
Thiophenic silicon phthalocyanines: synthesis, characterization, and photophysical properties†
Kuizhi
Chen
a,
Xia
Li
a,
Bingcheng
Huang
a,
Qiuhao
Ye
a,
Wenling
Xiao
a,
Xinqiao
Guan
a,
Limin
Chen
*b and
Yiru
Peng
 *a
*a
aCollege of Chemistry & Materials, Fujian Provincial Key Laboratory of Advanced Materials Oriented Chemical Engineering, Fujian Provincial Key Laboratory of Polymer Materials, Fujian Normal University, Fuzhou, 350007, China. E-mail: yirupeng@fjnu.edu.cn
bFujian Medical University, Affiliated Hospital of Fujian Medical University, Fuzhou 350004, China. E-mail: 156500406@qq.com
First published on 25th November 2020
Abstract
Three new thienyl/benzothienyl/dibenzothienyl groups axially substituted silicon(IV) phthalocyanines were synthesized. The structures of these thiophenic silicon(IV) phthalocyanines were characterized. The influence of different thiophenic substituents on the photophysical/photochemical properties of phthalocyanines was investigated by fluorescence and UV/Vis absorption spectroscopies. Among them, benzothienyl group substituted silicon(IV) phthalocyanine possessed the highest fluorescence intensity, lifetime, and singlet oxygen quantum yield. Benzothienyl group substituted silicon(IV) phthalocyanine could be transported into MCF-7 breast cancer cells and was not evenly distributed in the cytoplasm, and mediated photodynamic therapy to kill cancer cells.
Introduction
Cancer is one of the most serious diseases throughout the world. There are numerous strategies to combat cancer, such as radiotherapy, surgery, chemotherapy and photodynamic therapy.1 Photodynamic therapy (PDT) has many advantages such as non-invasiveness, inherent site selectivity, and little side effects.2 The PDT process involves three major components: light, photosensitizers (PSs), and oxygen.3 A PS is activated by light of an appropriate wavelength to produce reactive oxygen species (ROS), which leads to cellular damage and subsequent cell death. Phthalocyanines (Pcs) are a promising Type II mechanism PS for PDT owing to their strong absorption in the near-infrared region, chemical structure stability, low phototoxicity, and high singlet oxygen quantum yield.4,5 However, Pcs have limited solubility in solutions and easily aggregate in water due to their strong π–π interactions, which reduce the generation of ROS and subsequent decrease in the PDT efficacy. Moreover, the other limitation of phthalocyanines is their lack of specifically targeting to tumors/tissues. Therefore, grafting bulky functional substituents to the axial positions of phthalocyanines could suppress the aggregation to some extent and be a potential target to the organelles of cancer cells.6Heterocyclic compounds, which are bioactive molecules, have been used as multifunctional building blocks to construct bioactive compounds in medicinal fields. Thiophene and benzothiophene derivatives are privileged heterocyclic compounds because of their wide applications in agrochemicals, medicinal agents, chemical sensors, and materials.7 Moreover, some compounds containing thiophene or benzothiophene substitutions have been demonstrated to be able to selectively target organelles to improve their PDT therapeutic efficacy. For example, thiophenic-substituted diphenylamines exhibited specific targeting of the lysosome, leading to high PDT efficacy.8 Benzo[b]thiophene-substituted porphyrin could simultaneously accumulate in mitochondria and nucleus and damage these organelle function to induce MCF-7 breast cancer cell death.9
Moreover, using multifunctional building blocks to construct a molecule could enhance its bioactivity. The introduction of fluorinated substituents at the axial and peripheral positions of phthalocyanine has been demonstrated to reduce the aggregation behavior of phthalocyanine and endow special physicochemical and biological properties such as lipophilicity and membrane permeability, which eventually enhance the photodynamic therapy efficacy.10–14
In this study, we prepared three dithiophene benzyl/phenyl dibenzothienyl/fluorinated bisphenol-A benzothienyl-substituted silicon phthalocyanines, namely di-(3,5-di-((thiophene-2-carboxylate)propyloxy)benzyloxy) axially substituted silicon phthalocyanine (M2SiPc), di-(4-dibenzoarylthiophene-2-phenoxy) axially substituted silicon phthalocyanine (BBT-SiPc), and di-((3-chlorophenylthiophene-2-ester)hexafluorophenoxy) axially substituted silicon phthalocyanine (CBT-SiPc). We hoped these new thienyl/benzothienyl/dibenzothienyl fluorinated substituents could not only reduce the aggregation behavior of silicon phthalocyanines but also modulate their photophysical properties as well as endow specific targeting ability to cellular organelles. The structures of these novel silicon(IV) phthalocyanines were characterized and their photophysicochemical properties were compared.
Results and discussion
Synthesis and spectroscopic characterization
The synthesis route for the three new thiophenic silicon(IV) phthalocyanines was depicted in Scheme 1. The precursor M1 was obtained via nucleophilic substitution between 1,3-dibromopropane and 2-carboxyarylthiophene. M1 further reacted with 3,5-dihydroxybenzyl alcohol in the presence of K2CO3 to get M2. BBT-OH was synthesized via the Suzuki coupling reaction between 2-bromodibenzoarylthiophene and 4-hydroxyphenylboronic acid using tetrakis (triphenylphosphine)palladium(0) (Pd(PPh3)4) as a catalyst. CBT-OH was obtained via an esterification reaction between 3-chloro-benzoarylthiophene-2-carboxylic acid and hexafluorobisphenol-A using N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) and 4-dimethylaminopyridine (DMAP) as a condensing agent and condensation activator, respectively. M2, BBT-OH, or CBT-OH reacted with silicon phthalocyanine dichloride (SiPcCl2) via nucleophilic substitution to obtain three novel thiophenic silicon(IV) phthalocyanines (M2SiPc, BBT-SiPc, and CBT-SiPc), respectively.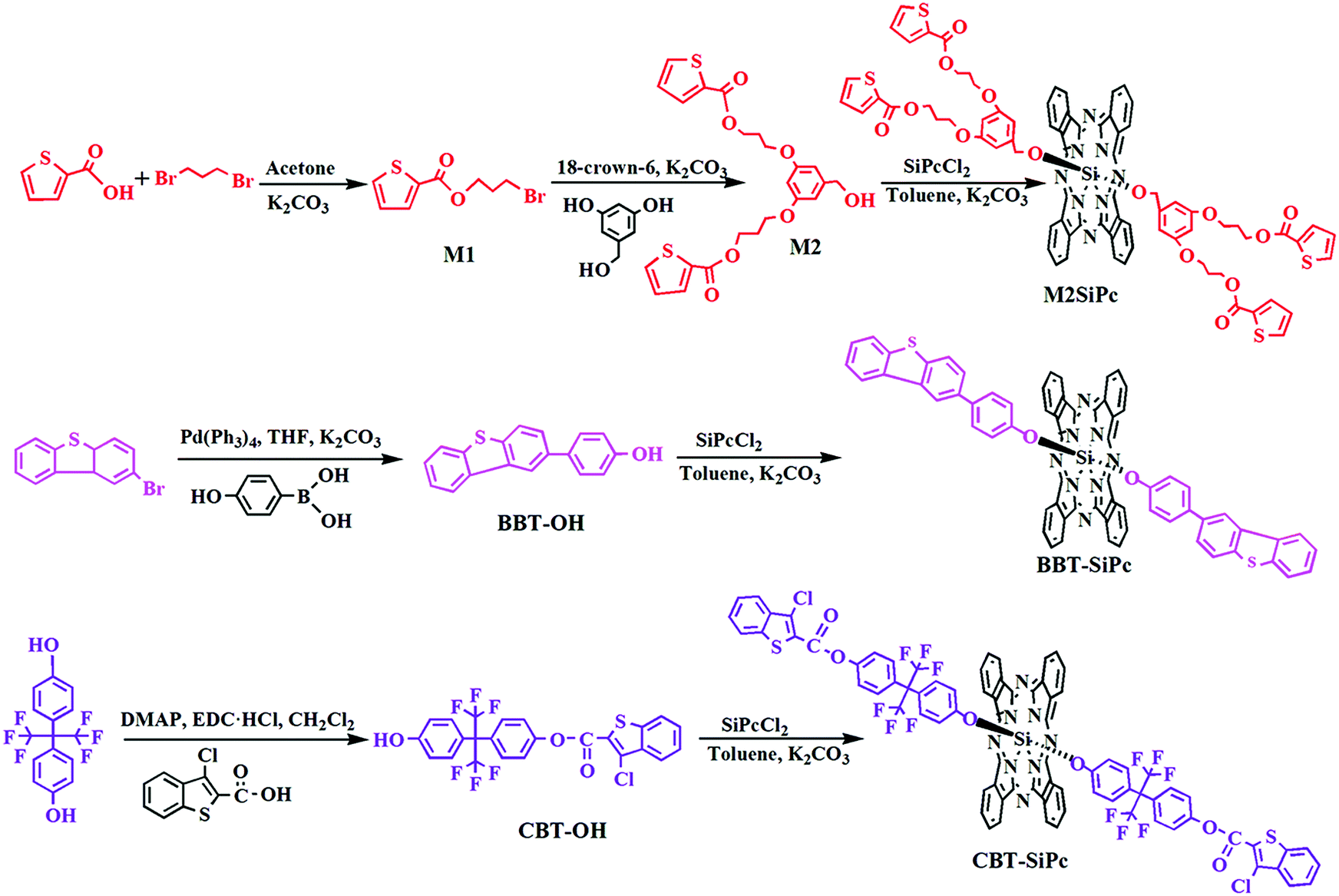 | ||
| Scheme 1 The scheme for the synthesis of the three thiophenic silicon(IV) phthalocyanines. | ||
CBT-SiPc and M2SiPc could be dissolved in some common solvents such as dichloromethane (CH2Cl2), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), DMF, ethyl acetate, and acetone. The structures of the three novel thiophenic silicon(IV) phthalocyanines were characterized via IR, 1H NMR, elemental analysis, ESI-MS, and MALDL-TOF mass spectroscopic methods (Fig. S1–S21, ESI†).
Singly charged molecular ion peaks were observed for M2SiPc, BBT-SiPc, and CBT-SiPc (Fig. S9, S15, and S21, ESI†), respectively. The above characterization results were consistent with the proposed structures.
The IR spectra of M2SiPc, BBT-SiPc, and CBT-SiPc were shown in Fig. S7, S13, and S19 (ESI†). The strong vibration absorptions for Si–O bonds were observed at 1080 cm−1. The vibration absorptions at 1416 and 1581 cm−1 were assigned to the submethyl group of M2SiPc. Besides, the stretching vibrations at 872 and 738 cm−1 belonged to the C–Cl bonds of CBT-SiPc.
The purity of M2SiPc, BBT-SiPc, and CBT-SiPc was confirmed by 1H NMR spectra (Fig. S8, S14, and S20, ESI†). Taking the 1H NMR spectrum of M2SiPc as an example, the resonances at 9.54 and 8.42 ppm were assigned to the proton signals of the phthalocyanine ring with 16 protons. The resonances at 7.92–7.79, 7.77–7.72, and 7.17 ppm belonged to the protons of thiophene substitutions with 12 protons. Two sets of resonances at 6.55–6.28 and 5.39–5.18 ppm were assigned to the protons of the benzoxyl group with 6 protons. The resonances at 4.35–4.32 ppm were assigned to the submethyl protons of –Si–O–CH2– with 4 protons. Three sets of resonances at 4.09, 4.01–3.92, and 2.07–1.95 ppm were attributed to the exterior submethyl protons with 24 protons. The proton resonances of BBT-SiPc and CBT-SiPc in the 1H NMR spectra were similar to that of M2SiPc.
Fig. 1(a) showed UV-Vis absorption spectra of M2SiPc, BBT-SiPc, and CBT-SiPc in DMF. They exhibited typical spectra for phthalocyanines with B bands at 353–356 nm and Q bands at 672–680 nm. In addition, the bands between 285 nm and 293 nm were assigned to the absorption of the thiophenic substituents. A typical intense non-aggregated Q-band was observed for M2SiPc, BBT-SiPc, and CBT-SiPc in different concentrations (Fig. S22–S24, ESI†), suggesting that molecular aggregation behavior was not obvious for these phthalocyanines in DMF (Fig. S22–S24, ESI†). It could be explained that the steric hindrance effect of thiophenic substituents at the axial positions lessened the aggregation behavior of phthalocyanines in DMF to some extent. The properties of thiophenic substituents influenced the intensity and position of the Q-bands of phthalocyanines. The Q bands were found to be at 677 nm for M2SiPc, 672 nm for BBT-SiPc, and 680 nm for CBT-SiPc. Compared with the Q band of SiPcCl2 (670 nm), the Q bands of M2SiPc, BBT-SiPc, and CBT-SiPc red-shifted by about 7 nm, 2 nm, and 10 nm, respectively. The thiophenic substituents were connected to the central silicon atom of phthalocyanine via the ether bond to form a larger conjugation system, leading to a decrease in the electronic density of the phthalocyanine ring, thereby lowering the energy gap between the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) and leading to the red-shift of Q bands.15 Because the fluorinated bisphenol-A conjugated with benzothienyl groups in CBT-SiPc possessed the largest conjugation system, it exhibited the largest red-shift for the Q band. However, the rotation of the carbon–carbon single bond between the dibenzothiazyl group and phenyl group in BBT-SiPc lowered the conjugation system, and it exhibited the smallest red-shift.16
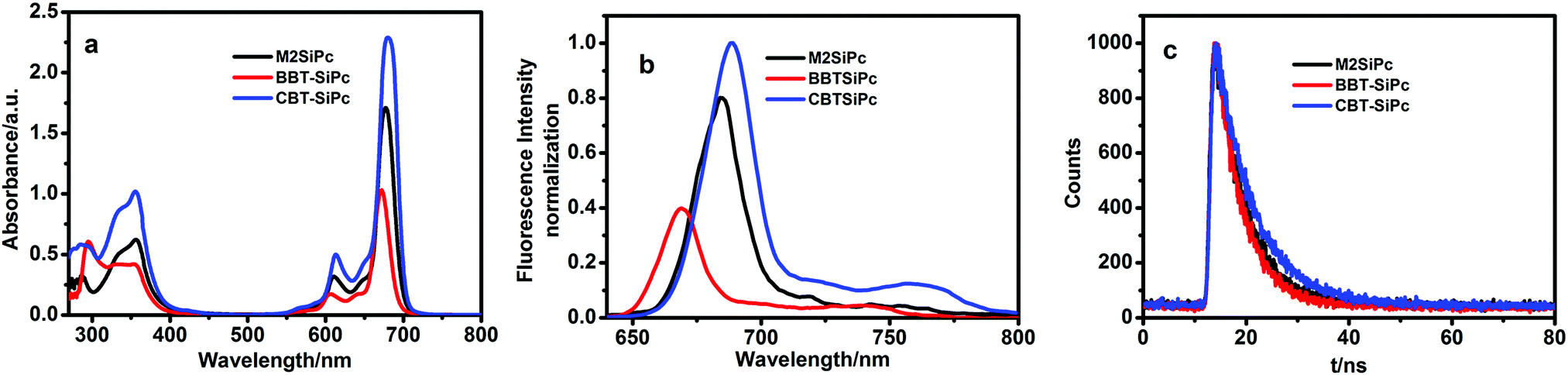 | ||
| Fig. 1 UV/Vis (a), fluorescence spectra (b), and fluorescence decay curves (c) of M2SiPc, BBT-SiPc, and CBT-SiPc in DMF (a: CM2SiPc = CBBT-SiPc = CCBT-SiPc = 1.0 × 10−5 mol L−1; b: CM2SiPc = CBBT-SiPc = CCBT-SiPc = 1.0 × 10−6 mol L−1, λex = 615 nm; c: CM2SiPc = CBBT-SiPc = CCBT-SiPc = 1.0 × 10−5 mol L−1, λex = 405 nm). | ||
The three thiophenic silicon(IV) phthalocyanines also had different fluorescence spectra, fluorescence lifetimes as well as quantum yields of fluorescence. The fluorescence emission spectra for M2SiPc, BBT-SiPc, and CBT-SiPc were observed at 684 nm, 669 nm, and 690 nm, respectively (Fig. 1(b)). Of the three thiophenic silicon(IV) phthalocyanines, the fluorescence intensity of CBT-SiPc was the strongest. The biggest steric hindrance effect of fluorinated bisphenol-A benzothienyl groups as well as the rigid structure of CBT-SiPc reduced the aggregation behavior, leading to the highest fluorescence intensity. The rotation of the carbon–carbon single bond between the dibenzothiazyl group and phenyl group lowered the rigid symmetry of BBT-SiPc, leading to the lowest fluorescence intensity.
The fluorescence quantum yields (ΦF) of M2SiPc, BBT-SiPc, and CBT-SiPc were listed in Table 1. The ΦF values of M2SiPc, BBT-SiPc, and CBT-SiPc were found to be 0.11, 0.03, and 0.12, respectively. The fluorescence lifetimes (τs) were found to be 5.77 ns for M2SiPc, 5.20 ns for BBT-SiPc, and 7.26 ns for CBT-SiPc (Table 1 and Fig. 1(c)).
Singlet oxygen quantum yields
1,3-Diphenylisobenzofuran (DPBF) was used as a scavenger to determine the quantum yields of singlet oxygen. The decrease in absorption at 414 nm for DPBF was due to the singlet oxygen generated by BBT-SiPc. No singlet oxygen generation in dark was observed for M2SiPc, BBT-SiPc, and CBT-SiPc (Fig. S28–S30, ESI†). However, upon irradiation, absorption at 414 nm for DPBF decreased, while the Q band intensities of BBT-SiPc did not change, indicating the generation of singlet oxygen by BBT-SiPc and no-degradation of BBT-SiPc (Fig. 2). The absorption spectra of the DPBF solution containing M2SiPc and CBT-SiPc upon irradiation were shown in Fig. S31 and S32, (ESI†), respectively. The quantum yields of singlet oxygen of M2SiPc, BBT-SiPc, and CBT-SiPc(Table 1) were 0.27, 0.23, and 0.29, respectively.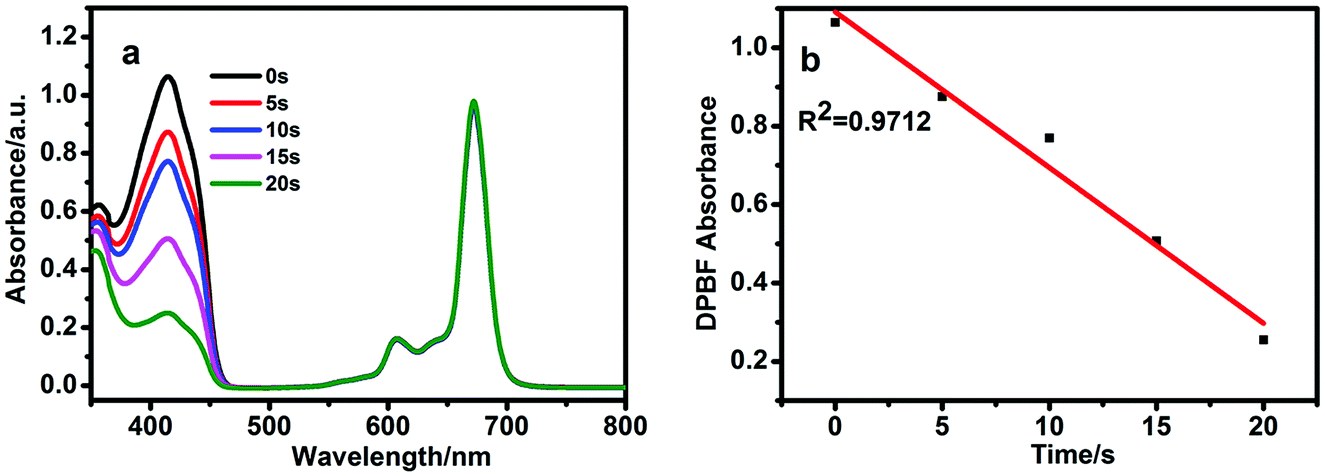 | ||
| Fig. 2 The spectra changes during the determination of the singlet oxygen quantum yield of BBT-SiPc in DMF using DPBF as a singlet oxygen quencher upon irradiation (670 nm, 10 mW cm−2) at a concentration of 1.0 × 10−5 mol L−1 (a); the plot of absorbance change of DPBF vs. time (b). | ||
In vitro photodynamic therapy efficacy of CBT-SiPc
Because CBT-SiPc possessed the highest fluorescence intensity, singlet oxygen quantum yield, and longest lifetime among the three thiophenic silicon phthalocyanines, it was selected as a photosensitizer to evaluate its PDT efficacy. MCF-7 breast cancer cells were selected as model cell lines. After CBT-SiPc incubation with MCF-7 breast cancer cells for 6 h, CBT-SiPc could be uptaken by cells. As shown in Fig. 3, red two-photonic fluorescence signals of CBT-SiPc could be visualized in the cytoplasm by confocal fluorescence microscopy and exhibited dotted fluorescence signals.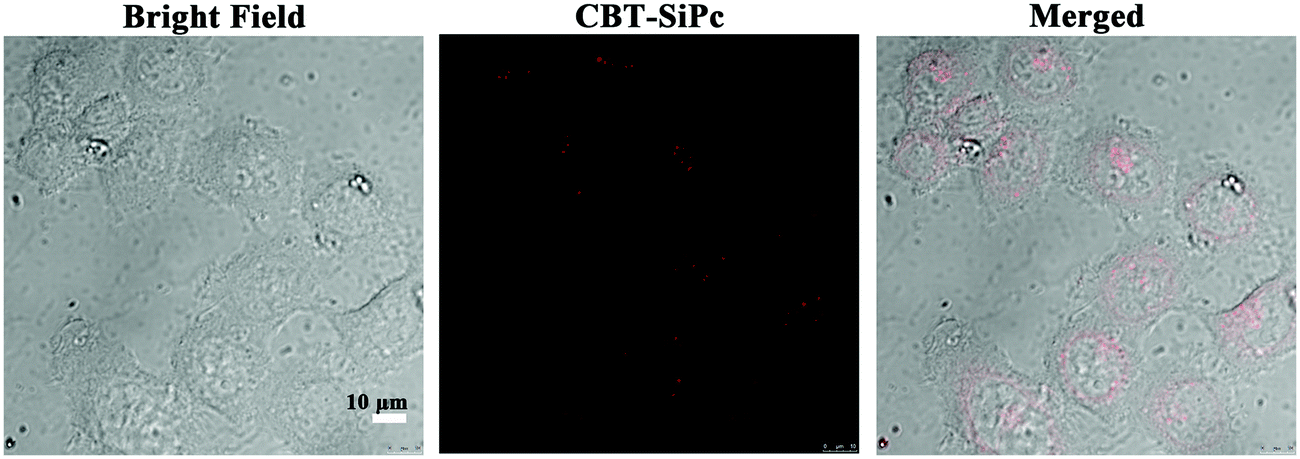 | ||
| Fig. 3 Confocal fluorescence microscopic images of MCF-7 cells before and after incubation with CBT-SiPc (two-photon laser excited at 860 nm, and monitored at 640–750 nm). | ||
The cytotoxicity and phototoxicity of CBT-SiPc against MCF-7 cells were evaluated via cell counting kit-8 (CCK-8) assay (Fig. 4). The results showed that CBT-SiPc exhibited non-cytotoxicity when the concentration of CBT-SiPc was up to 4 μM in the absence of light. However, upon irradiation with 670 nm laser (100 mW cm−2) for 10 min, CBT-SiPc showed phototoxicity.
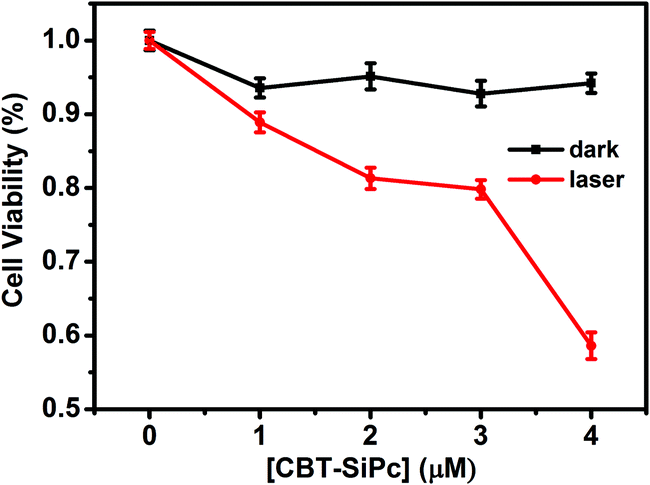 | ||
| Fig. 4 Concentration-dependent cytotoxicity of CBT-SiPc against MCF-7 breast cancer cells in the absence and presence of light (670 nm laser, 100 mW cm−2). | ||
Conclusions
The synthesis, characterization, and photophysicochemical properties of three novel thiophenic silicon(IV) phthalocyanines were described. They mainly existed as monomers in DMF. CBT-SiPc possessed the highest fluorescence intensity, longest lifetime, and singlet oxygen quantum yields, followed by M2SiPc, and the weakest one was BBT-SiPc. This may be related to the high steric hindrance of fluorinated bisphenol-A benzothienyl groups on the axial positions of silicon phthalocyanine that not only reduced the aggregation behavior but also lowered the HOMO–LUMO energy level gap. CBT-SiPc could be transported into MCF-7 breast cancer cells and mediated the in vitro photodynamic therapy efficacy against MCF-7 breast cancer cells.Experimental
Materials and methods
1,3-Dibromopropane, hexafluorobisphenol A, 2-carboxyarylthiophene, 18-crown-6, 3,5-dihydroxybenzyl alcohol, 4-hydroxyphenylboronic acid, Pd(PPh3)4, DPBF, 3-chloro-benzoarylthiophene-2-carboxylic acid, DMAP, and EDC·HCl were obtained from Energy Chemical Company (Shanghai, China). SiPcCl2 was synthesized as in the reported literature.17A PerkinElmer IR spectrometer was used for IR data collection. 1H NMR spectra were recorded on a Varian UNITY-400 NMR spectrometer. Mass spectra were recorded on a Bruker mass spectrometer. UV-Vis and fluorescence spectra were recorded on a Cary 50 UV-Vis spectrophotometer and a FL900/FS920 steady-state fluorescence spectrometer, respectively. Two-photonic fluorescence microscopy images were observed on a confocal laser scanning microscope (CLSM, Leica TCS SP8 Ltd).
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1514 (CC), 1426 (–CH2), 748 (C–S), 574 (C–Br). 1H NMR (DMSO-d6, 400 MHz, δ/ppm): 7.81 (m, 1H, ArH), 7.57 (m, 1H, ArH), 7.11 (m, 1H, ArH), 4.46–4.43 (t, 2H, COOCH2), 3.55–3.52 (t, 2H, CH2Br), 2.32–2.29 (m, 2H, CH2) (Fig. S2, ESI†). HR-MS (ESI) (C8H9BrSO2) [M + Na + 2]+: calcd: 274.1120; found: 274.2607.
C), 1010, 1070, 1172 and 1219 (CO), 846, 799, 745 and 702 (CH). 1H NMR (DMSO-d6, 400 MHz, δ/ppm): 8.32 (d, J = 8 Hz, 1H, ArH), 8.28–8.23 (m, 1H, ArH), 7.92–7.88 (m, 2H, ArH), 7.96–7.61 (m, 3H, PhH), 7.51–7.49 (m, 2H, ArH), 7.00–6.98 (m, 2H, PhH), 4.92 (s, 1H, OH) (Fig. S11, ESI†). HR-MS (ESI) (C18H12SO) [M + Na]+: calcd: 299.3360; found: 300.0610.
O), 1079 (SiO), 799 (PhH), 715 (PcH). 1H NMR (DMSO-d6, 400 MHz, δ/ppm): 9.59 (s, 8H, PcH), 8.59 (s, 8H, PcH), 8.51–8.49 (m, 2H, ArH), 8.06–8.02 (m, 2H, ArH), 7.77–7.74 (q, 4H, ArH), 7.68 (d, J = 8 Hz, 8H, PhH), 7.54–7.52 (m, 2H, ArH), 6.91 (d, J = 8 Hz, 4H, PhH) (Fig. S14, ESI†). HR-MS (ESI) (C68H38S2O2N8Si) [M + H]+: calcd: 1092.3020; found: 1092.2960.
O), 1610 and 1499 (Ph), 1192 and 1059 (C–O), 958 and 926 (C–O–C), 802, 744, 698 and 586 (C–Cl). 1H NMR (CDCl3, 400 MHz, δ/ppm): 13.37–13.28 (m, 2H, ArH), 12.96–12.87 (m, 2H, ArH), 12.80–12.71 (m, 4H, PhH), 12.48 (d, J = 8 Hz, 2H, PhH), 12.18–12.16 (m, 2H, PhH), 5.21 (s, 1H, OH) (Fig. S17, ESI†). HR-MS (ESI) (C24H13ClSO3F6) [M + K]+: calcd: 570.9720; found: 571.5790.
O), 1504 (Ph), 1272 and 1176 (C–O), 1081 (SiO), 1045 and 928 (C–O–C), 872 and 738 (C–Cl). 1H NMR (CDCl3, 400 MHz, δ/ppm): 9.67–9.63 (m, 8H, PcH), 8.42–8.38 (m, 8H, PcH), 8.12–8.10 (m, 2H, ArH), 7.96–7.89 (m, 2H, ArH), 7.65–7.60 (m, 4H, PhH), 7.35–7.18 (m, 4H, PhH), 6.81–6.77 (m, 4H, PhH), 6.58–6.52 (m, 4H, PhH), 5.63–5.61 (m, 4H, ArH) (Fig. S20, ESI†). ESI-MS: m/z 1599 [M]+. HR-MS (ESI) (C80H40O6F12N8S2Cl2Si) [M + H]+: calcd: 1599.3300; found: 1599.8870.
O), 1514 (CC), 1426 (–CH2), 748 (C–S), 574 (C–Br). 1H NMR (DMSO-d6, 400 MHz, δ/ppm): 7.81 (m, 1H, ArH), 7.57 (m, 1H, ArH), 7.11 (m, 1H, ArH), 4.46–4.43 (t, 2H, COOCH2), 3.55–3.52 (t, 2H, CH2Br), 2.32–2.29 (m, 2H, CH2) (Fig. S2, ESI†). HR-MS (ESI) (C8H9BrSO2) [M + Na + 2]+: calcd: 274.1120; found: 274.2607.
C), 1010, 1070, 1172 and 1219 (CO), 846, 799, 745 and 702 (CH). 1H NMR (DMSO-d6, 400 MHz, δ/ppm): 8.32 (d, J = 8 Hz, 1H, ArH), 8.28–8.23 (m, 1H, ArH), 7.92–7.88 (m, 2H, ArH), 7.96–7.61 (m, 3H, PhH), 7.51–7.49 (m, 2H, ArH), 7.00–6.98 (m, 2H, PhH), 4.92 (s, 1H, OH) (Fig. S11, ESI†). HR-MS (ESI) (C18H12SO) [M + Na]+: calcd: 299.3360; found: 300.0610.
O), 1079 (SiO), 799 (PhH), 715 (PcH). 1H NMR (DMSO-d6, 400 MHz, δ/ppm): 9.59 (s, 8H, PcH), 8.59 (s, 8H, PcH), 8.51–8.49 (m, 2H, ArH), 8.06–8.02 (m, 2H, ArH), 7.77–7.74 (q, 4H, ArH), 7.68 (d, J = 8 Hz, 8H, PhH), 7.54–7.52 (m, 2H, ArH), 6.91 (d, J = 8 Hz, 4H, PhH) (Fig. S14, ESI†). HR-MS (ESI) (C68H38S2O2N8Si) [M + H]+: calcd: 1092.3020; found: 1092.2960.
O), 1610 and 1499 (Ph), 1192 and 1059 (C–O), 958 and 926 (C–O–C), 802, 744, 698 and 586 (C–Cl). 1H NMR (CDCl3, 400 MHz, δ/ppm): 13.37–13.28 (m, 2H, ArH), 12.96–12.87 (m, 2H, ArH), 12.80–12.71 (m, 4H, PhH), 12.48 (d, J = 8 Hz, 2H, PhH), 12.18–12.16 (m, 2H, PhH), 5.21 (s, 1H, OH) (Fig. S17, ESI†). HR-MS (ESI) (C24H13ClSO3F6) [M + K]+: calcd: 570.9720; found: 571.5790.
O), 1504 (Ph), 1272 and 1176 (C–O), 1081 (SiO), 1045 and 928 (C–O–C), 872 and 738 (C–Cl). 1H NMR (CDCl3, 400 MHz, δ/ppm): 9.67–9.63 (m, 8H, PcH), 8.42–8.38 (m, 8H, PcH), 8.12–8.10 (m, 2H, ArH), 7.96–7.89 (m, 2H, ArH), 7.65–7.60 (m, 4H, PhH), 7.35–7.18 (m, 4H, PhH), 6.81–6.77 (m, 4H, PhH), 6.58–6.52 (m, 4H, PhH), 5.63–5.61 (m, 4H, ArH) (Fig. S20, ESI†). ESI-MS: m/z 1599 [M]+. HR-MS (ESI) (C80H40O6F12N8S2Cl2Si) [M + H]+: calcd: 1599.3300; found: 1599.8870.
Photophysical properties
Quantum yields of fluorescence (ΦF) and singlet oxygen (ΦΔ) of the three thiophenic silicon(IV) phthalocyanines were investigated by our group previously reported.18,19Intracellular uptake of CBT-SiPc in MCF-7 cells
MCF-7 cells were cultured in DMEM at 37 °C with 5% CO2 for 20 h. Then CBT-SiPc (10−3 mol L−1, 6 μL) was added to the cells and the cells were further cultured for 6 h. After washing with the PBS buffer three times, the cells were imaged by a confocal fluorescence microscope.In vitro photodynamic therapy efficacy of CBT-SiPc
The cytotoxicity and phototoxicity of CBT-SiPc against MCF-7 cells were evaluated by the cell counting kit-8 (CCK-8) assay method. MCF-7 cells with a density of 8 × 103 cells per well were cultured with 100 μL of culture medium. One day later, the cells were treated with CBT-SiPc (0 μM, 1 μM, 2 μM, 3 μM, and 4 μM), respectively, and cultured for 6 h. Five replicates were made for each measurement. Then, the cells were irradiated with a 671 nm laser (100 mW cm−2) for 10 min. Finally, 10 μL of the CCK-8 reagent was added into each well and the cells were cultured for 2 h. The OD value at 450 nm was measured by a multifunction microplate reader.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Joint Funds of Fujian Provincial Health and Education Research (No. 2019-WJ-2), Natural Science Foundation of Fujian Province (No. 2019Y0007, 2020J11202), Fujian Province Educational Project A (No. JAT190077), Science and Technology Fund project of Fujian province (No. 2018Y9081), and Special Funds of the Central Government Guiding Local Science and Technology Development (No. 2020L3008).References
- C. Huang, C. Chen and C. R. Shyr, Cancer Cell Int., 2018, 18, 9 CrossRef.
- X. Li, B. Zheng, X. Peng, S. Li, J. Ying, Y. Zhao and J. Yoon, Coord. Chem. Rev., 2019, 379, 147–160 CrossRef CAS.
- A. R. M. Soares, M. G. P. M. S. Neves, A. C. Tomé, M. C. I. L. Cruz, A. Zamarron, E. Carrasco, S. Gonzalez, J. A. S. Cavaleiro, T. Torres, D. M. Guldi and A. Juarranz, Chem. Res. Toxicol., 2012, 25, 940–951 Search PubMed.
- H. Ali and J. E. van Lier, Chem. Rev., 1999, 99, 2379–2450 CrossRef CAS.
- A. C. Tedesco, J. C. Rotta and C. N. Lunardi, Curr. Org. Chem., 2003, 7, 187–196 CrossRef CAS.
- X. Zhao, Y. Huang, G. Yuan, K. Zuo, Y. Huang, J. Chen, J. Li and J. Xue, Chem. Commun., 2019, 55(6), 866–869 RSC.
- S. Ke, Y. Wei, Z. Yang, K. Wang, Y. Liang and L. Shi, Med. Chem. Lett., 2013, 23, 5131–5134 CrossRef CAS.
- F. Baldassarre, F. Foglietta, V. Vergaro, N. Barbero, A. L. Capodilupo, L. Serpe, S. Visentin, A. Tepore and G. Ciccarella, J. Photochem. Photobiol., B, 2016, 158, 16–22 CrossRef CAS.
- S. Rangasamy, H. Ju, S. Um, D. C. Oh and J. M. Song, J. Med. Chem., 2015, 58, 6864–6874 CrossRef CAS PubMed.
- M. A. Revuelta-Maza, P. Gonzalez-Jimenez, C. Hally, M. Agut, S. Nonell, G. Torre and T. Torres, Eur. J. Med. Chem., 2019, 187, 111957 CrossRef PubMed.
- B. Pucelik, I. Gürol, V. Ahsen, F. Dumoulin and J. M. Dąbrowski, Eur. J. Med. Chem., 2016, 124, 284–298 CrossRef CAS.
- İ. Gürol, M. Durmus and V. Ahsen, Eur. J. Inorg. Chem., 2010, 1220–1230 CrossRef.
- S. Mori, H. Yoshiyama, E. Tokunaga, N. Iida, M. Hayashi, T. Obata, M. Tanaka and N. Shibata, J. Fluorine Chem., 2015, 174, 137–141 CrossRef CAS.
- S. Mori and N. Shibata, J. Org. Chem., 2017, 13, 2273–2296 CAS.
- D. Ma, S. Pan, T. Zhang, B. Huang, S. Xie, H. Yang and Y. Peng, Dyes Pigm., 2016, 127, 78–86 CrossRef CAS.
- M. Gadogbe, Y. Zhou, S. Zou and D. Zhang, J. Phys. Chem. B, 2016, 1–18 Search PubMed.
- M. K. Lowery, A. J. Starshak, J. N. Esposito, P. C. Krueger and M. E. Kenxey, Inorg. Chem., 1965, 4, 128 CrossRef CAS.
- X. Chen, S. Wu, D. Ma, J. Chen, Q. Guo, X. Han, K. Chen, H. Yang, Y. Huang and Y. Peng, Chem. Commun., 2018, 54, 13279–13282 RSC.
- D. Ma, X. Chen, Y. Wang, Q. Guo, Q. Ye, R. Guo, S. Xiao, Q. Ye, Y. Huang and Y. Peng, J. Lumin., 2019, 207, 597–601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04355a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |