DOI:
10.1039/D0NJ04585C
(Paper)
New J. Chem., 2021,
45, 45-52
Alkali complexes of non-steroidal anti-inflammatory drugs inhibit lung and oral cancers in vitro†
Received
16th September 2020
, Accepted 24th November 2020
First published on 24th November 2020
Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely used to treat pain, fever, and inflammation. In this paper, we report four new alkali metal complexes (1–4) containing 1,10-phenanthroline (phen), and two NASID drugs, that is, ibuprofen (ibu) and flurbiprofen (fibu). Complexes 1–4 were characterized by a variety of analytical techniques including infrared and UV-vis spectroscopy, 1H NMR, and single-crystal and powder X-ray diffraction analysis. In complexes 1–4, phen binds via an N,N′-chelate pocket, while the ibu and fibu ligands coordinate in a bidentate fashion in all four complexes. The complexes were evaluated for their anticancer activity against lung and oral cancer cell lines. The complexes displayed excellent activities against oral and lung cancer cells with the least toxicity toward normal cells when compared to the standard drug 5-fluorouracil.
Introduction
Metal complexes of drugs are an attractive area of research, where the goal is not only to enhance the bioactivity and bioavailability of the drugs but also to search for new targets.1–9 Several drug-based metal complexes have been prepared and, in some cases, exhibited remarkable performances.10 For instance, a Ru2+ complex of chloroquine, an antiviral drug, is reported to exhibit activity two to five times higher than chloroquine.11 Moreover, the interaction of drugs with metal ions may lead to a better understanding of metal ion antagonism.12 Cancer is a collection of diseases characterized by unconstrained multiplication of any of the various types of cells in the body.13 Cisplatin, an expensive drug and associated with severe side effects, is still the drug of choice for different types of cancers.14–16 One of the major side effects of cisplatin is binding with proteins at the positions occupied by zinc ions, which ultimately results in the loss of Zn2+ ions.17,18 Therefore, the search for new but cheap metal-based drugs with less side effects is a challenging task for bioinorganic chemists.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are an important class of drugs that are used to reduce pain, fever, and inflammation.19 Most NSAIDs possess carboxylic groups that can bind to metal ions through oxygen atoms. In this respect, several metal complexes of NSAIDs have been prepared and their biological properties have been explored.20–23 Among NSAIDs, ibuprofen (ibu) and flurbiprofen (fibu) (Fig. 1) possess analgesic and antipyretic properties,24 and are used in inflammatory and painful diseases of rheumatic and non-rheumatic origin.25 It is reported that a regular dose of ibu may be effective against breast26 and lung cancer.27 Both ibu and fibu have also been studied for their antimicrobial and antibacterial properties.28–31 Moreover, metal–organic frameworks derived from ibu/fibu with organic linkers have been evaluated for controlled drug release.32–42
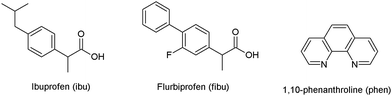 |
| | Fig. 1 Structural representation of ibuprofen (ibu), flurbiprofen (fibu) and 1,10-phenanthroline (phen). | |
We started a research program and aimed to synthesize and structurally characterize new alkali/alkaline earth metal complexes of biologically active compounds and to evaluate their biological activity for potentially useful pharmaceutical applications. We recently reported alkali/alkaline earth metal complexes of diclofenac, an NSAID, which exhibited excellent cytotoxic activity against lung and oral cancer cell lines.43 In the present work, we selected ibu and fibu as O-donor ligands and 1,10-phenanthroline (phen) as an N-donor ligand for complexation reactions with selected alkali metals (lithium, potassium, and caesium). Phen is not only a powerful predictable ligand44–46 but is also known for its enzyme inhibition properties.47,48 The alkali complexes of ibu/fibu with phen as a co-ligand are unknown (CSD 2019).52 There is only one example in the literature, where the luminescence properties of an inner transition metal (Re2+) complex containing ibu− and phen were reported.49 The bioactivities of coordination complexes of cheaper metals (i.e., alkali/alkaline earth metals) did not receive much attention compared to transition metals. Alkali metals play a vital role in many cellular pathways, apoptosis, homeostasis, adjusting electrolyte balance, and nerve impulses.50,51
In the current new study, we have investigated the reactions of phen and ibu/fibu with M2CO3 (where M = Li, K, and Cs) and described the structures of a mononuclear complex [Li(phen)(ibu)(H2O)] (1), a dinuclear complex [Li2(phen)2(fibu)(H2O)3] (2), and 1D polymer chains of [K(phen)(ibu)(ibuH)]n (3) and [Cs(phen)(fibu)]n (4). The structures of 1–4 were analysed by single-crystal X-ray diffraction and detailed descriptions of their crystal structures are given. The bulk compositions were confirmed by 1H NMR, UV-vis and IR spectroscopy, and powder X-ray diffraction analysis. The anticancer activities against lung and oral cancer cell lines were studied to check their therapeutic potential.
Synthesis of the complexes
Ligands ibu and fibu and metal carbonates were obtained commercially and were used without further purification. The reactions of phen, ibu, and fibu with M2CO3 (where M = Li, K, and Cs) in MeOH/H2O produced a mononuclear Li complex (1), a dinuclear Li complex (2), and 1D chains of both K (3) and Cs (4) complexes. All complexes were characterized by single-crystal and powder X-ray diffraction, and UV-vis, IR and 1H NMR spectroscopy. X-ray quality single crystals were grown directly from the mother liquor at room temperature.
Analytical data and spectral characterization
Complexes 1–4 were obtained in good yields (64–73%) in an analytically pure form. The complexes were soluble in MeOH and DMSO. The IR spectra of complexes 1–4 are presented in Fig. S1 of the ESI.† The broad peaks in the range 2920–3619 cm−1 are assigned to the O–H stretching frequency, which are broadened due to hydrogen-bonding. The peaks in the range of 1618–1722 cm−1 reflected the presence of the carbonyl group, while the peaks in the range of 1549–1621 cm−1 are attributed to the aromatic carbon–carbon stretching. The heterocyclic C–N (phen) bond stretching appeared in the range of 1505–1514 cm−1. All these assignments with their values are in good agreement with literature data.52,53 The UV-vis data collection for complexes 1–4 in non-coordinating solvents (such as CH2Cl2) was hindered by insolubility. Therefore, the spectral data for the complexes were recorded in MeOH. The UV-vis data for complexes 1–4 were measured in methanol and are presented in Fig. S2 of the ESI.† The UV-vis spectra are dominated by intense π–π* ligand-centred absorption bands which are attributed to ligand-based transitions by comparison with the free ligands (Fig. S2, ESI†). The increase in the absorption coefficient (ε) of complexes 1–4 (ε = 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 233–65604 M−1 cm−1) compared to the free ligand supports the suggestion that these ligands remained coordinated in the solution, with metal coordination enhancing the extinction coefficient. The 1H NMR spectra of complexes 1–4 were recorded in MeOD/DMSO-d6 (Fig. S3–S6, ESI†). The signals of the free ligands are present in the 1H NMR spectra of the complexes when compared to the free ligands. The signals for the phen protons are unchanged, while there is a small shift in the ibuH/ibu−/fibu− protons, indicating some binding of the anion to the metal ion in solution.
233–65604 M−1 cm−1) compared to the free ligand supports the suggestion that these ligands remained coordinated in the solution, with metal coordination enhancing the extinction coefficient. The 1H NMR spectra of complexes 1–4 were recorded in MeOD/DMSO-d6 (Fig. S3–S6, ESI†). The signals of the free ligands are present in the 1H NMR spectra of the complexes when compared to the free ligands. The signals for the phen protons are unchanged, while there is a small shift in the ibuH/ibu−/fibu− protons, indicating some binding of the anion to the metal ion in solution.
PXRD on complex 1–4
The experimental powder diffraction patterns of complexes 1–4 (Fig. S7–S10, ESI†) are in good agreement with those calculated based on the single-crystal data. Small differences between the intensity profiles may be accounted for by the texture (preferred orientation) of the samples.
Description of the crystal structures
Crystallographic details for 1–4 are listed Table 1.
Table 1 Crystal data for complexes 1–4
| Complexes |
1
|
2
|
3
|
4
|
| Chemical formula |
C25H27LiN2O3 |
C54H46F2Li2N4O7 |
C38H43KN2O4 |
C27H20CsFN2O2 |
|
M
r (g mol−1) |
410.42 |
914.83 |
630.84 |
556.36 |
| Crystal system, space group |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Monoclinic, I12/c |
Triclinic, P |
Monoclinic, P21/n |
|
a (Å) |
7.1787 (5) |
19.4525 (11) |
10.3951 (10) |
18.108 (3) |
|
b (Å) |
10.2822 (6) |
6.6516 (3) |
10.6836 (12) |
7.1063 (12) |
|
c (Å) |
16.1772 (10) |
35.989 (2) |
17.2357 (19) |
19.003 (3) |
|
α (°) |
96.057 (3) |
90 |
72.414 (5) |
90 |
|
β (°) |
101.245 (3) |
98.923 (3) |
77.519 (5) |
109.543 (5) |
|
γ (°) |
106.314 (3) |
90 |
88.313 (5) |
90 |
|
V (Å3) |
1107.59 (12) |
4600.3 (4) |
1780.2 (3) |
2304.5 (6) |
|
Z
|
2 |
4 |
2 |
4 |
|
μ (mm−1) |
0.08 |
0.09 |
0.19 |
1.64 |
|
T
min, Tmax |
0.630, 0.745 |
0.706, 0.745 |
0.599, 0.745 |
0.453, 0.745 |
| No. of measured, independent and observed [I > 2σ(I)] reflections |
18297, 4508, 2645 |
30472, 4655, 3566 |
35049, 6246, 4257 |
25136, 4721, 3667 |
|
R
int
|
0.059 |
0.040 |
0.064 |
0.059 |
| (sin θ/λ) max (Å−1) |
0.626 |
0.625 |
0.595 |
0.627 |
|
R[F2 > 2σ(F2)], wR(F2), S |
0.062, 0.184, 1.04 |
0.050, 0.151, 1.04 |
0.093, 0.293, 1.10 |
0.041, 0.108, 1.00 |
| No. of reflections |
4508 |
4655 |
6246 |
4721 |
| No. of parameters |
420 |
377 |
441 |
328 |
| No. of restraints |
436 |
63 |
42 |
14 |
| Δρmax, Δρmin (e Å−3) |
0.24, −0.19 |
0.24, −0.25 |
1.05, −0.37 |
0.63, −1.05 |
| CCDC |
2015737
|
2015738
|
2015739
|
2015740
|
[Li(ibu)(phen)(H2O)] (1).
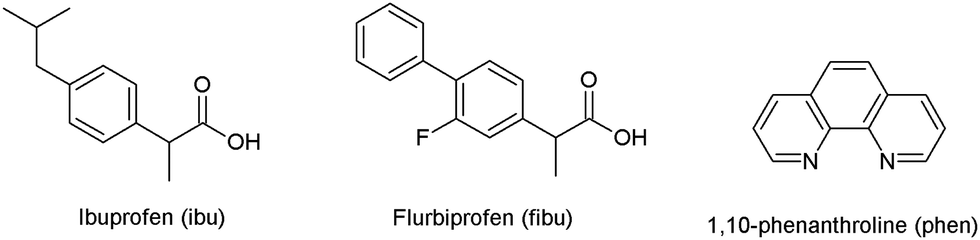
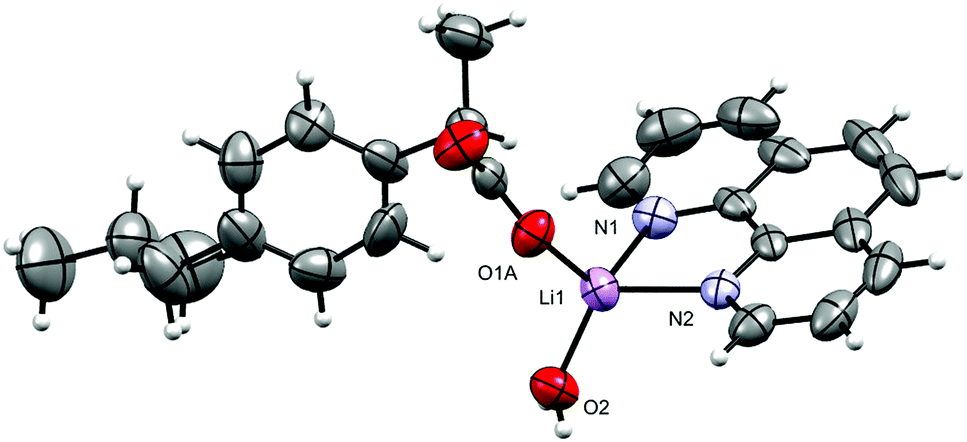
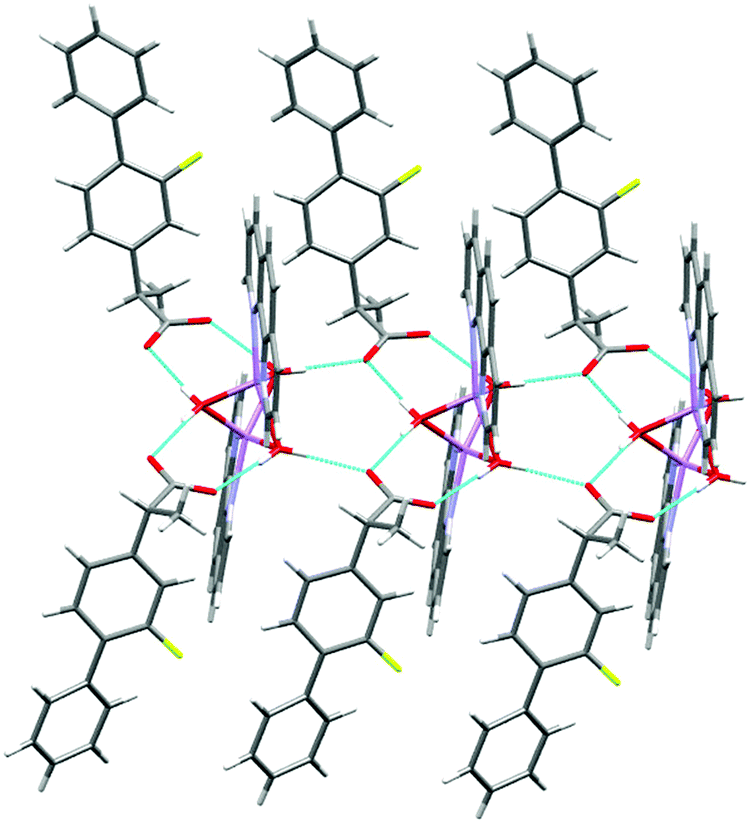
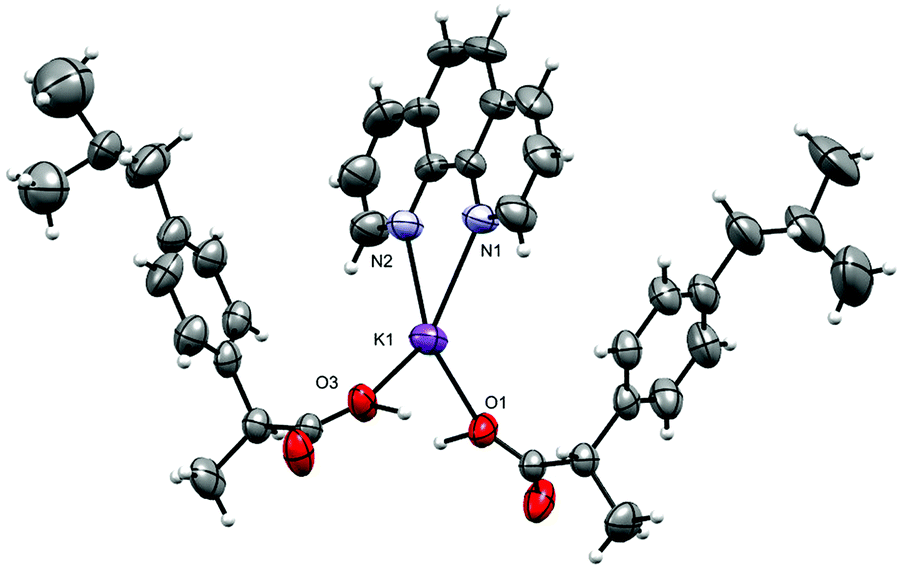
The molecular structure of 1 is shown in Fig. 2 and selected interatomic distances and angles are listed in Table 2. The complex crystallizes in the triclinic space group P, with one phen ligand, a deprotonated ibu− anionic ligand, a Li+ cation, and a coordinated water molecule in the asymmetric unit. The Li+ ion adopts a four-coordinated slightly disordered tetrahedral geometry (Fig. S11, ESI†) comprising a [N2O2] donor set. The phen ligand adopts an N,N′-chelate mode through the N1 and N2 atoms, making a five-membered ring, with Li–N distances of 2.077 (5) and 2.147 (5) Å. The Li–N bond lengths lie within the normal observed range of 2.077 (5)–2.29 Å for related lithium complexes with phen ligands.47 In complex 1, the ibu− anion is disordered and was modeled over two sites (Fig. S12, ESI†). The bond angles around Li+ are in the range 79.6 (2)–118.1 (2)°. The Li–Owater distance is 1.893 (5) Å, consistent with hydrated Li+ complexes reported by us54 and others.44 Lithium complexes of phen with carboxylate as co-ligands are rare. A CSD (2019) search resulted in only two hits,55,56 where the Li+ coordination geometry is similar to that of 1. Complex 1 exhibits hydrogen-bonding interactions which involve the carboxylate moiety and the water molecule. The ibu− carboxylate O atoms (O1A and O3A) form hydrogen bonds with water molecules, which are coordinated to neighboring molecules (O2–H2O⋯O1Ai, and O2–H3O⋯O3Aii), resulting in a supramolecular hydrogen-bonded 1D infinite chain parallel to the crystallographic a-axis (Fig. 3). The structure is additionally stabilized by intermolecular π⋯π stacking between phen ligands, with centroid–centroid (cg⋯cg) distances of 3.544 Å (Fig. S13, ESI†).
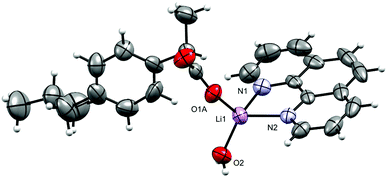 |
| | Fig. 2 The molecular structure of [Li(ibu)(phen)(H2O)] (1). Displacement ellipsoids are drawn at the 50% probability level. The disordered part of ibu− is truncated for clarity. | |
Table 2 Selected geometric parameters (Å, °) for 1
| Li1–O2 |
1.893 (5) |
Li1–N1 |
2.148 (5) |
| Li1–O1B |
1.72 (2) |
Li1–N2 |
2.077 (5) |
| Li1–O1A |
1.934 (7) |
|
|
|
|
|
|
|
| O1B–Li1–O2 |
121.0 (10) |
O1B–Li1–N1 |
100.5 (16) |
| O2–Li1–O1A |
113.5 (3) |
O2–Li1–N1 |
113.9 (2) |
| O1B–Li1–N2 |
114.0 (8) |
O1A–Li1–N1 |
118.0 (3) |
| O2–Li1–N2 |
118.1 (2) |
N2–Li1–N1 |
79.60 (16) |
| O1A–Li1–N2 |
109.8 (2) |
|
|
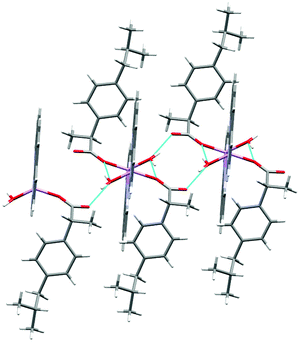 |
| | Fig. 3 The hydrogen-bonded polymeric arrangement of 1. The disordered part of ibu− is truncated for clarity. | |
[Li2(phen)2(H2O)3][fibu]2 (2).
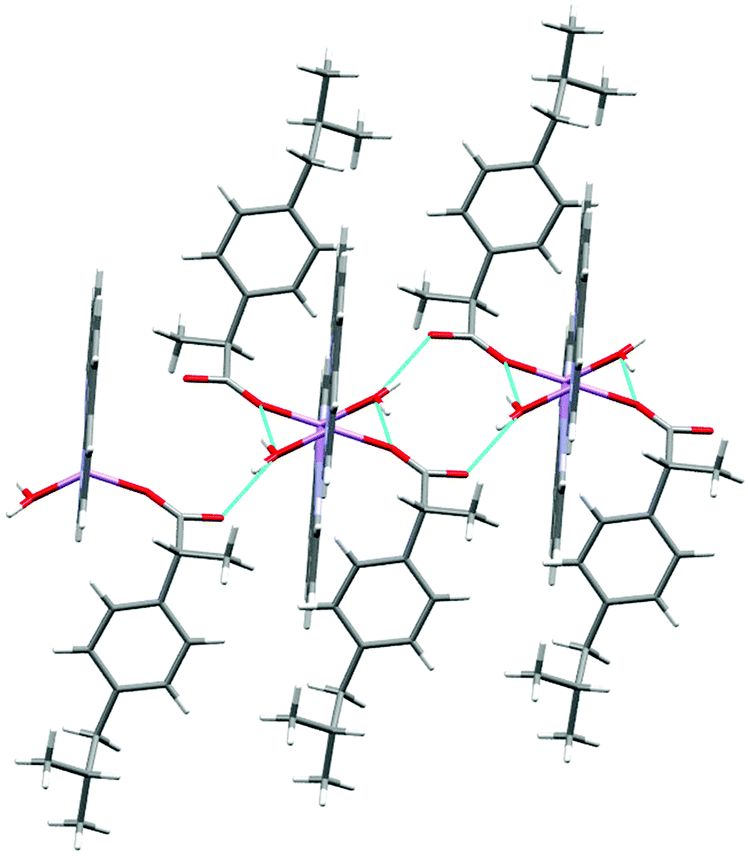
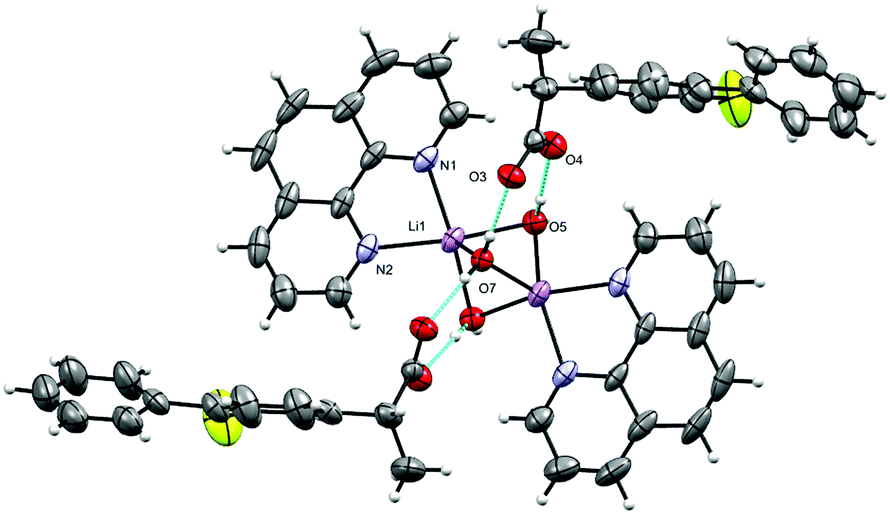
The molecular structure of 2 is shown in Fig. 4 and selected interatomic distances and angles are listed in Table 3. This complex is a binuclear centrosymmetric dimer with one half of the molecule comprising the asymmetric unit of the structure about an inversion centre. The Li+ cations are charge-balanced by two deprotonated fibu− anions to satisfy the overall charge balance on the complex. Each Li+ ion adopts a four coordinated distorted spherical square pyramid geometry (Fig. S14 and Table S2, ESI†) comprising a [N2O5] donor set. The bond angles around Li+ are in the range 76.9 (1)–114.70°. The phen ligand adopts an N,N′-chelate mode through the N1 and N2 atoms, making a five-membered ring, with Li–N distances of 2.174 (4) and 2.176 (3) Å. The Li–N bond lengths lie within the normal observed range of 2.04–2.29 Å for related lithium complexes with phen ligands.44 The Li–Owater distances are 2.027 (5) and 2.152 (3) Å, slightly larger than the hydrated Li+ complexes reported by others.44 The two crystallographically identical Li+ are connected to each other via three bridging water ligands leading to a Li⋯Li separation of 2.661 (4) Å. The un-coordinated carboxylate oxygen (O2) of fibu− forms hydrogen bonds with two water molecules, each of which are coordinated to neighboring molecules (dO–H⋯O = 2.732 and 2.822 (2) Å, and θO–H⋯O = 173.4 and 175.7°), resulting in a supramolecular hydrogen-bonded chain parallel to the crystallographic b axis (Fig. 5). Lithium complexes containing both phen and fibu mixed ligands are unknown (CSD 2019).
 |
| | Fig. 4 Molecular structure of [Li2(phen)2(H2O)3][fibu]2 (2). Displacement ellipsoids are drawn at the 50% probability level. | |
Table 3 Selected geometric parameters (Å, °) for 2
| Symmetry code: (i) −x, y, −z + 1/2 |
| O5–Li1i |
2.113 (3) |
O5–H5B |
0.8186 |
| O5–Li1 |
2.151 (3) |
O7–Li1i |
2.027 (3) |
| O5–H5A |
0.8281 |
O7–Li1 |
2.027 (3) |
|
|
|
|
|
| O7–Li1–O5i |
83.59 (11) |
O5–Li1–N2 |
162.66 (15) |
| O7–Li1–O5 |
82.63 (10) |
N1–Li1–N2 |
76.88 (10) |
| O5i–Li1–O5 |
85.83 (11) |
O7–Li1–Li1i |
48.98 (8) |
| O7–Li1–N1 |
119.66 (14) |
O5i–Li1–Li1i |
52.04 (10) |
| O5i–Li1–N1 |
156.62 (15) |
O5–Li1–Li1i |
50.76 (10) |
| O5–Li1–N1 |
94.36 (13) |
N1–Li1–Li1i |
141.1 (2) |
| O7–Li1–N2 |
114.70 (14) |
N2–Li1–Li1i |
141.4 (2) |
| O5i–Li1–N2 |
96.32 (13) |
|
|
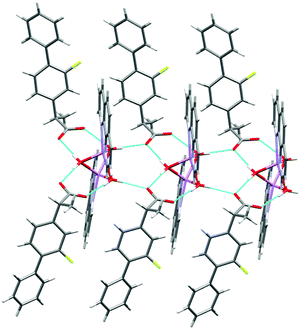 |
| | Fig. 5 The hydrogen-bonded polymeric arrangement of 2. | |
[K(phen)(ibu)(ibuH)]n (3).
Complex 3 crystallizes in the triclinic space group P; the asymmetric unit is shown in Fig. 6, and selected interatomic distances and angles in Table 4. The K+ adopt a distorted [N2O4] geometry with one phen ligand acting as an N2 donor (K–Nphen = 2.826 (4) and 2.830 (6) Å) and protonated and deprotonated ibu ligands adopting a 1,3-O,O′-bridging mode (K–O = 2.681 (3)–2.875 (4) Å). The oxygen atoms O3 and O3′ (and O1/O1′) are related via an inversion center and appeared to be very close to each other (O3⋯O3i = 2.464 (5) Å and O1⋯O1i = 2.473 (5) Å). An H-atom of half occupancy is associated with each O1⋯H+⋯O1 and O3⋯H+⋯O3 unit. The K–N bond lengths lie within the normal observed range of 2. 2.748 (4)–2.872 (2) Å for related potassium complexes with phen ligands.44 The K+ cation is charge-balanced by an ibu− anion, which satisfies the overall charge on complex 3. The six-coordinate coordination geometry around K+ is highly distorted and can be described as between a trigonal prism and a pentagonal pyramid (Fig. S15 and Table S3, ESI†). The phen ligand is virtually planar, and has similar bond distances and angles comparable to those found in the structure of free phen.57 The K+ ion lies almost planar to the phen mean plane (δK = 0.061 Å); the phen ligand tilts with respect to the K1–N1–N2 plane by a dihedral angle of 2.05°. The bridging mode of the ibu− (ibuH) ligands results in a 1-D infinite chain structure of 3 parallel to the crystallographic a-axis (Fig. 7). The K+ ions are connected to inversion related K+ ions via bridging of Ocarboxylates (O1, O2, O3, and O4) atoms resulting in a zig-zag arrangement, so that an eight-membered ring K(μ-O)4K is formed, in which the distance between the two K+ ions is 6.418 (2) Å. The structure is additionally stabilized by π⋯π interactions with cg⋯cg distances of 3.541 and 3.468 Å (depicted in Fig. S16, ESI†). Potassium complexes of phen are known; however, those with ibu as a co-ligand are not known (CSD 2019).
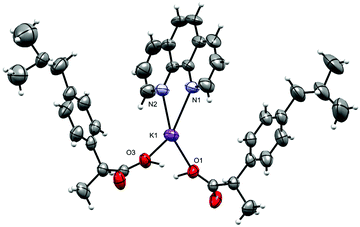 |
| | Fig. 6 The asymmetric unit in [K(phen)(ibu)(ibuH)]n (3): displacement ellipsoids are drawn at the 50% probability level. The disordered part of ibu− is truncated for clarity. | |
Table 4 Selected geometric parameters (Å, °) for 3
| Symmetry codes: (i) −x + 1, −y + 2, −z + 1; (ii) −x, −y + 2, −z + 1. |
| N1–K1 |
2.826 (4) |
K1–O1 |
2.695 (3) |
| N2–K1 |
2.830 (4) |
K1–O4i |
2.852 (3) |
| K1–O3 |
2.681 (3) |
K1–O2ii |
2.876 (3) |
|
|
|
|
|
| O3–K1–O1 |
105.77 (12) |
N1–K1–O4i |
77.99 (12) |
| O3–K1–N1 |
116.75 (13) |
N2–K1–O4i |
134.99 (12) |
| O1–K1–N1 |
127.59 (12) |
O3–K1–O2ii |
91.23 (11) |
| O3–K1–N2 |
133.89 (13) |
O1–K1–O2ii |
68.15 (10) |
| O1–K1–N2 |
111.12 (13) |
N1–K1–O2ii |
135.40 (12) |
| N1–K1–N2 |
57.33 (12) |
N2–K1–O2ii |
78.15 (12) |
| O3–K1–O4i |
69.05 (10) |
O4i–K1–O2ii |
146.56 (13) |
| O1–K1–O4i |
90.81 (11) |
|
|
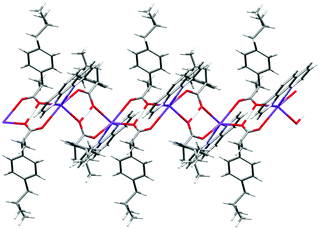 |
| | Fig. 7 The oxygen bridged polymer chain in 3. An H-atom of half occupancy is associated with each O1⋯H+⋯O1 and O3⋯H+⋯O3 unit. The disordered part of ibu is truncated for clarity. | |
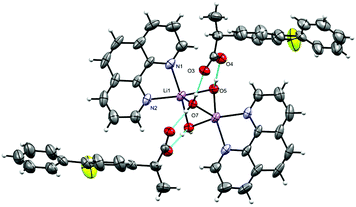
[Cs(phen)(fibu)]n (4).
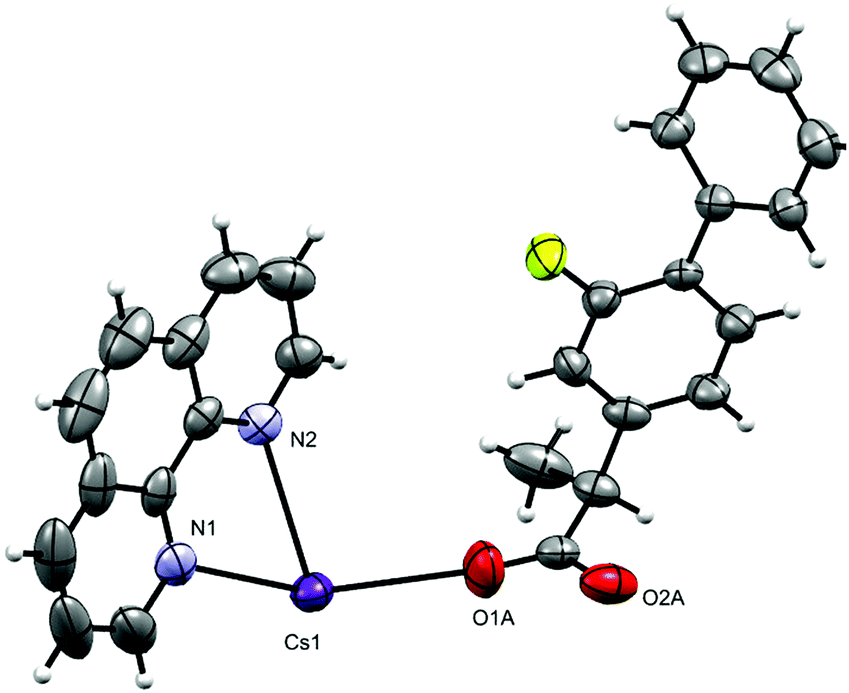
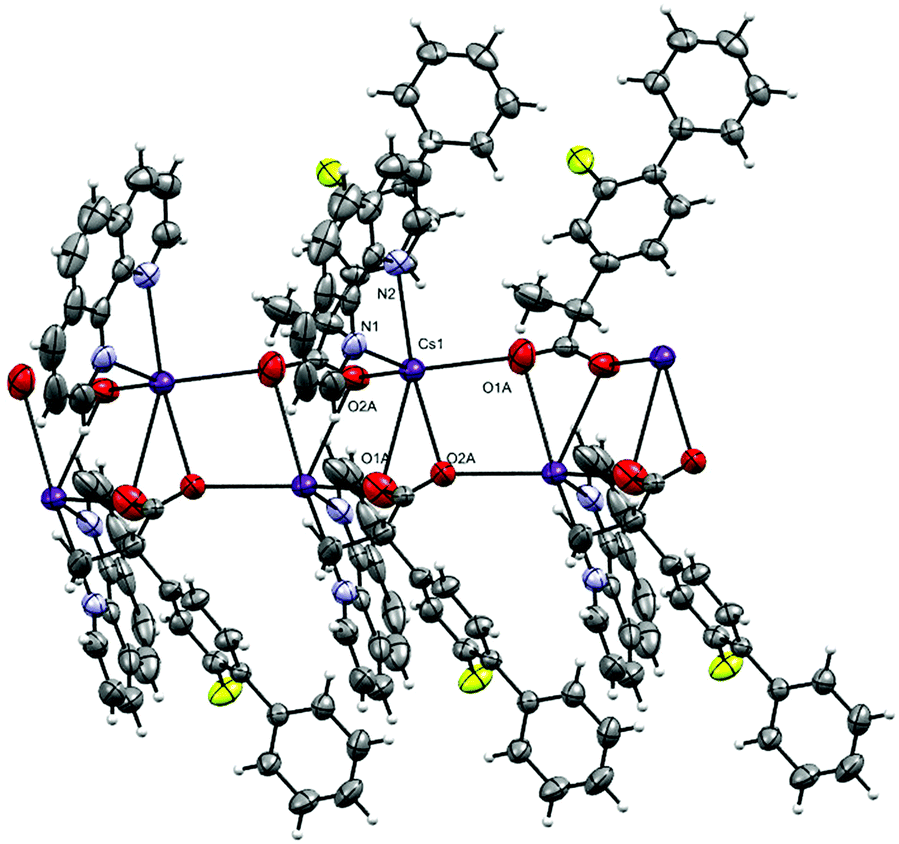
The complex crystallizes in the monoclinic space group P21/n with one phen, a fibu−, and a Cs+ cation in the asymmetric unit (Fig. 8). Selected interatomic distances and angles are listed in Table 5. The fundamental repeat units in 4 are binuclear [Cs(phen)(fibu)] motifs which are connected to each other via μ2-Ocarboxylate (O1A and O2A) bridging. The Cs+ ion lies almost planar to the phen mean plane. The Cs+ ion is six-coordinate and adopts a distorted [N2O4] trigonal prism (Fig. S17 and Table S4, ESI†) with one phen ligand acting as an N2 donor [Cs–Nphen (N1, N2) = 3.184 (5) and 3.220 (3) Å] and two fibu− ligands acting as μ2-O donors [μ2-O, Cs1–O1A = 3.24 (3) Å, and Cs1–O2A = 3.14 (3) Å]. The Cs+ ions are connected to each other via the bridging of Ocarboxylate atoms, resulting in a zig-zag arrangement (Fig. 9) The distance between the two Cs+ ions is 4.639 (7) Å. The complex is additionally stabilized by π⋯π interactions between phen rings with cg⋯cg distances in the range 3.693–3.721 Å (Fig. S18, ESI†). Cs+ coordination chemistry with phen and/or fibu ligands is unknown and complex 4 is the first example containing phen and/or fibu− ligands.
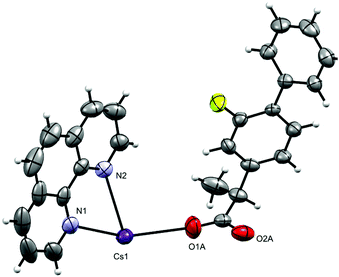 |
| | Fig. 8 Asymmetric unit in [Cs(phen)(fibu)]n (4). Displacement ellipsoids are drawn at the 50% probability level. | |
Table 5 Selected geometric parameters (Å, °) for 4
| Symmetry codes: (i) x, y − 1, z; (ii) −x + 1/2, y − 1/2, −z + 1/2; (iii) −x + 1, −y, −z + 1; (iv) −x + 1/2, y + 1/2, −z + 1/2; (v) x, y + 1, z. |
| Cs1–O2Bi |
2.821 (16) |
Cs1–N1 |
3.183 (5) |
| Cs1–O1A |
2.892 (11) |
Cs1–N2 |
3.221 (4) |
| Cs1–O1B |
2.974 (19) |
Cs1–O1Aii |
3.24 (2) |
| Cs1–O2Ai |
3.000 (17) |
Cs1–O2Bii |
3.31 (5) |
| Cs1–O1Bii |
3.05 (3) |
Cs1–C13ii |
3.468 (4) |
| Cs1–O2Aii |
3.14 (3) |
Cs1–C6iii |
3.907 (5) |
|
|
|
|
|
| O1A–Cs1–O2Ai |
119.9 (7) |
O1Bii–Cs1–N2 |
135.1 (6) |
| O1A–Cs1–O1Bii |
119.0 (4) |
O2Aii–Cs1–N2 |
125.7 (5) |
| O2Ai–Cs1–O1Bii |
81.0 (7) |
N1–Cs1–N2 |
50.62 (11) |
| O1A–Cs1–O2Aii |
81.9 (5) |
O1A–Cs1–O1Aii |
111.9 (5) |
| O2Ai–Cs1–O2Aii |
116.3 (7) |
O2Ai–Cs1–O1Aii |
78.6 (6) |
| O2Bi–Cs1–N1 |
113.8 (10) |
O2Aii–Cs1–O1Aii |
39.8 (3) |
| O1A–Cs1–N1 |
117.6 (5) |
N1–Cs1–O1Aii |
94.2 (4) |
| O1B–Cs1–N1 |
127.8 (6) |
N2–Cs1–O1Aii |
144.6 (4) |
| O2Ai–Cs1–N1 |
120.4 (6) |
O1A–Cs1–O2Bii |
80.4 (5) |
| O1Bii–Cs1–N1 |
84.9 (6) |
O2Ai–Cs1–O2Bii |
111.0 (6) |
| O2Aii–Cs1–N1 |
85.7 (5) |
O2Aii–Cs1–O2Bii |
7.1 (12) |
| O2Bi–Cs1–N2 |
106.4 (7) |
N1–Cs1–O2Bii |
92.5 (8) |
| O1A–Cs1–N2 |
91.2 (5) |
N2–Cs1–O2Bii |
132.5 (8) |
| O1B–Cs1–N2 |
101.1 (6) |
O1Aii–Cs1–O2Bii |
36.9 (4) |
| O2Ai–Cs1–N2 |
113.6 (4) |
|
|
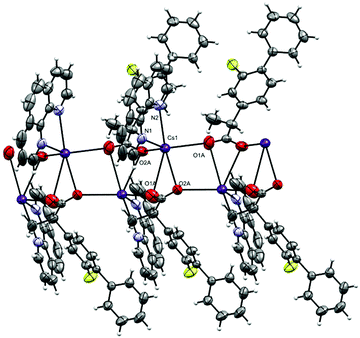 |
| | Fig. 9 The oxygen bridged polymer chain in 4. The disordered part of fibu− is truncated for clarity. | |
Anticancer activity
The cytotoxic potential of complexes 1–4 was evaluated against cancer cells [oral (CAL-27); lung (NCI-H460)], and a normal mouse fibroblast cell line (NIH-3T3). The synthesized alkali complexes showed potent anticancer activities against cancer cells and low toxicity toward normal cells, comparatively, Table 6. All complexes 1–4 showed potent anticancer potential against oral cancer with IC50 values in the range of 1.93 ± 0.18–6.83 ± 0.19 μM, as compared to 5-Fluorouracil. Complexes 3 and 4 were found to be the most active for oral cancer having IC50 values of 1.93 ± 0.18 and 2.74 ± 0.28 μM, respectively. Complexes 1 and 2 almost equally inhibited both cancer cells (oral and lung) with an IC50 of 5.92 ± 1.97 and 6.83 ± 0.19 μM against CAL-27 (oral cancer), and 3.75 ± 0.097 and 6.60 ± 0.46 μM against NCI-H460 (lung cancer), respectively. However, both complexes 3 and 4 also inhibited lung cancer cells but at higher concentrations i.e. IC50 values of 10.00 ± 4.93 and 46.60 ± 5.74 μM, respectively. All four complexes were found to be less cytotoxic against normal cells (NIH-3T3), which signifies their anticancer potential. Our results clearly suggested that complexes 1–4 have enhanced cytotoxic activity compared to free phen, ibu and fibu ligands or their equimolar mixtures of phen/ibu and phen/fibu (Table 6).
Table 6 Percent inhibitory concentration of the compounds on different cancer and normal cell lines
| Complexes |
Cytotoxic activities (μM) |
| CAL-27 |
NCI-H460 |
NIH-3T3 |
| IC50 ± S.D. |
IC50 ± S.D. |
IC50 ± S.D. |
|
phen (100 μM) |
>100 |
>100 |
>100 |
|
fibu (100 μM) |
>100 |
>100 |
>100 |
|
ibu (100 μM) |
>100 |
>100 |
>100 |
|
phen (50 μM) + fibu (50 μM) |
75.0 ± 1.90 |
68.00 ± 3 |
>100 |
|
phen (50 μM) + ibu (50 μM) |
85.0 ± 2 |
84.00 ± 4 |
>100 |
|
1
|
6.00 ± 2 |
3.80 ± 0.10 |
143.00 ± 0.20 |
|
2
|
6.85 ± 0.20 |
6.60 ± 0.46 |
131.30 ± 6.50 |
|
3
|
2.75 ± 0.30 |
10.00 ± 5.0 |
108 ± 4 |
|
4
|
2.00 ± 0.20 |
46.60 ± 5.74 |
102 ± 3 |
|
5-Florouracil
|
2.40 ± 0.20 |
4.54 ± 1.67 |
>100 |
Conclusions
We have successfully prepared four novel lithium, potassium and caesium complexes containing phen, and ibu/fibu co-ligands. In all four complexes, phen coordinated in an N,N-chelate mode, while the ibu-H/ibu− and fibu− ligands coordinated in a bidentate fashion. All complexes (1–4) showed significant anticancer activities against oral cancer (IC50 = 1.93 ± 0.18–6.83 ± 0.19 μM), while complexes 1 and 2 have excellent anti-lung cancer activity (IC50 = 3.75 ± 0.097 and 6.60 ± 0.46 μM), when compared to normal mouse fibroblast cells. The significant anticancer potential of these complexes clearly indicated that these complexes can be used as anti-cancer drug candidates for new drugs, but more detailed in vivo studies are required to understand their mechanism of action. Future reactions of the above mixed ligand systems with Group I and Group II metal ions are currently under investigation.
Experimental section
General considerations
Solvents, ligands and metal salts were obtained from commercial suppliers and used without further purification.
Physical measurements
Melting points were determined using a Stanford Research Systems MPA120 EZ-Melt automated melting point apparatus. UV-vis spectra were measured on an Agilent 8453 spectrophotometer using 10−5–10−6 M solutions in MeOH in the range 200–500 nm. IR spectra were obtained using a Bruker Alpha FT-IR spectrometer equipped with a Platinum single reflection diamond ATR module. The 1H NMR spectra were recorded on a Bruker 600 MHz in DMSO-d6. The degree of distortion of the coordination polyhedrons in complexes 1–4 with respect to an ideal polyhedron was calculated by continuous shape measure (CshM) theory utilizing SHAPE software.58
X-Ray crystallography
Crystal data are summarized in Table 1. Single crystals of 1–4 were mounted on a MiTeGen loop with grease and examined on a Bruker D8 Venture APEX diffractometer equipped with a Photon 100 CCD area detector at 296 (2) K using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Data were collected using APEX-II software,59 integrated using SAINT60 and corrected for absorption using a multi-scan approach (SADABS).61 The final cell constants were determined from full least squares refinement of all observed reflections. The structure was solved using intrinsic phasing (SHELXT).62 All non-H atoms were located in subsequent difference maps and refined anisotropically. H-atoms on carbons were added at calculated positions and refined with a riding model. O–H atoms were located in the difference map and refined isotropically with U(iso) riding on the O with DFIX constraints applied. The structures of 1–4 have been deposited with the CCDC (CCDC deposition numbers 2015737–2015740).† A powder X-ray diffraction (PXRD) scan was performed using a Bruker D8 Discover instrument operated at 40 kV with 2θ ranging from 5 to 50°.
Cell cytotoxicity assay
The cell cytotoxic potential of the synthesized complexes was evaluated by using 3-(4′,5′-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously against NCI-H460 (lung cancer), CAL-27 (oral cancer), and NIH-3T3 (mouse normal fibroblast) cell lines.63 The cell lines were obtained from our institute biobank facility (PCMD, ICCBS, University of Karachi). The cells were passaged and maintained in Dulbecco's modified Eagle's medium (Gibco, USA) as described previously. The respective cells (104 per well) were seeded in a 96 well microtiter plate and incubated overnight to form a uniform monolayer. The next day, the wells were washed and the monolayer was treated with different concentrations of alkali metal metallic complexes followed by incubation for a further 48 h. Following incubation, 10 μL of MTT dye (0.5 mg mL−1) was added in each well and incubated for 4 h. After incubation the dye was removed carefully and the formed insoluble formazan crystals were dissolved in DMSO. The absorbance of the plate was recorded at 570 nm, and the percent inhibition of cells was calculated. The inhibitory concentration i.e. IC50 was calculated by using Ez-Fit Enzyme Kinetics software (Perrella Scientific Inc., Amherst, NH, USA).
Preparation of [Li(phen)(H2O)(Ibu)] (1)
Li2CO3 (0.0369 g, 0.5 mmol) and ibu (0.206 g, 1.0 mmol) were stirred together in an MeOH/H2O (4:1) mixture. The reaction mixture was refluxed for ½ hour. Phen (0.198 g, 1 mmol) in 10 mL of MeOH solution was added dropwise to the above solution. The reaction mixture was further refluxed for ½ hour. The reaction mixture was then filtered and left undisturbed at room temperature. In two days small colourless transparent crystals appeared. The crystals were quite suitable for X-ray diffraction. Yield 0.289 g, 70%. Melting point 127–130 °C, selected IR data (cm−1): νOH = 3683–3110, νC–H = 2957, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1656, νCC = 1590, νCN = 1511. UV-vis (1.59 × 10−5 M, MeOH; λmax, nm; ε, M−1 cm−1): 225 (5.1 × 104), 263 (3.1 × 104). 1H NMR (600 MHz, MeOD) δ 9.11 (d, J = 3.8 Hz, 1H), 8.48 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 7.9 Hz, 1H), 3.57 (q, J = 7.1 Hz, 1H), 2.43 (d, J = 7.1 Hz, 1H), 1.83 (t, J = 13.5 Hz, 1H), 1.83 (d, J = 13.5 Hz, 1H), 1.41 (d, J = 7.1 Hz, 1H), 0.90 (d, J = 6.6 Hz, 1H).
O = 1656, νCC = 1590, νCN = 1511. UV-vis (1.59 × 10−5 M, MeOH; λmax, nm; ε, M−1 cm−1): 225 (5.1 × 104), 263 (3.1 × 104). 1H NMR (600 MHz, MeOD) δ 9.11 (d, J = 3.8 Hz, 1H), 8.48 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 7.9 Hz, 1H), 3.57 (q, J = 7.1 Hz, 1H), 2.43 (d, J = 7.1 Hz, 1H), 1.83 (t, J = 13.5 Hz, 1H), 1.83 (d, J = 13.5 Hz, 1H), 1.41 (d, J = 7.1 Hz, 1H), 0.90 (d, J = 6.6 Hz, 1H).
Preparation of [Li(phen)2(H2O)3(fibu)2] (2).
Li2CO3 (0.0369 g, 0.5 mmol) and fibu (0.244 g, 1.0 mmol) were refluxed for half an hour in 8 mL of MeOH. Phen (0.198 g, 1 mmol) in 5 mL of MeOH solution was added dropwise to the above reaction mixture. The reaction mixture was further heated and stirred for 30 minutes. The reaction mixtures were then cooled down, filtered and left undisturbed for crystallization at room temperature. In one week colorless transparent crystals were collected. Yield 0.312 g, 68%. Selected IR data (cm−1): νOH = 3587–3133, νC–H = 2957, νCO = 1722, νCC = 1621, νCN = 1514. UV-vis (1.19 × 10−5 M, MeOH; λmax, nm; ε, M−1 cm−1): 229 (3.8 × 104), 260 (4.5 × 104). 1H NMR (600 MHz, MeOD) δ 9.10 (d, J = 4.0 Hz, 1H), 8.47 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 12.3 Hz, 1H), 7.78 (d, J = 3.7 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 12.4 Hz, 1H), 3.64 (q, J = 7.1 Hz, 1H), 1.47 (d, J = 7.1 Hz, 1H).
Preparation of [K(phen)(ibu)(ibuH)]n (3).
K2CO3 (0.069 g, 0.5 mmol) and ibu (0.206 g, 1.0 mmol) were dissolved in 5 mL of an MeOH/H2O (4:1) mixture. The reaction mixture was refluxed for one hour. Phen (0.198 g, 1 mmol) in 10 mL of MeOH solution was added dropwise to the above solution. The reaction mixture was further refluxed for an additional 30 minutes. The reaction mixture was then cooled down and the solution was filtered and left undisturbed at room temperature. Colorless transparent crystals appeared in a week. Yield 0.327 g, 71%. Selected IR data (cm−1): νOH = 3646–3130, νC–H = 2951, νCO = 1701, νCC = 1618, νCN = 1505. UV-vis (9.80 × 10−5 M, MeOH; λmax, nm; ε, M−1 cm−1): 224 (6.5 × 104), 262 (3.6 × 104). 1H NMR (600 MHz, MeOD) δ 9.11 (d, J = 4.1 Hz, 1H), 8.48 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 4.3 Hz, 1H), 7.78 (d, J = 4.3 Hz, 1H), 7.26 (d, J = 7.8 Hz, 3H), 7.07 (d, J = 7.8 Hz, 3H), 3.62 (q, J = 7.1 Hz, 2H), 2.45 (d, J = 7.1 Hz, 3H), 1.85 (d, J = 13.4 Hz, 1H), 1.85 (t, J = 13.5 Hz, 2H), 1.43 (d, J = 7.1 Hz, 5H), 0.90 (d, J = 6.6 Hz, 10H).
Preparation of [Cs(phen)(fibu)]n (4).
Cs2CO3 (0.165 g, 0.5 mmol) and fibu (0.244 g, 1.0 mmol) were refluxed together in 12 mL of H2O for 30 min. To this solution, phen (0.198 g, 1 mmol) in 5 mL of MeOH was added dropwise and the reaction mixture was further refluxed for an additional 30 min. The reaction mixture was then cooled down, filtered and left undisturbed at room temperature. Colorless crystals appeared after several days. Yield 0.302 g, 64%. Selected IR data (cm−1): νC–H = 2964, νCO = 1626, νCC = 1580, νCN = 1508. UV-vis (1.48 × 10−5 M, MeOH; λmax, nm; ε, M−1 cm−1): 229 (5.5 × 104), 260 (4 × 104). 1H NMR (600 MHz, DMSO) δ 9.09 (d, J = 4.1 Hz, 1H), 8.49 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.30 (t, J = 8.3 Hz, 1H), 7.13 (d, J = 18.7 Hz, 1H), 3.22 (q, J = 7.1 Hz, 1H), 1.23 (d, J = 7.1 Hz, 1H).
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to thank the University of Nizwa and The Oman Research Council (TRC) for their generous support.
References
- Y. Shi, B.-Y. Zhang, R.-F. Zhang, S.-L. Zhang and C.-L. Ma, J. Coord. Chem., 2012, 65, 4125–4136 CrossRef CAS.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549–1560 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC.
- K. Ling, F. Men, W.-C. Wang, Y.-Q. Zhou, H.-W. Zhang and D.-W. Ye, J. Med. Chem., 2017, 61, 2611–2635 CrossRef.
- B. Albada and N. Metzler-Nolte, Chem. Rev., 2016, 116, 11797–11839 CrossRef CAS.
- M. Wenzel and A. Casini, Coord. Chem. Rev., 2017, 352, 432–460 CrossRef CAS.
- A. Merlino, Coord. Chem. Rev., 2016, 326, 111–134 CrossRef CAS.
- P. Jain, G. Pandey, D. Kumar and S. Chandra, Adv. Sci., Eng. Med., 2019, 11, 144–154 CrossRef CAS.
- R. A. Sánchez-Delgado, M. Navarro, H. Pérez and J. A. Urbina, J. Med. Chem., 1996, 39, 1095–1099 CrossRef.
- C. Dendrinou-Samara, G. Tsotsou, L. V. Ekateriniadou, A. H. Kortsaris, C. P. Raptopoulou, A. Terzis, D. A. Kyriakidis and D. P. Kessissoglou, J. Inorg. Biochem., 1998, 71, 171–179 CrossRef CAS.
- D. L. Eaton and E. P. Gallagher, Annu. Rev. Pharmacol. Toxicol., 1994, 34, 135–172 CrossRef CAS.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- S. Kumar, S. Garg, R. P. Sharma, P. Venugopalan, L. Tenti, V. Ferretti, L. Nivelle, M. Tarpin and E. Guillon, New J. Chem., 2017, 41, 8253–8262 RSC.
- L. Tabrizi, L. O. Olasunkanmi and O. A. Fadare, Dalton Trans., 2019, 48, 728–740 RSC.
- C. Abbehausen, Metallomics, 2019, 11, 15–28 RSC.
- Z. A. Radi and K. N. Khan, J. Toxicol. Sci., 2019, 44, 373–391 CrossRef CAS.
- K. Kafarska, D. Czakis-Sulikowska and W. Wolf, J. Therm. Anal. Calorim., 2009, 96, 617–621 CrossRef CAS.
- S. N. Omar and H. Abu Ali, J. Coord. Chem., 2017, 70, 2436–2452 CrossRef CAS.
- E. H. H. Chow, F. C. Strobridge and T. Friščić, Chem. Commun., 2010, 46, 6368–6370 RSC.
- L. C. Cunha, D. P. Lage, L. S. Ferreira, L. Saboia-Vahia, E. A. F. Coelho, V. S. Belo, R. G. Teixeira-Neto, L. F. Soares, R. C. R. Chagas and E. S. da Silva, Inorg. Chem. Commun., 2020, 113, 107756 CrossRef CAS.
-
S. H. Roth, Rheumatic therapeutics, McGraw-Hill Companies, 1985 Search PubMed.
- P. Kolhe, E. Misra, R. M. Kannan, S. Kannan and M. Lieh-Lai, Int. J. Pharm., 2003, 259, 143–160 CrossRef CAS.
- R. E. Harris, S. Kasbari and W. B. Farrar, Oncol. Rep., 1999, 6, 71–74 CAS.
- T. Hida, J. Leyton, A. N. Makheja, P. Ben-Av, T. Hla, A. Martinez, J. Mulshine, S. Malkani, P. Chung and T. W. Moody, Anticancer Res., 1998, 18, 775–782 CAS.
- S. K. Mohamed, M. Albayati, W. A. M. Omara, A. A. Abdelhamid, H. Potgeiter, A. S. Hameed and K. M. Al-Janabi, J. Chem. Pharm. Res., 2012, 4, 3505–3517 CAS.
- M. F. Zayed and M. H. Hassan, Saudi Pharm. J., 2014, 22, 157–162 CrossRef.
- P. Governa, M. G. Cusi, V. Borgonetti, J. M. Sforcin, C. Terrosi, G. Baini, E. Miraldi and M. Biagi, Biomedicines, 2019, 7, 73 CrossRef CAS.
- P. Zimmermann and N. Curtis, Antimicrob. Agents Chemother., 2017, 61, e02268-16 CrossRef.
- M. Paul and P. Dastidar, Chem. – Eur. J., 2016, 22, 988–998 CrossRef CAS.
- H.-Q. Hao, M.-X. Peng, L. Li, S.-J. Li and X.-R. Wen, Inorg. Chem. Commun., 2012, 20, 263–265 CrossRef CAS.
- X. Gao, R. Cui, M. Zhang and Z. Liu, Mater. Lett., 2017, 197, 217–220 CrossRef CAS.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS.
- A. B. Lago, A. Pino-Cuevas, R. Carballo and E. M. Vázquez-López, Dalton Trans., 2016, 45, 1614–1621 RSC.
- M. Sarker, S. Shin and S. H. Jhung, ACS Omega, 2019, 4, 9860–9867 CrossRef CAS.
- H. A. Ali, H. Fares, M. Darawsheh, E. Rappocciolo, M. Akkawi and S. Jaber, Eur. J. Med. Chem., 2015, 89, 67–76 CrossRef.
- J. E. Weder, C. T. Dillon, T. W. Hambley, B. J. Kennedy, P. A. Lay, J. R. Biffin, H. L. Regtop and N. M. Davies, Coord. Chem. Rev., 2002, 232, 95–126 CrossRef CAS.
- M. O’Connor, A. Kellett, M. McCann, G. Rosair, M. McNamara, O. Howe, B. S. Creaven, S. McClean, A. Foltyn-Arfa Kia and D. O’Shea, J. Med. Chem., 2012, 55, 1957–1968 CrossRef.
- I. Ott, K. Schmidt, B. Kircher, P. Schumacher, T. Wiglenda and R. Gust, J. Med. Chem., 2005, 48, 622–629 CrossRef CAS.
- I. Ott, B. Kircher, C. P. Bagowski, D. H. W. Vlecken, E. B. Ott, J. Will, K. Bensdorf, W. S. Sheldrick and R. Gust, Angew. Chem., Int. Ed., 2009, 48, 1160–1163 CrossRef CAS.
- S. R. Shah, Z. Shah, A. Khan, A. Ahmed, Sohani, J. Hussain, R. Csuk, M. U. Anwar and A. Al-Harrasi, ACS Omega, 2019, 4(25), 21559–21566 CrossRef CAS.
- J. H. N. Buttery, G. A. Koutsantonis, S. Mutrofin, N. C. Plackett, B. W. Skelton, C. R. Whitaker and A. H. White, Z. Anorg. Allg. Chem., 2006, 632, 1829–1838 CrossRef CAS.
- C. Ma, W. Wang, X. Zhang, C. Chen, Q. Liu, H. Zhu, D. Liao and L. Li, Eur. J. Inorg. Chem., 2004, 3522–3532 CrossRef CAS.
- A. E. Platero-Prats and S. de la Peña-O, J. Am. Chem. Soc., 2012, 134, 4762–4771 CrossRef CAS.
- H. Boumans, M. C. M. van Gaalen, L. A. Grivell and J. A. Berden, J. Biol. Chem., 1997, 272, 16753–16760 CrossRef CAS.
- C. Sartorius, M. F. Dunn and M. Zeppezauer, Eur. J. Biochem., 1988, 177, 493–499 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- J. Skiba, A. Kowalczyk, P. Stączek, T. Bernaś, D. Trzybiński, K. Woźniak, U. Schatzschneider, R. Czerwieniec and K. Kowalski, New J. Chem., 2019, 43, 573–583 RSC.
- N. J. Aburto, A. Ziolkovska, L. Hooper, P. Elliott, F. P. Cappuccio and J. J. Meerpohl, BMJ, 2013, 346, f1326 CrossRef.
- N. S. Awwad, K. A. Saleh, H.-A. S. Abbas, A. M. Alhanash, F. S. Alqadi and M. S. Hamdy, Mater. Res. Express, 2019, 6, 55403 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- M. Neykov, T. van Almsick and G. Dimitrov, Z. Anorg. Allg. Chem., 2006, 632, 1554–1559 CrossRef CAS.
- S. R. Shah, Z. Shah, N. Ullah, J. Hussain, R. Al-Harrasi, A. Khan, J. M. Rawson, A. Al-Harrasi and M. U. Anwar, Acta Crystallogr., Sect. C: Struct. Chem., 2019, C75, 294–303 CrossRef.
- M. S. Hundal, G. Sood, P. Kapoor and N. S. Poonia, J. Chem. Crystallogr., 1991, 21, 395–398 CrossRef CAS.
- S. Nishigaki, H. Yoshioka and K. Nakatsu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 875–879 CrossRef.
-
M. Llunell, M. Casanova, D. Cirera, J. Alemany, P. Alvarez and S. Shape, Version 2.1., Univ. Barcelona, Barcelona, Spain, 2013 Search PubMed.
- APEX-II, Bruker AXS, Madison, Wisconsin, USA, 2012.
-
SAINT, Bruker AXS, Madison, Wisconsin, USA, 2012.
-
SADABS, Bruker AXS, Madison, Wisconsin, USA, 2001.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef.
- B. Zehra, A. Ahmed, R. Sarwar, A. Khan, U. Farooq, S. A. Ali and A. Al-Harrasi, Cancer Manage. Res., 2019, 11, 1667 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Zarbad
Shah
b,
Ajmal
Khan
a,
Ayaz
Ahmed
c,
Shariqa
Khwaja
c,
Rene
Csuk
ab,
Zarbad
Shah
b,
Ajmal
Khan
a,
Ayaz
Ahmed
c,
Shariqa
Khwaja
c,
Rene
Csuk