z. Luo, Q. Chen, H. Cao, R. Huo, C. C. Xue, L. Sutrisno, L. Hao, Y. Cao, H. Ran, L. Lu, K. Li and K. Y. Cai, ACS Nano, 2018, 12, 2858–2872 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr06505a |
| This journal is © The Royal Society of Chemistry 2020 |