
Copper(I) ionic complexes based on imidazo[4,5-f][1,10]phenanthrolin diimine chelating ligands: crystal structures, and photo- and electroluminescence properties†
Anton V.
Rozhkov
 *a,
Svetlana N.
Eliseeva
a,
Sergey V.
Baykov
a,
Lev E.
Zelenkov
a,
Dmitry O.
Goriachiy
b and
Ilya V.
Taydakov
b
*a,
Svetlana N.
Eliseeva
a,
Sergey V.
Baykov
a,
Lev E.
Zelenkov
a,
Dmitry O.
Goriachiy
b and
Ilya V.
Taydakov
b
aInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, 199034 Saint Petersburg, Russian Federation. E-mail: iomcrozhkov@gmail.com
bP. N. Lebedev Physical Institute of the Russian Academy of Science, Leninsky pr., 53, 119991 Moscow, Russian Federation
First published on 25th November 2019
Abstract
Four new ionic diimine Cu(I) complexes with general formula [Cu(POP)(NN)]BF4 have been synthesized by convenient methods from readily available starting materials. All the complexes were characterized by a range of spectroscopic, electrochemical, and computational techniques and, in the cases of 1, 2, and 3, by single crystal X-ray diffraction. The complexes are emissive in the solid state at ambient temperature and the emission maxima are located in the orange range. Furthermore, the suitability of these complexes for application in organic light-emitting diodes (OLEDs) has been demonstrated in a solution-processed device.
Introduction
In recent times, luminescent Cu(I) complexes have been the object of intense studies for application in organic light emitting diodes (OLEDs).1–5 Cu(I) complexes possess significant advantages such as tunable phosphorescent emission bands, long excited state lifetimes, abundance of the metal in nature and easy mining, low cost, and environmental benignancy, which make them a tempting alternative to the commonly used triplet phosphorescent iridium complexes in electroluminescent device applications.6 Iridium and platinum complexes display strong spin–orbital coupling (SOC) leading to effective intersystem crossing from the lowest excited singlet to the lowest triplet state, which allows all injected excitons to be used for light generation in electroluminescent devices through the triplet harvesting effect.7 Yet another way to harvest excitons in OLEDs is to use temperature activated delayed fluorescence (TADF) emitters.8,9 Due to the small energy separation ΔE(S1–T1) between the first excited singlet state S1 and the triplet state T1, the S1 state can be thermally populated from the energetically lower lying T1 state. In an electroluminescent device, this mechanism allows utilizing all injected excitons for light generation. Accordingly, the excitation is harvested and emitted, essentially, via the singlet S1 state. As distinct from platinum and iridium complexes, copper(I) complexes have smaller SOC constants, which can result in an increase in decay time and a decrease in triplet emission and can affect the OLED performance. Fortunately, copper complexes can show TADF with short radiative lifetimes at room temperature.10–13 These TADF copper complex-based OLEDs present an internal quantum efficiency of 100% by harvesting both singlet and triplet excitons. Copper(I) complexes with diimine ligands have been successfully used as TADF emitters.11,14,15 In particular, the imidazo[4,5-f][1,10]phenanthrolin derivatives as ligands have been extensively utilized to prepare Cu(I) complexes with attractive luminescence features.16–19Herein, we describe the synthesis of a new series of cationic heteroleptic diimine–diphosphine copper(I) complexes with POP (bis[2-(diphenylphosphino)phenyl]-ether) and 1,10-phenimidazole (L1 = 2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol, L2 = 4-chloro-2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol, L3 = 4-bromo-2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol, L4 = 2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)-4-nitrophenol) ligands. These complexes demonstrated orange photoluminescence in the solid state at room temperature and were applied in solution-processed OLEDs using a host–guest system. We have found that the color of electroemission can be fine-tuned by varying the dopant concentration.
Results and discussion
Synthetic procedures
The diimine N^N ligands L1–L4 were prepared by condensation of 1,10-phenanthroline-5,6-dione with an appropriate aldehyde in the presence of ammonium acetate20–22 (Scheme 1).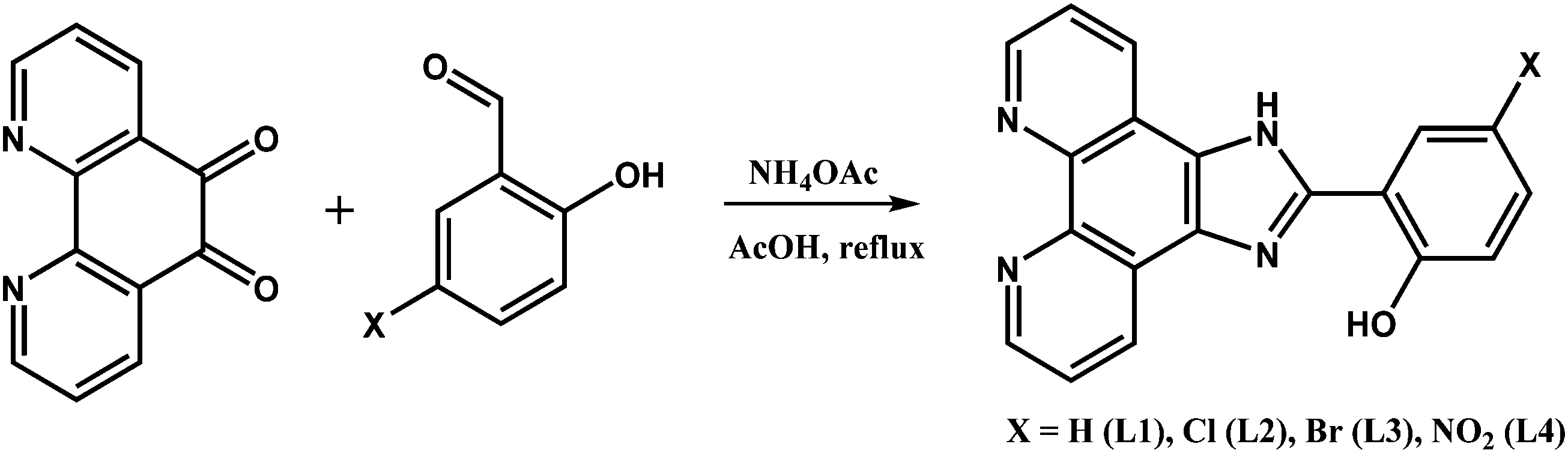 | ||
| Scheme 1 The synthetic route to the N^N ligands L1–L4. | ||
The synthetic route to the Cu(I) complexes is shown in Scheme 2. Complexes 1–4 (X = H, Cl, Br, NO2) were prepared by the conventional reaction of [Cu(CH3CN)4]BF4 with 1 equiv. of the POP ligand and 1 equiv. of the appropriate N^N ligand in dichloromethane at room temperature.
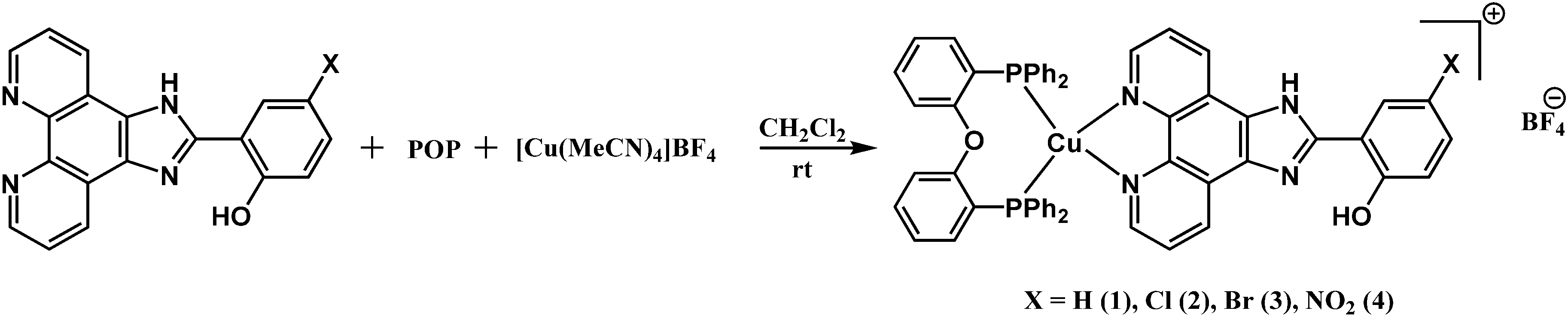 | ||
| Scheme 2 The synthetic route to the Cu(I) complexes. | ||
The complexes were obtained in good yields as air stable, yellow crystalline solids soluble in the majority of organic solvents, including CHCl3, CH2Cl2, MeOH, MeCN, DMF, and DMSO. The formation of the desired Cu(I) complexes was confirmed by 1H, 13C, 31P NMR and FTIR spectroscopy, as well as high-resolution mass spectrometry (see the Experimental section).
Structure determination by single crystal X-ray diffraction
Single crystals suitable for X-ray determination were obtained by slow evaporation of the solvent from a DMSO solution (for complex 1) or by slow diffusion of pentane to a saturated solution of the complex in CH2Cl2 at ambient temperature (for complex 2). The structures obtained are presented in Fig. 1 (complex 1), Fig. 2 (complex 2), and Fig. 3 (complex 3). Selected bond lengths and bond angles are given in Table 1.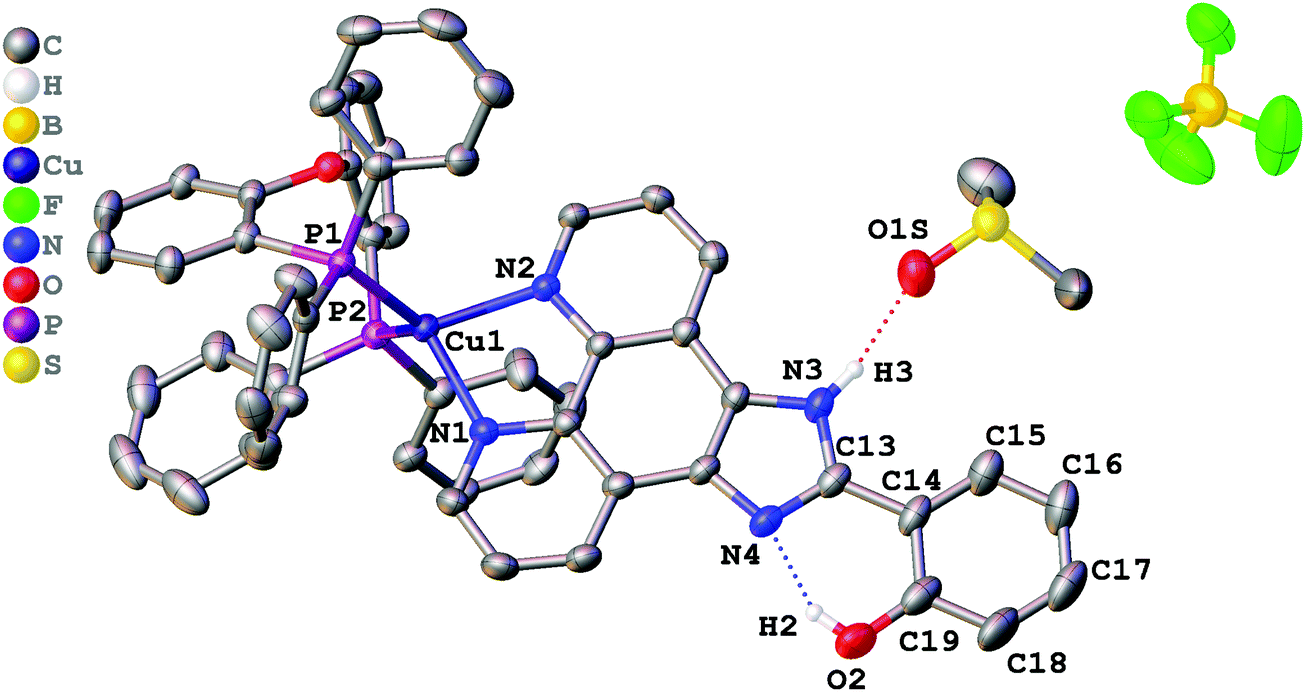 | ||
| Fig. 1 Crystal structure of 1·DMSO with thermal ellipsoids shown at the 30% probability level. Most of the hydrogen atoms are omitted for clarity. | ||
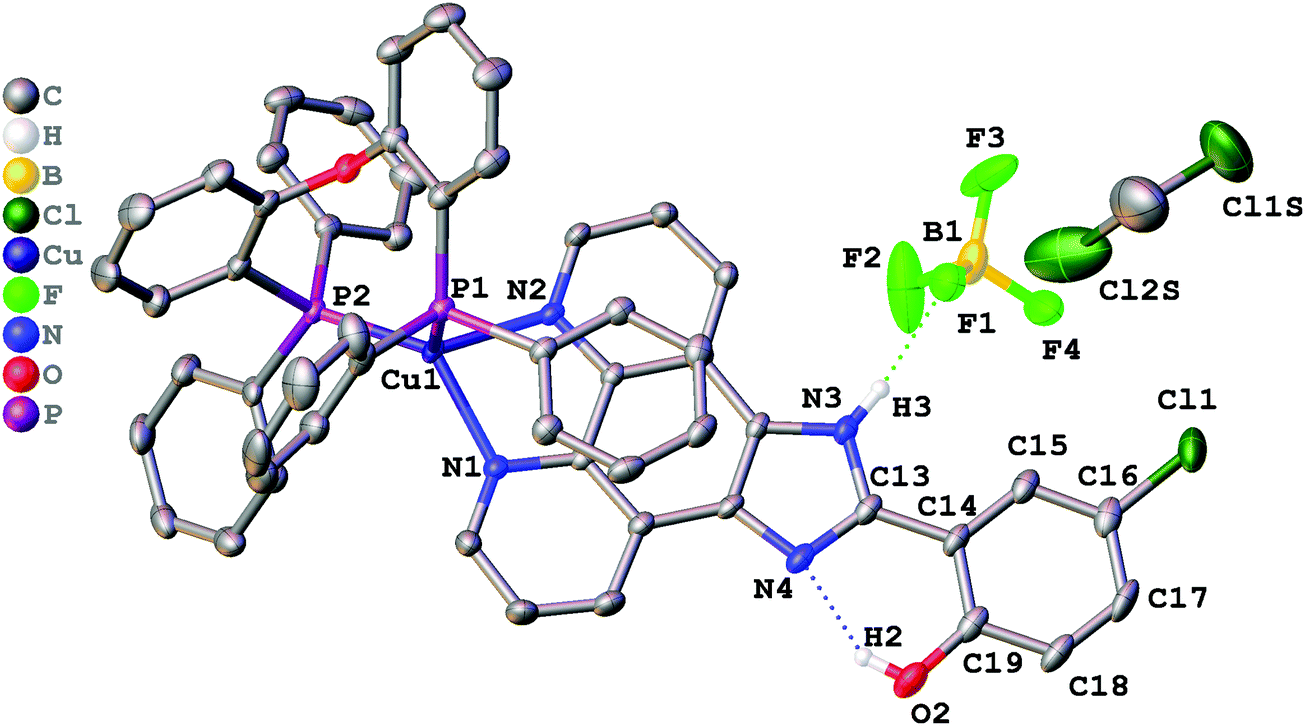 | ||
| Fig. 2 Crystal structure of 2·CH2Cl2 with thermal ellipsoids shown at the 30% probability level. Most of the hydrogen atoms are omitted for clarity. | ||
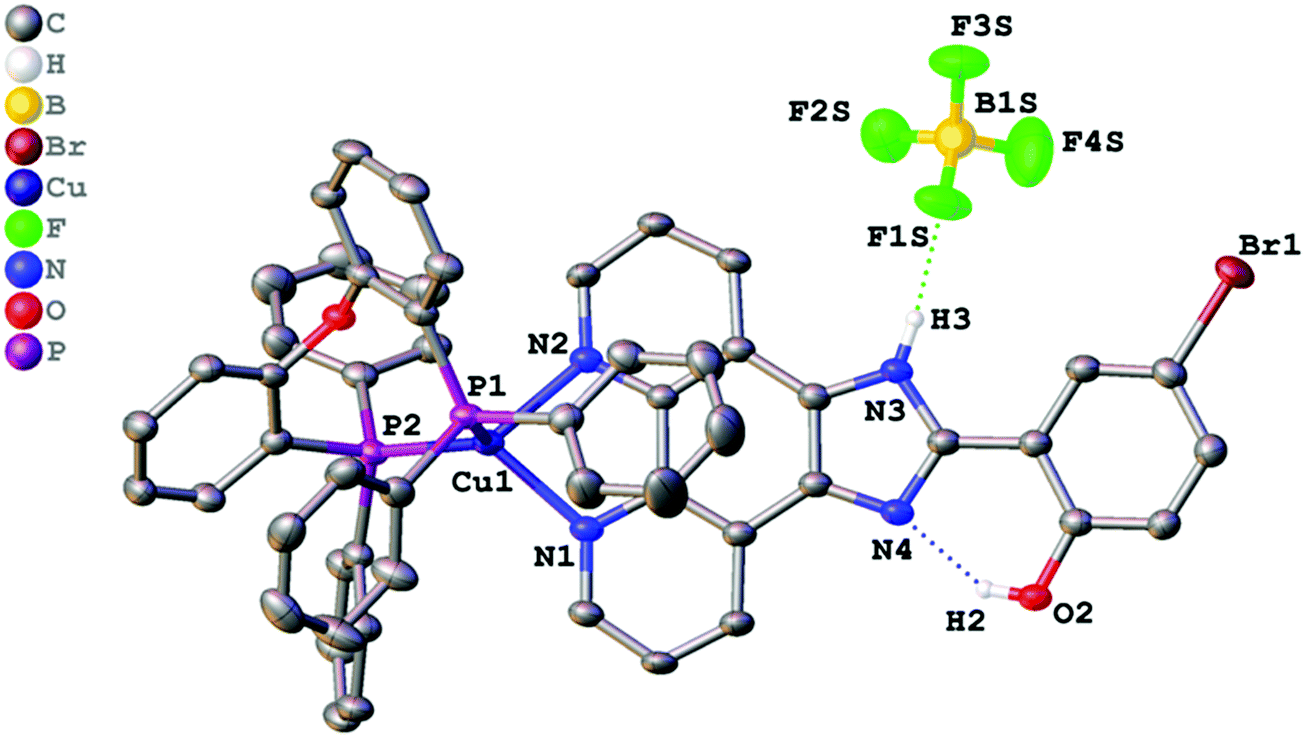 | ||
| Fig. 3 Crystal structure of 3 with thermal ellipsoids shown at the 30% probability level. Most of the hydrogen atoms are omitted for clarity. | ||
| 1·DMSO | 2·CH2Cl2 | 3 | |
|---|---|---|---|
| Cu1–N1 | 2.059(2) | 2.074(3) | 2.068(2) |
| Cu1–N2 | 2.081(2) | 2.054(3) | 2.057(3) |
| Cu1–P1 | 2.2410(7) | 2.2988(9) | 2.2825(5) |
| Cu1–P2 | 2.2663(8) | 2.2059(9) | 2.2066(9) |
| N1–Cu1–N2 | 80.92(8) | 81.18(9) | 81.10(9) |
| P1–Cu1–P2 | 112.93(3) | 114.69(3) | 115.89(4) |
| N1–Cu1–P1 | 118.12(7) | 98.33(7) | 106.13(7) |
| N1–Cu1–P2 | 115.73(7) | 132.53(8) | 123.00(9) |
| N2–Cu1–P1 | 118.13(6) | 101.57(7) | 100.10(8) |
| N2–Cu1–P2 | 106.90(6) | 121.06(7) | 124.11(7) |
All crystalized complexes display mononuclear structures. Complex 1 cocrystallized with one molecule of DMSO linked via the O1S⋯H3 hydrogen bond. Complex 2 cocrystallized with one molecule of dichloromethane. The Cl1S⋯Cl1 distance (3.36588(9) Å) is shorter than the corresponding sums of Bondi vdW radii (3.5 Å),23 and the contact angle C1S–Cl1S–Cl1 (154.2208(9)°) does not significantly deviate from 160°. Both facts allow us to assign the observed intermolecular interaction to halogen bonding (Fig. S1, ESI†).24 The Cu(I) center in all the complexes has a distorted tetrahedral geometry formed by two phosphorus atoms from the POP ligand and two nitrogen atoms from the diimine ligand. The Cu–N and Cu–P bond lengths are in the range of 2.05440(6)–2.081(2) Å) and 2.2059(9)–2.2988(9) Å, respectively, which are consistent with the previously studied copper(I) complexes with the same or similar ligands.16,18,25–27 The P–Cu–P bite angle is 112.93(3)° in 1, 114.69(3)° in 2, and 115.89(4)° in 3. The angle between Cu–P1 and the mean plane of imidazo[4,5-f][1,10]phenanthroline in 2 (99.8875(12)°) is smaller than in 1 (128.27(4)°) due to the intramolecular π⋯π interaction between the POP phenyl group and the phenanthroline moiety of L2. The centroid distance is 3.33295(8) Å. Intramolecular O–H⋯N hydrogen bonds are observed between the phenol –OH group and the other imidazole nitrogen atom in all X-ray structures. It prevents the rotation of the phenyl group around the C13–C14 bond and leads to flattening of the diimine ligand and an increase in the degree of its conjugation. The dihedral angles between the phenol ring (C14–C19) and the mean plane of imidazo[4,5-f][1,10]phenanthroline are 2.98(11)° in 1, 2.68540(8)° in 2, and 0.30(11)° in 3. Also, N–H⋯F hydrogen bonds are observed in 2 and 3 between the imidazole –NH group and the BF4− anion. In all obtained crystals, each molecule interacts with a neighbouring molecule through intermolecular π⋯π interactions (Fig. S2–S4, ESI†). π⋯π Overlapping formed through the stacking of imidazo[4,5-f][1,10]phenanthroline moieties with a π⋯π distance of 3.6118(17) Å in 1, 3.4092(16) Å in 2, and 3.6590(18) Å in 3.
Thermal stability
Thermogravimetric analysis (TGA) was performed on samples of 1–4 powders under an argon atmosphere to investigate their stability characteristics. The TGA traces are presented in Fig. 4.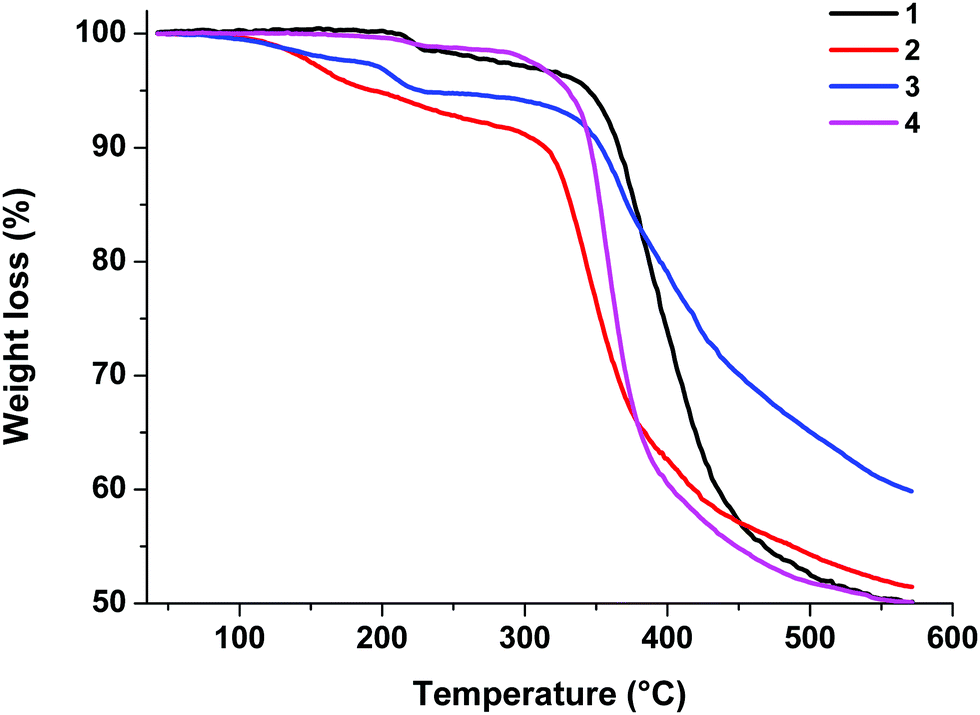 | ||
| Fig. 4 TGA curves of complexes 1–4. | ||
As shown by the TGA results, these complexes are thermally stable. The decomposition temperatures corresponding to a 5% loss of weight are 344 °C for 1, 193 °C for 2, 227 °C for 3, and 330 °C for 4.
Photophysical properties
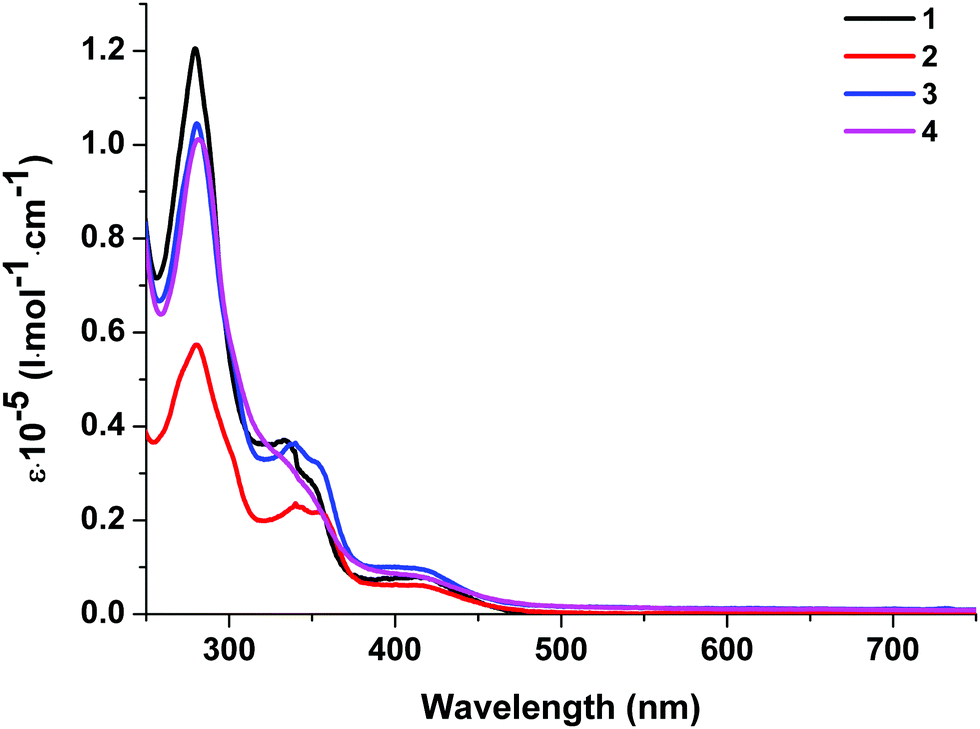 | ||
| Fig. 5 Absorption spectra of complexes 1–4 in dichloromethane solution at room temperature. | ||
| λ abs, nm (ε × 10−5 l mol−1 cm−1) (CH2Cl2) | λ em, nm | S1,a eV | T1,b eV | ΔES–T, eV | τ em, μs | QY, % | ΔEg,c eV | HOMOd/LUMO,e eV | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 293 K | 77 K | |||||||||
| a Estimated from the high-energy flank at 50% of the emission maximum intensity measured at 298 K. b Estimated from the high-energy flank at 50% of the emission maximum intensity measured at 77 K. c Calculated from the absorption edge. d Estimated by cyclic voltammetry in DMF with SCE (4.74 eV) as the reference.28 e Derived from the relation ΔE = HOMO–LUMO. | ||||||||||
| 1 | 280 (1.20), 334 (0.37), 350 sh (0.27), 420 (0.07) | 577 | 602 | 2.39 | 2.27 | 0.12 | 4.2 | 1.30 | 2.75 | −5.53/−2.78 |
| 2 | 280 (0.57), 340 (0.24), 356 (0.21), 420 (0.06) | 578 | 596 | 2.37 | 2.25 | 0.12 | 3.9 | 0.85 | 2.75 | −5.55/−2.80 |
| 3 | 280 (1.04), 339 (0.36), 353 (0.32), 420 (0.09) | 636 | 636 | 2.19 | — | — | 1.5 | 0.09 | 2.79 | −5.55/−2.76 |
| 4 | 281 (1.01), 338 sh (0.31), 419 (0.08) | 594 | 622 | 2.31 | 2.19 | 0.12 | 1.9 | 0.04 | 2.74 | −5.67/−2.93 |
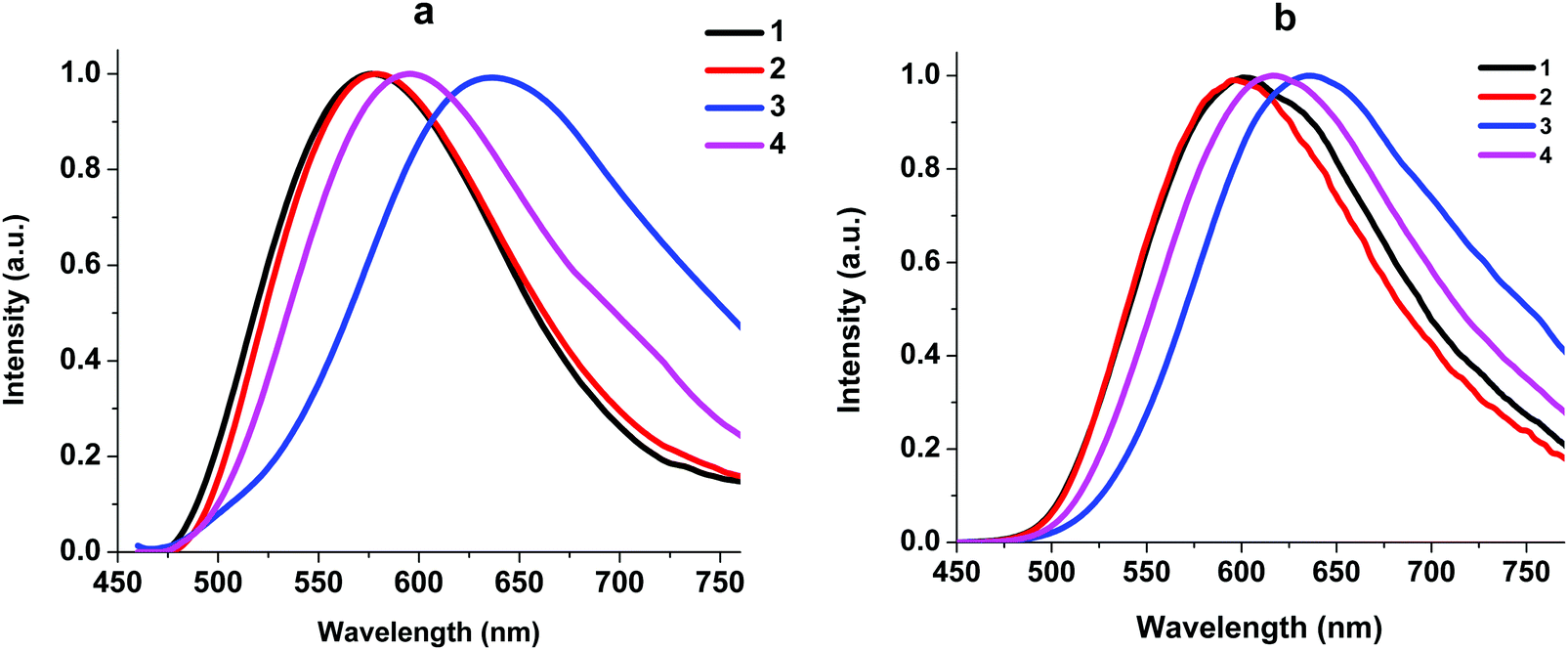 | ||
| Fig. 6 Normalized emission spectra of neat powders of complexes 1–4 at 293 K (a) and 77 K (b). λex = 450 nm. | ||
At 293 K, the complexes show broad and structureless intense emission bands with maxima at 577 nm (1), 578 nm (2), 636 nm (3) and 594 nm (4), which could be assigned to the metal to ligand charge transfer (MLCT) character.27,29 At 77 K, complexes 1, 2, and 4 exhibit slightly lower energy emission with the peaks at 602, 596, and 564 nm, respectively. The considerable difference in emission between 293 K and 77 K indicates that the luminescence of these complexes in the solid state might originate from two different excited states in thermal equilibrium, that is, S1 and T1. Small energy separations ΔE(S1–T1) of about 0.12 eV were observed for 1, 2, and 4. This behavior can be attributed to the TADF process.10 For complex 3 we did not detect a shift in the luminescence maxima upon a decrease in temperature to 77 K. The existence of Br in 3 led to spin–orbit coupling due to the heavy atom effect and we observed triplet emission at room temperature. The luminescence quantum yields of 1–4 at room temperature are in the range of 1.30–0.04% and the lifetimes are in the range of 1.5–4.2 μs (Table 2), which are close to those of other POP-based diimine copper complexes.16,27,30 It should be noted that replacement of H with Br at position 4 of the phenyl moiety in the N^N ligand leads to a decrease in the quantum yield from 1.30 to 0.09% due to the heavy atom effect.31 Incorporation of an NO2 group leads to a decrease in the quantum yield, too. Nitro group substituents that have acceptor properties may withdraw the π-electron density from the aromatic ring, thus increasing the ILCT and, accordingly, decreasing the MLCT processes.
Electrochemical properties
The electrochemical properties of complexes 1, 2, 3, and 4 were studied by cyclic voltammetry (Fig. 7).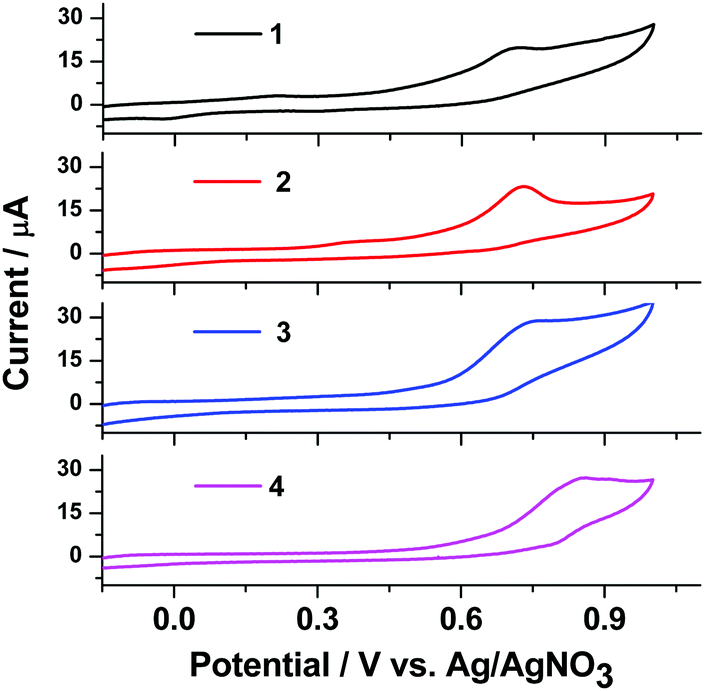 | ||
| Fig. 7 Cyclic voltammograms of complexes 1–4 in DMF solution. | ||
In cyclic voltammetry (CV) studies in the potential range from −0.2 to +1.0 V, complexes 1–4 show one oxidation peak at 0.72–0.86 V. Analysis of the CV dependence on the potential range at a scan rate of 50 mV s−1 shows that the peak potentials stabilize at 0.72–0.86 V for oxidation of complexes 1–4, respectively. The oxidation potential of complexes 1, 2, 3, and 4 in DMF solution is 0.72, 0.74, 0.74 and 0.86 V, respectively. In the CV of complex 4 we can see a wider oxidation peak at a higher potential (0.86 V) compared with the other complexes. All the Cu(I) complexes show an irreversible oxidation process, indicating a rapid decomposition of the Cu(II) species. Nevertheless, based on the start-of-oxidation potential, the highest occupied molecular orbital (HOMO) energy levels of these Cu(I) complexes can be calculated: −5.19 eV for 1, −5.21 eV for 2, −5.21 eV for 3 and −5.33 eV for 4. The lowest unoccupied molecular orbitals (LUMOs) were determined from EHOMO and the optical band gap ΔEg values, which were obtained from the UV absorption edge as reported elsewhere.32 The LUMOs (−2.64 eV for 1, −2.63 eV for 2, −2.62 eV for 3 and −2.59 eV for 4) of these complexes were obtained using the equation ELUMO = EHOMO − ΔEg. The corresponding data are summarized in Table 2 along with the optical band gap.
DFT calculations
In order to obtain insight into the optical spectroscopic results, TD-DFT (time-dependent DFT) calculations were performed. The computational methodology is described in detail in the Experimental section.The frontier molecular orbitals (HOMO/LUMO) together with simulated absorption spectra of complex 1 as a representative example are shown in Fig. 8.
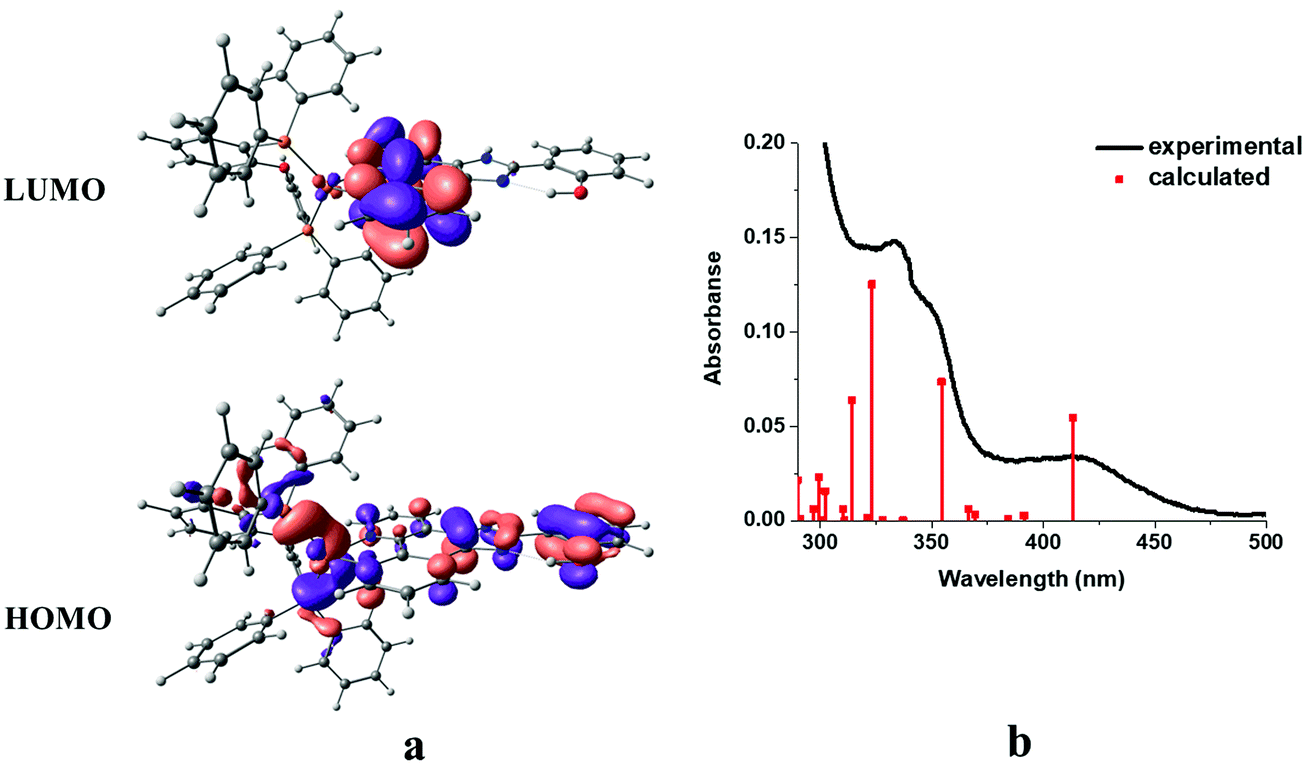 | ||
| Fig. 8 Frontier orbitals (a) and simulated absorption spectra from TD-DFT calculations in dichloromethane (red bar) compared to solution absorption spectra (black line) of complex 1 (b). | ||
A good match is observed between the experimental spectrum and calculated bars. As expected, the HOMO of the complex is mainly localized over the metal center and phenyl and imidazole moieties of the phenantroline-based ligand with little contribution of the triphenylphosphine coligand. On the other hand, the LUMO was mainly delocalized over the phenanthroline moiety of the diimine ligand (Fig. 8a). The main trends in the simulated spectra compare reasonably well with the corresponding solution spectra (Fig. 8b). The calculated lowest-energy absorption at 413 nm involves the HOMO → LUMO transition, which can be assigned as the mixture of metal to ligand (Cu → N^N) (MLCT), intra-ligand (ILCT), and ligand to ligand (POP → N^N) (LLCT) charge transfer characters. At higher energies, i.e. 354, 323, 314, 302 and 289, MLCT and ILCT are the main characters with minor contribution of the LLCT character. At higher energies, i.e. 354, 323, 314, 302 and 289, IL, MLCT and ILCT are the main characters with minor contribution of the LLCT character. The calculated transitions with their assignments are summarized in Table 3.
| Model structure | λ (nm) | Oscillator strength | Transitions | Assignment |
|---|---|---|---|---|
| 1 | 413 | 0.1750 | HOMO → LUMO | MLCT/ILCT/LLCT |
| 354 | 0.2353 | HOMO−1 → LUMO+1 | MLCT/ILCT/LLCT | |
| 323 | 0.4010 | HOMO → LUMO+2 | MLCT/IL/LLCT | |
| 314 | 0.2038 | HOMO−1 → LUMO+2 | ILCT/LLCT | |
| 2 | 413 | 0.1811 | HOMO → LUMO | MLCT/LLCT |
| 352 | 0.3350 | HOMO−1 → LUMO+1 | MLCT/IL | |
| 329 | 0.1537 | HOMO → LUMO+2 | LLCT | |
| 322 | 0.2443 | HOMO−1 → LUMO+2 | IL | |
| 318 | 0.0706 | HOMO−4 → LUMO | ILCT | |
| 3 | 413 | 0.1796 | HOMO → LUMO | MLCT/ILCT/LLCT |
| 353 | 0.3269 | HOMO−1 → LUMO+1 | MLCT/ILCT/LLCT | |
| 329 | 0.1659 | HOMO → LUMO+2 | IL/LLCT | |
| 322 | 0.1399 | HOMO−1 → LUMO+2 | IL/LLCT | |
| 320 | 0.1374 | HOMO−4 → LUMO | ILCT | |
| 4 | 415 | 0.1561 | HOMO → LUMO+1 | MLCT/LLCT |
| 368 | 0.0919 | HOMO−1 → LUMO | MLCT/ILCT | |
| 336 | 0.1261 | HOMO−3 → LUMO | LLCT | |
| 313 | 0.4852 | HOMO−1 → LUMO+3 | IL | |
Fig. S9–S12 (ESI†) depict the frontier molecular orbitals of complexes 1–4.
Electroluminescence properties
Multilayer devices can be made by vacuum deposition, and commercial OLEDs are currently made using this process. However, vacuum thermal evaporation has the drawbacks of low material utilization rates, poor scalability, high capital cost, and difficulty in patterning. Solution processing, in principle, provides a low-cost approach to fabricate OLEDs.33 To estimate the performance of the emitting compound upon electrical excitation, a solution-processed OLED test device was prepared with the following architecture: ITO/PEDOT:PSS/complex:CBP/TPBI/LiF/Al (Fig. 9). Water-soluble PEDOT:PSS was used as the hole-injection layer. A copper complex doped in the hole-transporting host (CBP (4,4′-bis(N-carbazolyl)-1,1′-biphenyl)) was spin-coated in CHCl3 over PEDOT:PSS to form a light-emitting layer. TPBI (2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)) was vacuum deposited and used as the electron-transporting and hole-blocking material.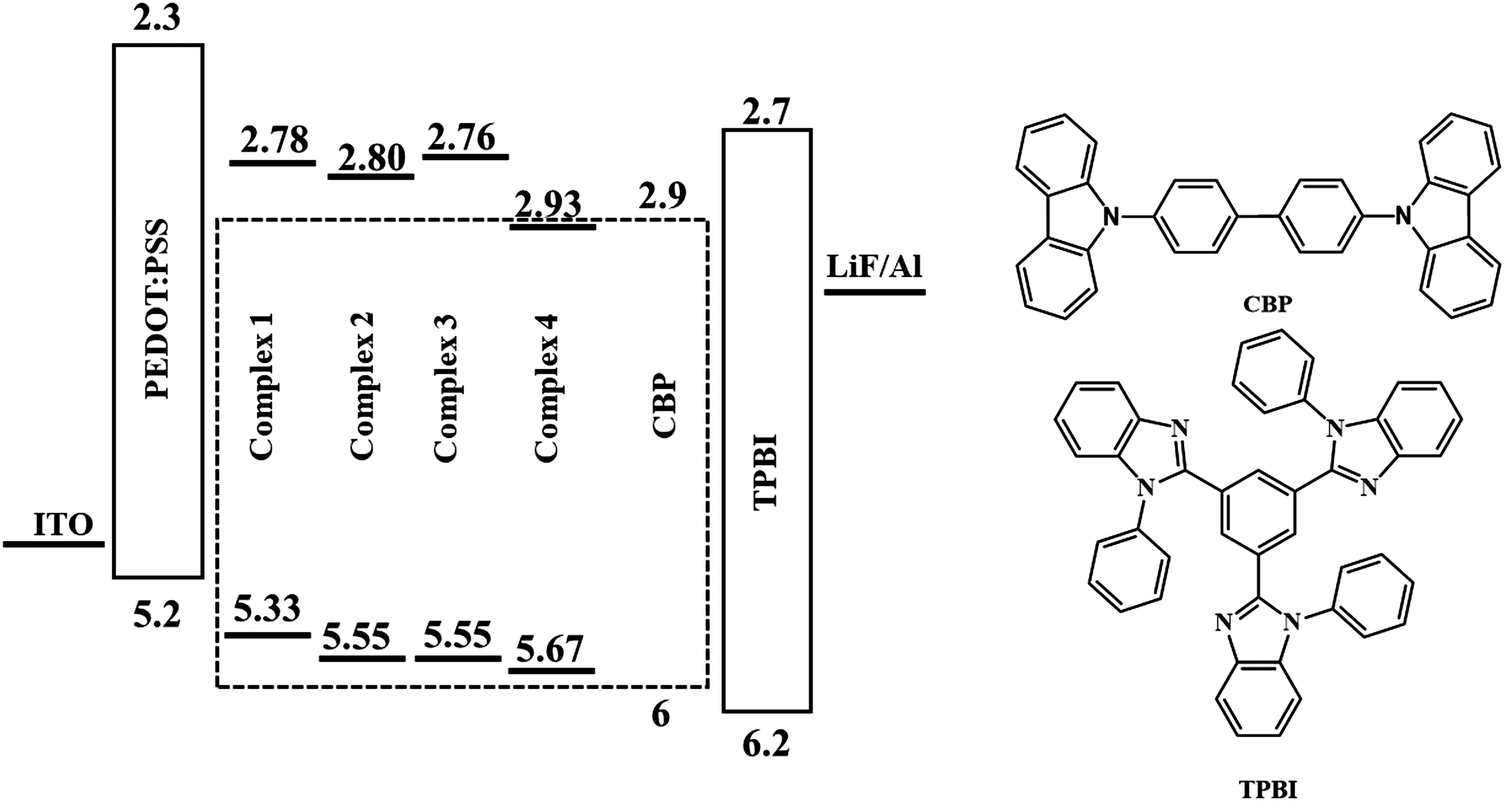 | ||
| Fig. 9 Device architecture and molecular structures of the materials used in the OLEDs. | ||
Complex 1 was used for a test device to optimize the guest concentration due to its higher quantum yield among the rest of the complexes. The EL spectra are presented in Fig. 10a.
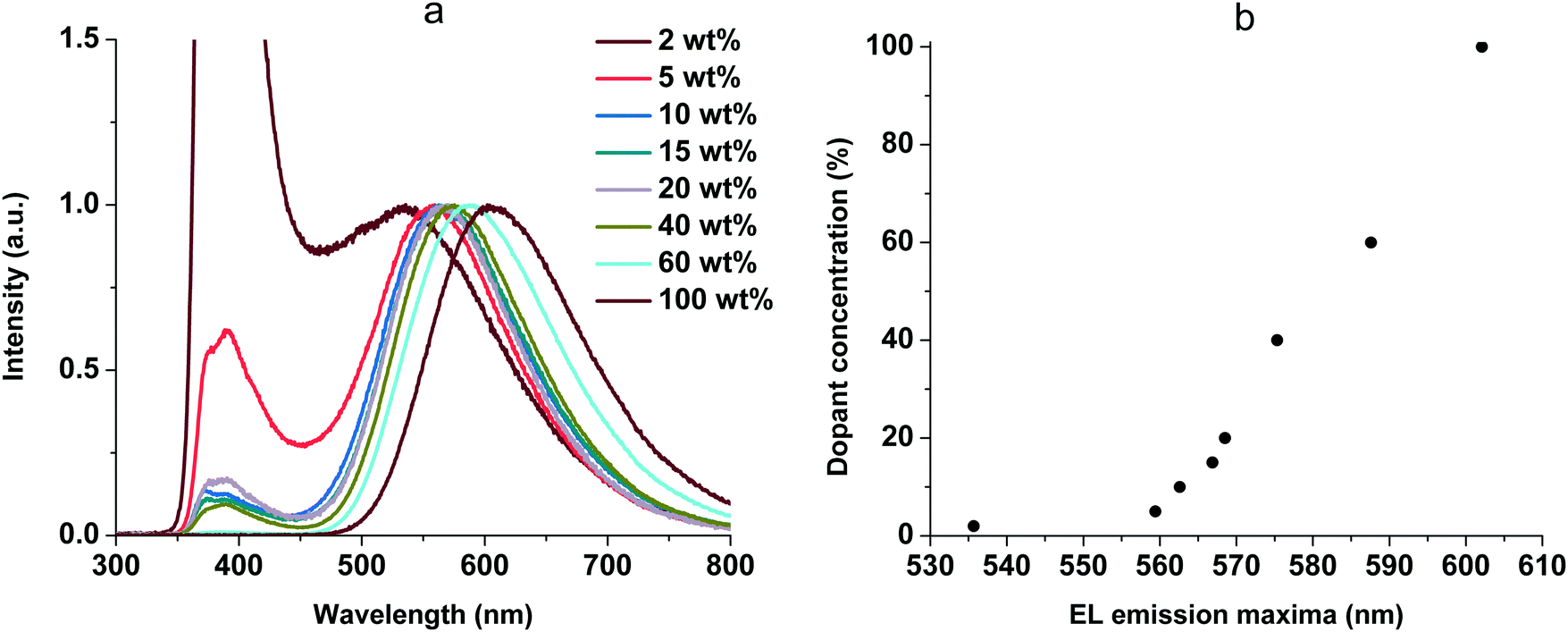 | ||
| Fig. 10 EL spectra of the OLEDs (a) and EL emission maxima shift (b) at various doping concentrations of 1. | ||
The EL spectra contain two types of emission bands: CBP emission (from 340 to 450 nm) and complex emission (from 450 to 800 nm). The CBP emission can be explained by incomplete energy transfer from host to guest molecules. In addition, we observed a red shift of the complex emission maxima with increasing guest–host ratio (Fig. 10b). It is possibly due to increased stabilization of CT states as the concentration of highly polar dopant molecules increases. This behavior is very similar to the solvatochromic effect as a result of intermolecular solute–polar solvent interaction. The same phenomena were studied earlier for 2,3,5,6-tetrakis(3,6-diphenylcarbazol-9-yl)-1,4-dicyanobenzene as a TADF dopant in a CBP:4CzTPN-Ph blend as the emissive layer in OLEDs.34 Additionally, the similarity of the peak shifting in various solvents and with different concentrations suggests that our complex suspended in a solid matrix of CBP undergoes an energy shift due to self-polarization.35 When we increased the concentration of the complex in non-polar CBP, the distance between nearest complex molecules decreased, thereby increasing the local polarization field, which leads to the red shift. Furthermore, with increasing dopant concentration the Dexter-type triplet–triplet energy transfer from the host to the dopant becomes more efficient.36 The EL spectra of the host-free OLED (100 wt%, Fig. 10a) are identical to the PL spectra at 77 K (Fig. 6b), and the phosphorescence (triplet emission) process was observed. The best performance was achieved at 15 wt% of 1 and this concentration was used for all the complexes. The EL spectra are shown in Fig. 11. The EL spectra and luminance–voltage and current density–voltage characteristics of the OLED for 1 at different host–guest ratios are shown in Fig. S5 (ESI†).
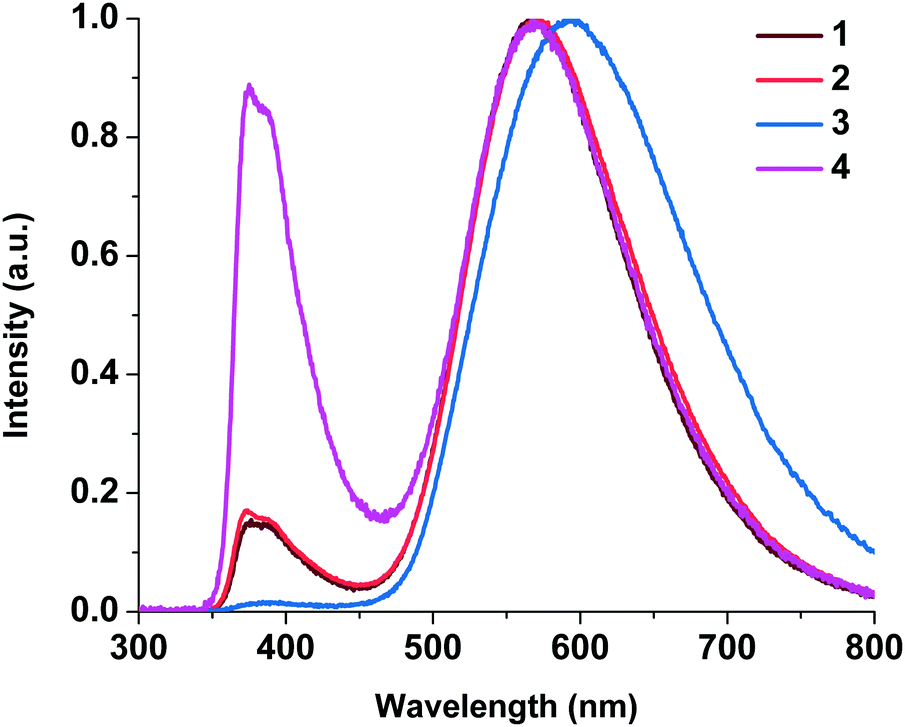 | ||
| Fig. 11 EL spectra of ITO/PEDOT:PSS/complex (15 wt%):CBP/TPBI/LiF/Al. | ||
The luminance–voltage and current density–voltage characteristics of the OLED are shown in Fig. S6 and S7 (ESI†). The EL data for complexes 1–4 are collected in Table 4.
| Complex | V turn-on (V) | L max (cd m−2) | η c (cd A−1) | λ em (nm) | CIE (x, y) |
|---|---|---|---|---|---|
| 1 | 7 | 110 | 0.04 (10 V) | 375, 386, 568 | 0.57, 0.39 |
| 2 | 7 | 50 | 0.02 (11 V) | 375, 386, 568 | 0.57, 0.39 |
| 3 | 9 | 23 | 0.02 (10 V) | 390, 595 | 0.60, 0.38 |
| 4 | 7 | 6 | 0.01 (7 V) | 374, 387, 569 | 0.56, 0.39 |
Conclusions
In summary, four mononuclear cationic Cu(I) orange emitting complexes 1, 2, 3, and 4 bearing different substituents at position 4 of the phenol ring in the 1,10-phenimidazole ligand have been prepared and comprehensively characterized, including spectroscopic, electrochemical, and computational studies. The described complexes are suitable for applying in solution-processed OLEDs. Their electroluminescence properties are strongly affected by the dopant concentration. The optimal dopant concentration was estimated as 15 wt% for the best performance. A maximum luminance of 110 cd m−2 was reached in the OLED based on complex 1. We believe that further optimization of the ligand structure and device architecture would allow enhanced OLED performance.Experimental
Measurement and characterization
1H, 13C, and 31P NMR spectra were measured on a Bruker 400 MHz WB Avance III spectrometer in DMSO-d6 at ambient temperature; residual solvent signals were used as the internal standard. The 31P chemical shifts are referenced to 85% H3PO4. Melting points were measured on a Stuart SMP30 apparatus at a heating rate of 2 °C min−1 in capillaries and were not corrected. Microanalyses (C, H, N) were carried out on a Euro EA3028-HT analyzer. Electrospray ionization mass spectra were obtained on a Bruker maXis spectrometer equipped with an electrospray ionization (ESI) source. The instrument was operated in positive ion mode using an m/z range of 50–3000. The capillary voltage of the ion source was set at ±70–150 V. The nebulizer gas pressure was 0.4 bar, and the drying gas flow was 4.0 L min−1. The most intense peak is recorded in the isotopic pattern. Infrared spectra (4000–400 cm−1) were recorded on a Shimadzu IRPrestige-21 instrument in KBr pellets. Ultraviolet-visible (UV-vis) absorption spectra were measured with a Shimadzu UV-1800 spectrophotometer. Emission spectra for solid-state samples at room temperature and at 77 K were measured on a Fluorolog 3 (Horiba Jobin Yvon) spectrofluorimeter. The photoluminescence quantum yield was defined as the number of photons emitted per photon absorbed by the system and was measured with an integrating sphere by a reported method.37 TGA was performed on ca. 2 mg samples of 1–4 by using a Netzsch TG 209 F1 Libra thermal analyzer. The samples were dried under a vacuum at 50 °C before being heated from 40 to 600 °C at a heating rate of 10 K min−1. A flow rate of 10 mL min−1 of dry argon was used to purge the samples at all times.Cyclic voltammetry
Electrochemical measurements were carried out with an Autolab PGSTAT30 (Eco Chemie, Netherlands) in a three-electrode cell consisting of a working electrode (glassy carbon 0.07 cm2), a counter electrode (platinum wire) and a non-aqueous Ag/Ag+ reference electrode (Ag wire immersed in acetonitrile containing 0.1 M AgNO3). All potentials in the text are referenced vs. the non-aqueous Ag/Ag+ electrode. The potential of the reference electrode was +0.07 V vs. saturated calomel electrode (SCE). Cyclic voltammetry measurements were performed at room temperature in argon purged anhydrous N,N-dimethylformamide solutions containing 1 × 10−3 M of a complex and 0.1 M of tetrabutylammonium tetrafluoroborate Bu4NBF4 (Aldrich) at a scan rate of 50 mV s−1. The three-electrode electrochemical cell was sealed during the experiments. Before measurements, the solutions were bubbled with dry argon for 20 minutes.Computational details
The DFT calculations were carried out with the B3LYP functional with inclusion of the CH2Cl2 solvent model (PCM) for studies of the electronic structures and the features of electronic transitions of the present complexes using the Gaussian-09 program package.38 The starting structures for the geometry optimization procedure were taken from the experimental X-ray diffraction data and no symmetry restrictions were applied. The calculations were performed using the LANL2DZ effective core potentials and the appropriate contracted basis sets for the copper atoms and the standard 6-31G* basis sets for other atoms. Vibrational analysis at the same level was performed to confirm that all the optimized structures do not have imaginary frequencies.OLED fabrication
Complex:CBP films were obtained by spin-coating on ITO/PEDOT-PSS from a 5 g l−1 solution in chloroform by applying 100 μL of the solution onto a rotating substrate at 1500 rpm rotation for 1 min.
A 15 nm TPBi (Lumtec LT-E302) film was thermally deposited in a vacuum less than 103 Pa from a quartz cuvette heated with a tantalum coil. Then 1 nm of LiF (Lumtec LT-E001) at a rate of 0.1 A s−1 and 40 nm of aluminium at a rate of 0.2 nm s−1 were thermally deposited through a mask to form four 12 mm2 pixels.
Materials and reagents
[Cu(CH3CN)4]BF4,39 1,10,-phenanthro-5,6-dione,40 2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol (L1),21 4-chloro-2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol (L2),20 and 4-bromo-2-(1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol (L3)22 were prepared according to the procedures reported in the literature. Bis[(2-diphenylphosphino)phenyl]ether (POP), poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS), 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP), and 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBI) were obtained from commercial sources and used without further purification. All experiments with air and moisture sensitive intermediates and compounds were carried out under an inert atmosphere of dry argon using standard Schlenk techniques. The solvents were purified and dried by conventional methods.1H NMR (400 MHz, DMSO-d6) δ: 13.98 (br s, 2H), 8.99–8.93 (m, 3H), 8.70 (dd, J = 8.1, 1.5 Hz, 2H), 8.08 (dd, J = 9.1, 2.8 Hz, 1H), 7.72 (dd, J = 8.1, 4.3 Hz, 2H), 6.98 (d, J = 9.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ: 163.3, 148.8, 148.6, 144.2, 140.0, 130.0, 126.7, 123.6, 120.9, 122.5, 118.4, 113.4. HRMS (ESI+, MeOH): calcd for C19H11N5O3 [M + H]+ 358.0935; found 358.0936. IR (KBr, selected bands, cm−1): 3068, 1594, 1481, 1411, 1342, 1301, 1126, 803, 738, 639.
Complex 1 was crystallized from CH2Cl2/pentane to yield the product as yellow crystals (0.66 g, 87%).
1H NMR (400 MHz, DMSO-d6) δ: 14.20 (s, 1H), 12.37 (s, 1H), 9.16 (d, J = 7.5 Hz, 2H), 8.87 (d, J = 4.3 Hz, 2H), 8.22 (d, J = 7.4 Hz, 1H), 7.94 (s, 2H), 7.47–7.42 (td, J = 7.9, 1.8 Hz, 3H), 7.30 (t, J = 7.4 Hz, 4H), 7.25–7.18 (m, 10H), 7.14–7.08 (m, 4H), 7.05–6.96 (m, 8H), 6.68 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 158.4 (t, JCP = 6.1 Hz), 157.6, 152.0, 148.3, 141.2, 134.3, 133.2 (t, JCP = 8.1 Hz), 132.8, 132.4, 132.0, 131.0 (t, JCP = 16.9 Hz), 130.6, 129.3 (t, JCP = 4.4 Hz), 127.0, 125.7, 123.6 (t, JCP = 14.6 Hz), 121.1, 120.0, 117.8, 113.3. 31P NMR (162 MHz, DMSO-d6) δ: −11.64 (s). HRMS (ESI+, MeOH): calcd for C55H40CuN4O2P2 [M]+ 913.1917; found 913.1915. IR (KBr, selected bands, cm−1): 3538, 3228, 3054, 1710, 1586, 1574, 1564, 1480, 1462, 1436, 1407, 1359, 1258, 1213, 1186, 1096, 1070, 732, 694.
Complex 2 was crystallized from CH2Cl2/pentane to yield the product as yellow crystals (82%).
1H NMR (400 MHz, DMSO-d6) δ: 14.23 (s, 1H), 12.45 (s, 1H), 9.14 (d, J = 8.3 Hz, 2H), 8.87 (d, J = 4.7 Hz, 2H), 8.27 (d, J = 2.4 Hz, 1H), 8.01–7.89 (m, 2H), 7.48–7.43 (m, 3H), 7.31 (d, J = 7.4 Hz, 4H), 7.24–7.16 (m, 10H), 7.12 (m, 3H), 6.99 (m, 8H), 6.70–6.66 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 158.4 (t, JCP = 5.9 Hz), 156.3, 150.5, 148.4, 141.3, 134.3, 133.2 (t, JCP = 8.0 Hz), 132.8, 132.0, 131.8, 131.0 (t, JCP = 16.9 Hz), 130.6, 129.3 (t, JCP = 4.5 Hz), 125.7, 123.6 (dd, JCP = 16.1 Hz, 13.0 Hz), 121.1, 119.7, 114.8, 55.4. 31P NMR (162 MHz, DMSO-d6) δ: −11.63 (s). HRMS (ESI+, MeOH): calcd for C55H39ClCuN4O2P2 [M]+ 947.1527; found 947.1517. IR (KBr, selected bands, cm−1): 3538, 3228, 3054, 1710, 1586, 1574, 1564, 1480, 1462, 1436, 1407, 1359, 1258, 1213, 1186, 1096, 1070, 732, 694.
Complex 3 was crystallized from CHCl3/pentane to yield the product as orange crystals (78%).
1H NMR (400 MHz, DMSO-d6) δ: 13.43 (br s, 2H), 9.05 (d, J = 8.1 Hz, 2H), 8.77 (d, J = 4.6 Hz, 2H), 8.42 (s, 1H), 7.83 (m, 2H), 7.44 (m, 3H), 7.30 (t, J = 7.3 Hz, 4H), 7.24–7.16 (m, 10H), 7.09 (t, J = 7.5 Hz, 2H), 7.02–6.95 (m, 9H), 6.69–6.63 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 158.4 (t, JCP = 6.2 Hz), 156.9, 147.2, 140.8, 134.3, 133.2 (t, JCP = 8.1 Hz), 132.8, 131.6, 131.2 (t, JCP = 16.7 Hz), 130.5, 129.2 (t, JCP = 4.5 Hz), 128.8, 125.6, 125.2, 123.7 (t, JCP = 14.4 Hz), 121.1, 119.6, 110.5, 79.8, 79.6, 79.4, 79.1. 31P NMR (162 MHz, DMSO-d6) δ: −11.88 (s). HRMS (ESI+, MeOH): calcd for C55H39BrCuN4O2P2 [M]+ 991.1022; found 991.1022. IR (KBr, selected bands, cm−1): 3540, 3070, 3055, 1587, 1574, 1479, 1462, 1435, 1398, 1257, 1211, 1161, 1094, 1055, 1000, 988, 873.
Complex 4 was crystallized from CH2Cl2/pentane to yield the product as orange crystals (81%).
1H NMR (400 MHz, DMSO-d6) δ: 14.02 (br s, 1H), 9.15 (s, 3H), 8.89 (s, 2H), 8.23 (s, 1H), 7.95 (s, 2H), 7.46 (t, J = 7.9 Hz, 3H), 7.31 (m, 4H), 7.25–7.16 (m, 8H), 7.11 (t, J = 7.5 Hz, 3H), 7.00 (m, 8H), 6.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 158.4 (t, JC–P = 5.8 Hz), 148.5, 141.4, 140.4, 134.3, 133.2 (t, JC–P = 8.1 Hz), 132.9, 131.0 (t, JC–P = 16.9 Hz), 130.6, 129.3, 125.7, 123.6 (t, JC–P = 14.6 Hz), 121.1, 55.4. 31P NMR (162 MHz, DMSO-d6) δ: −11.63 (s). HRMS (ESI+, MeOH): calcd for C55H39CuN5O4P2 [M]+ 958.1768; found 958.1791. IR (KBr, selected bands, cm−1): 3490, 3255, 3055, 1710, 1607, 1589, 1506, 1480, 1463, 1435, 1340, 1300, 1260, 1216, 1127, 1095, 1075, 1028, 998, 974.
X-Ray crystallography
The XRD experiments for complex 1 and 3 were performed on a SuperNova (Agilent Technologies/Oxford Diffraction) diffractometer, equipped with a dual microfocus Cu/Mo X-ray source and Atlas CCD detector, using monochromated Cu Kα (λ = 1.54184 Å) radiation. The XRD experiment for the 2 single crystal was performed on an Xcalibur (Agilent Technologies/Oxford Diffraction) diffractometer using monochromated Mo Kα (λ = 0.71073 Å) radiation. The temperature was kept at 100 K for 2 and 3, and 130 K for 1 because of possible crystal destruction at 100 K. The structures were solved by direct methods by means of the SHELX program41 incorporated in the OLEX2 program package.42 An empirical absorption correction was applied in the CrysAlisPro43 software package using spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm. Supplementary crystallographic data for this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC numbers 1941884, 1941885, and 1953636).†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by RSF (17-73-10078). Physicochemical studies were performed at the Centre for Magnetic Resonance, Centre for X-ray Diffraction Studies, Thermogravimetric and Calorimetric Research Centre, Centre for Optical and Laser Materials Research, and Centre for Chemical Analysis and Materials Research (all belong to Saint Petersburg State University).References
- F. Dumur, Org. Electron., 2015, 21, 27–39 CrossRef CAS.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601 CrossRef CAS PubMed.
- M. Xie, C. Han, Q. Liang, J. Zhang, G. Xie and H. Xu, Sci. Adv., 2019, 5, eaav9857 CrossRef.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- D. Volz, M. Wallesch, C. Fléchon, M. Danz, A. Verma, J. M. Navarro, D. M. Zink, S. Bräse and T. Baumann, Green Chem., 2015, 17, 1988–2011 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508–3535 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- C. Bizzarri, F. Hundemer, J. Busch and S. Bräse, Polyhedron, 2018, 140, 51–66 CrossRef CAS.
- L. Bergmann, G. J. Hedley, T. Baumann, S. Bräse and I. D. W. Samuel, Sci. Adv., 2016, 2, e1500889 CrossRef PubMed.
- F. Zhang, Y. Guan, X. Chen, S. Wang, D. Liang, Y. Feng, S. Chen, S. Li, Z. Li, F. Zhang, C. Lu, G. Cao and B. Zhai, Inorg. Chem., 2017, 56, 3742–3753 CrossRef CAS PubMed.
- F. Brunner, A. Babaei, A. Pertegás, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, Dalton Trans., 2019, 48, 446–460 RSC.
- R. Liu, M.-M. Huang, X.-X. Yao, H.-H. Li, F.-L. Yang and X.-L. Li, Inorg. Chim. Acta, 2015, 434, 172–180 CrossRef CAS.
- M.-M. Huang, Y.-M. Guo, Y. Shi, L. Zhao, Y.-W. Niu, Y. Shi and X.-L. Li, Inorg. Chim. Acta, 2017, 457, 107–115 CrossRef CAS.
- X. Liu, R. Li, L. Ma, X. Feng and Y. Ding, New J. Chem., 2016, 40, 619–625 RSC.
- C. Yi, S. Xu, J. Wang, F. Zhao, H. Xia and Y. Wang, Eur. J. Inorg. Chem., 2016, 4885–4890 CrossRef.
- N. M. Gabra, B. Mustafa, Y. P. Kumar, C. Shobha Devi, M. Shilpa, K. L. Reddy and S. Satyanarayana, Med. Chem. Res., 2014, 23, 224–235 CrossRef CAS.
- Z.-B. Cai, L.-F. Liu, Y.-Q. Hong and M. Zhou, J. Coord. Chem., 2013, 66, 2388–2397 CrossRef CAS.
- M. Gomleksiz, B. Erdem and C. Alkan, S. Afr. J. Chem., 2013, 66, 107–112 CAS.
- A. Bondi, J. Phys. Chem., 1966, 70, 3006–3007 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- Y. Shi, X. Liu, Y. Shan, X. Zhang, W. Kong, Y. Lu, Z. Tan and X.-L. Li, Dalton Trans., 2019, 48, 2430–2441 RSC.
- X. Liu, Y. Shan, J. Xu, X. Zhang, S. Shang and X.-L. Li, Polyhedron, 2019, 164, 152–158 CrossRef CAS.
- L. Shi and B. Li, Eur. J. Inorg. Chem., 2009, 2294–2302 CrossRef CAS.
- C. Q. Ma, L. Q. Zhang, J. H. Zhou, X. S. Wang, B. W. Zhang, Y. Cao, P. Bugnon, M. Schaer, F. Nüesch, D. Q. Zhang and Y. Qiu, J. Mater. Chem., 2002, 12, 3481–3486 RSC.
- D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6–7 CrossRef CAS PubMed.
- S.-X. Xu, J.-L. Wang, F. Zhao, H.-Y. Xia and Y.-B. Wang, Transition Met. Chem., 2015, 40, 723–732 CrossRef CAS.
- K. N. Solov'ev and E. A. Borisevich, Phys.-Usp., 2005, 48, 231–253 CrossRef.
- J. Chen, T. Teng, J.-Y. Wang, L. Kang, X.-L. Chen, L.-J. Xu, R. Yu and C.-Z. Lu, Eur. J. Inorg. Chem., 2016, 3036–3041 CrossRef CAS.
- P.-A. Will and S. Reineke, in Handbook of Organic Materials for Electronic and Photonic Devices, ed. O. Ostroverkhova, Woodhead Publishing, 2nd edn, 2019, pp. 695–726 Search PubMed.
- Y. R. Cho, S. J. Cha and M. C. Suh, Synth. Met., 2015, 209, 47–54 CrossRef CAS.
- V. Bulović, A. Shoustikov, M. A. Baldo, E. Bose, V. G. Kozlov, M. E. Thompson and S. R. Forrest, Chem. Phys. Lett., 1998, 287, 455–460 CrossRef.
- W. S. Jeon, T. J. Park, S. Y. Kim, R. Pode, J. Jang and J. H. Kwon, Org. Electron., 2009, 10, 240–246 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS.
- J. R. Hickson, S. J. Horsewill, C. Bamforth, J. McGuire, C. Wilson, S. Sproules and J. H. Farnaby, Dalton Trans., 2018, 47, 10692–10701 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- CrysAlisPro, Version 1.171.136.120, Agilent Technologies Ltd, release 127-106-2012.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1941884, 1941885 and 1953636. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj05109k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |