
Time-resolved SANS reveals pore-forming peptides cause rapid lipid reorganization†
Michael H. L.
Nguyen
a,
Mitchell
DiPasquale
 a,
Brett W.
Rickeard
a,
Caesar G.
Yip
a,
Kaity N.
Greco
a,
Elizabeth G.
Kelley
*b and
Drew
Marquardt
*ac
a,
Brett W.
Rickeard
a,
Caesar G.
Yip
a,
Kaity N.
Greco
a,
Elizabeth G.
Kelley
*b and
Drew
Marquardt
*ac
aDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario, Canada. E-mail: drew.marquardt@uwindsor.ca
bNIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD, USA. E-mail: egk@nist.gov
cDepartment of Physics, University of Windsor, Windsor, Ontario, Canada
First published on 7th December 2020
Abstract
Cells depend on proper lipid transport and their precise distribution for vital cellular function. Disruption of such lipid organization can be initiated by external agents to cause cell death. Here, we investigate two antimicrobial pore-forming peptides, alamethicin and melittin, and their influence on lipid intervesicular exchange and transverse lipid diffusion (i.e. flip-flop) in model lipid vesicles. Small angle neutron scattering (SANS) and a strategic contrast matching scheme show the mixing of two isotopically distinct dimyristoylphosphocholine (DMPC) vesicle populations is promoted upon the addition of high (1/40) and low (1/150, 1/1000) peptide-to-lipid (P/L) molar ratios. Parsing out the individual exchange and flip-flop rate constants revealed that alamethicin increases both DMPC flip-flop and exchange by ≈2-fold when compared to methanol alone (the carrier solvent of the peptides). On the other hand, melittin affected DMPC flip-flop by a factor of 1 to 4 depending on the concentration, but had little effect on inter-vesicle lipid exchange at low P/L ratios. Thermodynamic parameters measured at high protein concentrations (P/L = 1/40) yielded remarkable similarity in the values obtained for both peptides, indicating likeness in their mechanism of action on lipid motion despite differences in their proposed oligomeric pore structures. The entropic contributions to the free energy of activation became favorable upon peptide addition, while the enthalpy of activation remained the major barrier to lipid exchange and flip-flop.
1 Introduction
Antibiotic-resistant bacteria continue to increase in number, and along the way develop diverse mechanisms that threaten to make current treatment options obsolete.1 The search for novel antibiotic agents has thus become evermore important. Antimicrobial peptides (AMPs) are promising templates to generate bactericides.2 They are a varied class of conserved peptides used in organismal defense against pathogenic microbes. Conventional antibiotics often target bacterial proteins or biochemical processes to cause cell death. Many of these targets, however, vary between bacterial strains and are prone to mutation, which can render the antibiotics ineffective. Meanwhile, most AMPs exert their cytotoxic activity upon membranes, killing a wide range of bacterial species (from Gram-negative to positive bacteria).3 The cationicity of AMPs direct their selectivity towards negatively-charged bacterial membranes. To adapt to AMP activity, the bacteria must make time- and energy-taxing changes to the global membrane composition.4 The success of AMPs' membrane-targeted activity therefore stems from the difficulty bacteria face to adapt. But despite this inherent advantage to conventional antibiotics, the major impediment to successful AMP development is the challenge in elucidating their mode of action.3,5 Certain structural and/or mechanistic features make some AMPs more successful than others. Uncovering these features will allow researchers to exploit and fine-tune desirable actions into novel therapeutics.Current thought suggests many AMPs work by forming pores that disrupt the careful biochemical and electrochemical gradients of cells. It is also thought that the perturbations in local bilayer structure caused by the pores can lead to lipid reorganization.6 But despite intense study, the complete elucidation of many antimicrobial mechanisms remains contentious. Many questions surround key peptide–membrane events.3,5,7 For instance, the presence of peptidic pores and their relation to cell death has yet to be confirmed and properly characterized in vivo.5 Much greater concentrations of peptides are often needed to observe stable oligomeric pore formation8in vitro, yet they lyse erythrocytes and kill microbes in vivo at sub-mmol L−1 concentrations.9 This discrepancy points to a different mode of action of antimicrobial peptides other than oligomeric pore formation.
Of interest to this study, two key processes in lipid reorganization are transverse lipid diffusion (flip-flop) and interbilayer exchange (or transfer). Intrabilayer flip-flop occurs via the translocation of lipids between leaflets, which destroys the well-defined lipid asymmetry, i.e. the distinct lipid composition between leaflets, present in nearly all natural membranes. In the absence of additives, passive flip-flop in pure lipid membranes is slow (on the order of hours to days10). Through the inclusion of pore-forming peptides, the lipid asymmetry, crucial for normal membrane function as well as for the function of membrane-associated proteins, can be quickly destroyed.11 A variety of biochemical and biophysical techniques, such as fluorescence-quenching assays,6,12,13 sum frequency vibrational spectroscopy,14 neutron reflectometry,15 solutions nuclear magnetic resonance,16 and electrical capacitance-based measurements17 have been applied to measure peptide effects on lipid organization. These techniques are limited in that many require the use of extrinsic probes, which are bulky and bilayer perturbing,18 or use flat planar bilayers that can be riddled with defects that accelerate flip-flop.19
With respect to the transfer of lipid monomers between bilayers, it is a fundamental process by which membranes are constructed and maintained. Lipid transfer or exchange, therefore, is intrinsically linked to bilayer homeostasis. The possibility of a peptide-mediated lipid exchange process has been suggested before and is considered a measure of AMP potency.20,21 Typically, lipid exchange has been measured using radioisotope22,23 or fluorescent20,21,24 lipids. Radioisotopic experimentation requires the use of multiple vesicle populations that are physically dissimilar, differing in size22 or charge23 for separation, while the use of fluorophore-labeled lipids in lipid exchange is marred by their perturbing nature of bilayers and their disparity to natural lipids. Again, experimental artifacts can arise through these experimental set-ups, eliciting the need for probe-free procedures that measure the true rates of lipid exchange and flip-flop.
Here we study the effects of two well-studied AMPs, alamethicin (Alm) and melittin (Mel), on lipid flip-flop and exchange in model lipid vesicles using time-resolved small angle neutron scattering.25 Alamethicin has a 20 amino acid primary structure and forms membrane-spanning voltage-gated channels.26 Melittin is a 26 amino acid long peptide that permeabilizes cells.26 Both are amphiphilic and, owing to their non-polar face, bind strongly to membranes and fold into α-helical structures. At a concentration threshold, Alm and Mel transition from transient pore formation to stable equilibrium pores.27,28 This transition is also defined by lipid and sterol content, temperature, and solvent. Both peptides display similar effects on membrane-thinning,29 induction of phospholipid asymmetry,30 phospholipid scrambling27 and cholesterol reorganization,31,32 but form oligomeric pores with different putative structures. Our probe-free approach allows us to simultaneously investigate the effects of pore structure on lipid flip-flop and interbilayer exchange in free-floating vesicles (as seen in Fig. 1) and calculate their respective rate constants. Both peptides were found to accelerate lipid reorganization, even at low P/L below the threshold to form oligomeric pores, perhaps suggesting a different mechanism of action for these AMPs.
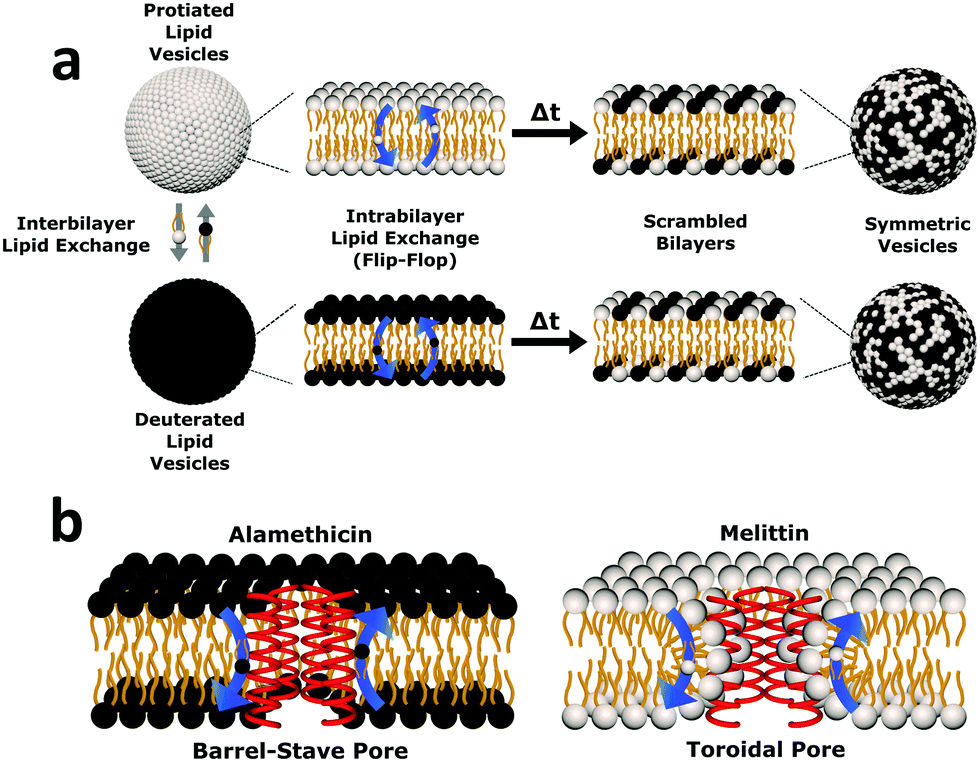 | ||
| Fig. 1 Cartoon schematic of the experimental protocol used in this study. (a) Uniform non-deuterated and deuterated DMPC LUV populations are mixed. Subsequent DMPC monomer transfer between and across bilayers occur (dubbed interbilayer exchange and intrabilayer exchange or flip-flop, respectively). Over time, the combination of such diffusive processes produces vesicles composed of both isotopic variants of DMPC, yielding symmetric LUVs with a distinct neutron scattering pattern from their isotopically-distinct precursors. (b) The transmembrane pore structure of alamethicin and melittin are illustrated. Alamethicin is shown to form pores lined solely by peptide monomers, while melittin is shown to display peptide and lipid components in its pore structure. The difference in pore structure has been postulated to influence lipid flip-flop6 but its role in interbilayer exchange has not been as well defined. | ||
2 Materials and methods
2.1 Materials
1,2-Dimyristoyl-sn-glycero-3-phosphoglycerol [14:0/14:0 PG, DMPG], 1,2-dimyristoyl-d54-sn-glycero-3-phosphocholine [14:0(d27)/14:0(d27) PC, d-DMPC] and 1,2-dimyristoyl-sn-glycero-3-phosphocholine [14:0/14:0 PC, h-DMPC] were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) as powders and used as received. Alamethicin and melittin were obtained from Sigma Aldrich (St. Louis, MO) and kept in deuterated methanol, Cambridge Isotope Laboratories, (Andover, MA) as stocks. D2O was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). All other reagents used were of ACS grade and purchased from Sigma Aldrich (St. Louis, MO).2.2 Large unilamellar vesicle sample preparation
Aliquots of h-DMPC and d-DMPC samples were prepared separately. Precise amounts of h-DMPC and d-DMPC were aliquoted and mixed with 5 mol% DMPG dissolved in chloroform. DMPG was added to favor the formation of unilamellar vesicles, as the presence of multilamellae would prevent the measurement of the true lipid exchange and flip-flop rates. Organic solvent was evaporated under a stream of nitrogen and via vacuum oven at 50 °C overnight. The lipid films were hydrated with 45% D2O by volume (the remainder being ultra-pure H2O), which corresponds to the desired contrast match point of uniformly mixed vesicles composed of h-DMPC and d-DMPC. The resulting lipid concentration was 17 mg mL−1. Subsequently, h-DMPC and d-DMPC samples underwent at least 5 freeze–thaw–vortex cycles, and then extruded separately through 100 nm diameter-sized filters at ≈35 °C, well above the transition temperature of DMPC to facilitate extrusion. Peptides in methanol were introduced to preformed populations of protiated and deuterated DMPC large unilamellar vesicles (h-LUV and d-LUV, respectively). Before introducing h-LUVs to the d-LUVs, both populations with peptides were incubated for one hour prior to mixing, which allowed for proper peptide binding and insertion to the membrane. Such a step eliminates the possible mechanistic artifacts that can hamper an investigation focused on the presence of pores and lipid flip-flop and exchange. Dynamic light scattering (DLS) on a Wyatt DynaPro NanoStar (Santa Barbara, CA, USA) was used to determine the mean particle diameter to ensure the peptide addition step did not produce morphological changes. Measurements conducted at 30 °C found a mean particle diameter of ≈140 nm before addition and after mixing, indicating vesicle integrity (as seen in Table S1 in the ESI†).2.3 Circular dichroism
Circular Dichroism (CD) measurements were performed using a Chirascan CD spectrometer (Applied Photophysics, Leatherhead, Surrey, UK). Samples were equilibrated at 37 °C for at least 15 min in a 1 mm path-length cell before data collection. Data were collected between 195 nm and 280 nm with a 1 nm bandwidth and 5 s averaging per point. The samples were diluted with D2O such that the peptide concentration in solution was between 0.02 and 0.6 mmol L−1. The background signal from the pure lipid controls at the corresponding sample concentration was subtracted from the peptide-containing samples. Raw data were converted to mean residue ellipticity by [θ] = θ/(10lc(N − 1)), where Θ is the measured raw data in mdeg, l is the path length of the cell in cm, c is the protein concentration in mol L−1, and N is the number of amino acids. The signal for the P/L 1/1000 was too low to collect spectra. CD spectra can be viewed in the ESI,† as Fig. S1.2.4 Measuring lipid flip-flop and exchange using SANS
SANS were performed on the Very Small Angle Neutron Scattering (VSANS) instrument located at the National Institute of Standards and Technology Center for Neutron Research (NIST-CNR, Gaithersburg, MD). The experiments took advantage of the high flux, white beam configuration using a neutron wavelength, λ, of 5.3 Å and a wavelength spread of Δλ/λ = 40%. Scattered neutrons were collected with two detector carriages with sample-to-detector distances of 4 m and 19 m which resulted in a q-range of 0.003 Å−1 to 0.12 Å−1. The scattering vector, q, is represented by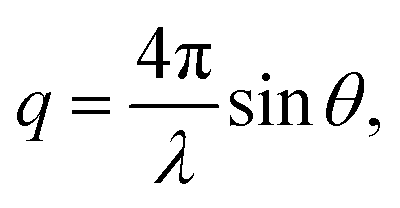 where 2θ is the scattering angle.
where 2θ is the scattering angle.
Aliquots of h-LUV and d-LUV were mixed at equal portions, pipetted into 1 mm or 2 mm path length quartz banjo cells and immediately placed into VSANS sample blocks temperature controlled by Peltier control systems and measured. Samples were not removed from the sample blocks until vesicle mixing was complete. Three minute measurements were periodically conducted to ensure adequate time-points. Over time, the distinct isotopic populations are lost due to mixing between the populations, eventually resulting in uniform LUVs composed of equal portions of h-DMPC and d-DMPC. As a result, the total scattering intensity produced by the samples slowly decreases or “decays” as the liposome neutron scattering length density approaches that of the 45% D2O buffer. The intensity scattering plots were calculated using the Igor Pro reduction software and VSANS macros provided by the NIST-CNR which subtracted scattering contributions from various background sources (empty cell, background radiation) as well as corrected for the detector pixel sensitivity and sample transmission.33 Plots from the two detector-carriages were stitched to produce the final intensity curves seen in Fig. 2a. The intensity decays were normalized assuming first order kinetics according to
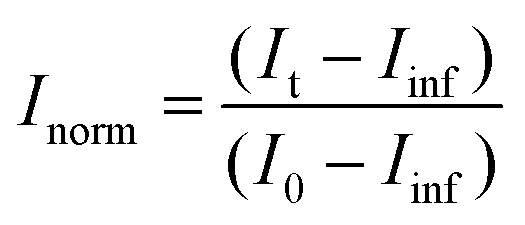 | (1) |
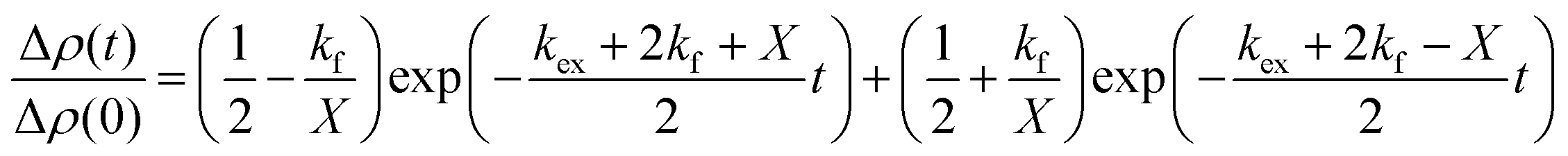 | (2) |
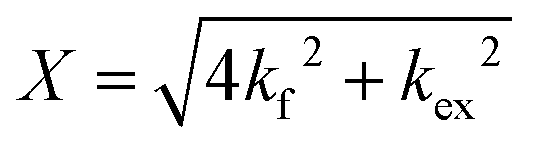 and
and 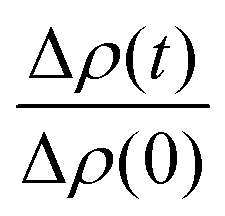 is the normalized intensity decay, models the interbilayer exchange and lipid flip-flop independently. The standard error is derived from fits to the data using OriginLab software. Rate constants (k) were converted to half-times (t1/2) using
is the normalized intensity decay, models the interbilayer exchange and lipid flip-flop independently. The standard error is derived from fits to the data using OriginLab software. Rate constants (k) were converted to half-times (t1/2) using| t1/2 = ln(2)/k | (3) |
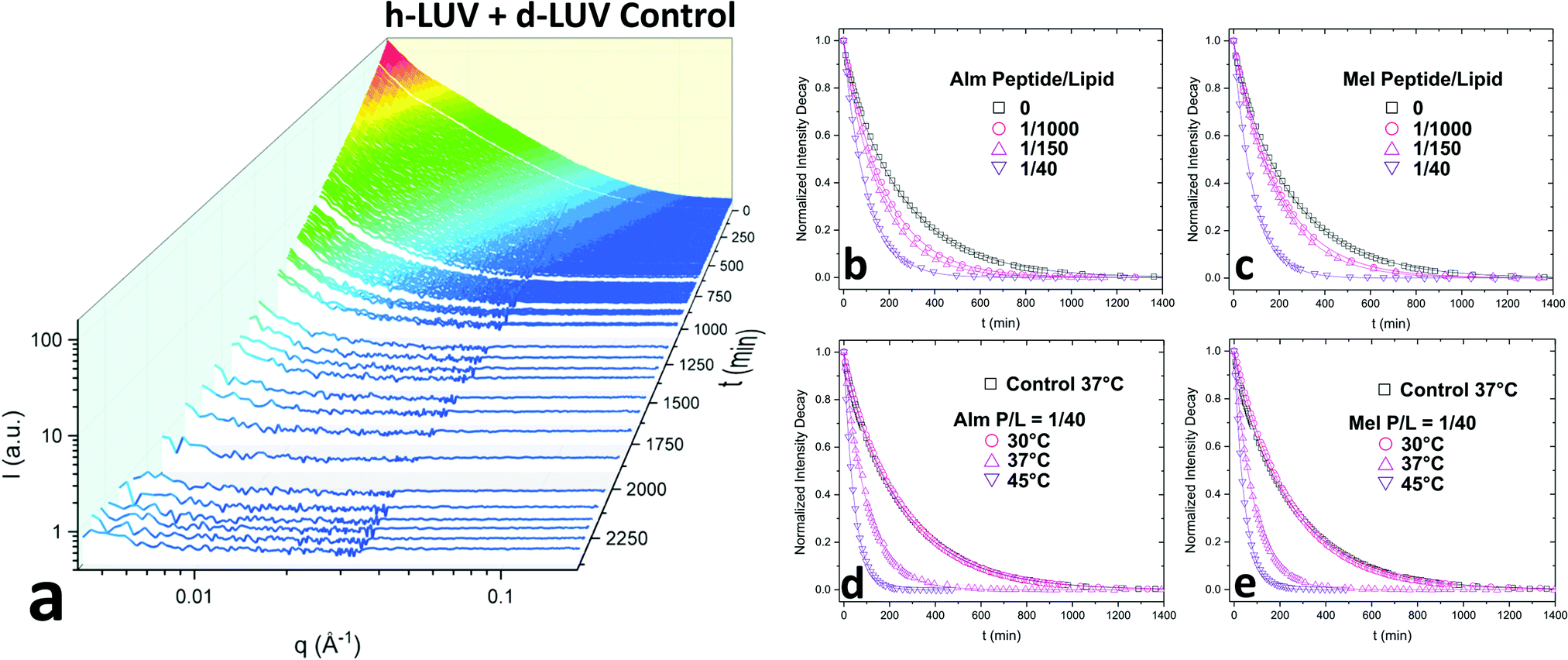 | ||
| Fig. 2 (a) I(q) scattering curves at various time points after mixing of h-LUVs and d-LUVs highlighting the time-resolved reduction in intensity. (b) and (c) Represents normalized total intensity decay curves of Alm and Mel, respectively. Measurements were of P/L ratios at 0, 1/1000, 1/150, and 1/40 and measured at physiological temperature, 37 °C. (d) and (e) Show a temperature series of data conducted at 30 °C, 37 °C, and 45 °C of Alm and Mel, respectively, at a set P/L ratio of 1/40. Included in the temperature decay plots is a h-LUV and d-LUV control sample without methanol for comparative purposes. The bold continuous lines in the normalized intensity decay curves are fits used to derive the flip-flop and exchange rate constants of DMPC. | ||
Measurements were taken at several temperatures (30 °C, 37 °C and 45 °C), which allowed for the thermodynamic analysis of both DMPC flip-flop and exchange rates. Activation energies (Ea) were derived from the linear fits of Arrhenius plots and used to calculate various thermodynamic quantities (i.e. ΔH‡, enthalpy of activation; ΔS‡, entropy of activation; ΔG‡, free energy of activation) using transition state theory as outlined by Homan and Pownall.34 In brief, the above thermodynamic quantities were calculated using the following:
| ΔH‡ = Ea − RT | (4) |
ΔS‡ = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(NhX/RT) ln(NhX/RT) | (5) |
| X = kexp(ΔH‡/RT) | (6) |
| ΔG‡ = ΔH‡ − TΔS‡ | (7) |
It is important to note that the lateral and transverse organization of peptides (e.g. aggregation) in vesicles can contribute to the measured scattered intensity.16,30 Such features can form small, but important, differences in the final, contrast-matched scattering spectra of different samples. This potential source of discrepancy is particularly exacerbated by the use of peptides that differ in aggregate structure and at varying concentrations. As these final curves are integral to further calculations and fitting, the most accurate data sets must be attained. For this reason, we allowed each sample to reach their scattering baseline, as exemplified in Fig. 2a. Instead of using already lipid scrambled control samples to measure the infinity time-point as typically done,25,35 we applied an exponential decay fit to find the asymptotic intensity values of each sample and used the resulting value to calculate normalized total intensities. Given the numerous conditions (e.g. methanol concentrations, peptide concentrations, temperatures and their different combinations) that may affect the final scrambled curves, it was logistically and temporally difficult to make and measure that many scrambled samples. Through the normalization method used in this study, we avoided using a single end-point for samples that may scatter differently. Still, a scrambled control was measured but was ill fit for many of the samples.
3 Results
To elucidate changes in lipid organization under peptidic conditions, time-resolved SANS was used to study lipid mixing in free-floating DMPC vesicles. In brief, chain perdeuterated d-DMPC LUVs were mixed with fully protiated h-DMPC LUVs. Over time, the continuous exchange of lipid monomers (i.e. between inner and outer leaflets and between isotopic populations) results in a single uniform LUV population of d- and h-DMPC (Fig. 1a). The neutron scattering length density contrast of the solvent is chosen to match this single LUV population – a technique often referred to as contrast matching. The transition from two isotopically distinct populations to a single uniform population is then tracked by measuring the decay in scattered intensity over time as the lipids mix and the contrast is lost. The application of contrast matching avoids the use of structural perturbing probes and has been applied successfully to study exchange in liposomes and peptidoliposomes.25,35–37Here we use the same concepts to study the effects of two pore-forming AMPs, Alm and Mel, on the lipid exchange. A comparison of these model peptides was conducted to elucidate the role of their putative oligomeric pore structure (illustrated in Fig. 1b) in facilitating lipid dynamics. Both the effects of peptide concentration and temperature dependence were investigated. The corresponding peptides were measured at the same peptide-to-lipid ratios (P/L = 1/1000, 1/150, and 1/40) to compare their potency in altering lipid organization, while temperature-dependent measurements were done to conduct a thermodynamic analysis of lipid dynamics at high P/L to better understand the effects of the different oligomeric pore structures.
Fig. 2a shows the collective scattering curves of a peptide-free sample that are typical of lipid mixing.25,37,38 At initial mixing (t = 0), the high intensity stems from the neutron scattering contrast between the deuterated and protiated lipid populations to the light/heavy water buffer. The intensity decay over time is indicative of the formation of LUVs with almost equal proportions of d-DMPC and h-DMPC due to intervesicular lipid exchange and lipid flip-flop. Intensity decays were quantified by calculating the scattering invariant (i.e. integration of the curve) over the measured q-range of 0.003–0.12 Å−1. The integrated intensities were normalized according to eqn (1). The resulting normalized intensity decay curves can be seen in Fig. 2b–d, where increasing peptide concentration and temperature accelerates liposomal mixing.
3.1 Exchange and translocation of DMPC in peptidoliposomes
Consistent with our initial expectations, the introduction of pore-forming peptides reduced the time needed to observe the minimum intensity baseline (Fig. 2b–e). In other words, Alm and Mel promoted lipid mixing, showing their potent ability to disrupt lipid organization in model membranes. Faster exchange was observed at all P/L concentrations (1/1000, 1/150, and 1/40) of Alm (Fig. 2b) and Mel (Fig. 2c). Comparing Fig. 2b and c suggests Alm is qualitatively better at promoting liposomal mixing at P/L = 1/1000 and 1/150 as the curves decay faster than the analogous Mel samples.As expected of thermodynamic processes, the lipid dynamics exhibit a temperature-dependency at the measured temperatures of 30 °C, 37 °C and 45 °C (Fig. 2d and e). As the temperature increases, so too does the mixing.
To determine whether flip-flop or interbilayer exchange was more affected, and to what extent, their respective rate constants and half-times were calculated using a double-exponential decay function, which assumes both processes follow first order kinetics.25 The half-time values of flip-flop and exchange for the DMPC control without methanol and peptide were measured for comparison. These peptide-free DMPC values are slightly faster than those previously reported for vesicular systems of pure DMPC.22,25 This difference is likely due to the small fraction of negatively-charged DMPG introduced, as also seen by Wah and colleagues.38 It is interesting to note that the rate they found increased with DMPG concentration, which was opposite to the trend reported by Brown et al. where neutral lipid flip-flop was not affected by increasing molar fraction of lipids with negatively-charged headgroups, phosphatidylserine.39
Since the incorporation of peptides used methanol as a carrier, the effects of methanol on lipid dynamics must be delineated to accurately assess the influence of Alm and Mel. We previously found methanol increased the rate of DMPC flip-flop exponentially and exchange linearly.37 Converting these rates into half-times, a methanol standard curve was constructed, as seen in Fig. 3.
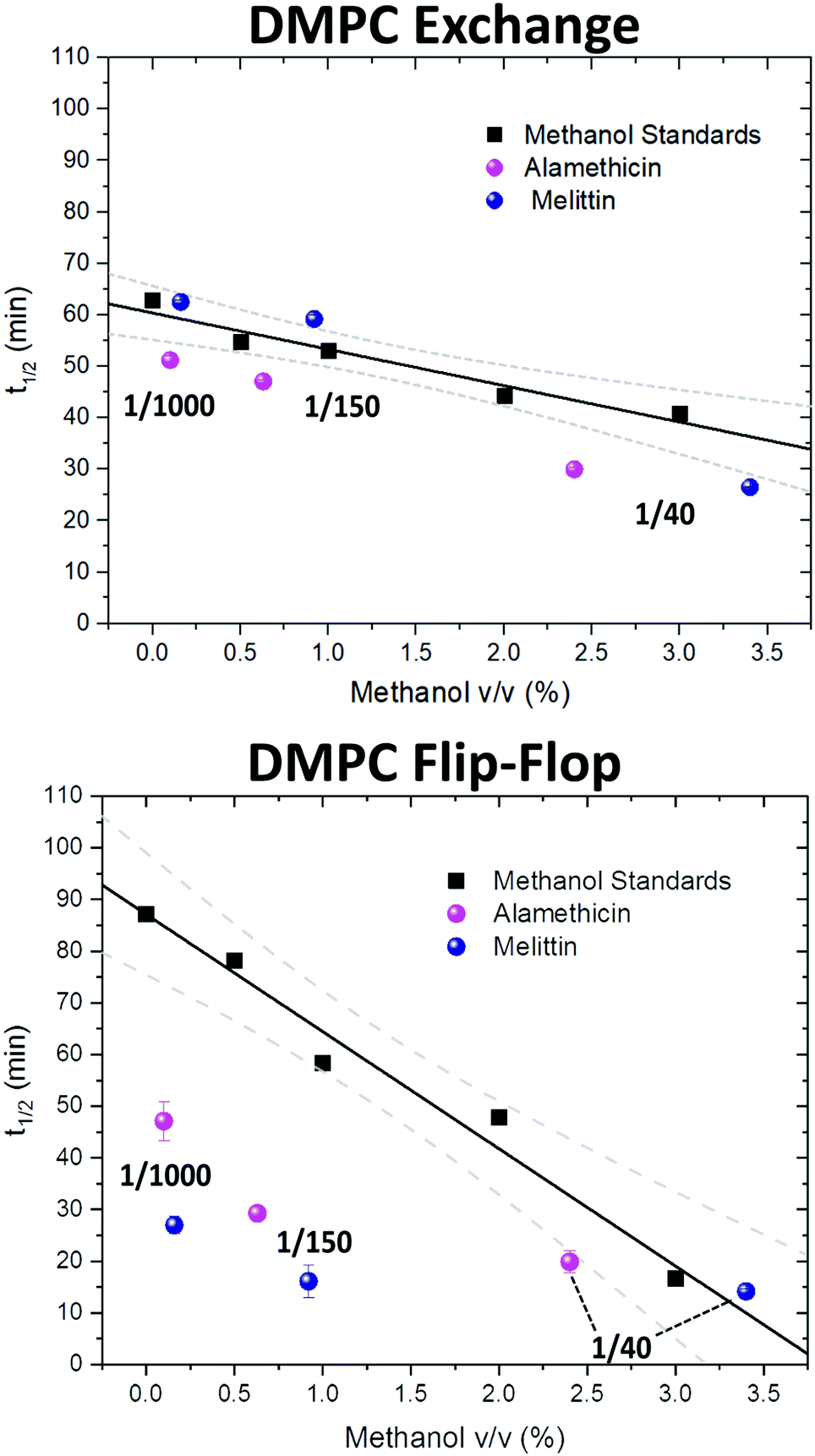 | ||
| Fig. 3 Half-times measured of DMPC flip-flop and DMPC exchange under various organic solvent and peptide conditions. The addition of each AMP required the introduction of methanol. Thus, a series of methanol values (black squares) and standard curve (solid black line) are shown to account for contributions by the carrier solvent.37 Each peptide concentration corresponds to a volume percent of methanol and was plotted accordingly. Top panel: DMPC exchange half-times at several P/L ratios of Alm (pink spheres) and Mel (navy spheres). The exact P/L ratios are found in the plots. Gray dashed lines represent a 95% confidence interval. Bottom panel: DMPC flip-flop half-times in the presence of various P/L of Alm (pink spheres) and Mel (navy spheres). | ||
AMP samples were plotted according to their final methanol volume percent and compared to the methanol standards (0–3%). Exchange half-times decreased with increasing Alm mole fraction. Compared against the methanol control (Fig. 3, top), Alm decreased the exchange half-time by a factor of ≈2. On the other hand, the relation between Mel and DMPC exchange is intricate, but on average the factor is ≈1, signifying a lack of change. On closer inspection, at P/L = 1/1000 and 1/150, Mel has no effect or slightly increases exchange, respectively (Fig. 3, top). However, at P/L = 1/40, Mel causes a decrease in exchange half-time.
Overall, DMPC flip-flop is slower than intervesicle exchange, but the rate of DMPC flip-flop increased in the presence of Mel and Alm compared to the control without peptide (Fig. 3, bottom). Mel is more potent at lower concentrations (P/L = 1/1000 and 1/150) which are not suspected to form oligomeric pore structures.8 At P/L = 1/40, the half-time (t1/2) values of DMPC flip-flop become comparable for both peptides. Overall, flip-flop t1/2 is reduced ≈2-fold by Alm and up to 4-fold (depending on P/L ratio) by Mel. Interestingly, the minimum t1/2 is suspected to be between 1/150–1/40 for both peptides, indicating a maximum rate of peptide-induced lipid flip-flop or that the time-scale of this accelerated lipid flip-flop is beyond the experimental set-up. Krauson and colleagues showed concentrations of 1/1000 of Alm amd Mel can cause calcein leakage,27 which we confirmed through our own calcein leakage assays (data not shown). In terms of the t1/2 minima, the potential of oligomeric pore formation at all three concentrations are discussed below.
Comparing the half-times suggests that both peptides had an overall greater affect on the rate of flip-flip compared to inter-vesicle exchange. However, the data in Fig. 3 show that Alm has a clear dual effect on both processes, influencing both lipid flip-flop and exchange; whereas, Mel's role is only clear with respect to flip-flop. This finding indicates membrane–peptide interactions differ depending on peptide identity, despite Alm and Mel possessing similar lytic abilities.
3.2 Thermodynamics of DMPC exchange and flip-flop under pore-forming peptides
Alm and Mel are traditionally thought to be barrel-stave and toroidal pore-formers, respectively (Fig. 1). Theoretically, the lipid- and peptide-lined toroidal pores formed by Mel should promote lipid translocation over peptide-only lined barrel-stave pores formed by Alm, as lipids can readily diffuse through the toroidal pore lining without a need to desolvate their headgroups, bypassing the large enthalpic penalty associated with passive lipid flip-flop. From a thermodynamic view, it may be possible to determine the extent of membrane perturbation by these AMPs and to compare their mechanisms of action. Thus, to investigate potential mechanistic differences in AMP pore-structure on lipid dynamics, Alm and Mel at sufficient oligomeric pore-forming concentrations (P/L = 1/40) were introduced to DMPC LUVs.Interbilayer exchange and flip-flop in DMPC vesicles exhibit Arrhenius behavior, as demonstrated by a linear dependence between ln(k) versus inverse temperature.25 Such behavior continues under conditions of methanol and AMP (P/L = 1/40) as seen in Fig. 4. Unperturbed DMPC exchange and flip-flop thermodynamic quantities (reported in Table 1 at 37 °C) are in agreement to values reported by Nakano et al.,25 despite the inclusion of 5 mol% DMPG to prevent multilamellae formation. This contrasts values determined for flip-flop thermodynamics by Wah et al.,38 where a near 2-fold increase in the Ea of DMPC flip-flop is seen in vesicles containing 6 mol% DMPG. We note Wah and colleagues monitored a temperature range of (48 °C, 53 °C and 58 °C) – outside the range of the present study. Therefore, the temperature dependence of DMPC flip-flop may not occur in a predictable, monotonic fashion (though it does in the present temperature range), which has been similarly observed with cholesterol in phosphatidylserine bilayers.40
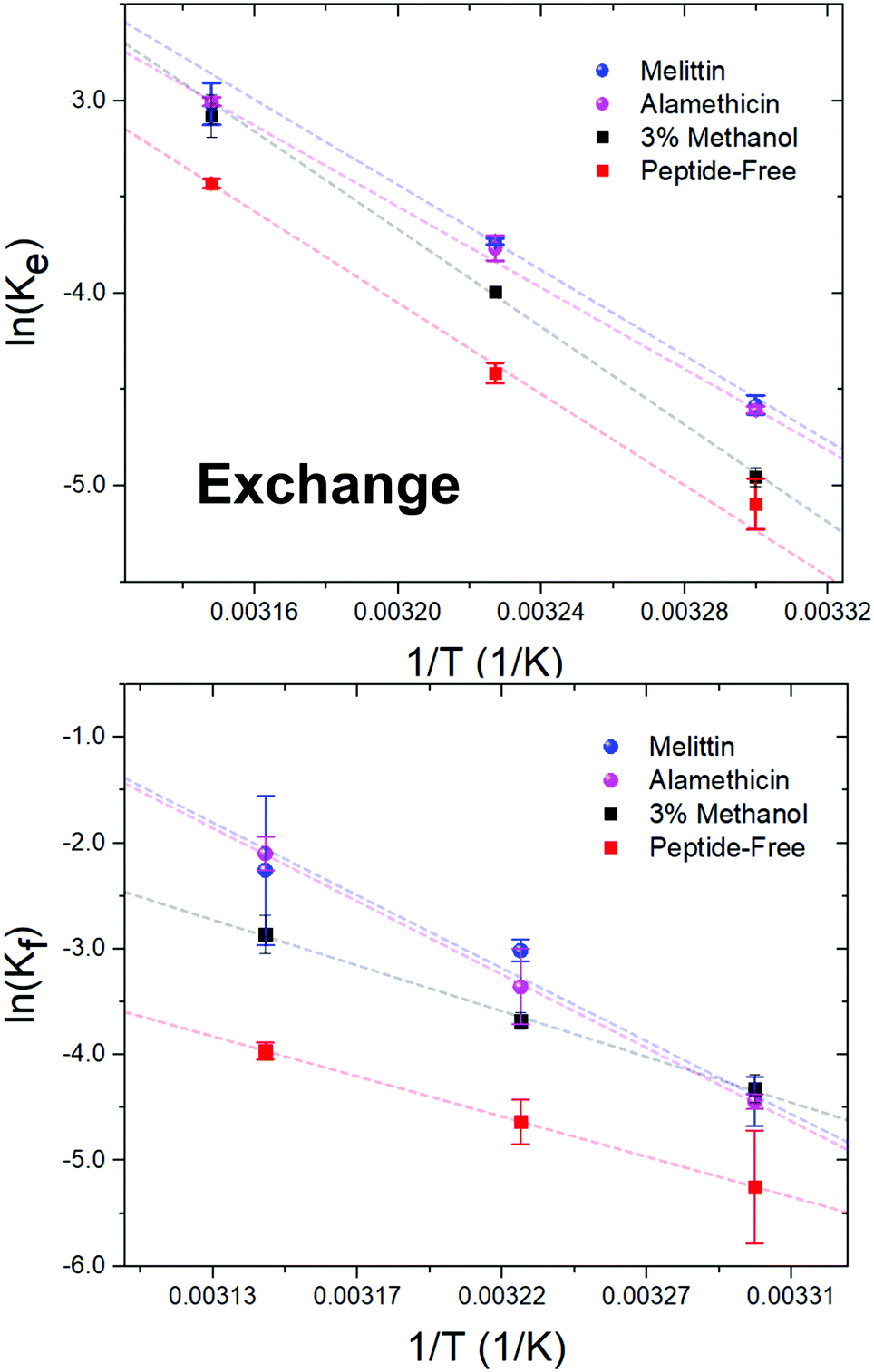 | ||
| Fig. 4 Arrhenius plots depicting the DMPC exchange (ke, top panel) and flip-flop (kf, bottom panel) rate constants as influenced by Alm and Mel at P/L ratios of 1/40, as well as 3% v/v methanol. | ||
| E a (kJ mol−1) | ΔH‡ (kJ mol−1) | TΔS‡ (kJ mol−1) | ΔG‡ (kJ mol−1) | ||
|---|---|---|---|---|---|
| DMPC | Exchange | 96.2 ± 1.3 | 93.6 | −4.4 | 97.9 |
| Flip-flop | 68.5 ± 3.8 | 65.9 | −32.6 | 98.5 | |
| DMPC + 3% methanol | Exchange | 103 ± 0.4 | 100 | 3.5 | 96.9 |
| Flip-flop | 76.9 ± 1.6 | 74.3 | −21.8 | 96.1 | |
| DMPC + Alm (1/40) | Exchange | 85.5 ± 0.8 | 83.0 | −13.3 | 96.3 |
| Flip-flop | 125 ± 8.5 | 123 | 27.3 | 95.2 | |
| DMPC + Mel (1/40) | Exchange | 89.9 ± 1.4 | 87.3 | −8.7 | 96.0 |
| Flip-flop | 124 ± 17 | 122 | 27.6 | 94.4 |
Activation energy calculations reveal the flip-flop energy of transition (Ea ≈ 124–125 kJ mol−1 in the presence of Alm and Mel) is much greater than during exchange (Ea = 85.5 kJ mol−1 and 89.9 kJ mol−1 for Alm and Mel, respectively), which is in contrast to methanol and methanol-free conditions where the Ea of exchange is greater than that of flip-flop. A greater Ea indicates a greater sensitivity to change in temperature (Fig. 4). Alm and Mel's self-association into higher-ordered structure is dependent on temperature,41,42 and the results here suggest that temperature changes peptide–membrane interactions. These changes are favorable for promoting intra-bilayer lipid flip-flop but less so with interbilayer exchange.
The Ea of lipid exchange lowers by ≈6–17 kJ mol−1 in the presence of either peptide, while the Ea of lipid flip-flop increased by ≈47–57 kJ mol−1 when compared to peptide-free conditions. Indeed, the energetics of flip-flop were significantly affected by high concentrations of pore-forming peptides, suggesting peptides indeed play a major role in influencing transverse lipid diffusion.
A near 2-fold increase is also observed for the ΔH‡ of DMPC flip-flop, while the ΔH‡ of exchange is reduced by ≈10 kJ mol−1 with peptide. Changes to the ΔH‡ corresponds to changes in the intermolecular bonding and interactions of the moving lipid with neighbouring particles. These results suggest peptides are stabilizing the lipid ground-state during the flip-flop process or are destabilizing the transition state. Such drastic changes to the ΔH‡ of flip-flop is in contrast to a previous study on Mel,43 where no change was detected. The total contribution by the entropy of activation to the free energy of activation is given as TΔS‡. The entropy of activation for DMPC exchange only slightly lowered from −4.4 kJ mol−1 (or 3.5 kJ mol−1 found in methanolic conditions) to −13.3 kJ mol−1 and −8.7 kJ mol−1 with Alm and Mel, respectively. Interestingly, the TΔS‡ for DMPC flip-flop under Alm and Mel is 27.3 kJ mol−1 and 27.6 kJ mol−1, respectively. Compared to −32.6 kJ mol−1 of pure DMPC flip-flop and −21.8 kJ mol−1 in 3% methanol conditions, adding peptides caused the entropic process of flip-flop to become favorable. As a result, the overall process of lipid flip-flop became much more disordered, as one would expect a system riddled with peptides and defects.
The free energy of activation (ΔG‡) represents the difference between the standard Gibbs free energy of the transition and ground states. The ΔG‡ of DMPC exchange corresponds closely to the Ea estimate, but does not for DMPC flip-flop (Table 1). By promoting these lipid dynamic processes, the introduced peptides lowered both ΔG‡, with Mel being slightly more effective. Specifically, a minor decrease of ≈3–4 kJ mol−1 is seen, likewise for flip-flop in a saturated lipid system (diC18:0-PC) with Mel.43 Further, the ΔG‡ of exchange and flip-flop remain similar, a finding consistent with other DMPC systems.22,25,44 With respect to ΔG‡, the enthalpic contributions dominate the free energy barrier for both flip-flop and exchange. Ultimately, the influence of methanol on the thermodynamics of DMPC motion is minimal, especially compared to the dramatic change that occur under peptidic conditions. Alm and Mel samples show similar kinetics and, in turn, thermodynamic quantities at the measured concentration, pointing to likeness in their mechanism of perturbation.
4 Discussion
Many membrane-active peptides destroy membrane integrity to cause bacterial cell death, as has been seen with both Alm and Mel.26 Their cytotoxicity thus is intimately tied to their interactions with the outer membrane. A key peptide–membrane interaction can involve the peptidic influence on movement and organization of lipids within these membranes. In order to understand and mimic the mechanism of actions of peptides, these lipid dynamics have to be studied and properly characterized. While eukaryotic membranes are composed mainly of neutral-charge phospholipids (NCP) and cholesterol, bacterial membranes possess not only NCPs, but also a significant amount of negatively-charged phosphatidylglycerol lipids.45 Bacterial NCPs have phosphatidylethanolamine (PE) as headgroups, but phosphatidylcholine (PC) systems are often used as a substitute.11,27,31,46 AMP activity on the two zwitterionic systems show similar trends as well.47 Due to possible multiple processes in PE systems,47 we reconstituted Alm and Mel into model DMPC membrane systems, which have been successfully used to measure external influence on lipid organization and dynamics.37,48,494.1 Pore-forming peptides promote DMPC exchange between vesicles
The present study suggests Alm enhances the exchangeability of zwitterionic lipids between bilayers at the studied concentrations, but Mel does not until a high concentration threshold is reached (P/L = 1/40). A process in which a peptide may aid lipid exchange is through the formation of contact points between vesicles, as seen by the antibacterial lipopeptide polymyxin B.20 These contact points permit exchange without vesicle fusion, in line with the present DLS data showing essentially no change in hydrodynamic diameter after peptidic treatment. This phenomenon has also been observed for Mel in a coupled interaction in which Mel facilitated the hydrolysis of lipids by phospholipase A2 by replenishing the supply of non-hydrolyzed phospholipids via exchange.50 It was posed that at low concentrations, Mel is in a transmembrane state, while at high concentrations it rests in a surface-bound state and can catalyze phospholipid exchange. This model details that membrane-bound peptide monomers can oligomerize with other monomers on adjacent vesicle bilayers and promote phospholipid exchange.It is known that the insertion and subsequent pore-formation of these peptides follow a concentration-dependent manner, whereby a critical P/L is used to describe when a majority of monomers transition from being surface-bound to an inserted state.51 In fully hydrated and fluid-phase DMPC bilayers, studies indicate that Alm and Mel are primarily in an inserted orientation at P/L ≥ 1/150.8,51–54 It is then expected that: at 1/1000, few pores should be present due to a low density of inserted peptide monomers; at 1/150, a moderate amount of pores should arise; and we are confident at 1/40 there exists significant pore formation. Thus it is reasonable to conclude that the aforesaid model cannot sufficiently describe Mel's incapacity to promote DMPC exchange at lower concentrations as both peptide's bilayer orientation are similar.
Many studies have looked at the exchange of amphipathic peptides between vesicles12,27,55–58 with several suggesting higher peptide hydrophobicity can lead to greater peptide exchange.27,57 In accordance, Alm has limited solubility in aqueous solutions, in contrast to water-soluble Mel, and consistently promotes lipid exchange at the examined P/L ratios. We cautiously speculate this to be the coupling of lipid and Alm monomers. Lipid amphiphiles can stabilize the otherwise insoluble Alm during aqueous diffusion to neighboring vesicles. With codiffusion transport, the observed enhancement of lipid exchange can be rationalized. Because Alm and Mel are themselves capable of exchanging between bilayers,27 the observed difference in DMPC exchange found here is most likely a result of the accompanied exchange of Alm with DMPC, while Mel should be able to exchange alone based on its water solubility. This is consistent with the low P/L (1/150, 1/1000) data. Krauson et al.,27 however, did not observe the coupled diffusion of Alm and lipids, albeit using fluorescently-labeled lipids. Such fluorescent lipid analogues possess physical and chemical properties that differ from natural lipids, which can influence its interaction with other biomolecules.
4.2 DMPC flip-flop is dictated by peptide contributions over methanol at low concentrations
Pore-forming peptides, including Alm and Mel,59 are known to induce lipid flip-flop depending on the P/L ratio and lipids used.27,30,59 Alm monomers are typically inserted at a greater fraction27 and are also better inducers of vesicle leakage as compared to Mel.60 These differences indicate that Alm forms either a greater number of pores or larger pores than Mel at a given concentration. Yet at all concentrations examined, our results indicate Mel is the better promoter of DMPC flip-flop. A surprising finding that, despite the monomeric orientation and relative pore number/size, suggests Mel has a greater effect on transversal bilayer organization, which agrees with a previous report.30 This is in contrast to a previous finding where several peptides, including Alm and Mel, at P/L ≥ 1/200 induced lipid flip-flop but only Alm was able to do so at lower concentrations (1/1000).27 It is known that lipid flip-flop depends on many factors, such as: headgroup polarity, charge, number of tails, saturation, chain length and even the bulk solvent.10,35,37 In the mentioned study, only Alm was introduced to the lipid system via a methanol stock, while Mel and the other peptides were through aqueous buffer. But as we found previously37 and show here, the enhanced rate under Alm found by Krauson et al. may be attributed to the addition of methanol. Combining these results, methanol may accentuate peptide influence on lipid flip-flop, and vice versa.The non-linear concentration-dependence of lipid flip-flop (Fig. 3) suggests the formation of pores, showing similar trends to calcein leakage assays of the same peptides.27 Calcein leakage studies indicate the potential of oligomeric pore-formation at concentrations lower than 1/40, which may help explain the observation of much faster DMPC flip-flop rates than the methanol controls. Though it may be easy to rush into such a conclusion, caution must be taken as the conditions are not quite the same to the SANS experimentation (i.e. peptides are not equilibrated with LUVs but added externally and measured immediately, which may enact the interfacial activity model3). Indeed, as one attempts to correlate biological and biomimetic systems, the present data affirm that membrane destabilization is caused long before oligomerization can be observed in vitro. Peptides conforming to the toroidal pore model (e.g. melittin) should permit diffusion of lipids across the bilayer normal, while lipids must still cross the hydrophobic core with barrel-stave pores. At low concentrations, Mel promotes a large degree of lipid flip-flop, almost 2× faster than Alm suggesting there indeed exists differences in their mechanisms. One such difference is the number of monomers required to form their pore structure; Mel forms tetrameric pores while Alm fluctuates between 4–11 monomers per oligomeric pore.61 These numbers suggest Mel has a greater pore density within the membranes, which would help explain the increase in DMPC flip-flop rates under the two peptidic conditions. Interestingly, though Alm had a smaller effect on lipid flip-flop, it was still quite potent. This may be further evidence that Alm forms toroidal pores instead of barrel-stave ones, as previously asserted by Wimley et al.3,59 Overall, we provide further evidence for the traditional line of thought that Mel forms higher ordered structures that better facilitate lipid translocation.
When flip-flop rates surpass that of intervesicular exchange, it becomes more complex to parse the two rates using the current method. Despite this, the 30 °C data of both peptides at P/L = 1/40 demonstrate similar kinetics and therefore a likeness in effect on lipid dynamics. This is important as in a slowed kinetic environment, where the mixing dynamics are not beyond the temporal resolution of the experimental set-up, the peptides still produce comparable rate constants that lay outside the 95% confidence interval produced by the methanol standard curve. Furthermore, kinetic rates plotted in Fig. 4 are extremely reminiscent of typical Arrhenius plots of other systems. The linear plots to such fits indicate typical Arrhenius behavior as well. In a qualitative sense, because the flip-flop rates at elevated temperatures are faster than the action of these two distinct AMPs, Alm and Mel, are more destructive to the compositional stability of bilayers than previously thought. Altogether, these points further prove a general pore-mediated mechanism that can scramble a lipid bilayer within minutes, a finding in agreement with what we have found in asymmetric liposomes.11 Therefore, our data unequivocally show that the presence of pore-forming peptides promote lipid flip-flop in bilayers in a far greater capacity than intervesicular exchange, even when accounting for contributions by methanol.
4.3 Lipid exchange and flip-flop thermodynamics reveal mechanism of peptides at high concentrations are similar
DMPC exchange and flip-flop rates under oligomeric pore-forming peptide conditions show a similar temperature dependency despite differences in pore structure (Fig. 4). Thermodynamic analysis of these rates revealed a very modest reduction to the flip-flop and exchange energetic barriers (ΔG‡) of roughly ≈1–4 kJ mol−1 with peptide, showing an initial likeness in effect. However, the change in DMPC exchange and flip-flop upon peptide addition are not immediately apparent when only accounting the ΔG‡. For instance, the activation enthalpies and entropies of DMPC flip-flop take on vast changes in value and sign. The activation enthalpy of flip-flop, for example in Table 1, almost doubles in value in the presence of either Alm or Mel. In contrast, the activation entropy of flip-flop changed signs completely and became a favorable process for flip-flop. Changes in exchange Ea, ΔH‡ and ΔS‡ are much less dramatic than changes in the flip-flop values: only slightly lowering in value (≈10–17 kJ mol−1). Interestingly, all general shifts in thermodynamic quantities are reflected in the same manner for both peptides. We believe that this hints at a generality in their mechanism of action at high concentrations. Alm and Mel are said to be rare among AMPs.3 Relative to other AMPs, they are highly potent at killing microbes, owing to their nonselectivity and true pore-forming behavior. Most other AMPs actually do not form such pores, instead broadly exerting their action upon the surface of the membrane (known as interfacial activity), disrupting the careful partitioning of the water–lipid and headgroup–chain interfaces.3 The peptides here are unlikely to exhibit this activity as they were incubated with the separate isotopic populations before mixing; in other words, the peptides were already equilibrated and thus could not cause a mass-action bilayer restructuring event that are often seen in experiments mimicking external antimicrobial peptide attack.11 But just as the interfacial activity is used to broadly describe the action of a majority of AMPs, we provide evidence that it is also possible to collectivize the action of pore-forming peptides in terms other than content leakage.Surprisingly, methanol has a dramatic effect on the kinetic half-times but not the thermodynamic quantities. To measure energetics is to measure the kinetic rates and their response to changes in temperature. From this perspective, it would appear that the basic reaction mechanisms of lipid exchange and flip-flop is unperturbed under the influence of methanol. While, the inclusion of peptides, biological entities whose properties and activity are sensitive to changes in temperature, into the system changed the energetics dramatically.
Our thermodynamic analysis suggests pores formed by Alm and Mel perturb membrane structure in similar manners. Interestingly, the presence of Alm and Mel at low concentrations (P/L = 1/1000 and 1/150) accelerated DMPC flip-flop by a factor of ≈2 to 3. At these low concentrations, our results support that lipid scrambling could be a major part in their antimicrobial action. Considering the design of small antibiotic peptides, researchers may need to consider the effects of novel therapeutics on lipid dynamics. These dynamics shape and define biological membranes, with cells placing great importance in maintenance of bilayer composition. Putative barrel-stave pore forming Alm can promote both lipid flip-flop and exchange, while the toroidal pore forming Mel only sufficiently enhances lipid flip-flop at lower doses. It is unclear exactly which aspect creates a more potent therapeutic: a peptide that influences both lipid dynamics or one that greatly influences a single dynamic process. In theory, AMPs that are able to freely exchange, especially after exhibiting their lytic activity, between bacterial outer membranes would likely exert greater potency. Despite this question, our results suggest the possibility of tuning a peptide structure to selectively target a single dynamical process to achieve a more potent agent.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. Access to the Very Small Angle Neutron Scattering (VSANS) instrument was provided by the Center for High Resolution Neutron Scattering, a partnership between the National Science Foundation and the National Institute of Standards and Technology under Agreement No. DMR-1508249. M. H. L. N. and M. D. are both supported by Ontario Graduate Scholarships (OGS). D. M. acknowledges the support of the Natural Sciences and Engineering Research Council of Canada (NSERC), [funding reference number RGPIN-2018-04841]. M. H. L. N. is the recipient of a NSERC CGS-D. The identification of any commercial products or trade names does not imply endorsement or recommendation by the National Institute of Standards and Technology.References
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M. P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. van der Meer, N. Van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- A. Falanga, L. Lombardi, G. Franci, M. Vitiello, M. R. Iovene, G. Morelli, M. Galdiero and S. Galdiero, Int. J. Mol. Sci., 2016, 17, 785 CrossRef PubMed.
- W. C. Wimley, ACS Chem. Biol., 2010, 5, 905–917 CrossRef CAS PubMed.
- M. Sinensky, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 522–525 CrossRef CAS PubMed.
- W. C. Wimley and K. Hristova, J. Membr. Biol., 2011, 239, 27–34 CrossRef CAS PubMed.
- E. Fattal, R. A. Parente, F. C. Szoka and S. Nir, Biochemistry, 1994, 33, 6721–6731 CrossRef CAS PubMed.
- W. C. Wimley, Biophys. J., 2018, 114, 251–253 CrossRef CAS PubMed.
- L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Biophys. J., 2001, 81, 1475–1485 CrossRef CAS PubMed.
- M. T. Tosteson, S. J. Holmes, M. Razin and D. C. Tosteson, J. Membr. Biol., 1985, 87, 35–44 CrossRef CAS PubMed.
- D. Marquardt, B. Geier and G. Pabst, Membranes, 2015, 5, 180–196 CrossRef CAS PubMed.
- M. H. L. Nguyen, M. DiPasquale, B. W. Rickeard, M. Doktorova, F. A. Heberle, H. L. Scott, F. N. Barrera, G. Taylor, C. P. Collier, C. B. Stanley, J. Katsaras and D. Marquardt, Langmuir, 2019, 35, 11735–11744 CrossRef CAS PubMed.
- B. Orioni, G. Bocchinfuso, J. Y. Kim, A. Palleschi, G. Grande, S. Bobone, Y. Park, J. I. Kim, K. S. Hahm and L. Stella, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1523–1533 CrossRef CAS PubMed.
- M. Hasan, M. A. S. Karal, V. Levadnyy and M. Yamazaki, Langmuir, 2018, 34, 3349–3362 CrossRef CAS PubMed.
- T. C. Anglin, J. Liu and J. C. Conboy, Biophys. J., 2007, 92, 01–3 CrossRef PubMed.
- T. K. Lind, L. Darré, C. Domene, Z. Urbanczyk-Lipkowska, M. Cárdenas and H. P. Wacklin, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2075–2084 CrossRef CAS PubMed.
- M. Doktorova, F. A. Heberle, D. Marquardt, R. Rusinova, R. L. Sanford, T. A. Peyear, J. Katsaras, G. W. Feigenson, H. Weinstein and O. S. Andersen, Biophys. J., 2019, 116, 860–873 CrossRef CAS PubMed.
- G. Taylor, M.-A. Nguyen, S. Koner, E. Freeman, C. P. Collier and S. A. Sarles, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 335–343 CrossRef CAS PubMed.
- J. Liu and J. C. Conboy, Biophys. J., 2005, 89, 2522–2532 CrossRef CAS PubMed.
- D. Marquardt, F. A. Heberle, T. Miti, B. Eicher, E. London, J. Katsaras and G. Pabst, Langmuir, 2017, 33, 3731–3741 CrossRef CAS PubMed.
- Y. Cajal, J. Rogers, O. G. Berg and M. K. Jain, Biochemistry, 1996, 35, 299–308 CrossRef CAS PubMed.
- J. T. Oh, Y. Cajal, E. M. Skowronska, S. Belkin, J. Chen, T. K. Van Dyk, M. Sasser and M. K. Jain, Biochim. Biophys. Acta, Biomembr., 2000, 1463, 43–54 CrossRef CAS.
- W. C. Wimley and T. E. Thompson, Biochemistry, 1991, 30, 1702–1709 CrossRef CAS PubMed.
- L. R. McLean and M. C. Phillips, Biochemistry, 1984, 23, 4624–4630 CrossRef CAS PubMed.
- R. E. Pagano, O. C. Martin, A. J. Schroit and D. K. Struck, Biochemistry, 1981, 20, 4920–4927 CrossRef CAS PubMed.
- M. Nakano, M. Fukuda, T. Kudo, H. Endo and T. Handa, Phys. Rev. Lett., 2007, 98, 238101 CrossRef PubMed.
- B. Bechinger, J. Membr. Biol., 1997, 156, 197–211 CrossRef CAS PubMed.
- A. J. Krauson, J. He and W. C. Wimley, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1625–1632 CrossRef CAS PubMed.
- M.-T. Lee, T.-L. Sun, W.-C. Hung and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14243–14248 CrossRef CAS PubMed.
- F. Y. Chen, M. T. Lee and H. W. Huang, Biophys. J., 2003, 84, 3751–3758 CrossRef CAS PubMed.
- S. Qian and W. T. Heller, J. Phys. Chem. B, 2011, 115, 9831–9837 CrossRef CAS PubMed.
- S. Qian and W. T. Heller, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2253–2260 CrossRef CAS PubMed.
- S. Qian, D. Rai and W. T. Heller, J. Phys. Chem. B, 2014, 118, 11200–11208 CrossRef CAS PubMed.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- R. Homan and H. J. Pownall, Biochim. Biophys. Acta, Biomembr., 1988, 938, 155–166 CrossRef CAS.
- M. Nakano, M. Fukuda, T. Kudo, N. Matsuzaki, T. Azuma, K. Sekine, H. Endo and T. Handa, J. Phys. Chem. B, 2009, 113, 6745–6748 CrossRef CAS PubMed.
- M. Kaihara, H. Nakao, H. Yokoyama, H. Endo, Y. Ishihama, T. Handa and M. Nakano, Chem. Phys., 2013, 419, 78–83 CrossRef CAS.
- M. H. Nguyen, M. DiPasquale, B. W. Rickeard, C. B. Stanley, E. G. Kelley and D. Marquardt, Biophys. J., 2019, 116, 755–759 CrossRef CAS PubMed.
- B. Wah, J. M. Breidigan, J. Adams, P. Horbal, S. Garg, L. Porcar and U. Perez-Salas, Langmuir, 2017, 33, 3384–3394 CrossRef CAS PubMed.
- K. L. Brown and J. C. Conboy, J. Phys. Chem. B, 2013, 117, 15041–15050 CrossRef CAS PubMed.
- S. Garg, Y. Liu, U. Perez-Salas, L. Porcar and P. D. Butler, Chem. Phys. Lipids, 2019, 223, 104779 CrossRef CAS PubMed.
- G. A. Woolley and B. A. Wallace, Biochemistry, 1993, 32, 9819–9825 CrossRef CAS PubMed.
- S. C. Quay and C. C. Condie, Biochemistry, 1983, 22, 695–700 CrossRef CAS PubMed.
- T. C. Anglin, K. L. Brown and J. C. Conboy, J. Struct. Biol., 2009, 168, 37–52 CrossRef CAS PubMed.
- W. C. Wimley and T. E. Thompson, Biochemistry, 1990, 29, 1296–1303 CrossRef CAS PubMed.
- S. Morein, A.-S. Andersson, L. Rilfors and G. Lindblom, J. Biol. Chem., 1996, 271, 6801–6809 CrossRef CAS PubMed.
- D. K. Rai, S. Qian and W. T. Heller, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2788–2794 CrossRef CAS PubMed.
- J. E. Nielsen, S. Prevost, J. Havard and R. Lund, Faraday Discuss., 2020 10.1039/D0FD00046A.
- T. Sugiura, C. Takahashi, Y. Chuma, M. Fukuda, M. Yamada, U. Yoshida, H. Nakao, K. Ikeda, D. Khan, A. H. Nile, V. A. Bankaitis and M. Nakano, Biophys. J., 2019, 116, 92–103 CrossRef CAS PubMed.
- H. Nakao, K. Ikeda, Y. Ishihama and M. Nakano, Biophys. J., 2016, 110, 2689–2697 CrossRef CAS PubMed.
- Y. Cajal and M. K. Jain, Biochemistry, 1997, 36, 3882–3893 CrossRef CAS PubMed.
- F.-Y. Chen, M.-T. Lee and H. W. Huang, Biophys. J., 2003, 84, 3751–3758 CrossRef CAS PubMed.
- H. Vogel, Biochemistry, 1987, 26, 4562–4572 CrossRef CAS PubMed.
- A. Naito, T. Nagao, K. Norisada, T. Mizuno, S. Tuzi and H. Saitô, Biophys. J., 2000, 78, 2405–2417 CrossRef CAS PubMed.
- H. Vogel and F. Jähnig, Biophys. J., 1986, 50, 573–582 CrossRef CAS PubMed.
- C. Mazzuca, B. Orioni, M. Coletta, F. Formaggio, C. Toniolo, G. Maulucci, M. De Spirito, B. Pispisa, M. Venanzi and L. Stella, Biophys. J., 2010, 99, 1791–1800 CrossRef CAS PubMed.
- T. I. Rokitskaya, E. A. Kotova, G. A. Naberezhnykh, V. A. Khomenko, V. I. Gorbach, A. M. Firsov, E. A. Zelepuga, Y. N. Antonenko and O. D. Novikova, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 883–891 CrossRef CAS PubMed.
- K. Lum, H. I. Ingólfsson, R. E. Koeppe and O. S. Andersen, Biophys. J., 2017, 113, 1757–1767 CrossRef CAS PubMed.
- F. D. Ablan, B. L. Spaller, K. I. Abdo and P. F. Almeida, Biophys. J., 2016, 111, 1738–1749 CrossRef CAS PubMed.
- W. C. Wimley and S. H. White, Biochemistry, 2000, 39, 161–170 CrossRef CAS PubMed.
- G. Wiedman, K. Herman, P. Searson, W. C. Wimley and K. Hristova, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 1357–1364 CrossRef CAS PubMed.
- M. Mihajlovic and T. Lazaridis, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1494–1502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04717a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |