
Real-time imaging of individual virion-triggered cortical actin dynamics for human immunodeficiency virus entry into resting CD4 T cells†
Wen
Yin‡§
ab,
Wei
Li‡
a,
Qin
Li
a,
Yuanyuan
Liu
ab,
Ji
Liu
ab,
Min
Ren
ab,
Yingxin
Ma
 a,
Zhiping
Zhang
a,
Xiaowei
Zhang
a,
Yuntao
Wu
c,
Shibo
Jiang
d,
Xian-En
Zhang
e and
Zongqiang
Cui
*a
a,
Zhiping
Zhang
a,
Xiaowei
Zhang
a,
Yuntao
Wu
c,
Shibo
Jiang
d,
Xian-En
Zhang
e and
Zongqiang
Cui
*a
aState Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan 430071, People's Republic of China. E-mail: czq@wh.iov.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
cNational Center for Biodefense and Infectious Diseases, Department of Molecular and Microbiology, George Mason University, Manassas, Virginia 22030, USA
dKey Laboratory of Medical Molecular Virology of Ministries of Education and Health, Shanghai Medical College and Institute of Medical Microbiology, Fudan University, Shanghai 200032, People's Republic of China
eNational Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, People's Republic of China
First published on 31st October 2019
Abstract
Real-time imaging of single virus particles allows the visualization of subtle dynamic events of virus–host interaction. During the human immunodeficiency virus (HIV) infection of resting CD4 T lymphocytes, overcoming cortical actin restriction is an essential step, but the dynamic process and mechanism remain to be characterized. Herein, by using quantum dot (QD) encapsulated fluorescent viral particles and single-virus tracking, we explored detailed scenarios of HIV dynamic entry and crossing the cortical actin barrier. The fine-scale temporal and spatial processes of single HIV virion interaction with the cortical actin were studied in depth during virus entry via plasma membrane fusion. Individual HIV virions modulate the subtle rearrangement of the cortical actin barrier to open a door to facilitate viral entry. The actin-binding protein, α-actinin, was found to be critical for actin dynamics during HIV entry. An α-actinin-derived peptide, actin-binding site 1 peptide (ABS1p), was developed to block HIV infection. Our findings reveal an α-actinin-mediated dynamic cortical actin rearrangement for HIV entry, and identify an antiviral target as well as a corresponding peptide inhibitor based on HIV interaction with the actin cytoskeleton.
1. Introduction
Live cell imaging of single virus particles allows the visualization of subtle molecular events in real time, such as viral entry, uncoating, and virus–host interactions.1,2 This powerful technology has been successfully used to elucidate the infection of viruses.3–5 Recently, semiconductor nanocrystal quantum dots (QDs), with their remarkable brightness and photostability, have been used to label viruses to investigate virus dynamics at the single-virus level by providing in situ and real-time evidence.6–9 The fine-scale temporal and spatial variations during viral infection can be studied in depth by QD-based single-virus tracking. To date, by employing single-virus tracking based on QD-labeling of viruses, some important information on virus infection and virus–host dynamics has been obtained.10–13CD4 T lymphocytes are the major targets of human immunodeficiency virus (HIV) infection. Resting T cells can be directly infected, and latently infected resting CD4 T cells can also serve as a major viral reservoir persisting in peripheral blood.14 However, unlike activated T cells, resting CD4 T cells are less permissive to productive HIV infection owing to several intracellular blocks, including a rigid layer of cortical actin, low levels of intracellular deoxyribonucleoside triphosphate (dNTP) pools, and the presence of restriction factors.15,16 In resting CD4 T lymphocytes, cortical actin has been suggested to represent a unique physical barrier to viral entry and nuclear migration.17 HIV must overcome the restriction of cortical actin for its entry and nuclear migration.18 There is evidence showing that actin dynamics are critical for the HIV latent infection of resting CD4 T cells.19,20 However, the dynamic process and mechanism by which HIV passes through the cortical actin layer remains to be characterized. The temporal and spatial interactions between HIV particles and cortical actin have not been studied in depth. In addition to HIV–actin interactions, the actual cellular fusion site remains controversial.21 In fact, the process of HIV entry and fusion in resting CD4 T cells has not been observed, and the involvement of actin dynamics in the HIV fusion process remains elusive.
In this study, using QD-based single-virus imaging, we tracked the dynamic processes of HIV fusion and interactions with the cortical actin in resting CD4 T cells. QD-encapsulated C–X–C chemokine receptor type 4 (CXCR4)-tropic HIV-1 (NL4-3) virion particles were constructed by site-specific labeling of the viral protein R (Vpr) during virus assembly and then used for imaging viral entry processes at the single particle level. The detailed scenarios, including the fine-scale temporal and spatial interactions of HIV with the cellular membrane and the cortical actin, were investigated using single particle imaging combined with inhibitor studies. Our results reveal HIV entry into resting CD4 T cells via an α-actinin-mediated cortical actin rearrangement following viral fusion at the plasma membrane. In addition, α-actinin, an actin-binding protein that cross-links actin filaments, was found to play an important role in regulating actin dynamics during HIV entry. An α-actinin-derived peptide was developed to block viral entry and HIV infection.
2. Materials and methods
2.1. Plasmids
Vpr-AP chimeric proteins were expressed by pcDNA3.1(+)-Vpr-AP. To construct pcDNA3.1(+)-Vpr-AP, the C-terminus of the Vpr sequence from plasmid pNL4-3 (CXCR4-tropic HIV-1 proviral DNA) was fused with the DNA sequence of the biotin acceptor peptide (AP) tag (DNA sequence: GGTCTGAACGATATCTTCGAAGCTCAGAAAATCGAATGGCACGAA, amino acid sequence: GLNDIFEAQKIEWHE) in pcDNA3.1(+) vectors (Invitrogen). The biotinylation of Vpr-AP was detected by western blotting. pcDNA3.1(+)-BirA was used to express Escherichia coli biotin ligase BirA. pNL4-3 was used to assemble the viral particles. pcDNA3.1(+)-iRFP-cortactin, pcDNA3.1(+)-iRFP-paxillin and pcDNA3.1(+)-iRFP-α-actinin were used to label cortactin, paxillin and α-actinin with near-infrared fluorescent protein (iRFP) in CD4 T cells, respectively. pEGFP-LifeAct and pECFP-LifeAct were used to label F-actin with green fluorescent protein (GFP) and cyan fluorescent protein (CFP) in CD4 T cells, respectively.2.2. Cell culture and cellular component labeling
HEK 293T (human embryonic kidney 293T) and TZM-bl (HeLa-derived indicator, obtained from ABSL-3 of Wuhan University) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco), supplemented with 10% fetal bovine serum (FBS) (Gibco), 50 U mL−1 of penicillin (Invitrogen) and 50 μg mL−1 of streptomycin (Invitrogen). CEM-SS and Rev-CEM cells were maintained in RPMI 1640 medium (Gibco), supplemented with 10% heat-inactivated FBS, 50 U mL−1 of penicillin and 50 μg mL−1 of streptomycin. Peripheral blood mononuclear cells (PBMCs) were purified from healthy donors using Ficoll-Paque Plus (GE Healthcare), through a Ficoll gradient centrifugation according to standard procedures. Resting CD4 T cells were isolated from PBMCs by using a CD4 T cell isolation kit (Miltenyi Biotec) according to the manufacturer's protocol. Purified resting CD4 T cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, penicillin (50 U mL−1), and streptomycin (50 μg mL−1). The cells were rested overnight before infection or treatment. All cell lines were maintained at 37 °C with 5% CO2.For virus entry imaging, F-actin, cortactin, α-actinin or paxillin in CD4 T cells were labeled using fluorescent proteins. Briefly, 5 × 106 CD4 T cells were nucleofected with 2.5 μg of pEGFP-LifeAct, 2.5 μg of pECFP-LifeAct, 2.5 μg of pcDNA3.1(+)-iRFP-cortactin, 2.5 μg of pcDNA3.1(+)-iRFP-paxillin or 2.5 μg of pcDNA3.1(+)-iRFP-α-actinin using an Amaxa® Human T cell Nucleofector® kit (Lonza). Nucleofection was carried out using a Nucleofector® device with the Nucleofector® Program V-024.
2.3. Peptide synthesis
Synthetic actin-binding site 1 peptide (ABS1p) (RKTFTAWCNSHLRKA), actin-binding site 3 peptide (ABS3p) (VEETSAKEGLLLWCQRKTAP) and Rhodamine B-labeled ABS1p (Rhodamine B-RKTFTAWCNSHLRKA) were synthesized, and purified by high performance liquid chromatography (HPLC) (Sangon Biotechnology Co.). The purity of the peptides was 95%. The peptides were dissolved in ultra-pure water to obtain a 10 mg mL−1 stock solution and stored at −80 °C.2.4. Virus preparation and infection
HIV-1-QDs were prepared by the transfection of 293T cells in 100 mm tissue culture dishes (Falcon; BD Biosciences) with plasmids using Lipofectamine® 2000 reagent (Invitrogen). Briefly, HIV-1-QD viruses were produced by cotransfection with 18.6 μg of pNL4-3, 2.5 μg of pcDNA3.1(+)-Vpr-AP and 2.5 μg of pcDNA3.1(+)-BirA. At 6 h post transfection, 50 μM biotin (Sigma-Aldrich) was added to the cells with DMEM containing 10% FBS. The cells were washed three times with phosphate-buffered saline (PBS, pH 7.4) after 12 h transfection, and 1 μL of Qdot™ 625 streptavidin conjugate (QDs-SA) (Invitrogen) with 20 μL of Lipofectamine® 2000 were added into DMEM without FBS. Virions in the supernatant were collected at 48 h post transfection, and filtered through an ethylene oxide sterilized 0.45 μm filter (Millipore).To generate DiO-labeled HIV-1-QDs, the QD-labeled HIV-1 (NL4-3) particles were stained with Vybrant™ 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) cell-labeling solution (Invitrogen) to label the viral envelope membrane for 2 h at 22 °C. To remove the free DiO dyes from the viral supernatant, a mixture of equal volumes of 20% polyethylene glycol (PEG) 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 solution in saline and DiO-labeled HIV-1-QDs was incubated at 4 °C for 16 h, and then the mixture was centrifuged at 17860g for 20 min to collect the viral pellets. To remove free QDs, the collected pellets were resuspended in PBS (pH 7.4) and filtered with 80 nm pore size filters (Whatman® Nuclepore). The fluorescence spectra of QDs and HIV-1-QDs were recorded on an LS 55 spectrofluorometer (PerkinElmer Co.).
000 solution in saline and DiO-labeled HIV-1-QDs was incubated at 4 °C for 16 h, and then the mixture was centrifuged at 17860g for 20 min to collect the viral pellets. To remove free QDs, the collected pellets were resuspended in PBS (pH 7.4) and filtered with 80 nm pore size filters (Whatman® Nuclepore). The fluorescence spectra of QDs and HIV-1-QDs were recorded on an LS 55 spectrofluorometer (PerkinElmer Co.).
For the infection of resting CD4 T cells, 1 × 106 cells were infected with HIV-1 (NL4-3) (225 ng p24) particles. For observing the drug-induced inhibition, the cells were pretreated with BMS-806 (10 μM), T20 (3 μM), C34 (3 μM), Jas (120 nM), ABS1p (0.1, 1, 10 μg mL−1) or ABS3p (0.1, 1, 10 μg mL−1) or left untreated for 1 h, and then the cells were infected with HIV-1 in the continuous presence of inhibitors for 2 h at 37 °C. For observing Lat-A inhibition, the cells were pretreated with 2.5 μM Lat-A for 5 min and then infected with HIV-1 for 2 h in the continuous presence of Lat-A. After infection, the cells were washed three times and then stimulated with 100 IU mL−1 interleukin-2 (IL-2) and 5 μg mL−1 phytohemagglutinin (PHA) for three days.
For the infection of CEM-SS cells and Rev-CEM cells, 1 × 106 cells were infected with HIV-1 (NL4-3) (500 ng p24). To observe drug-induced inhibition, the cells were pretreated with 10 μM of BMS-806, 3 μM of T20, 3 μM of C34, ABS1p (0.1, 1, 10 μg mL−1) or ABS3p (0.1, 1, 10 μg mL−1) or left untreated for 1 h and were then infected with HIV-1 for 2 h at 37 °C. Infected cells were washed three times and then resuspended in fresh RPMI-1640 medium supplemented with 10% FBS, penicillin (50 U mL−1), and streptomycin (50 μg mL−1), at a density of 1 × 106 cells per mL and incubated for two days.
The cytotoxicity of inhibitors was determined using a LIVE/DEAD® viability/cytotoxicity kit (Invitrogen), and was compared with that of the cells that were pretreated with 10 μM H2O2 for 1 h. Flow cytometry analysis was performed on a flow cytometer (BD LSRFortessa) and data were analyzed with FlowJo software.
2.5. Isolation of HIV-1 cores
The HIV-1 virus was concentrated by centrifugation at 130000g for 2 h at 4 °C, through a cushion of 20% (w/w) sucrose. The collected viral pellets were slowly resuspended in PBS (pH 7.4) and mixed with an equal volume of 200 mM NaCl/100 mM morpholinepropanesulfonic acid (MOPS; pH 7.0). The virions were lysed for 2 min by adding Triton X-100 to a final concentration of 0.5% at room temperature. Viral cores were collected by centrifugation at 138000g for 10 min at 4 °C and then washed with 100 mM NaCl/50 MOPS (pH 7.0). Next, the cores were resuspended in 8 μL of NaCl/50 MOPS (pH 7.0) and subjected to TEM analysis immediately.22
2.6. Luciferase activity assay
TZM-bl cells were placed in 96-well plates at 1 × 104 cells per well and incubated for 24 h. Then, the culture medium was replaced with fresh medium containing 15 μg mL−1 diethylaminoethyl-dextran (DEAE-dextran), and the cells were infected with a serial tenfold dilution of viruses. At 48 h post infection, the culture medium was aspirated, and 100 μL cell culture lysis reagent (Beyotime) was added to each well. Every 100 μL sample was mixed with 100 μL luciferase assay substrate (Beyotime). Luciferase activity was measured by using a Synergy Mx (BioTek). The titer was calculated based on the Reed and Muench method. A cut-off value of 2.5 times the background relative luciferase unit (RLU) was used to quantify positive samples.232.7. Immunofluorescence staining and colocalization analysis
For immunofluorescence staining, the HIV-1-QD particles or HIV-1-QD-DiO dual labeled viruses were adhered to a 35 mm glass-bottom dish (Shengyou Biotechnology) for 30 min at 37 °C. After that, HIV-1-QD particles were fixed with 4% paraformaldehyde for 15 min at room temperature, and permeabilized by treatment with 0.1% Triton-X100/PBS (pH 7.4) for 15 min. Subsequently, viruses were blocked with 10% FBS/PBS (pH 7.4) and incubated with mouse monoclonal antibody against HIV-1 p24 (Abcam) (1:200 dilution in 1% FBS/PBS) at 37 °C for 2 h, and were then incubated with Alexa Fluor® 488 rabbit anti-mouse IgG (Cell Signaling Technology) (1:1000) for 1 h at 37 °C. After removing the unincorporated antibody, the fluorescence was monitored with an UltraView VoX confocal system (PerkinElmer, Co.) using a 60×, 1.4 NA oil immersion objective lens. Volocity software was used for fluorescence colocalization analysis. Regions of interest (ROIs) were drawn around the viral particles and analyzed using Volocity software for quantitatively analyzing the colocalization between HIV-1-QDs and envelope-DiO or cellular components. The threshold of Pearson's Correlation Coefficients (PCC) for the entire ROI was calculated respectively. It has been stated that a threshold of PCC > 0.5 indicates a meaningful colocalization.24 In our experiments, when the PCC > 0.7, it was qualified as colocalization, which was a higher standard.
2.8. Transmission electron microscopy analysis
For the ultrathin section analysis of HIV-1 particle-producing cells, the 293T cells that packaged wild-type HIV-1 (NL4-3) and QD-labeled HIV-1 (NL4-3) were washed with PBS (pH 7.4) at 48 h post transfection, and fixed with 2.5% glutaraldehyde in PBS (pH 7.4) overnight at 4 °C. Subsequently, the cells were harvested and post-fixed in 1.0% osmium tetroxide for 2 h. After dehydration in a series of alcohol solutions and permeation, the samples were embedded in Araldite for polymerization. Followed by ultrathin sectioning and staining with uranyl acetate and lead citrate, the samples were analyzed under a 200 kV HITACHI-7000FA transmission electron microscope.For negative staining (NS) electron microscopy (EM) examination, HIV-1 virions and HIV-1 cores were fixed with 4% paraformaldehyde for 1 h at room temperature. Then, a carbon-coated copper grid was placed on the samples for 5 min. After the removal of the redundant droplets, the samples were negatively stained with 0.5% phosphotungstic acid (PTA, pH7.0) for 5 s. The images were acquired by using a 200 kV HITACHI-7000FA transmission electron microscope.
2.9. Single virus tracking
For confocal imaging HIV-1 entry into CD4 T or CEM-SS cells, aliquots of 1 × 106 cells were attached to a glass-bottom dish (Shengyou Biotechnology) coated with poly-L-lysine by centrifugation at 1000g at 12 °C for 15 min. Afterwards, viral particles corresponding to 250 ng p24 were inoculated in a glass-bottom dish and kept at 4 °C for 30 min, and then they were shifted to the cell culture chamber (Tokai Hit, 37 °C, 5% CO2) on the microscope to initiate fusion.Qdot™ 625 and Rhodamine B were excited with 561 nm lasers and detected with a emission wheel of 615 nm (W70), and Vybrant® DiO, EGFP, and Alexa Fluor® 488 were excited with 488 nm lasers and detected with a emission wheel of 525 nm (W50). iRFP was excited with a 640 nm laser and detected with an emission wheel of 705 nm (W90). ECFP was excited with a 440 nm laser and detected with an emission wheel of 485 nm (W60). Volocity software was used for imaging data analysis. Sequential images were obtained at 2.6–10 s intervals, and the video frame rate was 6 fps or 15 fps.
2.10. Virus infection assays
To determine HIV-1 infection, the infected cells were fixed with 4% paraformaldehyde for 1 h at room temperature, and then intracellular p24 was stained using an anti-HIV-1 p24 antibody and goat anti-mouse Ig G-R (Santa Cruz). After being washed with PBS (pH 7.4) containing 4% BSA twice and Hank's buffer twice respectively, the cells were resuspended in 1% paraformaldehyde. Analysis was performed on an FACS-Calibur™ platform (BD Biosciences).2.11. Real-time PCR
Viral total DNA was measured by real-time PCR. Viruses used for measuring were assembled in 293T cells by plasmid transfection. The viral supernatant was harvested at 48 h post-transfection and filtered through a 0.45 μm filter (Millipore). After being treated with Benzonase Nuclease (Sigma) (125 units per mL) at 37 °C for 15 min, the viruses were stored at −80 °C until use, with the avoidance of repeated freezing and thawing. DNA was isolated with a Wizard SV Genomic DNA purification system (Promega) according to the manufacturer's instructions. Quantitative real time PCR analyses of viral total DNA were carried out using the forward primer 5′-LTR-U5 (Seq: 5′-AGA TCC CTC AGA CCC TTT TAG TCA-3′), the reverse primer 3′-Gag (Seq: 5′-TTC GCT TTC AAG TCC CTG TTC-3′), and the probe FAM U5/Gag (Seq: 5′-(FAM)–TGT GGA AAA TCT CTA GCA GTG GCG CC–(BHQ)-3′).19 Real-time PCR reactions were performed on a CFX Connect Real-Time System (Bio-Rad, USA).2.12. Statistical analysis
All results were expressed as the mean ± standard deviation for each experiment. The statistical significance of differences between groups was analyzed by t-tests. GraphPad Prism 5 software or SPSS PASW statistics software version 18.0 were used for analysis. P < 0.05 was considered to indicate a statistically significant difference.3. Results
3.1. Construction and characterization of QD-encapsulated HIV-1 particles
To track viral entry in live resting CD4 cells, CXCR4-tropic HIV-1 (NL4-3) particles encapsulating QDs were assembled (Fig. 1a). QD-encapsulated HIV-1 (NL 4-3) particles were constructed by a modification of the labeling of CCR5-tropic HIV-1 (AD8) (Fig. S1a†).11 Since the insertion of Qdots-streptavidin conjugate (QDs-SA) might impact the infectivity of HIV-1 (NL-43), we determined the ratio of the plasmids used in transfection to obtain the optimal infectivity. The ratio of plasmids pNL4-3, pcDNA3.1(+)-BirA and pcDNA3.1(+)-Vpr-AP varied from 1:1:1, 3:2:1, 7:1:1, and 5:3:1 to 10:1:1. The capacity of QD-labeled HIV-1 (NL4-3) to enter target cells was assessed by measuring the luciferase activity in indicator TZM-bl cells. Compared with the original HIV-1 (NL4-3), the ratios of 1:1:1, 3:2:1, 7:1:1, 5:3:1 and 10:1:1 exhibited 77.9%, 81.6%, 84.1%, 75% and 74.7% infectivity, respectively (Fig. S1b†). The virus particles acquired using a plasmid ratio of 7:1:1 exhibited infectivity (84.1%) that was not significantly different from that of the pNL4-3 plasmid (1:0:0), and thus a ratio of 7:1:1 was chosen for the preparation of viral particles.
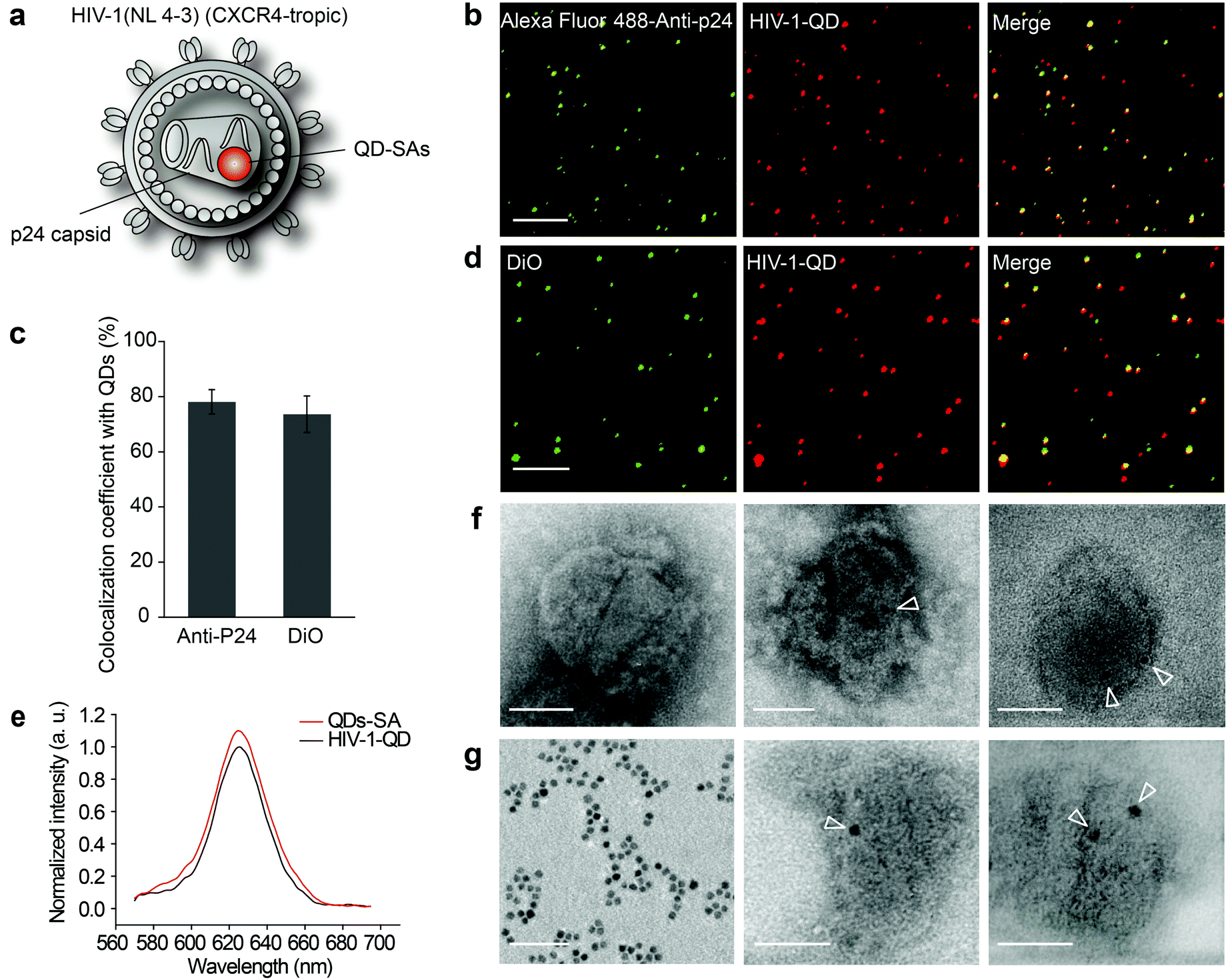 | ||
| Fig. 1 Characterization of HIV-1-QDs. (a) The scheme of QD-encapsulated HIV-1 (NL4-3) particles. (b) Colocalization fluorescence imaging of HIV-1-QDs that were immunostained with anti-p24 antibody (Alexa Fluor® 488). Scale bar, 1 μm. (c) Colocalization coefficiency assay of the QD signal and anti-p24 antibody (Alexa Fluor® 488) or DiO. (d) Fluorescence imaging of HIV-1-QDs that were stained with DiO. Scale bar, 1 μm. (e) Fluorescence emission spectra of HIV-1-QDs and free QDs-SA. The maximum fluorescence intensity of HIV-1-QDs is defined as 1. Both the fluorescence intensity of HIV-1-QDs and free QDs-SA are normalized by the maximum fluorescence intensity that is produced by HIV-1-QDs. (f) TEM image of unlabeled HIV-1 (left panel) and HIV-1-QD encapsulated with one QD-SA (middle panel) or two QDs-SA (right panel). Arrowheads indicate QDs-SA. Scale bar, 50 nm. (g) TEM image of free QDs-SA (left panel) and the viral core of an HIV-1 particle encapsulated with one QD-SA (middle panel) or two QDs-SA (right panel). Arrowheads indicate the QDs-SA. Scale bar, 50 nm. | ||
The assembled QD-encapsulated HIV-1 particles were exposed to anti-P24 immunofluorescence (IF) staining (Fig. 1b). Fluorescence detection revealed that 78.13% ± 4.42% of p24 colocalized with QDs (Fig. 1c). Furthermore, when the QD-encapsulated viruses were incubated with a lipophilic membrane dye, DiO, to label the lipid envelope, the QDs also co-localized with DiO (Fig. 1d). These analyses demonstrated that QDs were successfully incorporated into HIV-1 virions. As a control, no QD fluorescence was detected when pcDNA3.1(+)-Vpr-AP was absent during the co-transfection. We compared the fluorescence spectrum between HIV-1-QDs and free QDs-SA and the results demonstrated that the fluorescence properties of the QDs-SA were not altered after being incorporated into HIV-1 virions (Fig. 1e).
QD-encapsulated HIV-1 particles were also characterized by negative staining under transmission electron microscopy (TEM) (Fig. 1f and g). Pure QDs-SA showed a dark electron dense core of ∼7 nm in size (Fig. 1g, left panel). A dark electron dense core of the same size was detected inside QD-labeled HIV-1 particles (Fig. 1f, middle panel and right panel), while no such dots were observed in wild-type particles (Fig. 1f, left panel). These results confirmed that QDs-SA were specifically encapsulated into HIV-1. Moreover, the isolated viral core was imaged under TEM. The results clearly showed that the QDs-SA were located inside the HIV-1 core after the viral envelope membrane was stripped (Fig. 1g, middle panel and right panel). These results further demonstrated that the QDs-SA were encapsulated into the HIV-1 core. HIV-1-QD virus-producing cells were also analyzed at 48 h post-transfection by ultrathin section electron microscopy. TEM imaging showed that the assembled QD-encapsulated viral particles in the QD-labeling process had a similar morphology to that of the unlabeled wild-type virus (Fig. S1c and d†), indicating that labeling did not interfere with virus production or morphogenesis.
3.2. Single-virus tracking of HIV entry in CD4 T cells via fusion at the cell surface
The QD-encapsulated CXCR4-tropic HIV-1 (NL4-3) viruses were then used for imaging viral entry into human resting CD4 T cells. The CD4 T cells were isolated from healthy donors and attached to a glass-bottom dish with poly-L-lysine using a centrifugation procedure. To track the fusion events, the cells were incubated with QD-encapsulated HIV-1 viruses bearing DiO-labeled envelopes and were imaged in real time. First, the virus particle with co-localized QD and DiO signals (yellow) was adsorbed onto the cell surface. Then, we observed the separation of the QD signal (red) from the DiO signal (green) at the cell surface, indicating that the viral core was released from the envelope. This result suggested a membrane fusion process at the cell surface, which was followed by the disappearance of lipid DiO fluorescence (green) at the cell surface and retention of viral core QD fluorescence (red) for some time (Fig. 2a and Video S1†). Statistical analysis showed that following a short period of retention (66.5 ± 7.1 s, n = 10), the QD-viral core showed a high instantaneous speed, suggesting that it moved rapidly after separation from the cell membrane (Fig. 2c). When the cells were treated with the fusion inhibitor enfuvirtide (T20), the separation of the QD signal (red) and the DiO signal (green) could hardly be detected (Fig. 2b and Video S2†). In our experiment, 11 events of viral core release from the envelope, showing membrane fusion, were detected from 12000 randomly selected HIV-1-QD particles in resting CD4 T cells. This proportion of viral entry is consistent with the low specific infectivity of HIV-1 stocks, which has been reported in previous reports.25–27 In the cells treated with T20, almost no viral core release from the envelope was observed in 12000 individual HIV-1-QD particles (Fig. 2d). These data suggested that the HIV-1 (NL4-3) virus underwent a fusion process in the plasma membrane to enter the resting CD4 T cells.
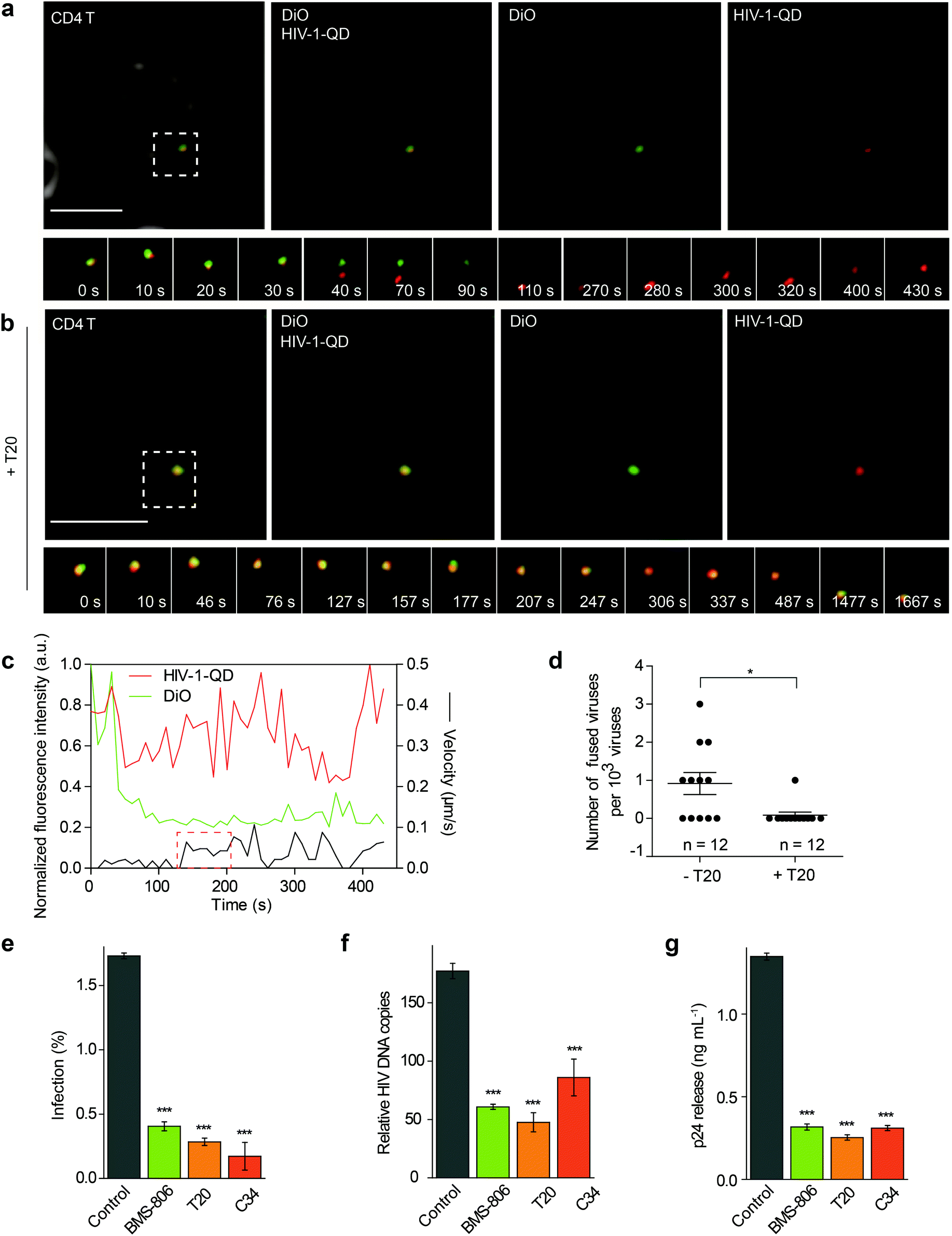 | ||
| Fig. 2 Real-time imaging of HIV-1 cellular membrane fusion in primary CD4 T cells. (a, b) Time-lapse images of dual-fluorescence-labeled HIV-1 particles (rectangular region) in primary CD4 T cells by treatment with medium (a) or 10 μg mL−1 T20 (b). Scale bar, 5 μm. (c) Velocity and fluorescence intensity analysis of the HIV-1 particle that is shown in (a). The fluorescence intensity of each indicator was normalized to the population peak. The red dashed rectangle marks the instantaneous velocity increase during viral separation. (d) Statistical analysis of the number of fused HIV-1 particles per 1 × 103 viruses under T20 treatment. *p < 0.05. (e–g) Inhibition of HIV-1 infection by BMS-806, T20 and C34 in CD4 T cells. The results represent the average ± standard deviation (SD) for three independent experiments. (e) Intracellular p24 was stained with an anti-p24 antibody and measured by flow cytometry. (f) Viral DNA synthesis was determined by real-time PCR. (g) The p24 release level in the supernatant was measured by p24 ELISA. Histograms display averages ± SD; n = 3; ***p < 0.001. | ||
The dynamic behavior of the fluorescent HIV-1 particles was further tracked in the T-lymphoblastoid cell line, CEM-SS. The separations of the QD signal (red) from the DiO signal (green) were tracked at the cell surface, showing that the viral core was released from the envelope (Fig. S2a and Video S3†). The lipid signal (green) disappeared after 140 s at the cell surface, which suggested an infinite dilution between the viral membrane and the cell membrane, and the viral core fluorescence (red) was retained in the cell. Fluorescence intensity analysis provided a quantification of the reduction in the DiO signals and the retention of QD-Vpr fluorescence signals. Statistical analysis showed that following a short period of retention (57.9 ± 14.1 s, n = 10), the red QD signal moved rapidly into the cytoplasm (Fig. S2b†). In CEM-SS, approximately 0.1% effective entry events were observed, which is also consistent with the infectivity of HIV-1 stocks. These results indicated that similar to the viral entry into CD4 T cells, HIV enters CEM-SS cells via a membrane fusion process at the cell surface.
3.3. Drug inhibition analysis verified the HIV fusion at the cell surface in CD4 T cells
Several inhibitors such as BMS-378806 (BMS-806), T20, and C34 were then used to treat the resting CD4 cells and test their effects on the HIV-1 infection. BMS-806 is a CD4-gp120 attachment inhibitor that binds to the gp120 exterior envelope glycoprotein of HIV-1.28,29 T20 and C34 are peptides that are based on the heptad repeat sequence of gp41 of HIV-1 and block gp41-mediated membrane fusion.30,31 All these inhibitors were used specifically at noncytotoxic dosages, which were monitored using control cells pretreated with 10 μM H2O2 for 1 h for toxicity control (Fig. S3a and b†).During drug inhibition analysis, resting CD4 T cells were pretreated with inhibitors for 1 h and then infected with HIV-1 for 2 h at 37 °C. After infection, we activated the infected cells. We first determined HIV-1 entry by intracellular p24 staining at 48 h after activation in drug-treated primary CD4 T cells (Fig. 2e). The p24 signal was significantly decreased when the cells were treated with BMS-806, T20 or C34. These results demonstrated that binding and fusion inhibitors diminished virus infection. The synthesis of total viral DNA was also examined at 24 h post-activation in the drug-treated primary CD4 T cells, and we observed a significant reduction in viral DNA synthesis in the group treated with BMS-806, T20, or C34 (Fig. 2f). Moreover, the amount of p24 released into the supernatants collected at 48 h post-activation was also largely reduced by BMS-806, T20, or C34 (Fig. 2g). These results were in agreement with the above-mentioned images showing that HIV-1 (NL4-3) entered CD4 T cells via fusion with the plasma membrane.
HIV-1 infection was also tested in drug-treated CEM-SS cells. When the cells were treated with BMS-806, T20, or C34, viral infection was significantly decreased demonstrating that binding and fusion inhibitors diminished viral infection (Fig. S2c and d†). We also performed drug inhibition analysis in a HIV-1 Rev-dependent GFP indicator cell line, Rev-CEM, which was derived from the CEM-SS T cell line being integrated with the Rev-dependent GFP reporter.32 Fluorescence was barely detectable in cells treated with BMS-806, T20, or C34 (Fig. S2e†). These data confirmed that the inhibitors of HIV-1 binding and fusion can inhibit HIV-1 entry into T lymphocytes.
3.4. Real-time visualization of HIV-1-mediated cortical actin rearrangement
Then, the actin dynamics were observed during the HIV-1 infection of resting CD4 T cells. LifeAct-EGFP was transiently expressed in CD4 T cells to visualize the fibrous-actin (F-actin). The cells were incubated with fluorescently tagged HIV-1 viruses for imaging. As shown in Fig. 3a, the real-time imaging showed that around the membrane regions, the HIV-1 binding triggered rapid actin polymerization and depolymerization (Video S4†). HIV-1-QD binding promoted the formation of a cap-like cluster of F-actin. Following viral entry, the F-actin cap cluster diffused gradually, and a pore-like channel (144 s) was formed in the cortical actin at the viral entry point. Subsequently, the channel was filled again by the polymerized F-actin following HIV-1-QD delivery. This time lapse imaging displays the sequential process by which an HIV particle remodels the F-actin and travels through the barrier of cortical actin. Fluorescence intensity analysis of the time lapse images showed that the green fluorescence signal of LifeAct-EGFP increased at the site of the virus attachment at 12–126 s, which represents the capping process (Fig. 3b). Then, the EGFP fluorescence signal decreased when the pore-like channel formed (126–240 s). Finally, the green fluorescence signal recovered after the viral entry. We also labeled HIV-1 (NL-43) by inserting a red fluorescent protein mCherry into the MA protein for real time imaging of actin rearrangement during HIV-1 infection. The result was consistent with that observed by QD tagged particles (Fig. S4 and Video S5†). Drugs that inhibit actin dynamics, such as jasplakinolide (Jas) and latrunculin A (Lat-A), were tested for their effects on the viral infection. The result showed that both Jas and Lat-A can inhibit HIV-1 entry when the viral infection was quantified by p24 staining in CD4 T cells at 48 h post activation (Fig. 3c). Moreover, the p24 release in the supernatants collected at 48 h post activation was also significantly decreased in the cells treated with Jas and Lat-A (Fig. 3d). These data support that actin dynamics are important for HIV-1 entry and infection in CD4 T cells.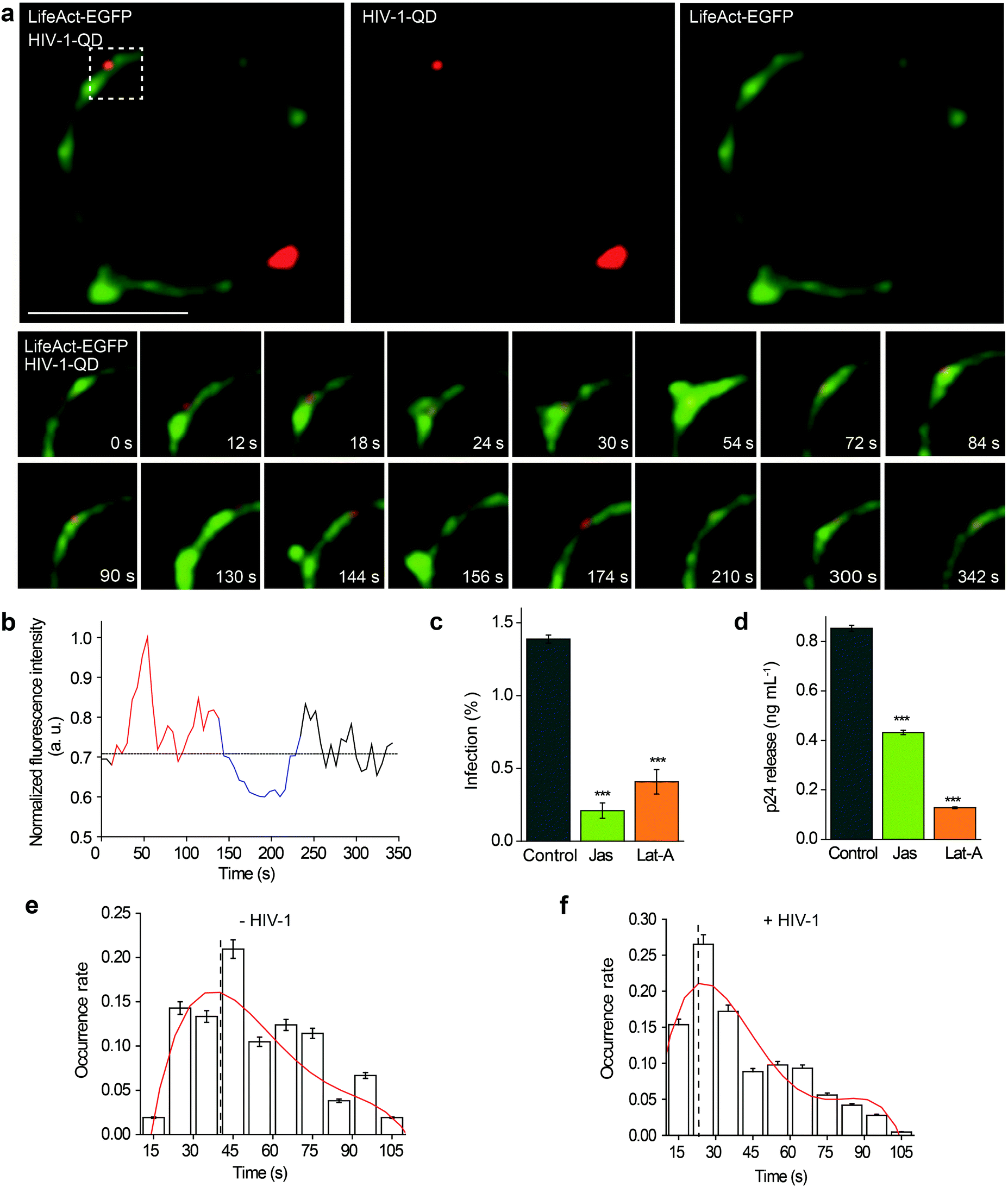 | ||
| Fig. 3 Real-time imaging of cortical actin rearrangement upon HIV-1 binding. (a) Real-time imaging of the HIV-1 entry triggered cellular actin polymerization and depolymerization. Scale bar, 5 μm. (b) Fluorescence intensity analysis of LifeAct-EGFP during HIV-1 entry that is shown in (a). Blue and red curves represent the rapid clustering and diffusion of F-actin, respectively. Black curve represents the normal arrangement of actin. The black dashed line indicates the baseline fluorescence intensity. (c) Intracellular p24 was examined by flow cytometry in the presence of Jas or Lat-A. (d) The p24 release level in the supernatant was monitored by p24 ELISA. Histograms display averages ± SD; n = 3; ***p < 0.001. (e, f) Statistical analysis of the duration time of actin polymerization and depolymerization triggered with or without HIV-1 binding for 75 different regions of CD4 T cells. The periods of fluorescence duration time, which we calculated from the F-actin fluorescence intensity peak, were compiled into different histograms and fit to the Gaussian distributions. | ||
The dynamic changes of the F-actin fluorescence signal duration of CD4 T cells with or without HIV-1 binding were statistically analyzed for 75 regions in 35 cells. As shown in Fig. 3e and f, we compiled the fluorescence signal lifetime into histograms and fit them into Gaussian distributions. The maximum of the Gaussian distribution was taken to compare fluorescence signal lifetimes either induced by HIV-1 binding or not.33,34 The results indicated that the maximum duration of fluorescence was approximately 40.0 ± 4.2 s when there was no HIV-1 binding. In contrast, in regions of viral attachment, the fluorescence duration become shorter and was 22.0 ± 3.1 s. These data demonstrate that the binding of HIV-1 affects F-actin dynamics, leading to a shorter fluorescence duration.
3.5. Real-time imaging of actin rearrangement during viral core release from the envelope
Next, we further investigated actin dynamics during the viral core release from the envelope in real time. F-actin was labeled using LifeAct-ECFP. The effect of actin rearrangement on DiO-labeled HIV-1-QD particle membrane fusion was imaged in live primary CD4 T cells. As shown in Fig. 4a, the colocalization of the three signals, the viral envelope DiO (green), the viral core QD (red), and LifeAct-ECFP labeled F-actin (cyan), was observed during the viral infection of CD4 T cells. During the tracking period, viral binding modulated F-actin to form a cap-like aggregation at the beginning, and the lipid DiO fluorescence (green) and QD fluorescence (red) separated from each other. Concomitant with the separation of the viral envelope from the core, the actin capping area diffused, and a pore like channel formed. Subsequently, the HIV-1 core passed though the cortical actin layer and was transported into the cytoplasm (Fig. 4b and Video S6†). These observations revealed the dynamic process of viral fusion and entry that is associated with actin remodeling.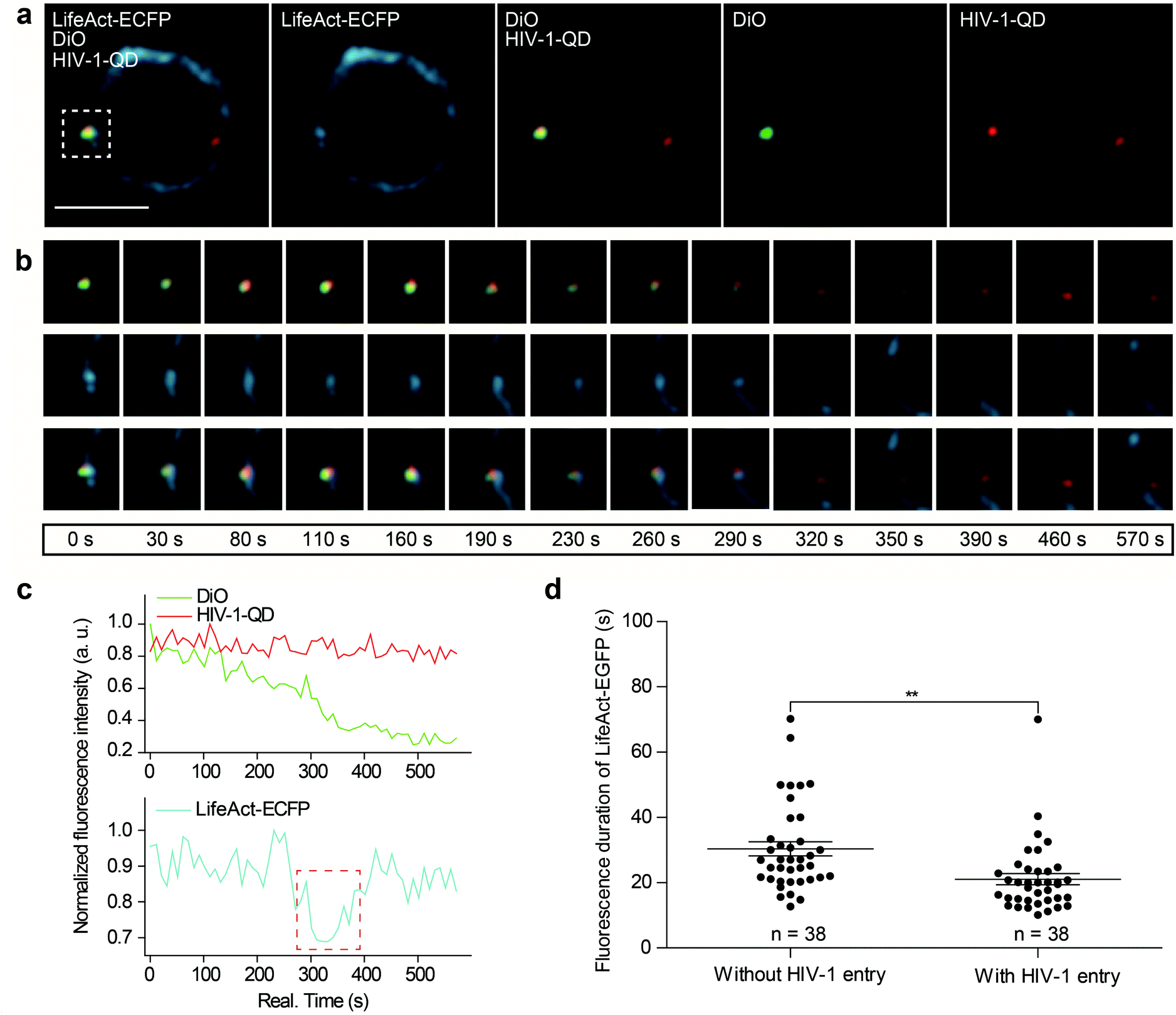 | ||
| Fig. 4 Real-time imaging of actin rearrangement during viral core release from the envelope. (a) A dual-labeled HIV-1-QD-DiO was colocalized with LifeAct-ECFP-marked F-actin in a CD4 T cell (rectangular region). Scale bar, 5 μm. (b) Sequential snapshots of the separation of QD-Vpr (red) from the DiO-labeled membrane (green) and the rapid actin arrangement in CD4 T cells. (c) The fluorescence profile of the viral core (red), viral envelope (green), and F-actin (cyan) that is shown in (b). The fluorescence intensity of each indicator was normalized to the population peak. The red dashed line marks the significant decrease in the cyan fluorescence signal during viral entry. (d) Statistical analysis of the average duration time of actin rearrangement in 38 cells with or without HIV-1 entry into CD4 T cells. The periods of fluorescence duration were calculated from the LifeAct-ECFP fluorescence intensity peak. Histograms display averages ± SD; **p < 0.01. | ||
The dynamic fluorescence intensities of the viral core, the viral envelope, and F-actin are depicted in Fig. 4c, which shows that the intensity of the green fluorescence signal was decreased and that the red fluorescence signal remained stable. In addition, to quantify actin rearrangement, we statistically analyzed the average duration of actin polymerization and depolymerization with or without HIV-1 infection, respectively. Statistical analysis showed that the maximum duration of actin rearrangement was approximately 22.0 ± 3.1 s at the point of HIV-1 binding. However, at the point of HIV-1 binding and entry, the average duration of actin rearrangement was approximately 21.1 ± 1.7 s, which was much shorter than that without HIV-1 entry, which was approximately 30.4 ± 2.2 s (Fig. 4d). These data further indicated that the binding of HIV-1 viral particles triggers actin dynamics and promotes actin rearrangement, which facilitates HIV-1 membrane fusion.
3.6. α-Actinin is involved in the actin dynamics during HIV-1 entry
Given that HIV-1 viral particles trigger actin dynamics and promote actin rearrangement, we further investigated the role of actin regulatory proteins during HIV-1 entry. There are several key actin-binding proteins regulating actin polymerization and depolymerization, including cortactin, paxillin and α-actinin. Cortactin is an F-actin stabilizer protein and activator of the Arp2/3 actin nucleation machinery;35 it binds F-actin and localizes to the site of dynamic actin assembly.36 Paxillin is a multidomain adaptor found at the interface between the plasma membrane and the actin cytoskeleton; it binds several proteins that contribute to the organization of the actin cytoskeleton at sites of integrin engagement with the extracellular matrix.37,38 α-Actinin is a ubiquitously conserved actin-binding protein that also interacts with many other protein partners such as cell signaling molecules and integrins and plays an important role in cell shape and motility.39,40 To test the effect of these proteins on F-actin polymerization during HIV-1 entry, LifeAct-EGFP in combination with either α-actinin-iRFP, cortactin-iRFP or paxillin-iRFP was transiently expressed in CD4 T cells. Then, the cells were infected with fluorescent HIV-1 viral particles for real-time imaging.We observed that α-actinin was recruited to the HIV-1 binding site during viral entry. As shown in Fig. 5a and b, at the site of HIV-1-QD entry, α-actinin aggregated dynamically during the first 85 s. With the aggregation of α-actinin, the cortical actin filaments appeared to be diffused and formed a channel at the viral entry site. Following the movement of virus across the cortical actin, the α-actinin dispersed, and the channel was filled by polymerized F-actin to return it to its original state (Video S7†). Kymograph analysis and the corresponding fluorescence intensity analysis clearly demonstrated this dynamic sequential process (Fig. 5c, the first row). Different from α-actinin, cortactin (Fig. 5c, the second row, and Video S8†) and paxillin (Fig. 5c, the third row, and Video S9†) did not exhibit this sequential process but showed a similar rearrangement mode to that of F-actin at the sites of HIV-1-QD entry (Fig. 5e). At the regions without HIV-1 binding, there was also no sequential process for α-actinin and F-actin, and they showed a consistent dynamic pattern (Fig. 5d and Video S10†). These results suggested that α-actinin may bridge the remodeling of the actin cortical network during HIV-1 entry. The virus may regulate actin dynamics via α-actinin.
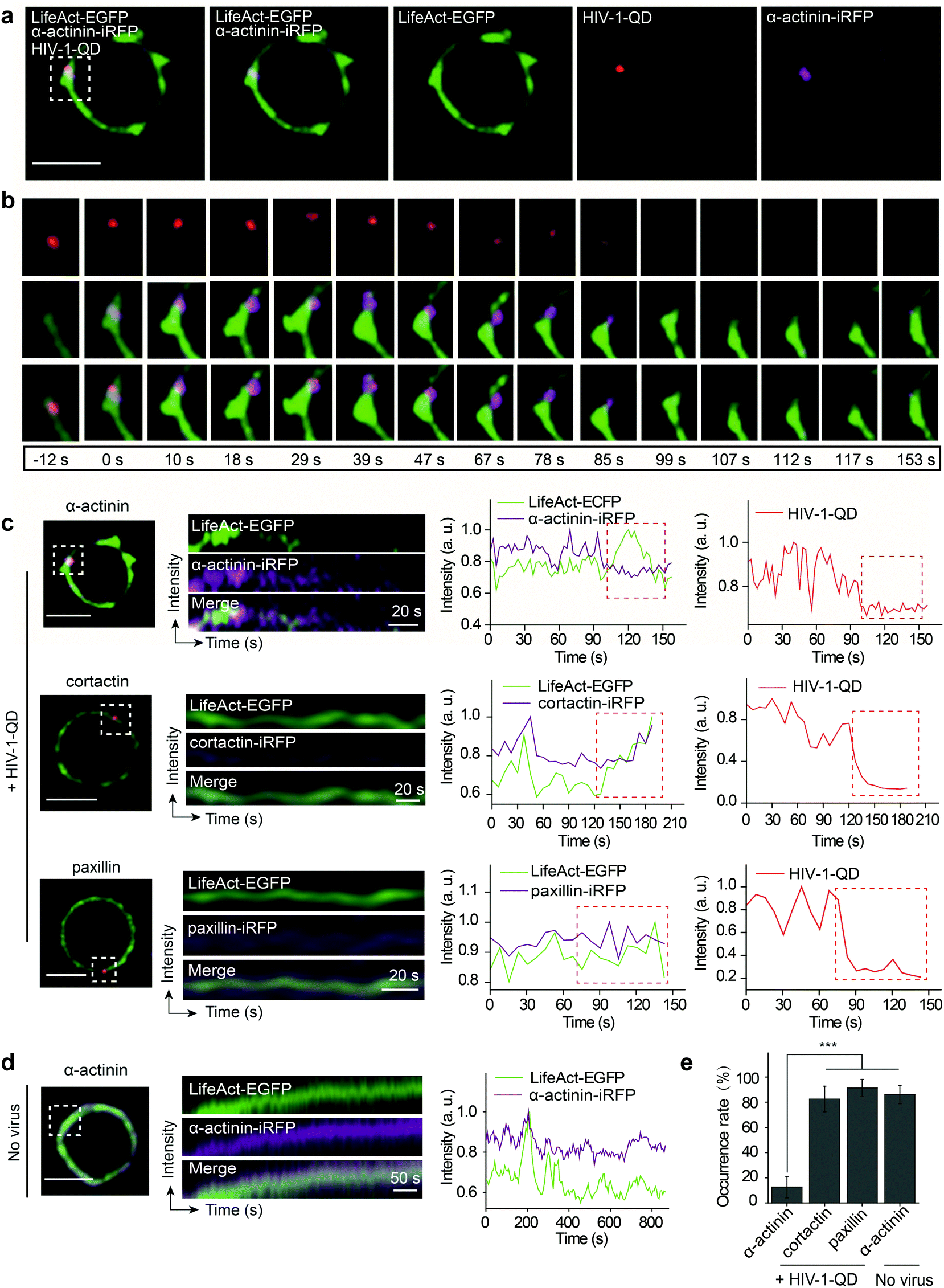 | ||
| Fig. 5 α-Actinin is involved in the actin dynamics during HIV-1 entry into resting CD4 T cells. (a) HIV-1-QD colocalized with LifeAct-EGFP-marked F-actin and α-actinin-iRFP-marked α-actinin in a CD4 T cell (rectangular region). Scale bar, 5 μm. (b) Sequential snapshots of a HIV-1-QD entering a cell accompanied by α-actinin aggregation and actin rearrangement. (c) Kymograph analysis and the corresponding fluorescence profile of LifeAct-EGFP (green), α-actinin-iRFP (purple), cortactin-iRFP (purple), and paxillin-iRFP (purple) with HIV-1-QDs (red) in CD4 T cells. The fluorescence intensity of each indicator was normalized to the population peak. The red dashed lines show the same time zones of actin dynamics accompanied by viral entry. (d) Kymograph and fluorescence intensity analysis of LifeAct-EGFP (green) and α-actinin-iRFP (purple) without HIV-1-QDs in CD4 T cells. (e) Statistical analysis of similar dynamic rearrangement modes of α-actinin, cortactin and paxillin with F-actin at 1000 sites of CD4 T cells binding with or without HIV particles. ***p < 0.001. | ||
3.7. α-Actinin-derived peptides can block HIV-1 entry and infection
Given that α-actinin plays roles in regulating actin rearrangement for HIV-1 entry, we further tested interfering with α-actinin activity to block the HIV-1 infection. We aimed to develop a peptide to competitively inhibit the interaction of α-actinin with F-actin. To design and select the peptide inhibitor, we modeled the 3D interaction structure of α-actinin and F-actin. The crystal structure of the actin-binding domain (ABD) of human α-actinin 1 at 1.7 angstrom resolution (PDB ID: 2EYI)41 and the F-actin structure (PDB ID: 2ZWH)42 were used to form a 3D complex by LeDock43 (Fig. 6a). The molecular docking analysis showed that the ABD of human α-actinin could recognize F-actin. The actin-binding regions exposed on the surface of the ABD were located and designated as actin-binding sites (ABSs) for α-actinin (Fig. 6b and c). | ||
| Fig. 6 α-Actinin-derived peptides block HIV-1 entry and infection. (a) Three-dimensional structure of F-actin and the ABD of α-actinin. (b) The ABS location of the ABD from Homo sapiens α-actinin. The F-actin, ABD, ABS1, ABS2 and ABS3 are colored in cyan, gray, magenta, blue and yellow, respectively. (c) The ribbon structure of the ABD from Homo sapiens α-actinin. (d) Intracellular p24 was analyzed by flow cytometry after treatment with ABS1p and ABS3p. Resting CD4 T cells were pretreated with ABS1p and ABS3p for 1 h and then infected with HIV-1 for 2 h at 37 °C. Intracellular p24 was detected 48 h after activation. Histograms display averages ± SD; n = 3; ***p < 0.001; ns: no significant difference. (e) Kymograph analysis of the relationship between actin dynamics during HIV-1 entry after treatment with ABS1p. Scale bar, 5 μm. (f) Sequential snapshots are shown for the entry of HIV-1 under treatment with ABS1p. Scale bar, 1 μm. (g) Statistical analysis of the number of fused HIV-1 per 1 × 103 viruses under ABS1p treatment. ***p < 0.001. | ||
We designed peptides derived from the ABSs that may competitively bind to actin and inhibit endogenous α-actinin binding to actin. Through sequence alignment of the ABSs in four types of human source α-actinins44 (Fig. S5†), some of the most conserved amino acid residues in ABSs were selected to be used to develop the inhibitory peptides. Here, three peptides, ABS1p, ABS2p and ABS3p, were selected to test their effects on the viral infection. During the experiment, only ABS1p and ABS3p could be synthesized. ABS2p could not be synthesized, likely due to its strong grand average of hydropathicity (GRAVY value: 0.928). Thus, ABS1p and ABS3p were used in the HIV-1 infection experiments.
We tested the inhibitory effect of the peptides at different concentrations against HIV-1 infection in CEM-SS and CD4 T cells. The results showed that ABS1p significantly inhibited the HIV-1 infection at a concentration of 1 μg mL−1 in CD4 T cells and CEM-SS cells. However, ABS3p had no significant impact on the HIV-1 infection (Fig. 6d and Fig. S6a†). Single-virus imaging was also carried out to detect the effect of ABS1p on HIV-1 entry. As shown in Fig. 6f and g, in CD4 T cells treated with ABS1p, the virus could not enter the cell. At the HIV-1-QD binding site, α-actinins did not aggregate to regulate F-actin rearrangement, in contrast to their aggregation in nontreated CD4 T cells. Kymograph analysis showed that the α-actinin dynamics was consistent with that of F-actin in the presence of ABS1p in CEM-SS (Fig. S6b and Video S11†) and CD4 T cells (Fig. 6e and Video S12†), and there was no typical sequential rearrangement at the virus binding site. In our experiment, we also showed that Rhodamine B-labeled ABS1p entered the cells and bound the F-actin (Fig. S7†). The peptides ABS1p and ABS3p showed no detectable cytotoxicity in the cells at our experimental concentration (Fig. S8†). Altogether, these results provide evidence that α-actinin plays an important role in regulating dynamic actin in T cells, which is critical for HIV-1 membrane fusion in the HIV-1 infection.
4. Discussion
Studying in depth and understanding HIV–cell interactions is crucial for the prevention of acquired immune deficiency syndrome (AIDS). Here, the subtle dynamic processes involved in the entry of individual HIV-1 particles into resting T cells and the passage of these particles across the cortical actin barrier were directly visualized and studied in depth. Using QD-encapsulated viral particles, we tracked viral fusion, actin rearrangements, and HIV–actin interactions at the single-virus-particle level in real time.Our single-virus imaging demonstrated that HIV virus entry into CD4 T cells occurs via fusion at the plasma membrane. As an enveloped virus, HIV reaches the cytoplasm by fusion of the viral and cellular membrane.45 However, because of the lack of direct visualization of the fusion process, it is difficult to pinpoint the exact fusion site of viral entry. It has long been thought that HIV-1 enters the cells via direct fusion with the plasma membrane.46,47 On the other hand, some reports argue that endocytosis contributes to productive HIV-1 entry.48 In this work, single-virus tracking revealed that the plasma fusion and viral core release occur on the cell surface in CD4 T cells and CEM-SS cells. Drug inhibition assays also support these results.
The dynamic interactions between single HIV particles and cortical actin were tracked in live resting CD4 T cells. During viral entry, viral particles encapsulating QDs were observed to attach to the cell surface and regulate the actin rearrangements. The HIV-1 binding sites presented acute actin polymerization and depolymerization and resulted in a cap-like structure formation (Fig. 4b). At the position of HIV-1 binding, the actin polymerization and depolymerization (21.1 ± 1.7 s) is faster than that at the position without viral entry (30.4 ± 2.2 s). HIV-1 entry requires serial engagement of the viral receptor, CD4, and of the coreceptor, CXCR4 or CCR5. This cap-forming process may lead to the recruitment and clustering of the receptor and coreceptor to facilitate HIV-1 fusion. Strikingly, our imaging revealed that a nick in the cortical actin is dynamically formed at the viral entry site, which may enable the viral particle to cross the cortical actin barrier. The process of viral core release from the envelope was also tracked and found to concur with the dynamic actin rearrangement. This means that viral fusion at the plasma membrane is coupled with the fine-scale spatiotemporal rearrangement of actin filaments. The interaction of HIV-1 with the cellular membrane triggers dynamic actin rearrangement to allow entry of the HIV core following viral fusion.
Through real-time imaging, the roles of several actin-binding proteins, including cortactin, paxillin, and α-actinin, in actin remodeling during viral entry were examined. The results showed that at the HIV-1 entry site, α-actinin aggregated, and then a nick in the cortical actin barrier formed. Following the viral penetration of the cortical actin, α-actinin dispersed, and the nick was filled by polymerized F-actin. By contrast, cortactin and paxillin did not show involvement in this sequential process. α-Actinin is a ubiquitous homodimer that cross-links actin filaments to form networks of loose bundles of actin filaments.39 α-Actinin also regulates the activity of receptors and connects the cytoskeleton to diverse signaling pathways, and the EWI-2-α-actinin complex regulates T cell immune synapses during HIV-1 infection.49 Our finding that HIV-1 recruits α-actinin to regulate cortical actin provides new insight into the role of α-actinin during viral entry, although further studies are needed to elucidate the exact mechanism.
Given that α-actinin is involved in cortical actin rearrangement during HIV-1 entry, we designed peptides derived from α-actinin to test their influence on the viral infection. This peptide, ABS1p, was designed from an analysis of the interaction between α-actinin and F-actin, and was found to block HIV-1 entry into the cell. Being derived from the ABD domain of α-actinin, ABS1p may act through disrupting the interaction between α-actinin and F-actin, and ABS1p may competitively bind to actin to inhibit endogenous α-actinin binding to actin. The inhibition of HIV-1 infection by ABS1p confirms that α-actinin plays a key role in HIV-1 entry. Our results also show that actin dynamics can serve as a target for the development of new treatments for HIV-1 infection.
Based on the findings via single-virus tracking, we proposed a model in which HIV virus particles modulate the subtle dynamics of cortical F-actin and the actin rearrangement to facilitate viral entry (Fig. 7). At the site of HIV entry, viral binding activates the actin dynamics to form a cap-like structure, and viral fusion occurs to release the viral core. Then, α-actinin aggregates at the viral entry site, and the actin cap becomes diffused. A nick in the cortical actin is formed, which facilitates the penetration of the viral core through the cortical actin barrier. The α-actinin-derived peptide, ABS1p, can interfere with the HIV-mediated actin process to block viral infection. Our findings clarify HIV entry into resting CD4 T cells and provide valuable insights into the cytoskeletal dynamics in the HIV infection, which identifies new targets for the development of novel anti-HIV inhibitors targeting HIV host-dependent factors.
 | ||
| Fig. 7 Model of HIV particles modulating the dynamics of cortical F-actin and the actin rearrangement to facilitate viral entry. At the site of HIV-1 entry, viral binding activates the actin dynamics to form a cap-like structure, and viral fusion occurs to release the viral core. Then, α-actinin aggregates at the viral entry site, and the actin cap becomes diffused. A nick in the cortical actin is formed, which facilitates the penetration of the viral core through the cortical actin barrier. The addition of the α-actinin-derived peptide, ABS1p, blocks virus entry by preventing the dynamics of F-actin. | ||
5. Conclusion
In conclusion, by using QD-encapsulated fluorescent viral particles and single-virus tracking, we reveal the process and mechanism of individual virion-triggered cortical actin dynamics for HIV entry into resting CD4 T cells. Our findings reveal an α-actinin-mediated cortical actin rearrangement for HIV entry, and identify an antiviral target as well as a corresponding peptide inhibitor based on HIV interaction with the actin cytoskeleton.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Huaidong Zhang for the structural analysis of α-actinin and Hao Tang for his excellent technical support in the BSL-3 Laboratory of Wuhan Institute of Virology. Many thanks to Pei Zhang, Ding Gao and Juan Min of the Core Facility and Technical Support, Wuhan Institute of Virology for their technical support in transmission electron microscopy and flow cytometry analysis. This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB29050201), the National Key Research and Development Program of China (2018ZX10301405), the National Natural Science Foundation of China (No. 31470269, No. 21727816, and No. 31470837), and the Youth Innovation Promotion Association of Chinese Academy of Sciences.Notes and references
- A. C. Francis, M. Marin, J. Shi, C. Aiken and G. B. Melikyan, PLoS Pathog., 2016, 12, e1005709 CrossRef.
- C. R. Chin, J. M. Perreira, G. Savidis, J. M. Portmann, A. M. Aker, E. M. Feeley, M. C. Smith and A. L. Brass, Cell Rep., 2015, 13, 1717–1731 CrossRef CAS.
- D. McDonald, M. A. Vodicka, G. Lucero, T. M. Svitkina, G. G. Borisy, M. Emerman and T. J. Hope, J. Cell Biol., 2002, 159, 441–452 CrossRef CAS.
- T. M. Desai, M. Marin, C. Sood, J. Shi, F. Nawaz, C. Aiken and G. B. Melikyan, Retrovirology, 2015, 12, 88 CrossRef PubMed.
- Y. Ma, Z. He, T. Tan, W. Li, Z. Zhang, S. Song, X. Zhang, Q. Hu, P. Zhou, Y. Wu, X. E. Zhang and Z. Cui, ACS Nano, 2016, 10, 6273–6282 CrossRef CAS PubMed.
- L. Wen, Y. Lin, Z. H. Zheng, Z. L. Zhang, L. J. Zhang, L. Y. Wang, H. Z. Wang and D. W. Pang, Biomaterials, 2014, 35, 2295–2301 CrossRef CAS.
- K. I. Joo, Y. Lei, C. L. Lee, J. Lo, J. Xie, S. F. Hamm-Alvarez and P. Wang, ACS Nano, 2008, 2, 1553–1562 CrossRef CAS.
- M. Howarth, K. Takao, Y. Hayashi and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7583–7588 CrossRef CAS PubMed.
- S. L. Liu, Z. Q. Tian, Z. L. Zhang, Q. M. Wu, H. S. Zhao, B. Ren and D. W. Pang, Biomaterials, 2012, 33, 7828–7833 CrossRef CAS.
- S. L. Liu, Z. L. Zhang, Z. Q. Tian, H. S. Zhao, H. Liu, E. Z. Sun, G. F. Xiao, W. Zhang, H. Z. Wang and D. W. Pang, ACS Nano, 2012, 6, 141–150 CrossRef CAS.
- Q. Li, W. Li, W. Yin, J. Guo, Z. P. Zhang, D. Zeng, X. Zhang, Y. Wu, X. E. Zhang and Z. Cui, ACS Nano, 2017, 11, 3890–3903 CrossRef CAS.
- Q. Li, W. Yin, W. Li, Z. Zhang, X. Zhang, X. E. Zhang and Z. Cui, Nano Lett., 2018, 18, 7457–7468 CrossRef CAS.
- C. Qin, W. Li, Q. Li, W. Yin, X. Zhang, Z. Zhang, X. E. Zhang and Z. Cui, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2577–2582 CrossRef CAS.
- M. L. Penn, J. C. Grivel, B. Schramm, M. A. Goldsmith and L. Margolis, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 663–668 CrossRef CAS.
- M. Spear, J. Guo and Y. Wu, Retrovirology, 2012, 9, 45 CrossRef CAS.
- M. Lehmann, D. S. Nikolic and V. Piguet, Viruses, 2011, 3, 1757–1776 CrossRef CAS.
- M. Spear and Y. Wu, Virol. Sin., 2014, 29, 139–147 CrossRef CAS.
- S. Padilla-Parra and M. L. Dustin, Trends Mol. Med., 2016, 22, 354–356 CrossRef CAS.
- W. Wang, J. Guo, D. Yu, P. J. Vorster, W. Chen and Y. Wu, J. Biol. Chem., 2012, 287, 35455–35469 CrossRef CAS.
- A. Yoder, D. Yu, L. Dong, S. R. Iyer, X. Xu, J. Kelly, J. Liu, W. Wang, P. J. Vorster, L. Agulto, D. A. Stephany, J. N. Cooper, J. W. Marsh and Y. Wu, Cell, 2008, 134, 782–792 CrossRef CAS.
- D. Yu, W. Wang, A. Yoder, M. Spear and Y. Wu, PLoS Pathog., 2009, 5, e1000633 CrossRef.
- R. Welker, H. Hohenberg, U. Tessmer, C. Huckhagel and H. G. Kraüsslich, J. Virol., 2000, 74, 1168–1177 CrossRef CAS.
- C. A. Todd, K. M. Greene, X. Yu, D. A. Ozaki, H. Gao, Y. Huang, M. Wang, G. Li, R. Brown, B. Wood, M. P. D'Souza, P. Gilbert, D. C. Montefiori and M. Sarzotti-Kelsoe, J. Immunol. Methods, 2012, 375, 57–67 CrossRef CAS PubMed.
- J. Adler and I. Parmryd, Cytometry, Part A, 2010, 77, 733–742 CrossRef PubMed.
- J. Kimpton and M. Emerman, J. Virol., 1992, 66, 2232–2239 CAS.
- V. Maréchal, F. Clavel, J. M. Heard and O. Schwartz, J. Virol., 1998, 72, 2208–2212 Search PubMed.
- P. Koch, M. Lampe, W. J. Godinez, B. Müller, K. Rohr, H. G. Kräusslich and M. J. Lehmann, Retrovirology, 2009, 6, 84 CrossRef PubMed.
- H. Azijn, I. Tirry, J. Vingerhoets, M. P. de Bethune, G. Kraus, K. Boven, D. Jochmans, E. Van Craenenbroeck, G. Picchio and L. T. Rimsky, Antimicrob. Agents Chemother., 2010, 54, 718–727 CrossRef CAS.
- B. Pacheco, N. Alsahafi, O. Debbeche, J. Prévost, S. Ding, J. P. Chapleau, A. Herschhorn, N. Madani, A. Princiotto, B. Melillo, C. Gu, X. Zeng, Y. Mao, A. B. Smith 3rd, J. Sodroski and A. Finzi, J. Virol., 2017, 91, e02219–e02216 CrossRef CAS.
- M. Armandugon, A. Gutierrez, B. Clotet and J. Este, Antiviral Res., 2003, 59, 137–142 CrossRef CAS.
- X. Wu, Z. Liu, X. Ding, D. Yu, H. Wei, B. Qin, Y. Zhu, H. Chong, S. Cui and Y. He, J. Virol., 2018, 92, e02044–e02017 CAS.
- Y. Wu, M. H. Beddall and J. W. Marsh, Curr. HIV Res., 2007, 5, 394–402 CrossRef CAS PubMed.
- J. B. Munro, J. Gorman, X. Ma, Z. Zhou, J. Arthos, D. R. Burton, W. C. Koff, J. R. Courter, A. B. Smith 3rd, P. D. Kwong, S. C. Blanchard and W. Mothes, Science, 2014, 346, 759–763 CrossRef CAS.
- Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pöhlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef CAS.
- H. Hering and M. Sheng, J. Neurosci., 2003, 23, 11759–11769 CrossRef CAS.
- A. M. Weaver, A. V. Karginov, A. W. Kinley, S. A. Weed, Y. Li, J. T. Parsons and J. A. Cooper, Curr. Biol., 2001, 11, 370–374 CrossRef CAS.
- C. E. Turner, Nat. Cell Biol., 2000, 2, 231–236 CrossRef.
- A. M. López-Colomé, I. Lee-Rivera, R. Benavides-Hidalgo and E. López, J. Hematol. Oncol., 2017, 10, 50 CrossRef.
- A. J. Ehrlicher, R. Krishnan, M. Guo, C. M. Bidan, D. A. Weitz and M. R. Pollak, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6619–6624 CrossRef CAS.
- S. L. Freedman, C. Suarez, J. D. Winkelman, D. R. Kovar, G. A. Voth, A. R. Dinner and G. M. Hocky, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16192–16197 CrossRef CAS.
- E. Borrego-Diaz, F. Kerff, S. H. Lee, F. Ferron, Y. Li and R. Dominguez, J. Struct. Biol., 2006, 155, 230–238 CrossRef CAS.
- T. Oda, M. Iwasa, T. Aihara, Y. Maeda and A. Narita, Nature, 2009, 457, 441–445 CrossRef CAS.
- Z. Wang, H. Sun, X. Yao, D. Li, L. Xu, Y. Li, S. Tian and T. Hou, Phys. Chem. Chem. Phys., 2016, 18, 12964–12975 RSC.
- H. Youssoufian, M. McAfee and D. J. Kwiatkowski, Am. J. Hum. Genet., 1990, 47, 62–72 CAS.
- G. B. Melikyan, Curr. Opin. Virol., 2014, 4, 1–7 CrossRef CAS.
- F. Clavel and P. Charneau, J. Virol., 1994, 68, 1179–1185 CAS.
- V. R. MaréChal, F. Clavel, J. M. Heard and O. Schwartz, J. Virol., 1998, 72, 2208–2212 Search PubMed.
- K. Miyauchi, Y. Kim, O. Latinovic, V. Morozov and G. B. Melikyan, Cell, 2009, 137, 433–444 CrossRef CAS.
- M. Gordon-Alonso, M. Sala-Valdes, V. Rocha-Perugini, D. Perez-Hernandez, S. Lopez-Martin, A. Ursa, S. Alvarez, T. V. Kolesnikova, J. Vazquez, F. Sanchez-Madrid and M. Yanez-Mo, J. Immunol., 2012, 189, 689–700 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr07359k |
| ‡ These authors contributed equally to this work. |
| § Present address: State Key Laboratory of Agricultural Microbiology, College of Life Science and Technology, Huazhong Agricultural University, Wuhan 430070, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2020 |