
Ligand effects on structural, protophilic and reductive features of stannylated dinuclear iron dithiolato complexes†
Hassan
Abul-Futouh
 *a,
Laith R.
Almazahreh
bc,
Sara J.
Abaalkhail
a,
Helmar
Görls
c,
Sven T.
Stripp
d and
Wolfgang
Weigand
*c
*a,
Laith R.
Almazahreh
bc,
Sara J.
Abaalkhail
a,
Helmar
Görls
c,
Sven T.
Stripp
d and
Wolfgang
Weigand
*c
aDepartment of Pharmacy, Al-Zaytoonah University of Jordan, P.O. Box 130, Amman 11733, Jordan. E-mail: h.abulfutouh@zuj.edu.jo
bERCOSPLAN Ingenieurbüro Anlagentechnik GmbH, Arnstädter Straße 28, 99096 Erfurt, Germany
cInstitut für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität Jena, Humboldt Str. 8, 07743 Jena, Germany. E-mail: wolfgang.weigand@uni-jena.de
dBioinorganic Spectroscopy, Department of Physics, Freie Universität Berlin, Arnimallee 14, 1495 Berlin, Germany
First published on 6th November 2020
Abstract
The synthesis and characterization of Fe2(CO)5(L){μ-(SCH2)2SnMe2} (L = PPh3 (2) and P(OMe)3 (3)) derived from the parent hexacarbonyl complex Fe2(CO)6{μ-(SCH2)2}SnMe2 (1) are reported. Whereas 1 exhibits a unique planar structure, X-ray crystallography showed that the apical orientation of L in complexes 2 and 3 results in a chair/boat conformation of the Fe2S2C2Sn fused six-membered rings, which is typical for diiron dithiolato complexes. In solution, NMR and FTIR spectroscopic techniques provide evidence for a dynamic process of apical–basal site exchange of the ligand L in 2 and 3. Protonation experiments on 2 and 3 in MeCN using CF3CO2H, HCl or HBF4·Et2O suggest enhanced protophilicity of the Fe–Fe bond due to the presence of the electron donor ligands L as well as the stannylation effect. While the carbonyl ligands in 2 stretch at lower wavenumbers ν(CO) than those in 3, the cyclic voltammetric reduction of 2 unpredictably occurs at less negative potential than that of 3. In contrast to 1, the presence of PPh3 and P(OMe)3 in 2 and 3, respectively, allows protonation prior to reduction as shown by FTIR spectroscopy and cyclic voltammetry.
Introduction
During the past two decades, considerable attention has been paid to search for viable alternatives for clean and renewable energy sources. Therefore, hydrogen has been gaining importance due to its affordability and environmental friendliness.1–4 The high ability of [FeFe]-hydrogenases in catalyzing the production of hydrogen through reduction of protons makes their active site an interesting target for biomimetic modeling.5–10 The active site of these enzymes, the so-called H-cluster (Fig. 1A), features a binuclear iron sub-site that is coordinated by an azadithiolato bridging moiety as well as carbon monoxide and cyanide ligands.11–14 In addition, a canonical iron–sulfur cluster ([4Fe4S]) is linked to one iron atom through a cysteine ligand.11–13 The reaction mechanism of H2 oxidation and H+ reduction at the H-cluster is under discussion.14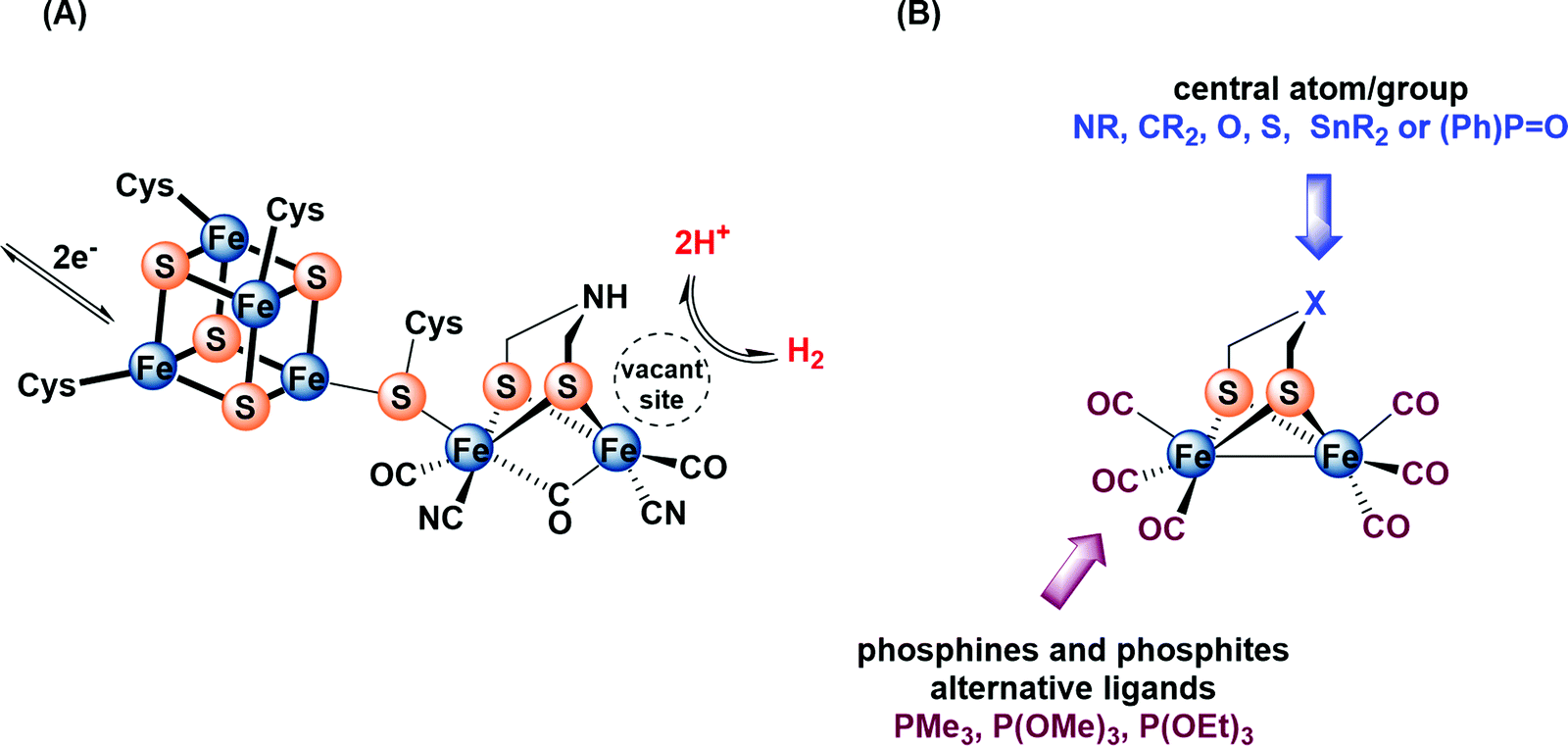 | ||
| Fig. 1 (A) Structure of the H-cluster. (B) Synthetic model of the active site of [FeFe]-hydrogenase. | ||
For several years, extensive efforts have been devoted to designing structural and functional models so as to mimic the protonation and redox properties of the H-cluster.15–24 These models include a variety of dithiolato ligands, μ-(SCH2)2X, in which the central atom/group X could be NR, CR2, O, S, SnR2 or (Ph)P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Fig. 1B).25–39 Moreover, several research groups have reported approaches for introducing strong σ-donor ligands, such as phosphines and phosphites, at the diiron core to enhance the protophilicity of the model complexes (Fig. 1B).40–58 In fact, multisubstituted complexes can be fully protonated affording bridging hydride (μ-H) products using strong or moderately strong acids.55,56 In comparison, monosubstituted complexes have been little investigated in terms of protonation features. To the best of our knowledge, only one recent article has been published, which describes the protonation features of monosubstituted complexes Fe2(CO)5(EPh3){μ-(SCH2)2} (E = P, As, Sb) containing only the Fe–Fe bond as a plausible basic site.58d This study has shown that the addition of ca. 5 equiv. HBF4·Et2O to a solution of Fe2(CO)5(EPh3){μ-(SCH2)2} (E = P, As, Sb) in CD2Cl2 leads to the formation of small amount (ca. 5%) of bridging hydride [Fe2(μ-H)(CO)5EPh3{μ-(SCH2)2}][BF4] (E = P, As, Sb) complexes.
O (Fig. 1B).25–39 Moreover, several research groups have reported approaches for introducing strong σ-donor ligands, such as phosphines and phosphites, at the diiron core to enhance the protophilicity of the model complexes (Fig. 1B).40–58 In fact, multisubstituted complexes can be fully protonated affording bridging hydride (μ-H) products using strong or moderately strong acids.55,56 In comparison, monosubstituted complexes have been little investigated in terms of protonation features. To the best of our knowledge, only one recent article has been published, which describes the protonation features of monosubstituted complexes Fe2(CO)5(EPh3){μ-(SCH2)2} (E = P, As, Sb) containing only the Fe–Fe bond as a plausible basic site.58d This study has shown that the addition of ca. 5 equiv. HBF4·Et2O to a solution of Fe2(CO)5(EPh3){μ-(SCH2)2} (E = P, As, Sb) in CD2Cl2 leads to the formation of small amount (ca. 5%) of bridging hydride [Fe2(μ-H)(CO)5EPh3{μ-(SCH2)2}][BF4] (E = P, As, Sb) complexes.
In our previous investigation, we have shown that the introduction of Sn atom into the dithiolato moiety of [FeFe]-hydrogenase model complexes, e.g. Fe2(CO)6{μ-(SCH2)2SnMe2} (1), increases the electron density of the μ-S atoms and the Fe–Fe bond owing to a geometry-dependent σ(Sn–C)↔3p(μ-S) filled–filled interaction.38 Moreover, we have revealed that further enhancement of the protophilicity of the Fe–Fe bond is established by replacing the μ-S in 1 by μ-Se, and hence the use of moderately strong CF3CO2H was sufficient to protonate the Fe–Fe bond.39 In the present study, we investigate the influence of substituting one CO in 1 by stronger electron donating ligands, PPh3 and P(OMe)3, toward the protonation properties of the resulting complexes, namely Fe2(CO)5(L){μ-(SCH2)2SnMe2} (L = PPh3 (2) and P(OMe)3 (3). Herein, the synthesis and characterization as well as the molecular structures of complexes 2 and 3 are described. Furthermore, the protonation properties of the complexes have been investigated by means of IR, 1H and 31P{1H} NMR spectroscopic techniques. The cyclic voltammetry of these complexes is employed to gain insights into reduction features in the absence and presence of acid.
Results and discussion
Synthesis and characterization
A solution of complex 1 in MeCN was treated with 1 equivalent of trimethylamine N-oxide (Me3NO·2H2O) at room temperature (r.t.) for 40 minutes to afford the in situ acetonitrile complex via oxidative abstraction of CO (decarbonylation) as shown in Scheme 1.42c Subsequent addition of PPh3 or P(OMe)3 affords the monosubstituted complexes Fe2(CO)5(L){μ-(SCH2)2SnMe2} (L = PPh3 (2) and P(OMe)3 (3)) in moderate yields after stirring for 20 hour at r.t. (Scheme 1). Complexes 2 and 3 have been characterized by spectroscopic methods (1H NMR, 13C{1H} NMR, 31P{1H} NMR and IR), mass spectrometry, elemental analysis, and X-ray crystallography.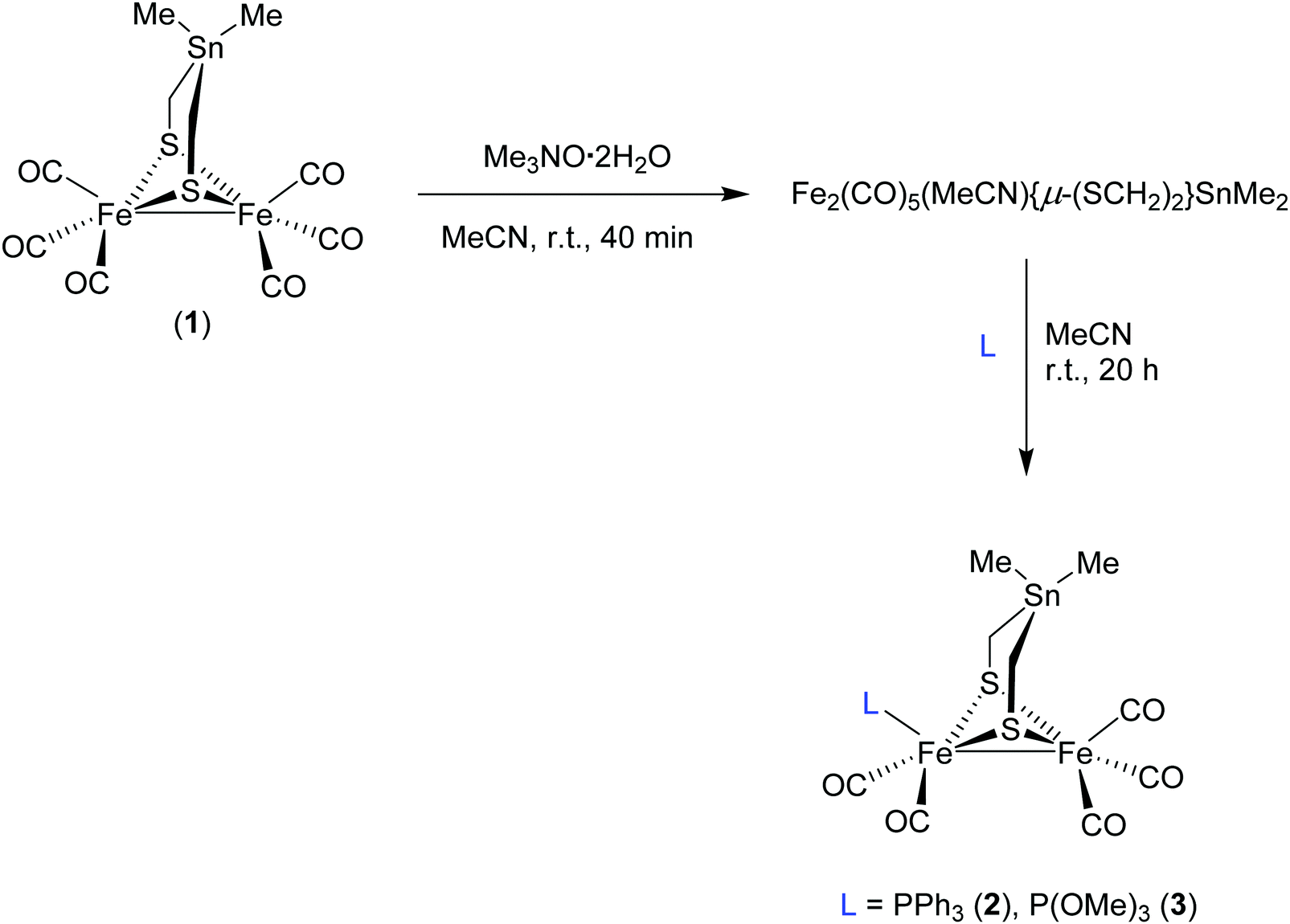 | ||
| Scheme 1 Reaction pathway toward monosubstituted complexes 2 and 3. | ||
The IR spectrum of 2 in MeCN displays four C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O absorption bands, ν(CO), at 2040, 1980, 1952 and 1918 cm−1 while those of 3 stretch at 2045, 1990, 1972 and 1935 cm−1. These frequencies are markedly shifted toward lower values relative to those of 1 (2070, 2032, 1997 and 1989 cm−1)38 by an average of 49 cm−1 and 36 cm−1 for 2 and 3, respectively. This difference in the shift of ν(CO) reflects the stronger electron donor ability of PPh3 in comparison to P(OMe)3. In comparison to 2, the analogues complex Fe2(CO)5(PPh3){μ-(SCH2)2CMe2} exhibits CO absorption bands at higher wavenumbers; ν(CO) = 2045, 1981, 1961 and 1927 cm−1.30 The disparity in the ν(CO) wavenumbers could be ascribed to an orbital interaction between the σ C–Sn bond and the S lone pair, which brings about an increased electron richness at the [2Fe2S] core.38 The 1H NMR spectrum of 2 exhibits two doublets (AB spin system) for the diastereotopic methylene protons centered at 1.74 (2J{1H–1H} = 12.0 Hz). In this 1H NMR spectrum, the protons of the two CH3 groups resonate as a singlet at 0.21 ppm with 119Sn satellites (2J{119Sn–1H} = 27.2 Hz). Additional signals are also detected in the range of 7.67–7.43 ppm for the phenyl protons. Moreover, the diastereotopic methylene protons of 3 resonate as two doublets (AB spin system) centered at 1.74 ppm (2J{1H–1H} = 16.0 Hz). The methyl protons of the P(OMe)3 ligand show a doublet at 3.76 ppm (3J{31P–1H} = 12.0 Hz). The 1H NMR spectrum of 3 shows two singlets at 0.23 and 0.16 ppm with Sn satellites (2J{119Sn–1H} = 26.8 Hz) assigned to the CH3 groups attributing to a slower apical–basal ligand exchange in 3 than that in 2. Indeed, the 31P{1H} NMR spectrum (at r.t.) of 2 displays a sharp singlet at 63.52 ppm for the PPh3 ligand while that of 3 shows a broad signal at 175.09 ppm for the P(OMe)3 ligand. This observation provides also an evidence for the apical–basal site exchange such that this process is faster for the bulkier PPh3.30 This broadness could be explained in terms of the fluxionality of the Fe(CO)2P(OMe)3 unit such that the P(OMe)3 substituent exchanges between the apical and basal positions. Upon cooling the sample to −50 °C this signal splits into singlets (182.7 and 171.0 ppm) indicating the presence of the basal and apical isomers (Fig. S7, ESI†). The 13C{1H} NMR spectra of both complexes display a singlet at 209.0 ppm for the carbonyl carbon atoms of the Fe(CO)3 unit whereas those carbons in the Fe(CO)2P(OMe)3 unit resonate as a doublet centered at 211.0 ppm. Furthermore, other signals are also detected as a doublet at −6.6 and −6.8 ppm for the CH3 group of 2 and 3, respectively, as well as a singlet at 2.5 and 2.2 ppm for the methylene groups in 2 and 3, respectively. The signals observed in the region of 128.5–135.5 ppm are attributed to the aromatic carbon atoms in 2 and a doublet centered at 52.2 ppm for the carbon atoms in P(OMe)3 substituent of 3.
O absorption bands, ν(CO), at 2040, 1980, 1952 and 1918 cm−1 while those of 3 stretch at 2045, 1990, 1972 and 1935 cm−1. These frequencies are markedly shifted toward lower values relative to those of 1 (2070, 2032, 1997 and 1989 cm−1)38 by an average of 49 cm−1 and 36 cm−1 for 2 and 3, respectively. This difference in the shift of ν(CO) reflects the stronger electron donor ability of PPh3 in comparison to P(OMe)3. In comparison to 2, the analogues complex Fe2(CO)5(PPh3){μ-(SCH2)2CMe2} exhibits CO absorption bands at higher wavenumbers; ν(CO) = 2045, 1981, 1961 and 1927 cm−1.30 The disparity in the ν(CO) wavenumbers could be ascribed to an orbital interaction between the σ C–Sn bond and the S lone pair, which brings about an increased electron richness at the [2Fe2S] core.38 The 1H NMR spectrum of 2 exhibits two doublets (AB spin system) for the diastereotopic methylene protons centered at 1.74 (2J{1H–1H} = 12.0 Hz). In this 1H NMR spectrum, the protons of the two CH3 groups resonate as a singlet at 0.21 ppm with 119Sn satellites (2J{119Sn–1H} = 27.2 Hz). Additional signals are also detected in the range of 7.67–7.43 ppm for the phenyl protons. Moreover, the diastereotopic methylene protons of 3 resonate as two doublets (AB spin system) centered at 1.74 ppm (2J{1H–1H} = 16.0 Hz). The methyl protons of the P(OMe)3 ligand show a doublet at 3.76 ppm (3J{31P–1H} = 12.0 Hz). The 1H NMR spectrum of 3 shows two singlets at 0.23 and 0.16 ppm with Sn satellites (2J{119Sn–1H} = 26.8 Hz) assigned to the CH3 groups attributing to a slower apical–basal ligand exchange in 3 than that in 2. Indeed, the 31P{1H} NMR spectrum (at r.t.) of 2 displays a sharp singlet at 63.52 ppm for the PPh3 ligand while that of 3 shows a broad signal at 175.09 ppm for the P(OMe)3 ligand. This observation provides also an evidence for the apical–basal site exchange such that this process is faster for the bulkier PPh3.30 This broadness could be explained in terms of the fluxionality of the Fe(CO)2P(OMe)3 unit such that the P(OMe)3 substituent exchanges between the apical and basal positions. Upon cooling the sample to −50 °C this signal splits into singlets (182.7 and 171.0 ppm) indicating the presence of the basal and apical isomers (Fig. S7, ESI†). The 13C{1H} NMR spectra of both complexes display a singlet at 209.0 ppm for the carbonyl carbon atoms of the Fe(CO)3 unit whereas those carbons in the Fe(CO)2P(OMe)3 unit resonate as a doublet centered at 211.0 ppm. Furthermore, other signals are also detected as a doublet at −6.6 and −6.8 ppm for the CH3 group of 2 and 3, respectively, as well as a singlet at 2.5 and 2.2 ppm for the methylene groups in 2 and 3, respectively. The signals observed in the region of 128.5–135.5 ppm are attributed to the aromatic carbon atoms in 2 and a doublet centered at 52.2 ppm for the carbon atoms in P(OMe)3 substituent of 3.
Molecular structures
The diffusion of pentane into a CH2Cl2 solution of 2 or 3 at −20 °C gave suitable single crystals for X-ray diffraction studies. Fig. 2 displays the molecular structures of 2 and 3 with ellipsoids drawn at the 50% probability level.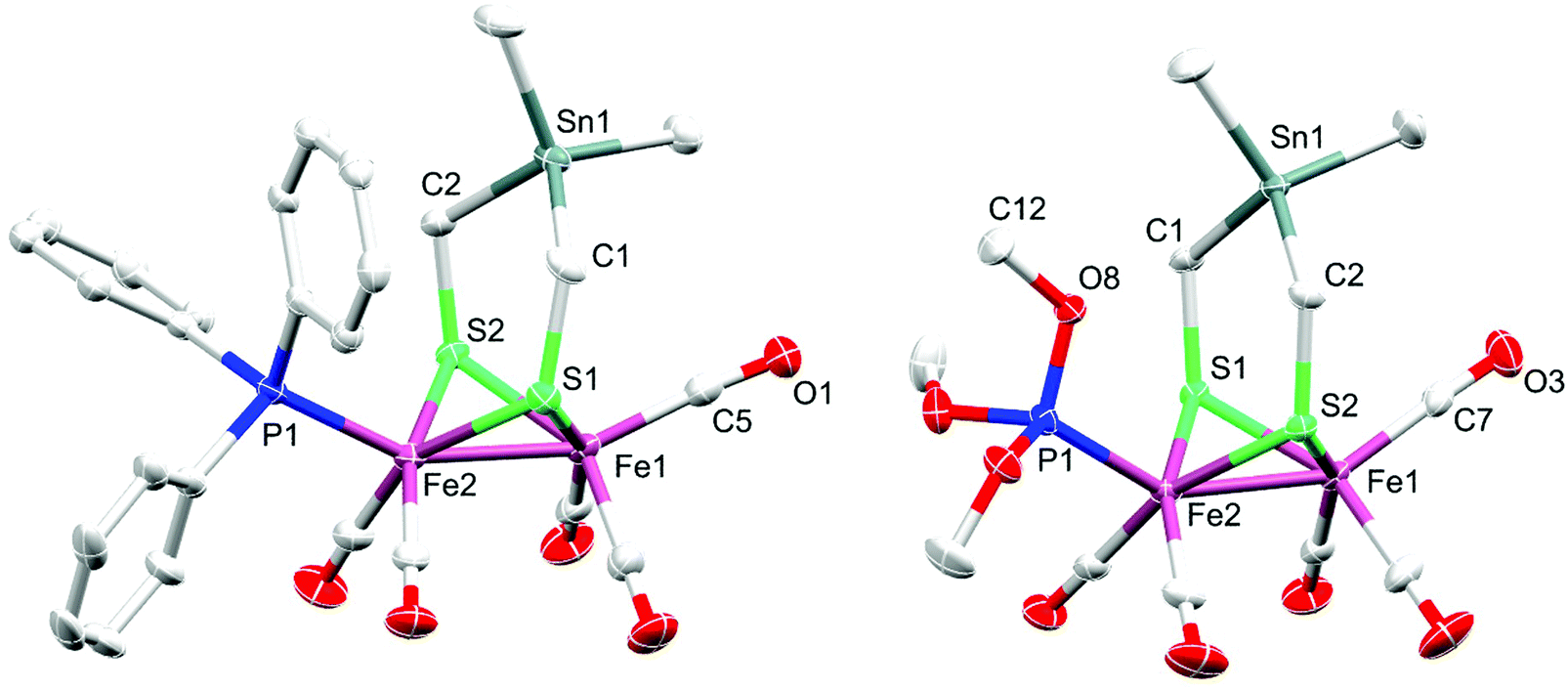 | ||
| Fig. 2 Molecular structures (50% probability) of 2 (left) and 3 (right). Hydrogen atoms are omitted for clarity. | ||
The molecular structure of both complexes reveals the typical butterfly conformation of [Fe2S2]-cluster core (Fig. 2). In each complex, the two Fe atoms are bridged by the dithiolato (SCH2)2SnMe2 ligand in which the bridgehead Sn atom is encircled by atoms in distorted tetrahedral fashion. The geometry of coordination around each iron center in both complexes can be best described as Fe(CO)3S2 (or Fe(CO)2PR3S2) square pyramid, with Fe slightly positioned outside the basal plane. The angle formed from the intersection between the C2Sn and S2C2 planes, so-called flap angle α, equals 146.1° or 160.3° in 2 or 3, respectively. These angles are smaller than that found in 1 (α = 173.6°), which exhibits an almost planar S2C2Sn moiety (Fig. S1, ESI†).38 The fact that the flap angle in 3 deviates from α(1) = 173.6° more than the deviation in the case of 2 is attributed to the higher steric bulkiness of PPh3 in comparison to that of P(OMe)3. The Fe–Fe bond length in complex 2 (2.5146(4) Å) is slightly shorter than that in 1 (2.5249(5) Å), which is in turn shorter than that in 3 (2.5322(4) Å). The Fe2–P1 bond length in 2 (2.2342(6) Å) is longer than that in 3 (2.1576(6) Å), owing to the higher π-acidity of P(OMe)3 compared to that of PPh3.59 Furthermore, the average Fe–CO bond lengths in 2 (1.78742(2) Å) and 3 (1.78898(2) Å) are slightly shorter than that in complex 1 (1.802(3) Å). These differences in Fe–CO bond lengths could be attributed to the higher Fe → CO π-backbonding in 2 or 3 than that in 1.38 The average Fe–S bond lengths in 2 (2.2737(6) Å) and 3 (2.2633(6) Å) are longer than that in 1 (2.2561(8) Å).38
Protonation study
The protophilicity of 2 and 3 towards CF3CO2H (pKMeCNa = 12.65),60 HCl (pKMeCNa = 10.4)60 and HBF4·Et2O (pKMeCNa = 0.20)60 has been investigated by spectroscopic techniques. Complexes 2 and 3 were analyzed by attenuated total reflection Fourier-transform infrared (ATR FTIR) spectroscopy at ambient temperature and pressure. All experiments were conducted under anoxic conditions (N2 atmosphere) and in the dark. Prior to the experiment, both complexes were dissolved in either pure MeCN and one equivalent of CF3CO2H, HBF4·Et2O or HCl. 3 μL of the solution were placed on the silicon crystal of the ATR cell and covered by a lid to minimize evaporation and protect the sample from stray light. Fig. 3 shows the FTIR absorbance spectra in the CO region (2200–1800 cm−1) as well as second derivative spectra for an assignment of individual contributions. Under aprotic conditions (pure MeCN), complexes 2 and 3 show a fairly similar band pattern with a single high-frequency band around 2040 cm−1 and a set of three CO bands between 1990–1918 cm−1 (see Table 1). In protic solvent, the presence of up to 10 equiv. CF3CO2H or HCl did not affect the ν(CO) bands of 2 and 3, which means that no protonation reaction took place. In the presence of 1 equiv. HBF4·Et2O, however, complexes 2 and 3 showed mean spectral up-shifts of 82 ± 12 cm−1 and 22 ± 7 cm−1, respectively. Despite the four-fold stronger up-shift of the CO frequencies in comparison to 3, the spectral pattern of 2 appeared to be well conserved. Both in the presence and absence of HBF4·Et2O, a strong band (s) is followed by another strong band with a spectral difference of ∼60 cm−1, a medium strong band (ms, spectral difference of ∼25 cm−1), and a weak band (w, spectral difference of ∼35 cm−1). The band pattern of 3 in protic solvent clearly differs from that of this complex in pure MeCN (Table 1).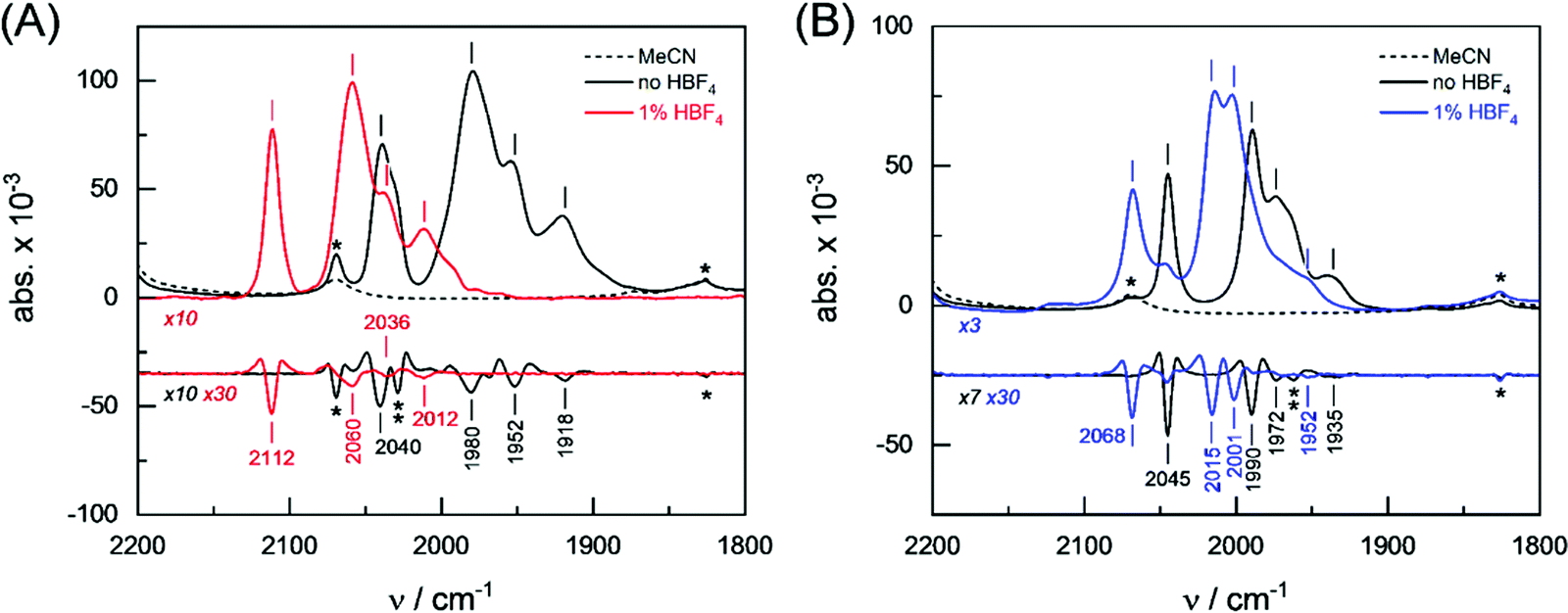 | ||
| Fig. 3 FTIR absorbance spectra of complexes 2 and 3. The CO frequency region is shown. (A) Complex 2 in solution with MeCN in the presence and absence of HBF4·Et2O (red and black traces) and (B) complex 3 in solution with MeCN in the presence and absence of HBF4·Et2O (blue and black traces). The second derivate of absorbance spectra facilitated an unambiguous frequency assignment. Asterisk: spectral contribution from MeCN. Double asterisk in (A): unassigned shoulder at 2028 cm−1 of the strong CO band at 2040 cm−1 in the absence of HBF4·Et2O. Double asterisk in (B): unassigned shoulder at 1962 cm−1 of the strong CO band at 1972 cm−1 in the absence of HBF4·Et2O. Dashed spectra: MeCN reference. | ||
| Complex | Solvent | v CO/cm−1 (band intensity) | |||
|---|---|---|---|---|---|
| a The band showed a shoulder at 2028 cm−1. b The band showed a shoulder at 1962 cm−1. | |||||
| 2 | MeCN | 2040 (s)a | 1980 (s) | 1952 (ms) | 1918 (w) |
| HBF4·Et2O | 2112 (s) | 2060 (s) | 2036 (ms) | 2012 (w) | |
| Δ | 72 | 79 | 84 | 94 | |
| 3 | MeCN | 2045 (s) | 1990 (s) | 1972 (ms)b | 1935 (w) |
| HBF4·Et2O | 2068 (s) | 2015 (s) | 2001 (s) | 1952 (w) | |
| Δ | 23 | 25 | 29 | 17 | |
In conclusion, the ATR FTIR investigation of CO band frequencies suggests protonation (or hydride formation) at complexes 2 and 3 in the presence of the strong acid HBF4·Et2O. Analysis of the spectral region from 2600–2400 cm−1 indicated no specific differences for 2 and 3 between aprotic and protic solvent, which argues against sulfur protonation, at least under steady-state conditions (Fig. S2, ESI†). Indeed, this approach has been supported by testing the in situ protonation reactions of 2 and 3 with 1 equiv. HBF4·Et2O via1H and 31P NMR techniques. The high-field region of the 1H NMR spectra of 2 and 3 in the presence of HBF4·Et2O show typical signals due to μ-hydride species 2(μ-H)+ and 3(μ-H)+. Unfortunately, decomposition accompanies the protonation of 2, which makes it complicated to interpret the spectra. The 1H NMR spectrum of 3 in the presence of 1 equiv. HBF4·Et2O is characterized by two sets of high-field doublets at −13.9 ppm (J{1H–31P} = 6.0 Hz) and −14.3 ppm (J{1H–31P} = 24.0 Hz) with ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 relative intensity. Since the coupling between the apical PR3 ligand and the μ-hydride is typically weak or might not be observed,25,58d we assign the signals at −13.9 ppm and −14.3 ppm to the apical and basal isomers, respectively. Similarly, the 31P NMR spectrum of the protonated complex 3(μ-H)+ displays two signals at −154.8 ppm and −145.3 ppm for the apical and basal isomers with relative intensity of ca. 1:2, respectively. Whereas Fe2(CO)5EPh3{μ-(SCH2)2} (E = P, As, Sb) required excess amount of HBF4·Et2O to protonate the Fe–Fe bond,58d the presence of Sn atom in the dithiolato ligand of the complex makes the Fe–Fe bond more susceptible to protonation.
:2 relative intensity. Since the coupling between the apical PR3 ligand and the μ-hydride is typically weak or might not be observed,25,58d we assign the signals at −13.9 ppm and −14.3 ppm to the apical and basal isomers, respectively. Similarly, the 31P NMR spectrum of the protonated complex 3(μ-H)+ displays two signals at −154.8 ppm and −145.3 ppm for the apical and basal isomers with relative intensity of ca. 1:2, respectively. Whereas Fe2(CO)5EPh3{μ-(SCH2)2} (E = P, As, Sb) required excess amount of HBF4·Et2O to protonate the Fe–Fe bond,58d the presence of Sn atom in the dithiolato ligand of the complex makes the Fe–Fe bond more susceptible to protonation.
Electrochemistry
Fig. 4 shows the cyclic voltammetric reduction of 2 and 3 in comparison to that of 1 at a scan rate of 0.2 V s−1. The cyclic voltammograms of 2 and 3 show irreversible cathodic peaks at Epc = −1.90 V (for 2) and Epc = −2.15 V (for 3). These reduction potentials are shifted to more negative values by 220 mV (for 2) and 470 mV (for 3) in comparison to that of 1 (Epc = −1.68 V) due to the presence of the strong electron donating PPh3 and P(OMe)3 ligands. The less negative reduction potential of 2 compared to that of 3 is unexpected because ν(CO) wavenumbers are lower for 2. Furthermore, this order of reduction potentials disagrees with the ligand electrochemical parameters (EL) determined by Lever for PPh3 (EL = 0.39 V) and P(OMe)3 (EL = 0.42 V).61 In contrast to 1, increasing the scan rate achieves slight chemical reversibility for the reduction of 2 and 3 (Fig. S12–S14, ESI†), suggesting a very fast follow-up decomposition. Indeed, the enhanced chemical reversibility of the reduction of 2 at higher scan rates is accompanied with appearance of small reduction even at ca. −2.0 V (Fig. S12, ESI†), which might be related to reduction of the monoanionic species 2− into dianionic one 22−.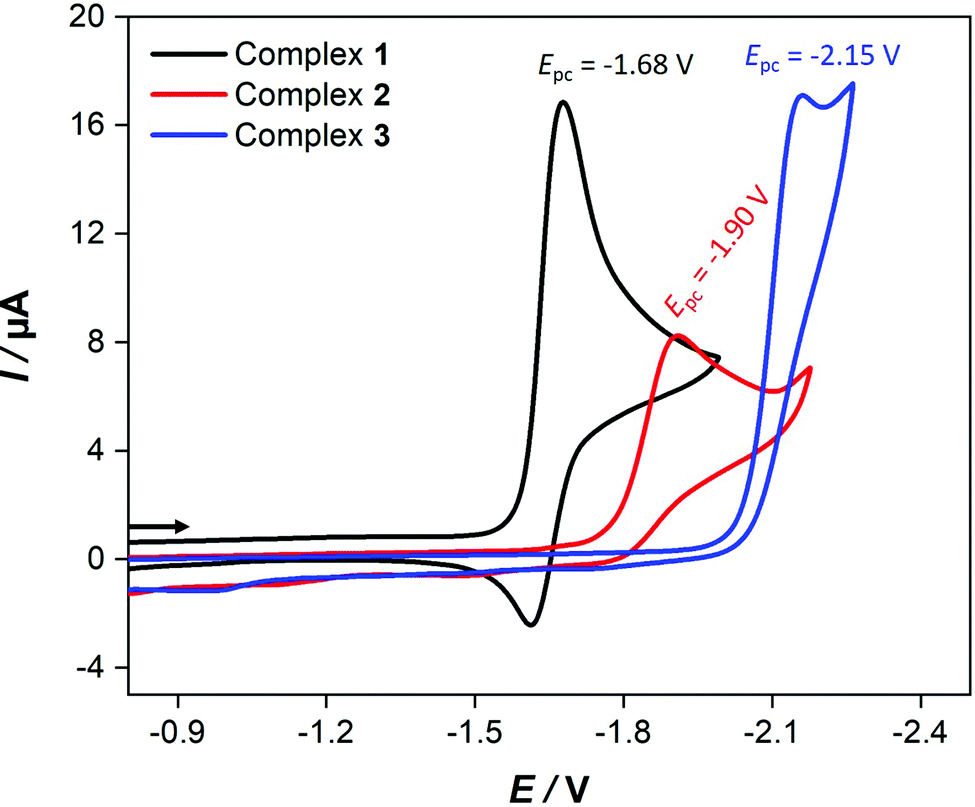 | ||
| Fig. 4 Cyclic voltammetry of 1.0 mM 1–3 in CH2Cl2–[n-Bu4N][BF4] (0.1 M) solutions at 0.2 V s−1 scan rate. The arrow indicates the scan direction. The potential E is given in V and referenced to the Fc+/Fc couple. | ||
Evidence for the number of electrons involved in the reduction of the complexes could be obtained from studying the scan rate dependence of the current function Ip/c·ν1/2 (Ip = peak current, c = concentration, ν = scan rate).62–70 While Ip/c·ν1/2 decreases towards a constant value as the scan rate increases in the cases of 138 and 3, the current function is found to be scan rate independent for 2 (Fig. 5). These observations suggest an ECE cathodic process (E = electron transfer and C = chemical process) in the case of 1 and 3 while the reduction of 2 involves simple transfer of one electron. The intervening chemical process in the ECE mechanism is suppressed by increasing the scan rate.
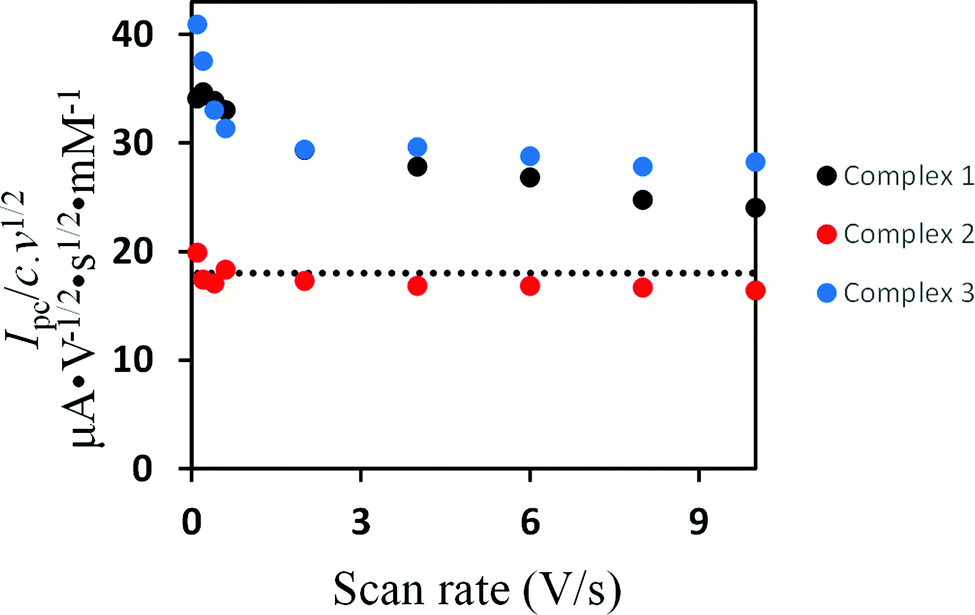 | ||
| Fig. 5 The scan rates dependence of the current function of the primary reduction peaks of 1.0 mM 1–3 in CH2Cl2–[n-Bu4N][BF4] (0.1 M) solutions. The dashed line represents the current function expected for a one electron process assuming D ≈ 9 × 10−6 cm2 s−1, a value calculated for various [FeFe]-hydrogenase models. | ||
In the presence of HBF4·Et2O, the cyclic voltammograms display new reduction peaks at 960 mV and 990 mV less negative potentials in comparison to the primary reduction waves of 2 and 3, respectively, observed in the absence of acid. These new reduction waves are attributed to the reduction of the protonated form of 2 and 3. Similar shifts have been reported and attributed to the protonation of the Fe–Fe bond in various complexes.55 In contrast, the presence of strong acids (HBF4·Et2O or CF3SO3H) has only led to small anodic shift of the reduction wave of 1, which is typical thermodynamic effect resulting from protonation of the reduced species of 1.38 Indeed, direct reduction of HBF4·Et2O at the electrode is significant at high concentrations and hence the measurements were conducted only at low acid concentrations. While the cyclic voltammetry of 3 (Fig. 6) displays three distinct reduction waves (Ep = −1.17 V, −1.64 V and −2.14 V) in the presence of 2–4 equiv. HBF4·Et2O, the situation is more complicated in the case of 2 (Fig. S15, ESI†) as an overlap of reduction waves is observed. The current of these three reduction events increases very slightly as the equiv. [HBF4·Et2O]/[3] increases up to 4.
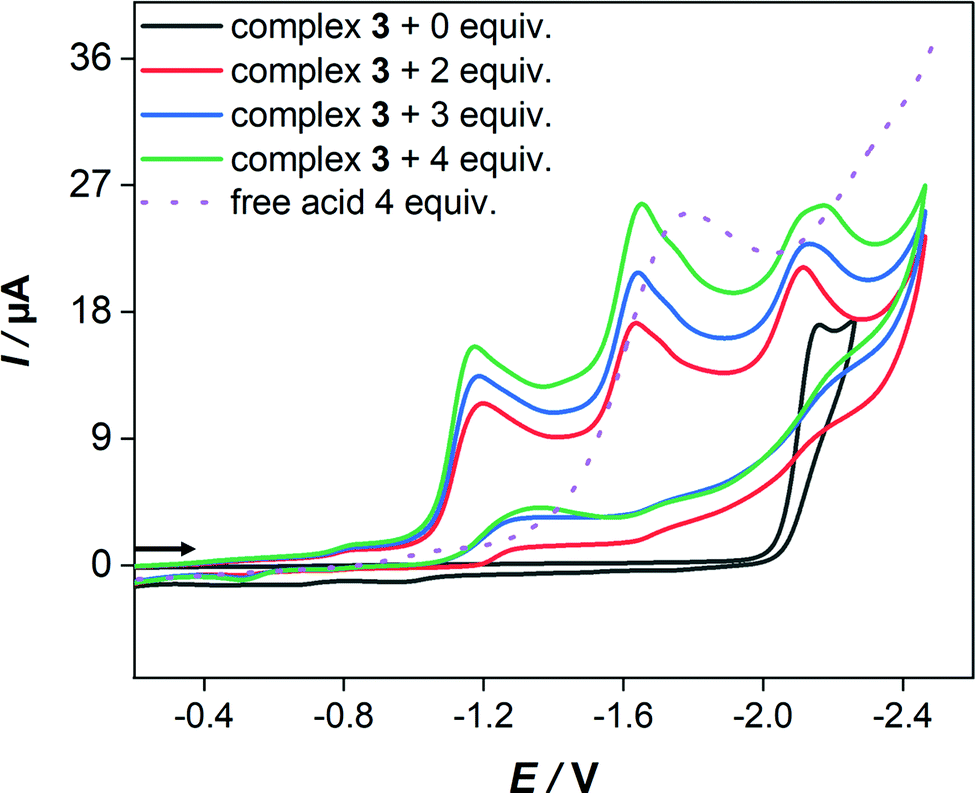 | ||
| Fig. 6 Cyclic voltammetry (0.2 V s−1) of 1.0 mM Fe2(CO)5(P(OMe)3){μ-(SCH2)2}SnMe2 (3) in CH2Cl2–[n-Bu4N][BF4] (0.1 M) at [HBF4·Et2O]/[3] = 0–4. Potential E is given in volts (V) and referenced to Fc+/Fc couple. The arrows indicate the scan direction. | ||
Conclusions
We have reported on the synthesis of Fe2(CO)5(L){μ-(SCH2)2}SnMe2 (L = PPh3 (2) and P(OMe)3 (3)) via oxidative abstraction of CO from Fe2(CO)6{μ-(SCH2)2}SnMe2 (1) followed by addition of PPh3 or P(OMe)3 (Scheme 1). The variable temperature 1H and 31P{1H} NMR spectroscopic techniques reveals the presence of two isomers of 3 at −50 °C with apically and basally oriented P(OMe)3 ligand. In contrast, 2 and 3 exhibit only the apical orientation of PPh3 or P(OMe)3 in the solid state (Fig. 2). While 1 exhibits an almost planar molecular structure (α = 173.6°), the introduction of PPh3 or P(OMe)3 into the apical position in 2 or 3, respectively, results in a deviation from planarity. The smaller flap angle measured in 3 (α = 146.1°) compared to that in the case of 2 (α = 160.3°) is attributed to the higher steric demand of the PPh3 with respect to that of P(OMe)3. Although PPh3 is stronger electron donor ligand than P(OMe)3 as evident from the ν(CO) wavenumbers of 2 and 3, the reduction potentials of these complexes show an unexpected trend; Epc = −1.90 V (for 2) and Epc = −2.15 V (for 3). This unexpected reductive behavior might be attributed to chemical processes intervening in the electron transfer. In contrast to the case of 1, the cyclic voltammetry of 2 or 3 show that protonation precedes electron transfer due to the presence of the strong electron donating ligands. By means of spectroscopic techniques, the protonation of 2 and 3 is proven to take place at the Fe–Fe bond leading to μ-hydride species. Only 1 equiv. of HBF4·Et2O is sufficient to protonate 2 and 3 at the Fe–Fe bond forming 2(μ-H)+ and 3(μ-H)+ species that is triggered not only by the presence of the PR3 ligands, but also via the filled-filled interaction between the C–Sn bond and the neighboring lone pair of the μ-S atoms.Experimental part
Materials and techniques
All reactions were performed using standard Schlenk and vacuum-line techniques under an inert gas (nitrogen). The 1H, 13C{1H} and 31P{1H} spectra were recorded with a Bruker Avance 400 MHz spectrometer. Chemical shifts are given in parts per million with references to internal SiMe4 (1H, 13C). External standard 85% H3PO4 was used as a reference for 31P{1H} spectral measurements. The mass spectrum was recorded with Finnigan MAT SSQ 710 instrument. Elemental analysis was performed with a Leco CHNS-932 apparatus. TLC was performed by using Merck TLC aluminum sheets (Silica gel 60 F254). Solvents from Fisher Scientific and other chemicals from Across and Aldrich were used without further purification. All solvents were dried and distilled prior to use according to standard methods. Complex 1 was synthesized according to the known literature method.38Infrared spectroscopy
Fourier-transform infrared (FTIR) spectroscopy was performed in attenuated total reflection (ATR) configuration. For this, the FTIR spectrometer (Tensor27, Bruker) was equipped with a three reflections ZnSe/Si mircrocrystal (DuraDisc, Smiths Detection). All experiments were performed in a protective N2 atmosphere, at dim light, and ambient temperatures. With 50 scans per spectrum and a scanning velocity of 80 MHz, a time resolution of 10 s was achieved that allowed probing the sample volume (1–3 μL) prior solvent evaporation. All absorbance spectra are shown as recorded (no baseline treatment or atmospheric corrections). The second derivative spectra were calculated in OPUS.Electrochemistry
Corrections for the iR drop were performed for all experiments. Cyclic voltammetric measurements were conducted in three-electrode technique [glassy carbon disk (diameter = 1.6 mm) as working electrode, Ag/Ag+ in MeCN as reference electrode, Pt wire as counter electrode] using a Reference 600 Potentiostat (Gamry Instruments). All experiments were performed in CH2Cl2 solutions (concentration of the complexes 1.0 mM) containing 0.1 M [n-Bu4N][BF4] at room temperature. The solutions were purged with N2 and a stream of it was maintained over the solutions during the measurements. The vitreous carbon disk was polished on a felt tissue with alumina before each measurement. All potential values reported in this paper are referenced to the potential of the ferrocenium/ferrocene (Fc+/Fc) couple.X-ray crystal structure analysis
The intensity data for the compounds were collected on a Nonius KappaCCD diffractometer using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans.71–73 The structures were solved by direct methods (SHELXS)74 and refined by full-matrix least squares techniques against Fo2 SHELXL-2018).75 The hydrogen atoms bonded to the methine-groups C1 and C2 of compound 3 were located by difference Fourier synthesis and refined isotropically. All other hydrogen atoms were included at calculated positions with fixed thermal parameters. All non-hydrogen atoms were refined anisotropically.75 MERCURY was used for structure representations.76![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.7998(2), b = 10.5057(2), c = 16.7575(3) Å, α = 106.986(1), β = 91.930(1), γ = 114.973(1)°, V = 1470.99(5) Å3, T = −140 °C, Z = 2, ρcalcd = 1.704 g cm−3, μ(Mo-Kα) = 20.47 cm−1, multi-scan, transmin: 0.6677, transmax: 0.7456, F(000) = 752, 9131 reflections in h(−12/12), k(−13/13), l(−21/18), measured in the range 2.272° ≤ Θ ≤ 27.362°, completeness Θmax = 99.2%, 6587 independent reflections, Rint = 0.0142, 6284 reflections with Fo > 4σ(Fo), 345 parameters, 0 restraints, R1obs = 0.0235, wRobs2 = 0.0517, R1all = 0.0254, wRall2 = 0.0533, GOOF = 1.070, largest difference peak and hole: 0.388/−0.491 e Å−3.
, a = 9.6675(3), b = 10.2531(3), c = 12.5768(3) Å, α = 102.077(1), β = 103.608(1), γ = 109.654(1)°, V = 1082.95(5) Å3, T = −140 °C, Z = 2, ρcalcd = 1.891 g cm−3, μ(Mo-Kα) = 27.65 cm−1, multi-scan, transmin: 0.6397, transmax: 0.7456, F(000) = 608, 6883 reflections in h(−12/12), k(−13/8), l(−16/16), measured in the range 2.438° ≤ Θ ≤ 27.101°, completeness Θmax = 99.2%, 4731 independent reflections, Rint = 0.0149, 4560 reflections with Fo > 4σ(Fo), 256 parameters, 0 restraints, R1obs = 0.0200, wRobs2 = 0.0467, R1all = 0.0211, wRall2 = 0.0474, GOOF = 1.099, largest difference peak and hole: 0.457/−0.389 e Å−3.
, a = 9.7998(2), b = 10.5057(2), c = 16.7575(3) Å, α = 106.986(1), β = 91.930(1), γ = 114.973(1)°, V = 1470.99(5) Å3, T = −140 °C, Z = 2, ρcalcd = 1.704 g cm−3, μ(Mo-Kα) = 20.47 cm−1, multi-scan, transmin: 0.6677, transmax: 0.7456, F(000) = 752, 9131 reflections in h(−12/12), k(−13/13), l(−21/18), measured in the range 2.272° ≤ Θ ≤ 27.362°, completeness Θmax = 99.2%, 6587 independent reflections, Rint = 0.0142, 6284 reflections with Fo > 4σ(Fo), 345 parameters, 0 restraints, R1obs = 0.0235, wRobs2 = 0.0517, R1all = 0.0254, wRall2 = 0.0533, GOOF = 1.070, largest difference peak and hole: 0.388/−0.491 e Å−3.
, a = 9.6675(3), b = 10.2531(3), c = 12.5768(3) Å, α = 102.077(1), β = 103.608(1), γ = 109.654(1)°, V = 1082.95(5) Å3, T = −140 °C, Z = 2, ρcalcd = 1.891 g cm−3, μ(Mo-Kα) = 27.65 cm−1, multi-scan, transmin: 0.6397, transmax: 0.7456, F(000) = 608, 6883 reflections in h(−12/12), k(−13/8), l(−16/16), measured in the range 2.438° ≤ Θ ≤ 27.101°, completeness Θmax = 99.2%, 4731 independent reflections, Rint = 0.0149, 4560 reflections with Fo > 4σ(Fo), 256 parameters, 0 restraints, R1obs = 0.0200, wRobs2 = 0.0467, R1all = 0.0211, wRall2 = 0.0474, GOOF = 1.099, largest difference peak and hole: 0.457/−0.389 e Å−3.
General procedure for the synthesis of Fe2(CO)5(L){μ-(SCH2)2SnMe2} (L = PPh3 and P(OMe)3)
To a solution of 1 in MeCN (25 mL), 2 equiv. Me3NO2·H2O were added to give the respective nitrile complex within 40 min, visible by darkening of the red solution. Subsequently, 1 equiv. L was added, and the reaction mixture was stirred at r.t. for 20 h. The resulting red solution was then treatment with small amount of silica and the solvent was evaporated using vacuum transfer line. The residue was purified by column chromatography using CH2Cl2/hexane (1/5) as eluent. The complexes were collected from the first red-orange band and the solvent was evaporated to give the complexes as red-orange solids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. W. and H. A.-F. are thankful to Deutsche Forschungsgemeinschaft (DFG) for supporting this work (WE 1179/11-1). H. A.-F. thanks the deanship of research, Al-Zaytoonah University of Jordan for financial support (Grant No. 18/2018-2019). S. T. S. acknowledges funding through the DFG priority program 1927 (grant agreement No. 1554/5-1).References
- D. J. Wuebbles and A. K. Jain, Fuel Process. Technol., 2001, 71, 99 CrossRef CAS.
- S. Dunn, Int. J. Hydrogen Energy, 2002, 27, 235 CrossRef CAS.
- M. Momirlan and T. N. Veziroglu, Int. J. Hydrogen Energy, 2005, 30, 795 CrossRef CAS.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. M. Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572 CrossRef CAS.
- J. G. Canadell, C. Le Quere, M. R. Raupach, C. B. Field, E. T. Buitenhuis, P. Ciais, T. J. Conway, N. P. Gillett, R. A. Houghton and G. Marland, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18866 CrossRef CAS.
- M. Sensi, C. Baffert, C. Greco, G. Caserta, C. Gauquelin, L. Saujet, M. Fontecave, S. Roy, V. Artero, P. Soucaille, I. Meynial-Salles, H. Bottin, L. de Gioia, V. Fourmond, C. Léger and L. Bertini, J. Am. Chem. Soc., 2016, 138, 13612 CrossRef CAS.
- M. W. Adams, Biochim. Biophys. Acta, 1990, 1020, 115 CrossRef CAS.
- M. Frey, ChemBioChem, 2002, 3, 153 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853 CrossRef CAS.
- Y. Nicolet, A. L. de Lacey, X. Vernede, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596 CrossRef CAS.
- (a) A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592 RSC; (b) G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66 CrossRef CAS.
- (a) M. Haumann and S. T. Stripp, Acc. Chem. Res., 2018, 51, 1755 CrossRef CAS; (b) H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069 CrossRef CAS.
- Y. Li and T. B. Rauchfuss, Chem. Rev., 2016, 116, 7043 CrossRef CAS.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127 CrossRef CAS.
- S. Gao, Y. Liu, Y. Shao, D. Jiang and Q. Duan, Coord. Chem. Rev., 2020, 402, 213081 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS.
- H. Abul-Futouh, L. R. Almazahreh, M. K. Harb, H. Görls, M. El-khateeb and W. Weigand, Inorg. Chem., 2017, 56, 10437 CrossRef CAS.
- H. Abul-Futouh, A. Q. Daraosheh, J. Windhager, H. Görls and W. Weigand, Polyhedron, 2019, 174, 114155 CrossRef CAS.
- J. D. Lawrence, H. Li, T. B. Rauchfuss, M. Bénard and M. Rohmer, Angew. Chem., Int. Ed., 2001, 40, 1768 CrossRef CAS.
- M. Watanabe, K. Goto, T. Miyazaki, M. Shibahara, Y. J. Chang, T. J. Chow and T. Ishihara, New J. Chem., 2019, 43, 13810 RSC.
- H. Abul-Futouh, A. Skabeev, D. Botteri, Y. Zagranyarski, H. Görls, W. Weigand and K. Peneva, Organometallics, 2018, 37, 3278 CrossRef CAS.
- H. Abul-Futouh, Y. Zagranyarski, C. Müller, M. Schulz, S. Kupfer, H. Görls, M. El-khateeb, S. Gräfe, B. Dietzek, K. Peneva and W. Weigand, Dalton Trans., 2017, 46, 11180 RSC.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843 CrossRef CAS.
- J. Hou, X. Peng, J. Liu, Y. Gao, X. Zhao, S. Gao and K. Han, Eur. J. Inorg. Chem., 2006, 4679 CrossRef CAS.
- W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung and L.-Z. Wu, Dalton Trans., 2009, 2712 RSC.
- Y. Si, C. Ma, M. Hu, H. Chen, C. Chen and Q. Liu, New J. Chem., 2007, 31, 1448 RSC.
- R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. De Goia and G. Zampelli, J. Am. Chem. Soc., 2012, 134, 19260 CrossRef CAS.
- M. L. Singleton, R. M. Jenkins, C. L. Klemashevich and M. Y. Darensbourg, C. R. Chim., 2008, 11, 861 CrossRef CAS.
- B. E. Barton, M. T. Olsen and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 16834 CrossRef CAS.
- H. X. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726 CrossRef CAS.
- L.-C. Song, Z.-Y. Yang, H.-Z. Bian, Y. Liu, H.-T. Wang, X.-F. Liu and Q.-M. Hu, Organometallics, 2005, 24, 6126 CrossRef CAS.
- L.-C. Song, Z.-Y. Yang, Y.-J. Hua, H.-T. Wang, Y. Liu and Q.-M. Hu, Organometallics, 2007, 26, 2106 CrossRef CAS.
- J. Windhager, M. Rudolph, S. Brautigam, H. Görls and W. Weigand, Eur. J. Inorg. Chem., 2007, 2748 CrossRef CAS.
- L.-C. Song, A.-G. Zhu and Y.-Q. Guo, Dalton Trans., 2016, 45, 5021 RSC.
- L. R. Almazahreh, U.-P. Apfel, W. Imhof, M. Rudolph, H. Görls, J. Talarmin, P. Schollhammer, M. El-khateeb and W. Weigand, Organometallics, 2013, 32, 4523 CrossRef CAS.
- H. Abul-Futouh, L. R. Almazahreh, T. Sakamoto, N. Y. T. Stessman, D. L. Lichtenberger, R. S. Glass, H. Görls, M. El-khateeb, P. Schollhammer, G. Mloston and W. Weigand, Chem. – Eur. J., 2017, 23, 346 CrossRef CAS.
- H. Abul-Futouh, M. El-khateeb, H. Görls, K. J. Asali and W. Weigand, Dalton Trans., 2017, 4, 2937 RSC.
- L. Duan, M. Wang, P. Li, Y. Na, N. Wang and L. Sun, Dalton Trans., 2007, 1277 RSC.
- L.-C. Song, Z.-Y. Yang, H.-Z. Bian, Y. Liu, H.-T. Wang, X.-F. Liu and Q.-M. Hu, Organometallics, 2005, 24, 6126 CrossRef CAS.
- (a) C. M. Thomas, O. Rudiger, T. Liu, C. E. Carson, M. B. Hall and M. Y. Darensbourg, Organometallics, 2007, 26, 3976 CrossRef CAS; (b) L. R. Almazahreh, W. Imhof, J. Talarmin, P. Schollhammer, H. Görls, M. El-khateeb and W. Weigand, Dalton Trans., 2015, 44, 7177 RSC; (c) J. Windhager, U.-P. Apfel, T. Yoshino, N. Nakata, H. Görls, M. Rudolph, A. Ishii and W. Weigand, Chem. – Asian J., 2010, 5, 1600 CrossRef CAS.
- P. Li, M. Wang, C. He, X. Liu, K. Jin and L. Sun, Eur. J. Inorg. Chem., 2007, 3718 CrossRef CAS.
- W. Gao, J. Ekstrom, J. Liu, C. Chen, L. Eriksson, L. Weng, B. Åkermark and L. Sun, Inorg. Chem., 2007, 46, 1981 CrossRef CAS.
- A. Jablonskytė, J. A. Wright and C. J. Pickett, Eur. J. Inorg. Chem., 2011, 1033 CrossRef.
- Y.-C. Liu, C.-H. Lee, G.-H. Lee and M.-H. Chiang, Eur. J. Inorg. Chem., 2011, 1155 CrossRef.
- H. Abul-Futouh, H. Görls and W. Weigand, Z. Anorg. Allg. Chem., 2017, 643, 1615 CrossRef CAS.
- A. Q. Daraosheh, H. Abul-Futouh, H. Görls and W. Weigand, Inorg. Chim. Acta, 2020, 503, 119377 CrossRef.
- F. Gloaguen, J. D. Lawrence, M. Schmidt, S. R. Wilson and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 12518 CrossRef CAS.
- C. M. Thomas, T. Liu, M. B. Hall and M. Y. Darensbourg, Inorg. Chem., 2008, 47, 7009 CrossRef CAS.
- S. Pullen, S. Maji, M. Stein and S. Ott, Dalton Trans., 2019, 48, 5933 RSC.
- Z. Wang, J. He, S. Lü, W.-D. Jiang, Y. Wu, J. Jiang, Y. Xie, C. Mu, A. Li, Y.-L. Li and Q.-L. Li, Appl. Organomet. Chem., 2019, 33, e5184 CAS.
- M.-Y. Hu, P.-H. Zhao, J.-R. Li, X.-L. Gu, X.-B. Jing and X.-F. Liu, Appl. Organomet. Chem., 2020, 34, e5523 CAS.
- H.-M. Lin, J.-R. Li, C. Mu, A. Li, X.-F. Liu, P.-H. Zhao, Y.-L. Li, Z.-Q. Jiang and H.-K. Wu, Appl. Organomet. Chem., 2019, 33, e5196 CAS.
- S. Tschierlei, S. Ott and R. Lomoth, Energy Environ. Sci., 2011, 4, 2340 RSC.
- N. Wang, M. Wang, L. Chen and L. Sun, Dalton Trans., 2013, 42, 12059 RSC.
- K. Fauvel, R. Mathieu and R. Poilblanc, Inorg. Chem., 1976, 15, 976 CrossRef CAS.
- (a) A. Jablonskyte, J. A. Wright and C. J. Pickett, Dalton Trans., 2010, 39, 3026 RSC; (b) X. Zhao, I. P. Georgakaki, M. L. Miller, R. Mejia-Rodriguez, C.-Y. Chiang and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 3917 CrossRef CAS; (c) X. Zhao, Y.-M. Hsiao, C.-H. Lai, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 699 CrossRef CAS; (d) S. Ghosh, A. Rahaman, G. Orton, G. Gregori, M. Bernat, U. Kulsume, N. Hollingsworth, K. B. Holt, S. E. Kabir and G. Hogarth, Eur. J. Inorg. Chem., 2019, 4506 CrossRef CAS; (e) J.-R. Li, M.-Y. Hu, S. Lü, X.-L. Gu, X.-B. Jing and P.-H. Zhao, Appl. Organomet. Chem., 2020, 34, e5929 CAS.
- W. Lin Su, P. H. Huang, W.-T. Chen, W.-Y. Hsu, H.-Y. Chang, S.-Y. Ho, S.-P. Wanga and S.-G. Shyu, J. Chin. Chem. Soc., 2011, 58, 163 CrossRef.
- R. H. Morris, Chem. Rev., 2016, 116, 8588 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271 CrossRef CAS.
- L. Schwartz, P. S. Singh, L. Eriksson, R. Lomoth and S. Ott, C. R. Chim, 2008, 11, 875 CrossRef CAS.
- J. Chen, A. K. Vannucci, C. A. Mebi, N. Okumura, S. C. Borowski, M. Swenson, L. T. Lockett, D. H. Evans, R. S. Glass and D. L. Lichtenberger, Organometallics, 2010, 29, 5330 CrossRef CAS.
- S. P. Best, S. J. Borg, J. M. White, M. Razavet and C. J. Pickett, Chem. Commun., 2007, 4348 RSC.
- F. Gloaguen and T. B. Rauchfuss, Chem. Soc. Rev., 2009, 38, 100 RSC.
- H. Abul-Futouh, M. El-khateeb, H. Görls and W. Weigand, Heteroat. Chem., 2018, 29, e21446 CrossRef.
- G. Qian, W. Zhong, Z. Wei, H. Wang, Z. Xiao, L. Long and X. Liu, New J. Chem., 2015, 39, 9752 RSC.
- R. S. Nicholson and I. Shain, Anal. Chem., 1965, 37, 178 CrossRef CAS.
- M. K. Harb, H. Alshurafa, M. El-khateeb, A. Al-Zuheiri, H. Görls, H. Abul-Futouh and W. Weigand, ChemistrySelect, 2018, 3, 8867 CrossRef CAS.
- R. Trautwein, H. Abul-Futouh, H. Görls, W. Imhof, L. R. Almazahreh and W. Weigand, New J. Chem., 2019, 43, 12580 RSC.
- COLLECT, Data Collection Software, Nonius B.V., Netherlands, 1998.
- Z. Otwinowski and W. Minor, Processing of X-Ray Diffraction Data Collected in Oscillation Mode, in Macromolecular Crystallography, Part A, ed. C. W. Carter and R. M. Sweet, Methods in Enzymology, Academic Press, San Diego, USA, 1997, vol. 276, p. 307 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data (excluding structure factors). CCDC 2026923 for 2 and 2026924 for 3. All NMR spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nj04790b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |