
Theoretical study of chloride complexes with hybrid macrocycles†
Éder Henrique da
Silva
a,
Renato Pereira
Orenha
 *a,
Alvaro
Muñoz-Castro
b,
Giovanni Finoto
Caramori
c,
Matheus Cachoeira
Colaço
c,
Graziele Capatto Guerra
Silva
a and
Renato Luis Tame
Parreira
*a
*a,
Alvaro
Muñoz-Castro
b,
Giovanni Finoto
Caramori
c,
Matheus Cachoeira
Colaço
c,
Graziele Capatto Guerra
Silva
a and
Renato Luis Tame
Parreira
*a
aNúcleo de Pesquisas em Ciências Exatas e Tecnológicas, Universidade de Franca, Franca, SP 14404-600, Brazil. E-mail: rpo9@hotmail.com; renato.parreira@unifran.edu.br
bLaboratorio de Química Inorgánica y Materiales Moleculares, Facultad de Ingenieria, Universidad Autonoma de Chile, Llano Subercaceaux 2801, San Miguel, Santiago, Chile
cDepartamento de Química, Universidade Federal de Santa Catarina, Campus Universitário Trindade, CP 476, Florianópolis, SC 88040-900, Brazil
First published on 23rd November 2020
Abstract
Anions show relevant roles in biological routes. The supramolecular chemistry investigates the chemical bonding between two or more molecules and/or ions. Herein, the nature of the bond between chloride anions and macrocycle receptors elaborated from (i) pyridines, (ii) pyrroles, (iii) borazines, (iv) triazines, and (v) 1,2,3-triazole rings are studied. The energy decomposition analysis (EDA) shows that the receptors that predominantly establish non-covalent interactions with the Cl− anions proportionate a preferable bond than the macrocycles that mostly form a covalent interaction with the Cl− anions. The substitution of pyridine by borazine rings in the macrocycles or the protonation of the receptors increases the interaction with the Cl− anions since there is an increase in the number of –BH or –NH groups available to establish hydrogen bonds with the Cl− anions. In addition, the pyridine → borazine substitution decreases the number of repulsive  interactions. The substitution of pyrrole by 1,2,3-triazole rings does not relevantly favor the interaction with the Cl− anions. The substitution of pyridine by the triazine rings or the addition of electron-withdrawing groups (–OH, –F and –NO2) in the receptor structures increases the acidity of the cavity of the macrocycles and, therefore, favors the interaction with the Cl− anions. The addition of electron-donating groups (–NH2) to the receptor structure promotes the opposite effect. Accordingly, the present investigation brings relevant information for the design of new hybrid macrocycles with the potential for anionic recognition.
interactions. The substitution of pyrrole by 1,2,3-triazole rings does not relevantly favor the interaction with the Cl− anions. The substitution of pyridine by the triazine rings or the addition of electron-withdrawing groups (–OH, –F and –NO2) in the receptor structures increases the acidity of the cavity of the macrocycles and, therefore, favors the interaction with the Cl− anions. The addition of electron-donating groups (–NH2) to the receptor structure promotes the opposite effect. Accordingly, the present investigation brings relevant information for the design of new hybrid macrocycles with the potential for anionic recognition.
1. Introduction
The supramolecular chemistry is characterized by the association of molecular species for the obtention of a determined property or functionality.1,2 Frequently defined as the chemistry beyond the molecule,2 supramolecular chemistry is related to the organized units with the relevant complexities, established by interactions between two or more chemical entities such as molecules and/or ions.3–8 The effect of these interactions is essential for a complete description of the supramolecular systems and further design of functional assemblies.In the host–guest supramolecular complexes, the guest structure is accommodated inside the host cavity to maximize the overlap related to the host–guest bond. This complementary fitting occurs at multiple sites of interaction. It allows for a strong interaction between the host and guest fragments. This bonding path also avoids strong repulsions concerning host–guest interactions. Thus, the fundamental requisite for the molecular recognition is the complementary principle.9,10
Anions have an important role in a large range of chemical, biological and environmental processes.11 Chloride ions are usually found in the extracellular fluid. The deregulation of the concentration of this anion can promote diseases such as, for example, cystic fibrosis.12 The iodide anion is necessary for the biosynthesis of hormones by the thyroid gland,13 while the fluoride is considered essential for the growth of healthy bones and teeth.14 Therefore, the diversity and importance of the anionic species request the investigation of structures capable of detecting these ions.
Hybrid macrocycle receptors generically called cycle[m]pyridine[n]pyrrole showed a large potential to anionic recognition.15 Such compounds in an acidic medium result in the protonation of two pyridine rings, which promote the increase in the conjugation of π-electrons in the macrocycle. Furthermore, the protonated receptors interact with anions of the acidic medium in a stoichiometry of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (host/guest).
:2 (host/guest).
One strategy typically employed for the design of receptors capable of recognizing anions is the use of Lewis acid centers or rings with electron deficiency, such as macrocycles containing groups that are derivatives of compounds such as hexafluorobenzene, triazine and borazine.16–19 Alternatively, 1,2,3-triazole exhibited to be a versatile unit for the recognition of cations and anions. This structure is able to bind anions supported by hydrogen bonds.20
Herein, the bonding mechanism between the chloride anions and the macrocycle receptors based in: (i) pyridines, (ii) pyrroles, (iii) borazines, (iv) triazines, and (v) 1,2,3-triazole structures is investigated (Scheme 1). It is important to mention that this study was based on the experimental structures of cycle[m]pyridine[n]pyrrole, reported by Zhang et al.15 The main objectives are to evaluate the influence of the: (1) nature of the aromatic rings, (2) protonation, and (3) electron-withdrawing or -donating groups in the receptor structures in relation to the host–guest interaction. This study was performed using the energy decomposition analysis (EDA), non-covalent interactions (NCI) and quantum theory of atoms in molecule (QTAIM) methodologies, as well as, of the molecular electrostatic potential (MEP) surfaces, in order to rationalize the underlying features leading to efficient anion recognition.
 | ||
| Scheme 1 Structure of the complexes investigated: (1–16)⋯2Cl−. | ||
2. Theoretical methods
The calculations of geometry optimization and vibrational frequencies of all investigated complexes were performed using the B3LYP21 method along with the SVP22,23 basis set using the Gaussian16 software.24 Vibrational analyses for all optimized geometries confirmed that they are all energy minima at the level of theory applied here. The host–guest bonding mechanism was investigated by the energy decomposition analysis (EDA)25 using the BLYP26–28 functional and Grimmes D3 dispersion correction with Becke–Johnson damping function (D3(BJ))29–33 along with the TZ2P basis set.34 The BLYP method was typically used in EDA calculations involving anions, providing results with good agreement to the experimental results.35,36 These calculations were done using the ADF2019 package.37,38 The host–guest systems were also investigated from the non-covalent interaction (NCI) methodology,39 using the NCIPLOT software,39 and from the quantum theory of atoms in molecules (QTAIM) method,40,41 using the AIMAll (Version 17.01.25) software.42 The wavefunctions used to perform the NCI and QTAIM methods were obtained from the B3LYP/SVP theory level using the Gaussian16 software.Induced magnetic field contour plots and 1H-NMR shifts were obtained via gauge-independent atomic orbital (GIAO)-DFT NMR calculations within the ADF2019 package, with the OPBE43 functional and all-electron TZ2P basis sets, useful for the evaluation of nuclear magnetic shifts.
3. Results and discussion
3.1 Accuracy
To validate the computational approach chosen (B3LYP/SVP) to optimize the geometry of the complexes investigated, the values of some geometric parameters of structure 4 were compared to X-ray data, reported by Zhang et al.15 Thus, the root mean square deviation (RMSd) relative to C–C and C–N bond lengths between the calculated and experimental results shows low values (0.013 and 0.021 Å, respectively). It attests the efficacy of the theory level used in this study to optimize the geometry of the complexes investigated.In addition, to validate the B3LYP/SVP computational approach in relation to B97D333,44 (method that includes dispersion corrections)/Def2-TZVP45 (more diffuse basis set regarding to SVP) theory level, the geometry of the complex 4⋯2Cl− was also optimized from the B97D3/Def2-TZVP computational approach. The RMSd concerning C–C and C–N bond lengths between the B3LYP/SVP and B97D3/Def2-TZVP results shows low values (0.006 and 0.004 Å, respectively). The EDA data were obtained using the BLYP-D3(BJ)/TZ2P computational model from the geometries optimized with B3LYP/SVP and B97D3/Def2-TZVP theory levels (Table 1). The energy and the percentage of each stabilizing contribution practically show the same values considering the different approaches for the complex 4⋯2Cl− (RMSd = 3.09 kcal mol−1 and 0%, respectively). The NCI and MEP surfaces obtained from the B3LYP/SVP and B97D3/Def2-TZVP computational models also show the same characteristics (Fig. S1, ESI†). The topological maps of the electron density obtained from the QTAIM method using both approaches show six bond critical points (BCPs) related to each: (i) N–H⋯Cl−; and (ii) N⋯Cl−, chemical interactions (Fig. S2, ESI†). In both theory levels, there are positive values of the Laplacian of the electron density, ∇2ρb, in the respective BCPs (Table S3, ESI†). Furthermore, there are values between 0.5 and 1.0 of the ration between the kinetic energy Gb and potential Vb energy, −Gb/Vb, and values larger than 1.0 of −Gb/Vb for the N–H⋯Cl− and N⋯Cl− BCPs, respectively. It shows that both methodologies indicate the N–H⋯Cl− bonds as partially covalent, while that the N⋯Cl− interactions appear with a predominantly non-covalent nature. Lastly, there are close values of ρb from the B3LYP/SVP and B97D3/Def2-TZVP data (Table S3, ESI†), RMSd = 0.002 a.u.
| Host | ΔEint | ΔVelstat | ΔEPauli | ΔEoi | ΔEdisp |
|---|---|---|---|---|---|
| a ΔEint = ΔVelstat + ΔEPauli + ΔEoi + ΔEdisp. b EDA calculation realized from the geometry optimized using the B97D3/Def2-TZVP computational model. | |||||
| 1 | −83.10 | −56.26 (35) | 78.87 | −93.25 (58) | −12.46 (8) |
| 2a | −103.81 | −83.76 (44) | 86.71 | −91.20 (48) | −15.57 (8) |
| 2b | −126.19 | −84.90 (43) | 71.66 | −97.20 (49) | −15.75 (8) |
| 3 | −102.03 | −69.35 (39) | 77.72 | −96.14 (53) | −14.26 (8) |
| 4 | −391.24 | −354.17 (71) | 105.98 | −125.59 (25) | −17.46 (4) |
| 4 | −388.87 | −349.96 (71) | 102.50 | −122.43 (25) | −18.98 (4) |
| 5 | −398.68 | −359.65 (71) | 106.81 | −126.72 (25) | −19.12 (4) |
| 6 | −411.78 | −370.87 (71) | 109.06 | −130.76 (25) | −19.21 (4) |
| 7 | −438.16 | −407.24 (74) | 115.03 | −127.12 (23) | −18.83 (3) |
| 8 | −491.90 | −448.10 (74) | 113.90 | −138.40 (23) | −19.30 (3) |
| 9 | −411.09 | −377.07 (71) | 117.69 | −134.26 (25) | −17.45 (3) |
| 10 | −459.83 | −432.96 (74) | 123.51 | −133.34 (23) | −17.04 (3) |
| 11 | −510.69 | −471.10 (74) | 127.60 | −149.67 (23) | −17.52 (3) |
| 12 | −87.35 | −64.26 (46) | 52.98 | −60.87 (43) | −15.19 (11) |
| 13 | −79.90 | −51.93 (39) | 52.65 | −65.40 (49) | −15.22 (11) |
| 14 | −92.06 | −67.22 (46) | 53.11 | −62.28 (43) | −15.68 (11) |
| 15 | −103.91 | −80.53 (54) | 55.74 | −63.79 (40) | −15.33 (10) |
| 16 | −143.81 | −115.78 (55) | 67.54 | −78.38 (37) | −17.20 (8) |
3.2 The bonding mechanism behind of the host–guest interaction
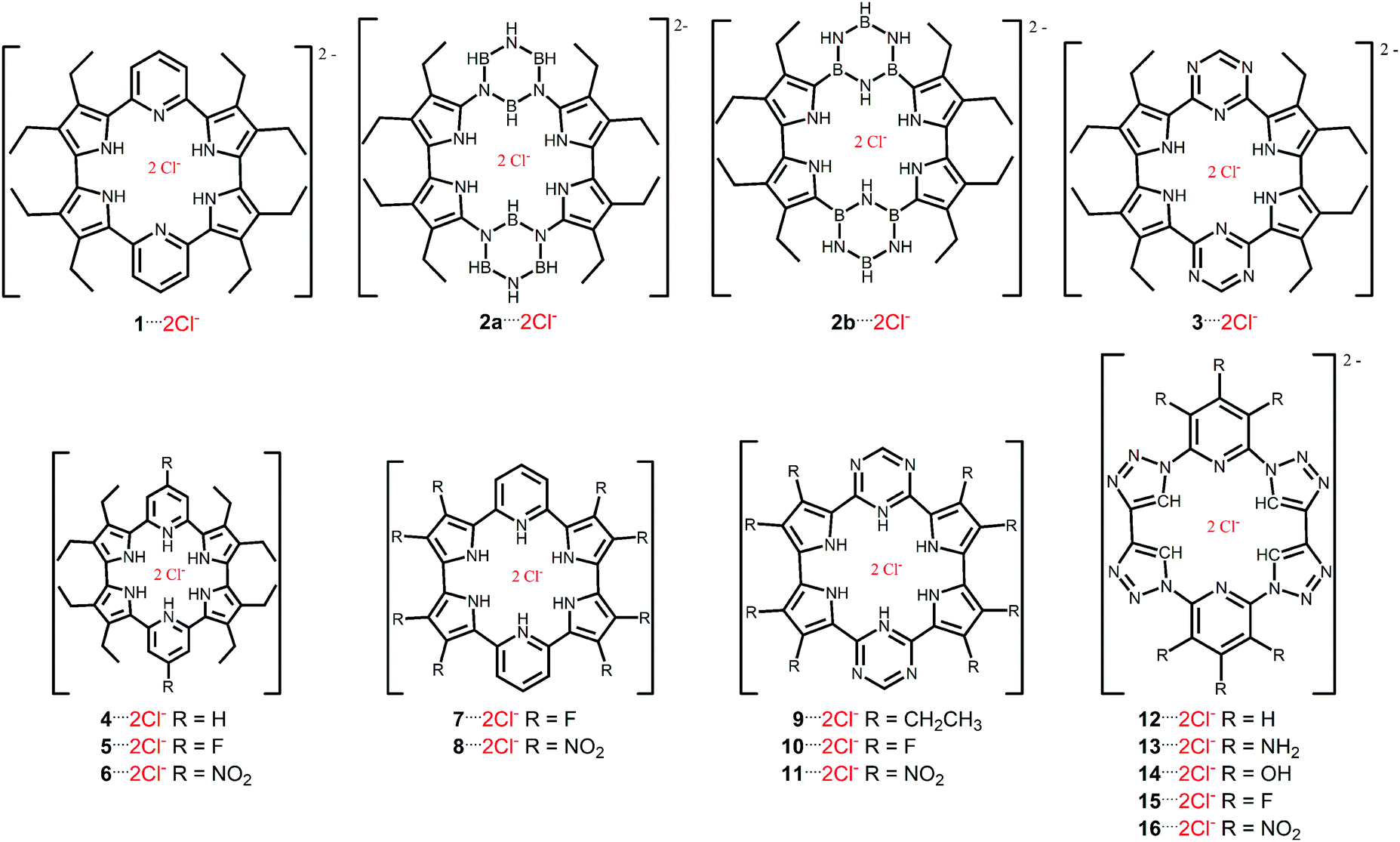
The receptor–chloride interactions occur in a stoichiometry of 1:2 (host/guest). There are two or three hydrogen atoms of the –NH groups pointed for one Cl− above the medium macrocycle plane, while there are other two or three hydrogen atoms of the –NH groups pointed for one Cl− below the medium macrocycle plane. Furthermore, it is possible to visualize in some receptors two –BH or four –CH groups pointed for Cl− anions. In host–guest systems containing two anionic guests, one of the hardest challenges compensates the electrostatic repulsion energy between the anions. The substitution of pyridine by the borazine rings (1 → 2b) increases the number of the –NH groups available for the interaction with the Cl− anions. It explains the decrease in the distance between the Cl− anions, r(Cl−⋯Cl−) (Fig. 1). Similarly, the protonation of pyridine (1 → 4–8) and triazine (3 → 9–11) rings promotes the decrease in the r(Cl−⋯Cl−) parameter. However, the presence of the –CH groups (12–16) and, mainly –BH (2a) groups, in the receptor structures in position to interact with the Cl− anions does not change significantly or increase the value of r(Cl−⋯Cl−). These results indicate the importance of the hydrogen bonds to stabilize the receptor–chloride interactions.
 | ||
| Fig. 1 (a) Geometric parameter related to distance between the Cl− anions, r(Cl−⋯Cl−); and (b) graphical representation of the value of r(Cl−⋯Cl−)/Å depending on the complex investigated. | ||
To elucidate the main features associated with the host–guest bond, the EDA investigation was made considering the macrocycle receptor as one fragment, while the two Cl− anions were attributed as the second fragment. The EDA results are presented in Table 1. First, the negative values of the total interaction energy, ΔEint, show that all receptor–chloride interactions are attractive. Interestingly, the anionic recognition performed from the receptors 1, 2a, 2b, 3 and 13 occurs from a predominantly covalent interaction. It can be observed because the largest contribution of the orbital interactions energy, ΔEoi, to sum of all attractive energetic components: ΔVelstat + ΔEoi + ΔEdisp, in the interactions (1, 2a, 2b, 3 and 13)⋯2Cl− (48–58%). However, it is important to highlight that the electrostatic energy, ΔVelstat (energetic term associated to non-covalent character of the interaction), which also shows a relevant contribution to these bonds (35–44%). Furthermore, the (4–12 and 14–16)⋯2Cl− interactions show a primarily non-covalent character (contribution of ΔVelstat to ΔVelstat + ΔEoi + ΔEdisp is equal to 46–74%). In this case, is possible visualize an important covalent contribution (weight of ΔEoi to ΔVelstat + ΔEoi + ΔEdisp is equal to 23–43%). The interactions between the receptors 1, 2a, 2b, 3 and 12–16, and 2Cl− also show a relevant contribution of the dispersion energy, ΔEdisp, (8–11%).
Importantly, the receptors that recognize the Cl− anions from a predominantly non-covalent interaction (4–12 and 14–16)⋯2Cl−, appear as the best candidates to perform the anionic recognition since they show the lowest values of the ΔEint energy.
The substitutions of the pyridine rings in 1 by: (i) borazine rings in 2; and (ii) triazine rings in 3, provide more stabilizing values of the ΔEoi, ΔEdisp and, mostly, ΔVelstat energy terms, despite the increase in the repulsive values of the ΔEPauli energy. As exception, the value of the ΔEoi energetic component increases from 1⋯2Cl− to 2a⋯2Cl−. These changes promote more attractive values of the ΔEint energy in the complexes 2⋯2Cl− and 3⋯2Cl− regarding structures 1⋯2Cl−. The largest decreases in the ΔVelstat energy from the molecule 1⋯2Cl− to compound 2⋯2Cl− can be justified by a larger number of hydrogen atoms available for interaction with the Cl− anions in receptor 2 (from four N–H plus two B–H groups, Fig. 1) concerning compound 1 (from four N–H groups, Fig. 1) or a due decrease in the number of  groups close to Cl− anions from 1 to 2 (2 and 0
groups close to Cl− anions from 1 to 2 (2 and 0  groups, respectively, Fig. 1). The anionic recognition realized from the receptor 2b is more efficient than the performed from the receptor 2a because of the more attractive ΔEoi energy and, mainly, less repulsive ΔEPauli component in the bond 2b⋯2Cl− regarding interaction 2a⋯2Cl−. It can be explained because of the lower values of the
groups, respectively, Fig. 1). The anionic recognition realized from the receptor 2b is more efficient than the performed from the receptor 2a because of the more attractive ΔEoi energy and, mainly, less repulsive ΔEPauli component in the bond 2b⋯2Cl− regarding interaction 2a⋯2Cl−. It can be explained because of the lower values of the  groups close to Cl− anions in the receptor 2b regarding receptor 2a. The presence of a more acid cavity in receptor 3 regarding structure 1 can be used as an argument to justify the more stable interaction 3⋯2Cl− in relation to bond 1⋯2Cl−. This will be better discussed from MEP results. The substitution of the pyrrole rings of 1 by 1,2,3-triazole rings in 12 does not significantly favor the interaction with the Cl− anions. It occurs because the more favorable values of the ΔVelstat and ΔEdisp energies, and the less repulsive ΔEPauli energy are almost overcome by the less stabilizing value of the ΔEoi energy in interaction 12⋯2Cl− concerning interaction 1⋯2Cl−.
groups close to Cl− anions in the receptor 2b regarding receptor 2a. The presence of a more acid cavity in receptor 3 regarding structure 1 can be used as an argument to justify the more stable interaction 3⋯2Cl− in relation to bond 1⋯2Cl−. This will be better discussed from MEP results. The substitution of the pyrrole rings of 1 by 1,2,3-triazole rings in 12 does not significantly favor the interaction with the Cl− anions. It occurs because the more favorable values of the ΔVelstat and ΔEdisp energies, and the less repulsive ΔEPauli energy are almost overcome by the less stabilizing value of the ΔEoi energy in interaction 12⋯2Cl− concerning interaction 1⋯2Cl−.
The protonation of the pyridine (1 → 4) and triazine rings (3 → 9) promotes a sizable stabilization of the interaction with the Cl− anions due to more favorable values of the ΔEoi, ΔEdisp and, mainly, ΔVelstat energies, which overcome the more repulsive value of the ΔEPauli energy. These results indicate that the more favorable interaction of the protonated receptors with the chloride anions occurs, probably, with an increase in the number or intensity of the N–H⋯Cl− hydrogen bonds, and the acidity in the cavity of the macrocycle. In fact, the more favorable electrostatic energy in 4⋯2Cl− regarding 1⋯2Cl− is associated with the spectroscopic characteristics observed experimentally, where the protonation of 1 to produce 4 results in an increase in the delocalization of the positive charge on adjacent pyrrolic heterocycles.15
The addition of electron-withdrawing groups (–F, –NO2 or –OH) in the (i) pyridine rings (4 → 5 and 6; and 12 → 14–16); and (ii) pyrrole rings (4 → 7 and 8; and 9 → 10 and 11) favors the interaction with the Cl− anions. In general, the substituted receptors have more attractive interactions with the Cl− anions than non-substituted compounds due to more favorable values of the ΔVelstat and ΔEoi energies, despite the more repulsive values of the ΔEPauli term. The more attractive dispersion energy, ΔEdisp, contributes to interactions (5, 6, 7 and 8)⋯2Cl− related to 4⋯2Cl−. The values of the ΔEdisp component are essentially the same comparing (10 and 11)⋯2Cl− and (14–16)⋯2Cl− concerning to 9⋯Cl− and 12⋯2Cl−, respectively. As an exception, the ΔEdisp energy decreases in 12⋯2Cl− → 16⋯2Cl−. These changes are more pronounced when the substitutions occur at the pyrrole rings. Nevertheless, the addition of an electron-donating group (–NH2) in the pyridine rings (12 → 13) produces a less stabilizing value of the ΔEint energy associated with chloride recognition proportioned by the less stabilizing value of the ΔVelstat energy term, despite the more stabilizing value of the ΔEoi energy in the interaction 13⋯2Cl− regarding interaction 12⋯2Cl−. The values of the ΔEPauli and ΔEdisp are practically constant in the interactions 12⋯2Cl− → 13⋯2Cl−. These data can be justified from the MEP surfaces.
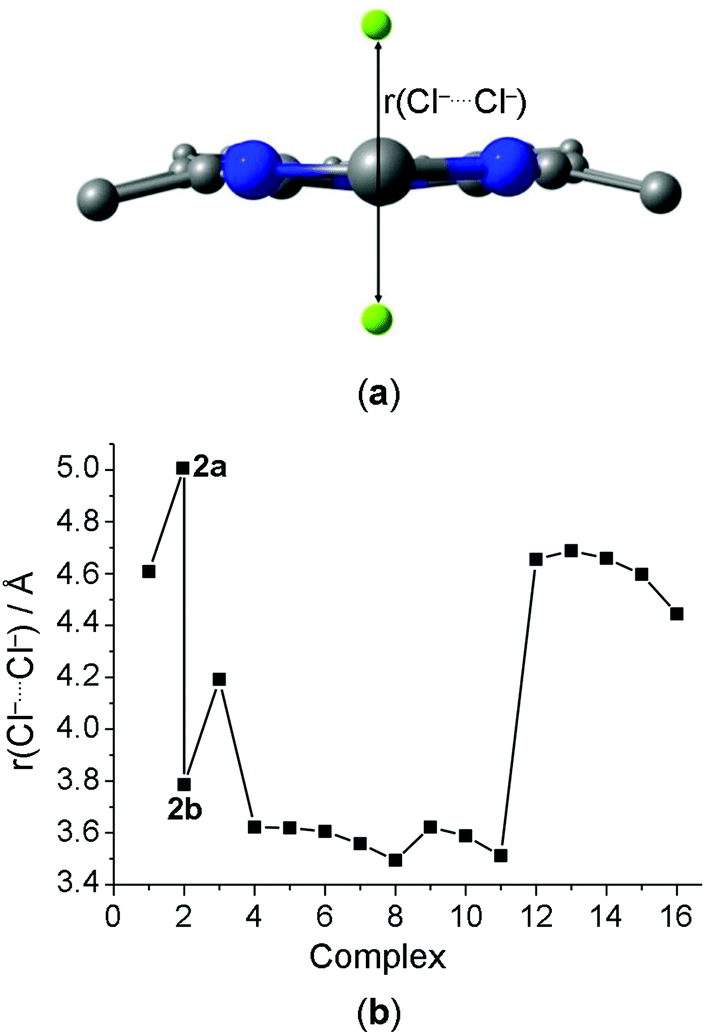
The NCI analysis shows the presence of attractive interactions (blue or light blue NCI surfaces) between the macrocycle receptors, or more specifically –NH and –CH groups of 1–11 and 12–16, respectively, and the chloride anions (Fig. 2; similar NCI surfaces shown in Fig. 2, involving atoms of the same nature, but in different positions, are presented in Fig. S3–S6, ESI†). The C–H⋯Cl− interactions (light blue NCI surfaces) appear with lower intensity relative to N–H⋯Cl− interactions (blue NCI surfaces). In addition, the structures 2a and 2b also provide –BH groups that can establish weak van der Waals interactions (green NCI surface) with the chloride anions: B–H⋯Cl−. In the center of the aromatic rings, it is possible visualize repulsion interactions (red NCI surface).
 | ||
| Fig. 2 Front (left) and top (right) views of the NCI topology for the systems: (2a, 4, 11 and 12)⋯2Cl−. Atoms color: H = silver; B = yellow; C = black; N = blue; O = red; and Cl = green. | ||
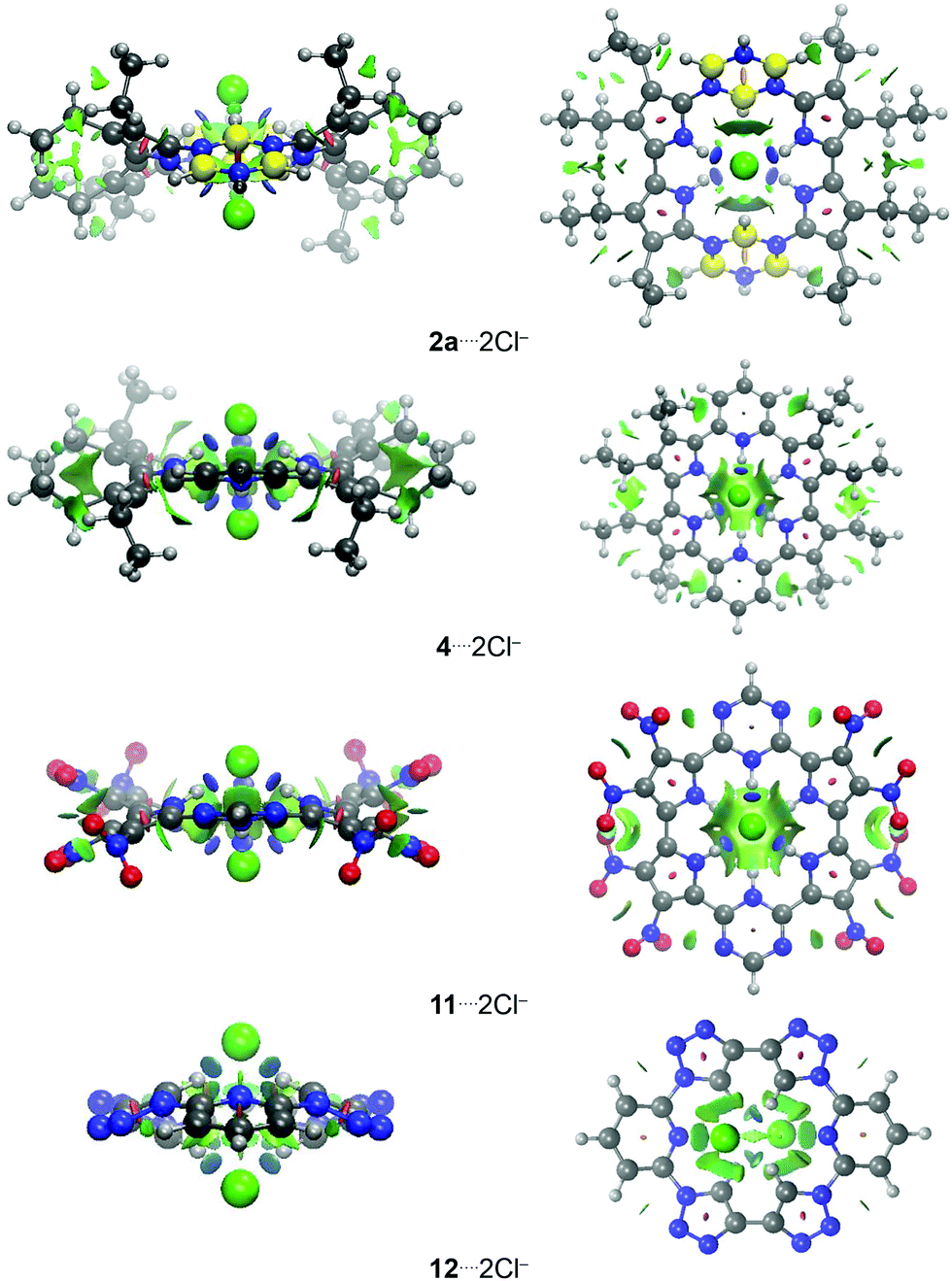
The molecular electrostatic potential (MEP) surfaces were generated for all complexes investigated to evaluate the main coulombic interactions present in the host–guest bond (Fig. 3; similar MEP surfaces shown in Fig. 3, involving atoms of same nature, but in different positions, are presented in Fig. S7, ESI†). In all the structures, there are areas localized at the Cl− anions that have high electron concentrations (primarily red color). It allows these atoms to interact with the hydrogen atoms, from the –NH, –BH and –CH groups, which show electron-poor regions (preponderance of blue color).46 Therefore, the substitution of the pyridine rings, in 1, by borazine rings, in 2a and 2b, increases the number of –BH and –NH groups, respectively. In addition, from 1 to 2, there is a decrease in the number of  groups (red MEP surface) in the receptor structure. It explains the more attractive value of the ΔVelstat energy from 1⋯2Cl− to 2⋯2Cl−. The substitution of the pyridine rings in 1 by triazine rings, in 3, and 1,2,3-triazole rings, in 12, helps to decrease the red surface area in the carbon and nitrogen atoms closer to Cl− anions present in the receptors 3 and 12 regarding 1, and supports the more favorable values of the ΔVelstat energy from 1⋯2Cl− to 3⋯2Cl− and 12⋯2Cl−.
groups (red MEP surface) in the receptor structure. It explains the more attractive value of the ΔVelstat energy from 1⋯2Cl− to 2⋯2Cl−. The substitution of the pyridine rings in 1 by triazine rings, in 3, and 1,2,3-triazole rings, in 12, helps to decrease the red surface area in the carbon and nitrogen atoms closer to Cl− anions present in the receptors 3 and 12 regarding 1, and supports the more favorable values of the ΔVelstat energy from 1⋯2Cl− to 3⋯2Cl− and 12⋯2Cl−.
 | ||
| Fig. 3 MEP surfaces for the complexes analyzed in this study. The isovalue used to represent the surfaces is equal to 0.050 a.u, while the values of the scales (red – blue a.u.) are (1–3, 12 and 13)⋯2Cl− (−0.100 to 0.100 a.u.); (4 and 5)⋯2Cl− (0.100–0.250 a.u.); and 8⋯2Cl− (0.150–0.300 a.u.). | ||
The protonation of the structures 1 and 3 to form the complexes 4 and 9, respectively, promotes an increase in the MEP scale (Fig. 3 and Fig. S7, ESI†). It allows us to infer that there is a cavity more acid in the receptors 4 and 9 regarding 1 and 3, respectively. Besides, there is an increase in the number of the –NH groups from 1 and 3 (four –NH groups) to 4 and 9 (six –NH groups, Fig. 1), respectively, which can favorably interact with the Cl− anions. This result agrees with the more stabilizing values of the ΔVelstat term in the complexes 4⋯2Cl− and 9⋯2Cl− regarding structures 1⋯2Cl− and 3⋯2Cl−, respectively. In general, the addition of electron-withdrawing groups to pyridine and, chiefly, to the pyrrole rings of the receptor structures increases the blue area in the MEP surface of the macrocycle receptor or increases the MEP scale (increases the acidity of the macrocycle cavity) and supports the more attractive values of the ΔVelstat energy in the interactions between the substituted receptors (5–8, 10, 11 and 14–16) and Cl− anions than between the non-substituted receptors (4, 9 and 12) and Cl− ions. However, the addition of electron-donating groups (–NH2) in the 1,2,3-triazole rings (12 → 13) increases the red MEP surface area in the carbon atoms close to Cl− anions. It justifies the less stabilizing value of the ΔVelstat energy from the complex 12⋯2Cl− to molecule 13⋯2Cl−.
Moreover, the topological analysis of the electron density of the complexes investigated was performed from the QTAIM method, and the atom–atom interactions can be evaluated from the bond critical points (BCPs). The topological maps of the complexes investigated are present in Fig. 4 (similar topological maps shown in Fig. 4, involving atoms of same nature, but in different positions, are presented in Fig. S8 and S9, ESI†). First, it is possible to note that the receptors can interact with the Cl− anions from the (i) N–H⋯Cl−, (ii) C–H⋯Cl−, (iii) B–H⋯Cl−, (iv) N⋯Cl−, (v) C⋯Cl−, and (vi) B⋯Cl− interactions. All these interactions show positive values of the Laplacian of the electron density, ∇2ρb, in the respective BCPs (Tables S2–S6, ESI†). However, the (i) N–H⋯Cl−, (ii) C–H⋯Cl−, and (vi) B⋯Cl− interactions show values between 0.5 and 1.0 of −Gb/Vb in the BCPs related to these interactions, which indicate interactions with a partially covalent character. The (iii) B–H⋯Cl−, (iv) N⋯Cl− and (v) C⋯Cl− interactions show values of −Gb/Vb larger than 1.0, which shows bonds with chiefly ionic nature. The N–H⋯Cl− and C–H⋯Cl− hydrogen bonds show larger values of ρb than the N⋯Cl− and C⋯Cl− interactions, respectively. However, the B⋯Cl− interactions show larger values of ρb concerning B–H⋯Cl− hydrogen bonds. Besides, the N–H⋯Cl− bonds show, in general, larger values of ρb than C–H⋯Cl− and, mainly, B–H⋯Cl− hydrogen bonds. Nevertheless, the B⋯Cl− interactions show, as a whole, larger values of ρb than N⋯Cl− and, principally, C⋯Cl− bonds.
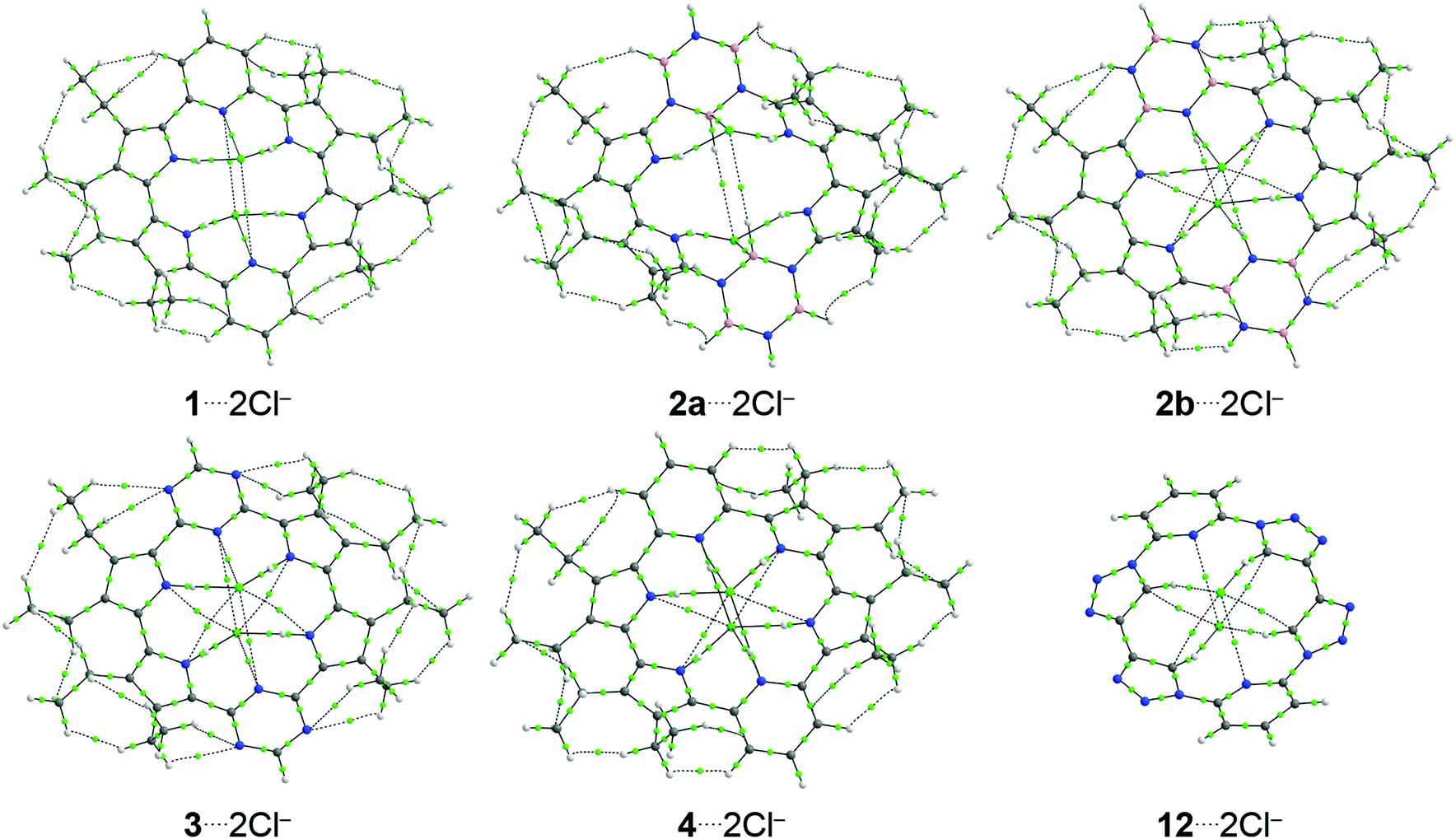 | ||
| Fig. 4 Topological map containing the bond paths (continuous or dashed lines connecting the cores) and bond critical points (light green points) for the structures (1–4 and 12)⋯2Cl−. Atoms color: H = white; B = rose; C = gray; N = blue; and Cl = light green. | ||
The substitution of the pyridine rings in 1 by borazine rings, in 2, and triazine rings, in 3, does not significantly change the values of ρb present in the interactions between receptors and chloride anions. However, from 1⋯2Cl− to 2b⋯2Cl−, there is an increase in the number of the N–H⋯Cl− and N⋯Cl− interactions, while from 1⋯2Cl− to 3⋯2Cl−, there is an increase in the number of the N⋯Cl− chemical bonds. It justifies the more stabilizing values of the ΔEoi energy in the complexes 2b⋯2Cl− and 3⋯2Cl− concerning the structure 1⋯2Cl−. The substitution of the pyrrole rings of 1 by 1,2,3-triazole rings, in 12, changes the nature of the interactions between the receptor and the chloride anions. The structure 1 interacts with the Cl− anions from the N–H⋯Cl− and N⋯Cl− interactions, while compound 12 interacts with the Cl− anions from the C–H⋯Cl− and C⋯Cl− interactions. Since those bonds involving the nitrogen atom show larger values of ρb than the bonds established through the carbon atoms, it explains the more attractive value of the ΔEoi energy in the interaction 1⋯2Cl− regarding the bond 12⋯2Cl−.
The protonation of the pyridine rings (1 → 4) increases the number of the N–H⋯Cl− and N⋯Cl− bonds (4 → 6, to each interaction class) and explains the more favorable ΔEoi energy from the structure 1⋯2Cl− to complex 4⋯2Cl−. Interestingly, the protonation of the triazine rings (3 → 9) produces a more attractive ΔEoi energy from compound 3⋯2Cl− to molecule 9⋯2Cl− due to the increase in the sum of the values of ρb in the N–H⋯Cl− and N⋯Cl− BCPs in 3⋯2Cl− → 9⋯2Cl−. The presence of (i) electron-withdrawing groups (–F, –NO2 or –OH) or (ii) electron-donating groups (–NH2) does not promote changes in the number of interactions between the receptor and the Cl− anions. In addition, the values of ρb are also practically constant in the BCPs related to receptor–chloride interactions investigated when electron-withdrawing or -donating groups are incorporated into the receptor structures (Tables S2–S6, ESI†).
Lastly, the role of the chloride anion encapsulation in the chemical environment of the inner-hydrogen atoms is evaluated from simplified models replacing ethyl groups with hydrogen atoms. The calculation of the free host shows a deshielded region at the center of the macrocycle, denoting the non-aromatic characteristics of the studied hosts (Fig. 5). The incorporation of Cl− anions induces a sizable decrease in the central deshielded region as observed from the induced magnetic field contour plots from Fig. 5, owing to the shielding contribution from each Cl− anion. The calculated chemical shielding of the hydrogen atoms nearby the Cl− ions sizably shielded in about −6 ppm (average), which is a useful pattern for further analyses of binding constants from 1H-NMR titration experiments (Table S7, ESI†).47,48
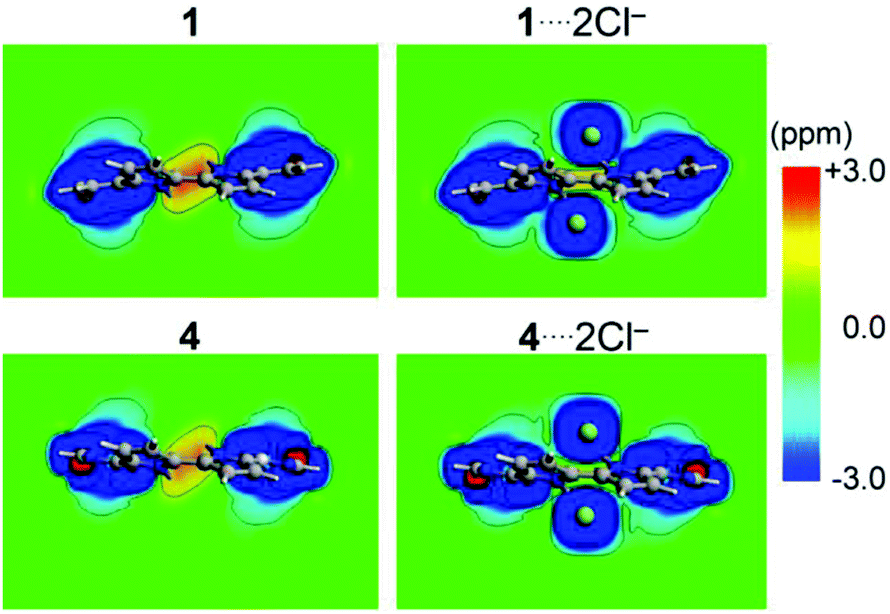 | ||
| Fig. 5 Induced magnetic field contour plots for 1⋯2Cl− and 4⋯2Cl−, and the related free host parents. Note the decrease in the central deshielded region for free host (yellow–red region) occurs upon the formation of the host–guest complex. | ||
Conclusions
Therefore, the interactions between the macrocycle receptors and Cl− anions investigated are attractive. Besides, the anionic recognition supported by a predominantly non-covalent character is more favorable than the interactions that show a chiefly covalent nature. The substitution of pyridine by borazine or triazine rings favors Cl− recognition due, principally, to the more stabilizing ΔVelstat energy. The stabilization associated with pyridine → borazine substitution is justified by the: (i) increase in the number of attractive hydrogen bonds and (ii) decrease in the number of repulsive interactions. The more attractive anionic recognition proportioned by the pyridine → triazine substitution is explained by the increase in the acidity in the macrocycle cavity, as illustrated from the MEP surfaces.
interactions. The more attractive anionic recognition proportioned by the pyridine → triazine substitution is explained by the increase in the acidity in the macrocycle cavity, as illustrated from the MEP surfaces.
The protonation of the receptors favors the interaction with the Cl− anions from a more attractive ΔVelstat energy. It can be clarified because of the increase in the number or electron density of the N–H⋯Cl− hydrogen bonds, and more acidity in the cavity of the protonated macrocycles relative to non-protonated receptors. The presence of electron-withdrawing groups (–OH, –F and –NO2) in the receptor structure promotes a more favorable interaction with the Cl− anions supported, largely, from a more attractive ΔVelstat energetic term. In an opposite way, the addition of electron-donating groups (–NH2) decreases the interaction of the receptor with the Cl− anions. These data are justified because the electron-withdrawing groups increase the acidity of the macrocycle receptor, while the donor group decreases the acidity in the cavity of the receptor, making it difficult to interact with the Cl− anions.
The NCI methodology indicates the presence and strength of the hydrogen bonds between receptors and anions in the following order: (1) N–H⋯Cl−; (2) C–H⋯Cl−; and (3) B–H⋯Cl−. The QTAIM method also shows the presence of the (i) N⋯Cl− (non-covalent), (ii) C⋯Cl− (non-covalent), and (iii) B⋯Cl− (partially covalent) chemical bonds. In addition, the 1H-NMR shifts show a shielding shift upon chloride anion incorporation.
Thus, the discussion in this manuscript besides elucidating the host–guest bonding mechanism between the hybrid macrocycles and Cl− anions provides information that can be used as the basis to design new receptor structures with large capability to recognize anions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) Finance Code 001. R. L. T. P., R. P. O. and G. C. G. S. thank grants 2011/07623–8, 2017/24856–2 and 2019/00543–0 São Paulo Research Foundation (FAPESP) for the financial support. A. M.-C. acknowledges Fondecyt ANID grant 1180683. G. F. C. and R. L. T. P. thank the National Council for Scientific and Technological Development (CNPq, grants 311963/2017–0 and 313648/2018–2) for the research fellowship.References
- K. Araki and H. E. Toma, Quim. Nova, 2002, 25, 962–975 CrossRef CAS.
- J.-M. Lehn, Science, 1985, 227, 849–856 CrossRef CAS PubMed.
- P. D. Beer, P. A. Gale and D. K. Smith, Supramolecular Chemistry, Oxford University Press, New York, 1999 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry: A Concise Introduction, Wiley, Chichester, 2009 Search PubMed.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2007, 111, 13287–13290 CrossRef CAS PubMed.
- S. Li, V. R. Cooper, T. Thonhauser, B. I. Lundqvist and D. C. Langreth, J. Phys. Chem. B, 2009, 113, 11166–11172 CrossRef CAS.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- D. J. Cram and G. M. Lein, J. Am. Chem. Soc., 1985, 107, 3657–3668 CrossRef CAS.
- M. L. C. Montanari, C. A. Montanari, D. Piló-Veloso, A. E. Beezer and J. C. Mitchell, Quím. Nova, 1998, 21, 470–476 CrossRef CAS.
- Q. He, P. Tu and J. L. Sessler, Chem, 2018, 4, 46–93 CAS.
- S. M. Rowe, S. Miller and E. J. Sorscher, N. Engl. J. Med., 2005, 352, 1992–2001 CrossRef CAS PubMed.
- F. Delange, Thyroid, 1994, 4, 107–128 CrossRef CAS PubMed.
- M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC.
- Z. Zhang, J. M. Lim, M. Ishida, V. V. Roznyatovskiy, V. M. Lynch, H.-Y. Gong, X. Yang, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 4076–4079 CrossRef CAS PubMed.
- A. Frontera, F. Saczewski, M. Gdaniec, E. Dziemidowicz-Borys, A. Kurland, P. M. Deyà, D. Quiñonero and C. Garau, Chem. – Eur. J., 2005, 11, 6560–6567 CrossRef CAS PubMed.
- S. Bagheri and H. R. Masoodi, Chem. Phys. Lett., 2015, 629, 46–52 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Org. Lett., 2003, 5, 2227–2229 CrossRef CAS PubMed.
- R. Miao, G. Yang, C. Zhao, J. Hong and L. Zhu, J. Mol. Struct.: THEOCHEM, 2005, 715, 91–100 CrossRef CAS.
- M. Alfonso, A. Tárraga and P. Molina, Tetrahedron Lett., 2016, 57, 3053–3054 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- F. M. Bickelhaupt and E. J. Baerends, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, pp. 1–86 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- A. N. Petelski and C. Fonseca Guerra, J. Phys. Chem. C, 2020, 124, 3352–3363 CrossRef CAS.
- R. P. Orenha, V. B. da Silva, G. F. Caramori, F. S. S. Schneider, M. J. Piotrowski, J. Contreras-Garcia, C. Cardenas, M. B. Gonçalves, F. Mendizabal and R. L. T. Parreira, New J. Chem., 2020, 44, 17831–17839 RSC.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. T. Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- R. F. W. Bader, Atoms and Molecules – A Quantum Theory, Claredon Press, Oxford, New York, 1994 Search PubMed.
- G. F. Caramori, R. L. T. Parreira and A. M. da Costa Ferreira, Int. J. Quantum Chem., 2012, 112, 625–646 CrossRef CAS.
- T. A. Keith, AIMAll, Revision 17.01.25, TK Gristmill Software, Overland Park KS, USA, 2017 Search PubMed.
- Y. Zhang, A. Wu, X. Xu and Y. Yan, Chem. Phys. Lett., 2006, 421, 383–388 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. A. Ford, Mol. Pharmacol., 2013, 10, 1171–1182 CrossRef CAS PubMed.
- A. J. Lowe, F. M. Pfeffer and P. Thordarson, Supramol. Chem., 2012, 24, 585–594 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized Cartesian coordinates, data from the NCI, MEP and QTAIM methods, as well as, calculated 1H-NMR chemical shift for the complexes investigated. See DOI: 10.1039/d0nj05234e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |