
Lutetium and yttrium complexes supported by an anilido-oxazoline ligand for polymerization of 1,3-conjugated dienes and ε-caprolactone†
Wenqiang
Li
a,
Xinxin
Jiang
a,
Yat-Ming
So
b,
Gaohong
He
 a and
Yu
Pan
*a
a and
Yu
Pan
*a
aState Key Laboratory of Fine Chemicals, School of Petroleum and Chemical Engineering, Dalian University of Technology, Panjin 124221, China. E-mail: ypan@dlut.edu.cn
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 26th November 2019
Abstract
Lutetium and yttrium dialkyl complexes supported by an anilido-oxazoline ligand were synthesized, and their catalytic behaviors toward polymerization of isoprene (IP), β-myrcene (Myr) and ε-caprolactone (CL) have been explored. The treatments of [Ln(CH2SiMe3)3(THF)2] (Ln = Lu, and Y) with the anilido-oxazoline pro-ligands 2-(2,6-R2PhN)-phenyl-4-Me2-oxazoline (HLa: R = Me, and HLb: R = iPr) afforded the lutetium and yttrium dialkyl complexes [(La,b)Ln(CH2SiMe3)2THF] (La: Ln = Lu (1a), Y (2a); Lb: Ln = Lu (1b), Y (2b)). All the complexes not only exhibited high cis-1,4-selectivity for IP polymerization (87.0–95.9%) and Myr polymerization (82.3–94.5%), but were also found to be active in initiating ring-opening polymerization (ROP) of CL with outstanding activity (i.e., more than 97% yield within 1 min). Gratifyingly, when CL was added to a 100% conversion IP polymerization system catalyzed by 2b, diblock copolymer polyisoprene-b-polycaprolactone (PIP-b-PCL) could be isolated. By increasing the content of PCL in the copolymers, the static contact angle of water of the copolymers changed from 108.9° to 93.3°.
Introduction
The development of high-performance organometallic catalysts for polymerization with high activity and selectivity is an important aspect for both academic and industrial fields.1 In this regard, rare-earth (Ln) based organometallic complexes have been developed flourishingly in recent decades and showed their unique catalytic performance on coordination polymerization of styrene, 1,3-conjugated dienes and some polar monomers.2–5 In particular, for polymerization of 1,3-conjugated dienes, Ln catalysts could produce polymers with different regularities (e.g. cis-1,4, and trans-1,4, and 3,4 (or 1,2) regularities) and versatile properties,6–9 and have also been successfully applied in the synthetic rubber industry to catalyze the production of cis-1,4-regular polyisoprene (PIP, also called isoprene rubber (IR)), which can be used as an alternative to natural rubber (NR).10 In general, Ln complexes supported by chelating spectator ligands are well demonstrated to facilitate the cis-1,4-polymerization of 1,3-conjugated dienes.11,12 Hou and coworkers reported that, upon activation with [PhMe2NH][B(C6F5)4], Ln dialkyl complexes containing a bis(phosphinophenyl)amido PNP-pincer ligand could induce living polymerization and copolymerization of isoprene (IP) and butadiene (BD) with high cis-1,4-selectivity (up to 99.6%).13 Ln dichloride complexes bearing NCN-pincer aryldiimine ligands were also found to exhibit high activity and outstanding cis-1,4-selectivity for polymerization of BD (up to 99.9%) and IP (up to 98.8%) upon activation with aluminum alkyls and organic borates.14 Ln complexes supported by other chelating ligands, such as bis(carbene)phenyl (CCC-pincer),15 β-diketiminato,16,17 aminopyridinato,18 bis(phosphino)carbazolide (PNP-pincer),19,20 bis(oxazolinymethylidene)isoindoline (NNN-pincer),21 indole-based ligands,22,23etc., have also been developed for cis-1,4-polymerization of 1,3-conjugated dienes and showed their good controllability.Ring-opening polymerization (ROP) of a wide variety of cyclic esters has been developed to provide renewable materials that can be used for packaging, the polyurethane industry, medical materials, tissue engineering, etc.24–27 Towards this area, Ln complexes show various advantages, such as nontoxicity, high activity and good controllability of molecular weight and stereochemistry, for catalyzing ROP of lactide (LA), ε-caprolactone (CL), and other cyclic esters.28,29 Amino-bis(phenolate)-supported Ln complexes with modified side-arm and initiating groups could effectively initiate ROP of rac-LA with excellent heteroselectivity (Pr up to 0.99).30–36 On the other hand, Y and Lu complexes bearing multidentate ligands could catalyze the production of polylactide (PLA) with high isotacticity.37–39 Highly active catalytic systems based on Ln complexes have also been developed for ROP of CL to obtain poly(ε-caprolactone) (PCL).40–44 Recently, ROPs of γ-butyrolactone (γ-BL) and its derivatives (e.g., 4,5-trans six-membered ring-fused γ-BL) were successfully achieved by Ln complexes to generate polymers with repeatable chemical recyclability.45–48
We are particularly interested in developing high-performance Ln complexes for coordination polymerization and ROP. For example, Sc complexes bearing N-heterocyclic carbene ligands were isolated and found to show high activity for α-olefin polymerization and copolymerization with α,ω-dienes;49,50 meanwhile, the highly isotactic poly(α-olefin)s (mmmm > 99%) could be achieved using a simple catalyst system ScCl3(THF)3/AliBu3/[Ph3C][B(C6F5)4] under mild conditions.51 Moreover, Ln dichloride complexes bearing the bis(oxazolinyl)phenyl ligand could initiate isoprene polymerization with high activity and high cis-1,4-selectivity.52 Recently, we also found that the anilido-oxazoline-ligated Sc and Y dialkyl complexes exhibited high activity and heteroselectivity for ROP of rac-LA, and high cis-1,4-selectivity for isoprene polymerization upon activation with organic borates.53,54 As an extension of this work, herein we isolated Lu complexes bearing the anilido-oxazoline ligands; the catalytic performance of these Lu complexes and the previously reported Y complexes on polymerization of IP and β-myrcene (Myr) and ROP of CL will also be reported.
Results and discussion
Synthesis and characterization of lutetium complexes
The Lu complexes [(La,b)Lu(CH2SiMe3)2THF] (1a,b) were prepared following the previous procedure by treating the anilido-oxazoline pro-ligands (HLa,b) with an equimolar amount of [Lu(CH2SiMe3)3(THF)2] at room temperature (Scheme 1).53 The 1H NMR spectra of 1 showed that the ratio of anilido-oxazoline ligand, trimethylsilylmethyl and coordinated THF was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1. The methylene protons in Lu-CH2SiMe3 resonated at δ = −0.84 to −0.78 and −0.52 to −0.46 ppm as a characteristic double doublet.55,56 According to the 13C NMR spectra, the resonance signals of C
:2:1. The methylene protons in Lu-CH2SiMe3 resonated at δ = −0.84 to −0.78 and −0.52 to −0.46 ppm as a characteristic double doublet.55,56 According to the 13C NMR spectra, the resonance signals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the oxazoline moiety were observed at δ = 168.92–169.52 ppm, which shifted to the low-field region compared with that of free ligand at δ = 162.26–162.30 ppm (Fig. S1–S4, ESI†). Meanwhile, red shifts were observed for ν(CN) stretches of 1 at 1607 cm−1 in the IR spectra (Fig. S5 and S6, ESI†), in comparison with that in free anilido-oxazoline at 1626–1630 cm−1. This indicated that the nitrogen atom in the oxazoline moiety coordinates to the central Lu3+. Similar to the reported Y complexes [(La,b)Y(CH2SiMe3)2THF] (2), the UV-Vis spectra of 1a and 1b exhibited two absorption peaks at 350 and 399 nm, and 352 and 406 nm, respectively. Moreover, the blue-green fluorescence emissions were observed at the emission maxima of 472 nm for 1a and 475 nm for 1b when they were excited at 400 nm (Fig. S7–S9, ESI†).
N in the oxazoline moiety were observed at δ = 168.92–169.52 ppm, which shifted to the low-field region compared with that of free ligand at δ = 162.26–162.30 ppm (Fig. S1–S4, ESI†). Meanwhile, red shifts were observed for ν(CN) stretches of 1 at 1607 cm−1 in the IR spectra (Fig. S5 and S6, ESI†), in comparison with that in free anilido-oxazoline at 1626–1630 cm−1. This indicated that the nitrogen atom in the oxazoline moiety coordinates to the central Lu3+. Similar to the reported Y complexes [(La,b)Y(CH2SiMe3)2THF] (2), the UV-Vis spectra of 1a and 1b exhibited two absorption peaks at 350 and 399 nm, and 352 and 406 nm, respectively. Moreover, the blue-green fluorescence emissions were observed at the emission maxima of 472 nm for 1a and 475 nm for 1b when they were excited at 400 nm (Fig. S7–S9, ESI†).
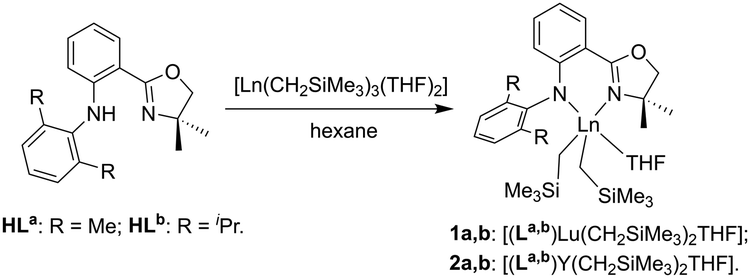 | ||
| Scheme 1 Synthesis of anilido-oxazoline supported Lu and Y complexes 1 and 2. | ||
The crystal structures of 1a and 1b were characterized by single crystal X-ray diffraction and are shown in Fig. 1. The Lu centres of both 1a and 1b were penta-coordinated, adopting a distorted trigonal bipyramidal configuration. The oxygen in THF and nitrogen in oxazoline are the vertices of the trigonal bipyramidal configuration in both 1a and 1b. The N1–Lu–O2 angles are 169.58° for 1a and 178.19° for 1b, which are larger than those in 2a (168.85°) and 2b (175.95°). The Lu–N1 and Lu–N2 bond distances in 1a (2.352 and 2.272 Å) and 1b (2.339 and 2.235 Å) are shorter than those in the Y complexes 2 (2.4064–2.4270 and 2.3148–2.3220 Å) and longer than those in the analogous Sc complexes (2.1650–2.2641 and 2.0631–2.1572 Å),53 which are consistent with the order of the ionic radii (Y3+ > Lu3+ > Sc3+).57 The 2,6-disubstituted phenyls are virtually perpendicular to the central phenyls with the respective dihedral angles of 77.34° and 80.90° for 1a and 1b. Because of the crowded coordination environment, the central metals are not in the coordinated planes (NCPhCPhCN plane) with distances of 0.5949 and 0.4793 Å for 1a and 1b, respectively. The out-of-plane distances of Lu complexes are between those of Sc (0.5538 Å) and Y complexes (0.6086 and 0.6135 Å) having the same ligands and analogous structures.53
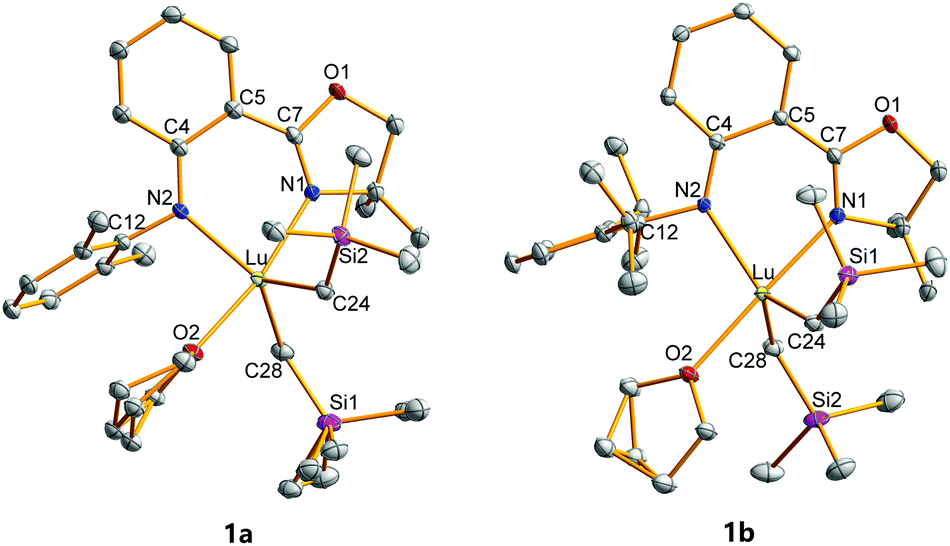 | ||
| Fig. 1 Geometric structures of 1a and 1b. Hydrogen atoms are omitted for clarity. The ellipsoids are drawn at 30% probability level. Selected bond lengths (Å) and angles (°) of 1a: Lu–N1, 2.352(2); Lu–N2, 2.272(2); Lu–O2, 2.3361(19); Lu–C24, 2.346(3); Lu–C28, 2.340(3); N1–C7, 1.292(3); N2–C4, 1.369(3); N1–Lu–O2, 169.58(7); N2–Lu–C24, 130.43(9); N2–Lu–C28, 117.70(9); C24–Lu–C28, 111.87(10). Selected bond lengths (Å) and angles (°) of 1b: Lu–N1, 2.3392(19); Lu–N2, 2.2353(17); Lu–O2, 2.3674(16); Lu–C24, 2.348(3); Lu–C28, 2.372(2); N1–C7, 1.298(3); N2–C4, 1.368(3); N1–Lu–O2, 178.19(6); N2–Lu–C24, 118.71(8); N2–Lu–C28, 117.02(8); C24–Lu–C28, 124.09(9). | ||
Polymerization of 1,3-conjugated dienes
The catalytic activity of 1 and 2 towards polymerization of 1,3-conjugated dienes has been explored and the representative results are summarized in Table 1. When IP was used as a monomer, the Lu complexes 1 could provide 51.8–65.1% yield of PIPs in 40 min under activation with [Ph3C][B(C6F5)4] (Table 1, runs 1 and 2). On the other hand, a sluggish polymerization was observed when [PhNHMe2][B(C6F5)4] or B(C6F5)3 was used as the activator (Table 1, runs 3 and 4). According to the NMR spectra, the resultant PIPs have high cis-1,4 regularity as suggested by 87.0% for 1a and 89.8% for 1b (Table 1, runs 1 and 2). In comparison, the Y complexes 2 exhibited higher activities that achieved complete conversion within 30 min under activation with [Ph3C][B(C6F5)4] (Table 1, runs 5 and 6). It is probably attributed to the larger ionic radius of Y3+, which is beneficial to the coordination of isoprene with η4-mode. Furthermore, higher cis-1,4-selectivity was observed by using the Y complexes (90.2% for 2a and 95.9% for 2b) compared to the corresponding Lu complexes with the same ligand. Regardless of the central metal, the bulkier steric hindrance led to higher cis-1,4-selectivity for IP polymerization.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Run | Cat. | Mon.b | t (min) | Conv. (%) | M n (×104) | M w/Mnc | Microstructured (%) | ||
| cis-1,4 | trans-1,4 | 3,4 | |||||||
| a Conditions: Cat. (10 μmol), [Ph3C][B(C6F5)4] (10 μmol), [Mon.]/[Ln] = 1000, toluene (5 mL), and T = 30 °C. b IP = isoprene, and Myr = β-myrcene. c Determined by means of gel permeation chromatography (GPC) against polystyrene standards. d Determined by the 1H and 13C NMR spectra of the polymers. e [PhNHMe2][B(C6F5)4] (10 μmol). f B(C6F5)3 (10 μmol). g n.d.: not determined. h Toluene (10 mL). | |||||||||
| 1 | 1a | IP | 40 | 51.8 | 17.61 | 1.87 | 87.0 | 1.6 | 11.4 |
| 2 | 1b | IP | 40 | 65.1 | 10.31 | 1.84 | 89.8 | 1.1 | 9.1 |
| 3e | 1b | IP | 60 | 36.7 | 7.33 | 1.58 | 78.2 | 6.4 | 15.4 |
| 4f | 1b | IP | 120 | Trace | n.d.g | n.d. | n.d. | n.d. | n.d. |
| 5 | 2a | IP | 30 | 100 | 10.82 | 1.27 | 90.2 | 3.0 | 6.8 |
| 6 | 2b | IP | 30 | 100 | 10.73 | 1.29 | 95.9 | 0 | 4.1 |
| 7 | 1a | Myr | 60 | 45.9 | 15.20 | 2.02 | 82.3 | 4.0 | 13.7 |
| 8 | 1b | Myr | 120 | 35.4 | 17.84 | 2.05 | 83.5 | 1.0 | 15.5 |
| 9h | 2a | Myr | 40 | 70.9 | 7.92 | 1.80 | 94.2 | 0 | 5.8 |
| 10h | 2b | Myr | 40 | 66.6 | 6.91 | 1.87 | 94.5 | 0 | 5.5 |
β-Myrcene (Myr), a 1,3-conjugated diene monomer with a long side-substituent, is derived from plants such as dill, thyme, and rosemary and can be used as an alternative for petroleum-based 1,3-diene monomers (e.g., butadiene, and isoprene) in polymer synthesis.58–67 Based on the study of IP polymerization, it is therefore of interest to study the catalytic performance of the Lu and Y complexes on Myr polymerization. The Lu complexes 1 exhibited moderate activity for Myr polymerization with the yield of 45.9% for 1a in 60 min and 35.4% for 1b in 120 min (Table 1, runs 7 and 8). This is probably caused by a long side-substituent in Myr that hinders the coordination of monomer to some extent, and the bulkier substituents in 1b showed a negative effect on catalytic activity. Similar to IP polymerization, the Y complexes 2 exhibited higher activities for Myr polymerization than the Lu complexes 1. According to the 1H NMR spectra of the resultant polymers, the resonance signals at δ = 4.80–4.76 ppm are assigned to the olefinic protons in 3,4-enchainments and the signals at δ = 5.12–5.09 ppm can be assigned to those in 1,4-enchainments and the olefinic side-chain (Fig. S14–S17, ESI†).68 The 1,4-regioregularity of 84.5–94.5% for the resultant polymyrcenes (PMys) could be calculated according to the integration of the characteristic peaks. The microstructure of the resultant PMys could be further identified by the 13C NMR spectra.68 It is shown that the high cis-1,4 regularity (82.3–94.5%) for PMys could be achieved by all the complexes according to the resonance signal regions at ca. δ = 37.0 ppm (Fig. S18–S21, ESI†). Similar to the IP polymerization, the Y complexes 2 were more favourable for cis-1,4 selectivity than the Lu complexes 1.
Ring-opening polymerization of ε-caprolactone
Besides the polymerization of 1,3-conjugated dienes, the catalytic performance of complexes 1 and 2 towards ROP of CL has also been studied, and the representative results are summarized in Table 2. Both Lu and Y complexes exhibited very high activities for initiating the ROP of CL, and almost complete conversions of 500 equivalents of CL could be achieved within 1 min (Table 2, runs 1–4). The molecular weights of the resultant PCLs were close to the calculated theoretical values that are based on a double-site initiation, suggesting that both alkyls in 1 and 2 are responsible for initiating the ROP of CL. Choosing 2b as a representative initiator, more than 91% conversion could be achieved within 1 min even at the feed ratio of 1000 to 1 (Table 2, run 6). The polymerization showed a bit of sluggishness when polar solvents were used (Table 2, runs 7 and 8).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Run | Cat. | [CL]/[Cat.] | Conv. (%) | M n,t,1 (×104) | M n,t,2 (×104) | M n (×104) | M w/Mnd |
| a Conditions: Cat. (10 μmol), toluene as solvent, [CL]0 = 1.0 mol L−1, T = 30 °C, and time = 1 min. b Calculated by ([CL]0/[Cat.]) × 114.13 × yield. c Calculated by ([CL]0/2[Cat.]) × 114.13 × yield. d Determined by means of gel permeation chromatography (GPC) against polystyrene standards, multiplied by 0.56. e Chlorobenzene as solvent. f Dichloromethane as solvent. | |||||||
| 1 | 1a | 500 | 98.2 | 5.60 | 2.80 | 2.98 | 1.78 |
| 2 | 1b | 500 | 97.3 | 5.55 | 2.78 | 2.73 | 1.79 |
| 3 | 2a | 500 | 100 | 5.71 | 2.85 | 3.04 | 1.71 |
| 4 | 2b | 500 | 98.9 | 5.64 | 2.82 | 2.63 | 1.77 |
| 5 | 2b | 300 | 100 | 3.42 | 1.71 | 2.12 | 1.66 |
| 6 | 2b | 1000 | 91.2 | 10.41 | 5.20 | 8.74 | 1.69 |
| 7e | 2b | 500 | 84.6 | 4.83 | 2.41 | 3.68 | 1.91 |
| 8f | 2b | 500 | 72.3 | 4.13 | 2.06 | 3.26 | 1.62 |
Copolymerization of isoprene and ε-caprolactone
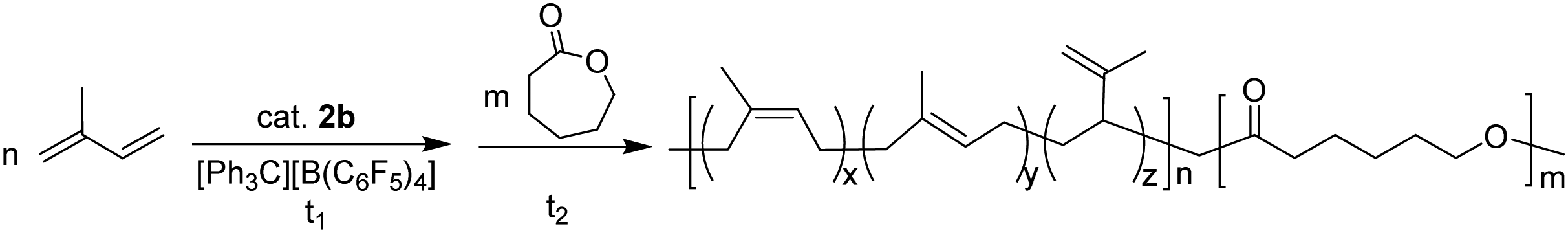
Our previous study showed that the catalyst system 2b/[Ph3C][B(C6F5)4] exhibited high activity and good controllability for IP polymerization.53 Combining its catalytic performance on ROP, we attempted to copolymerize IP and CL via adding the two monomers sequentially and the results are shown in Table 3. After finishing the IP polymerization by 2b/[Ph3C][B(C6F5)4], CL was added to the catalytic system to initiate ROP, and the copolymer was isolated by precipitation in alcohol. The GPC traces showed that the molecular weight of the copolymer increased after CL addition, yet the molecular weight distribution remained monomodal (Fig. S22–S24, ESI†). The IP and CL contents in the copolymers were found to be close to their feed ratio according to the 1H NMR spectra. Moreover, with an increase of the CL contents in the copolymers, the Tm values appeared at ca. 58 °C and the degree of crystallinity turned higher as suggested by the increased ΔHm values ranging from 19.58 to 39.68 J g−1 (Fig. 2). In comparison, the ΔHm value of 45.15 J g−1 was observed for homopolymer PCL. This indicated the formation of block copolymers rather than a mixture of homopolymers.19 The PIP block in the copolymer maintained a high cis-1,4 regularity (95.2–96.2%), which is similar to the result of IP homopolymerization by 2b/[Ph3C][B(C6F5)4] (Table 1, run 6). The proposed mechanistic pathway for copolymerization of IP and CL is shown in Scheme 2. After IP insertion and propagation, the Y active species with the η3-π-prenyl moiety is generated.69,70 CL is then added to the system and coordinated to the central Y3+via a carboxyl oxygen. The nucleophilic attack of the Y–prenyl bond on the carboxyl carbon obtains a new active species with a Y–OR bond, that can initiate ROP of CL continuously to produce the copolymer.71|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Run | Feed ratio [IP]:[CL] |
t 1/t2 (min) | Conv. (%) | cis-1,4b (%) | trans-1,4b (%) | 3,4b (%) | M n (×104) | M w/Mnc | IP contentb (mol%) | T g (°C) | T m (°C) | ΔHmd (J g−1) |
| a Conditions: complex 2b (10 μmol), [Ph3C][B(C6F5)4] (10 μmol), toluene (10 mL), and T = 30 °C. b Determined by the 1H and 13C NMR spectra of polymers. c Determined by means of gel permeation chromatography (GPC) against polystyrene standards. d Determined by differential scanning calorimetry (DSC). | ||||||||||||
| 1 | 700:300 |
50/10 | 94.0 | 96.2 | 0 | 3.8 | 9.91 | 1.64 | 68.9 | −59.8 | 57.1 | 19.58 |
| 2 | 500:500 |
45/5 | 93.2 | 96.2 | 0 | 3.8 | 10.80 | 1.70 | 48.5 | −60.0 | 58.5 | 28.13 |
| 3 | 300:700 |
40/5 | 93.4 | 95.2 | 1.9 | 2.9 | 13.70 | 1.36 | 28.5 | −60.7 | 59.2 | 39.68 |
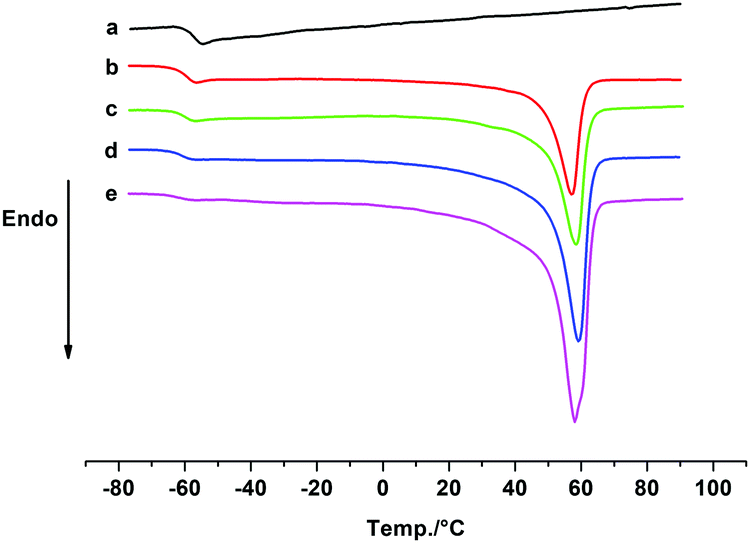 | ||
| Fig. 2 DSC thermograms for PIP, copolymers and PCL: (a) PIP; (b) the copolymer prepared with the feed ratio of IP and CL 700:300 (Table 3, run 1); (c) the copolymer prepared with the feed ratio of IP and CL 500:500 (Table 3, run 2); (d) the copolymer prepared with the feed ratio of IP and CL 300:700 (Table 3, run 3); (e) PCL. | ||
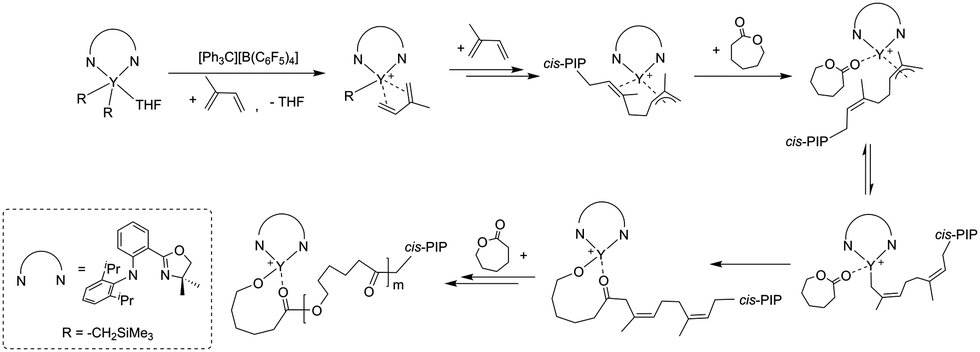 | ||
| Scheme 2 Probable mechanistic pathway for copolymerization of IP and CL. | ||
Introduction of polar functional groups into nonpolar polymers, such as polyolefin, polystyrene, PIP, etc., is an efficient strategy to modify their surface properties. Therefore, the surface properties of the resultant copolymers were tested and evaluated by the static contact angle of water.50,72 It is found that the homopolymer PIP is hydrophobic according to a relatively larger static contact angle of water (121.3°). By increasing the content of PCL from ca. 30% to 70% in the copolymers, the static contact angles of water decreased from 108.9° to 93.3°, indicating the improvement of their hydrophilicity (Fig. 3). A static contact angle of 69.6° was observed for the homopolymer PCL under the same conditions. This demonstrated that the surface properties of the copolymers could be adjusted by changing the contents of PCL in the copolymers.
 | ||
| Fig. 3 The static contact angle of water on: (a) PIP; (b) the copolymer prepared with the feed ratio of IP and CL 700:300 (Table 3, run 1); (c) the copolymer prepared with the feed ratio of IP and CL 500:500 (Table 3, run 2); (d) the copolymer prepared with the feed ratio of IP and CL 300:700 (Table 3, run 3); (e) PCL. | ||
Conclusions
In summary, we have synthesized and isolated anilido-oxazoline-ligated Lu complexes 1a and 1bvia a convenient one-pot reaction of [Lu(CH2SiMe3)3(THF)2] with the pro-ligands. The analysis of their crystal structures showed that both of the complexes adopted a distorted trigonal bipyramidal configuration with one molecule THF. Under activation with [Ph3C][B(C6F5)4], the Lu complexes could catalyze IP polymerization with moderate activity to obtain the PIP with 87.0–89.8% cis-1,4 regularity. The organic borate exhibited an effect on activity and selectivity. In comparison, the analogous Y complexes 2a and 2b showed higher activity and cis-1,4 selectivity for IP polymerization, indicating the influence of the central metal. Polymerization of Myr could also be achieved by the Lu and Y catalytic systems to produce the PMys with high cis-1,4 regularity (82.3–94.5%). Moreover, complexes 1 and 2 were found to be highly active in initiating ROP of CL showing almost complete conversion within 1 min. When CL was added to a 100% conversion IP polymerization system catalyzed by 2b, diblock copolymer polyisoprene-b-polycaprolactone (PIP-b-PCL) could be isolated. By increasing the content of PCL, the degree of crystallinity of the copolymers increased according to the increased ΔHm values (19.58–39.68 J g−1), in addition, the static contact angle of water on the copolymers changed from 108.9° to 93.3°, indicating the improvement of their hydrophilicity.Experimental section
General methods and materials
All air- and moisture-sensitive reactions were carried out using standard Schlenk techniques or in a glove box under argon atmosphere. Toluene, n-hexane, and tetrahydrofuran (THF) were dried with sodium/benzophenone under argon. Anhydrous LnCl3 (Ln = Lu, and Y), [Ph3C][B(C6F5)4], [PhMe2NH][B(C6F5)4] and B(C6F5)3 were purchased from Jilin University and used as received. IP was purchased from TCI and Myr was purchased from Sigma Aldrich, and both of them were dried with CaH2, degassed and stored in a glove box at −30 °C. CL was obtained from Energy Chemistry and purified by distillation over CaH2 under reduced pressure. [Ln(CH2SiMe3)3(THF)2] (Ln = Lu, and Y)73 and anilido-oxazoline ligands (HLa,b)74 were synthesized according to the literature. Deuterated solvents and CDCl3 (99.8 atom% D) and C6D6 (99.6 atom% D) were purchased from the Cambridge Isotope Laboratory.1H and 13C NMR spectra were recorded using a Bruker AVANCE-500 NMR spectrometer and referenced to SiMe4 (TMS) or residual solvent resonances. The molecular weight and molecular weight distribution of the polymers were tested with a Hitachi Chromaster 2000 (column: PL gel 5 μm MIXED-C) at 35 °C using THF as an eluent (flow rate is 1.0 mL min−1) against polystyrene standards. DSC measurements were performed on a NETZSCH DSC 214 Polyma instrument equipped with an IC70 intra cooler. For the measurements, samples weighing about 10 mg were heated or cooled at 10 °C min−1. The Tg was taken as the midpoint of the inflection tangent, and Tg and Tm were based on the third heating scan. Elemental analysis was performed on an Elementar Vario EL cube. UV-visible spectra were recorded using a Macy UV-1800A spectrophotometer at room temperature. Fluorescence measurements were carried out using a Lengguang Tech F97Pro fluorescence spectrophotometer.
A typical procedure for IP or Myr polymerization
A typical procedure for IP polymerization (Table 1, run 2) was described as follows. In a glovebox, complex 1b (10 μmol) and [Ph3C][B(C6F5)4] (10 μmol) were mixed in 5 mL toluene, and then IP (0.68 g, 10 mmol) was added to the mixture in one portion. Polymerization was initiated and the mixture stirred vigorously for a desired time. Afterward, the polymerization was quenched by ethanol and poured into a large amount of ethanol (50 mL) containing butylated hydroxytoluene (BHT, 0.1% w/w). The resultant polymer was precipitated, washed several times with ethanol and dried at 35 °C under vacuum to a constant weight. A typical procedure for Myr polymerization was very similar to the above-mentioned procedure for IP polymerization.A typical procedure for CL polymerization
A typical procedure for CL polymerization (Table 2, run 2) is described as follows. In a glovebox, complex 1b (10 μmol) was dissolved in 5 mL toluene, and then CL (0.57 g, 5 mmol) was added to the above-mentioned solution in one portion. Polymerization was initiated immediately and quenched after 1 min by ethanol and poured into a large amount of ethanol (50 mL). The resultant polymer was collected, washed several times with ethanol and dried at 45 °C under vacuum to a constant weight.A typical procedure for copolymerization of IP and CL
A typical procedure for copolymerization of IP and CL (Table 3, run 2) is described as follows. In a glovebox, complex 2b (10 μmol) and [Ph3C][B(C6F5)4] (10 μmol) were mixed in 10 mL toluene, and then IP (0.34 g, 5 mmol) was added to the mixture to initiate the polymerization. Then the mixture was stirred vigorously for a desired time and CL (0.57 g, 5 mmol) was added to the above-mentioned mixture in one portion. The polymerization was quenched by ethanol after a desired reaction time, and poured into a large amount of ethanol (50 mL) containing butylated hydroxytoluene (BHT, 0.1% w/w). The resultant polymer was precipitated, washed several times with ethanol and dried at 45 °C under vacuum to a constant weight.Crystal structure measurement
X-ray quality crystals were generated from concentrated n-hexane solutions at −30 °C in a glove box for ca. 1 week. The X-ray diffraction data of complexes 1a and 1b were collected at 150 K on a Bruker D8 QUEST diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The crystal structures were solved by direct methods and refined by full-matrix least squares on F2. All calculations were performed using the SHELXTL crystallographic software packages.75 The details of the crystal data, data collections, and structure refinements are summarized in Table 4. CCDC 1956486 (for 1a) and 1956487 (for 1b) contain the supplementary crystallographic data for this paper.†| 1a | 1b | |
|---|---|---|
| Formula | C31H51N2O2Si2Lu | C35H59N2O2Si2Lu |
| Formula weight | 714.90 | 771.01 |
| Cryst. system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a (Å) | 17.8355(6) | 11.2601(8) |
| b (Å) | 9.5060(3) | 17.6609(11) |
| c (Å) | 20.6821(7) | 19.5210(13) |
| α (deg) | 90.00 | 90.00 |
| β (deg) | 99.911(1) | 102.384(2) |
| γ (deg) | 90.00 | 90.00 |
| V (Å3) | 3454.2(2) | 3791.7(4) |
| Z | 4 | 4 |
| R int | 0.0308 | 0.0299 |
| R 1/wR2 [I > 2σ(I)] | 0.0239/0.0550 | 0.0216/0.0495 |
| R 1/wR2 [all, data] | 0.0298/0.0571 | 0.0253/0.0520 |
| Goof | 1.034 | 1.101 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank Prof. Wei Gao in Jilin University for his help in analysis of single crystal data, and Prof. Guang-Peng Wu and Dr Guan-Wen Yang in Zhejiang University for their help in DSC and MALDI-TOF analyses. This work was supported by the National Natural Science Foundation of China (21971029, and 21404018), the Natural Science Foundation of Liaoning Province, China (20180550505), Fundamental Research Funds for the Central Universities (DUT19LK27) and State Key Laboratory of Fine Chemicals (KF1711).Notes and references
- D. Astruc, Organometallic Chemistry and Catalysis, Springer, Berlin, Germany, 1st edn, 2007, pp. 5–20 Search PubMed.
- M. Nishiura and Z. Hou, Nat. Chem., 2010, 2, 257–268 CrossRef CAS.
- M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209–2220 CrossRef CAS.
- J. Jothieswaran, S. Fadlallah, F. Bonnet and M. Visseaux, Catalysts, 2017, 7, 378–391 CrossRef.
- J. Huang, Z. Liu, D. Cui and X. Liu, ChemCatChem, 2018, 10, 42–61 CrossRef CAS.
- L. Friebe, O. Nuyken and W. Obrecht, Adv. Polym. Sci., 2006, 204, 1–154 CrossRef CAS.
- A. Fischbach and R. Anwander, Adv. Polym. Sci., 2006, 204, 155–281 CrossRef CAS.
- C. O. Hollfelder, L. N. Jende, D. Diether, T. Zelger, R. Stauder, C. Maichle-Mössmer and R. Anwander, Catalysts, 2018, 8, 61–81 CrossRef.
- Y. Pan, X. Zhao, X. Xu and G. He, Mini-Rev. Org. Chem., 2016, 13, 349–362 CrossRef CAS.
- Z. Zhang, D. Cui, B. Wang, B. Liu and Y. Yang, Struct. Bonding, 2010, 137, 49–108 CrossRef CAS.
- T. Li, J. Jenter and P. W. Roesky, Struct. Bonding, 2010, 137, 165–228 CrossRef CAS.
- F. Yang and X. Li, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2271–2280 CrossRef CAS.
- L. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2007, 46, 1909–1913 CrossRef CAS PubMed.
- W. Gao and D. Cui, J. Am. Chem. Soc., 2008, 130, 4984–4991 CrossRef CAS.
- K. Lv and D. Cui, Organometallics, 2010, 29, 2987–2993 CrossRef CAS.
- D. Li, S. Li, D. Cui and X. Zhang, Organometallics, 2010, 29, 2186–2193 CrossRef CAS.
- L. Li, C. Wu, D. Liu, S. Li and D. Cui, Organometallics, 2013, 32, 3203–3209 CrossRef CAS.
- Y. Yang, K. Lv, L. Wang, Y. Wang and D. Cui, Chem. Commun., 2010, 46, 6150–6152 RSC.
- L. Wang, D. Cui, Z. Hou, W. Li and Y. Li, Organometallics, 2011, 30, 760–767 CrossRef CAS.
- L. Li, S. Li and D. Cui, Macromolecules, 2016, 49, 1242–1251 CrossRef CAS.
- C. Yu, D. Zhou, X. Yan, F. Gao, L. Zhang, S. Zhang and X. Li, Polymers, 2017, 9, 531–541 CrossRef PubMed.
- G. Zhang, Y. Wei, L. Guo, X. Zhu, S. Wang, S. Zhou and X. Mu, Chem. – Eur. J., 2015, 21, 2519–2526 CrossRef CAS PubMed.
- G. Zhang, B. Deng, S. Wang, Y. Wei, S. Zhou, X. Zhu, Z. Huang and X. Mu, Dalton Trans., 2016, 45, 15445–15456 RSC.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–808 CrossRef CAS.
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- Y. Sarazin and J.-F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS.
- C.-X. Cai, A. Amgoune, C. W. Lehmann and J.-F. Carpentier, Chem. Commun., 2004, 330–331 RSC.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem. – Eur. J., 2006, 12, 169–179 CrossRef CAS PubMed.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- K. Nie, X. Gu, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2010, 39, 6832–6840 RSC.
- M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem. – Eur. J., 2011, 17, 1872–1883 CrossRef CAS PubMed.
- K. Nie, L. Fang, Y. Yao, Y. Zhang, Q. Shen and Y. Wang, Inorg. Chem., 2012, 51, 11133–11143 CrossRef CAS PubMed.
- Y. Chapurina, J. Klitzke, O. Casagrande, M. Awada, V. Dorcet, E. Kirillov and J.-F. Carpentier, Dalton Trans., 2014, 43, 14322–14333 RSC.
- C. Bakewell, T.-P.-A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS PubMed.
- T.-Q. Xu, G.-W. Yang, C. Liu and X.-B. Lu, Macromolecules, 2017, 50, 515–522 CrossRef CAS.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef.
- W. Gao, D. Cui, X. Liu, Y. Zhang and Y. Mu, Organometallics, 2008, 27, 5889–5893 CrossRef CAS.
- H. Ouyang, D. Yuan, K. Nie, Y. Zhang, Y. Yao and D. Cui, Inorg. Chem., 2018, 57, 9028–9038 CrossRef CAS PubMed.
- F. Wang, X. Zhao, X. Meng and S. Wang, Dalton Trans., 2019, 48, 12193–12198 RSC.
- Y. Liu, W. Huang, N. Zhu and K. Guo, RSC Adv., 2017, 7, 37412–37418 RSC.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS PubMed.
- J.-B. Zhu and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2018, 57, 12558–12562 CrossRef CAS PubMed.
- J.-B. Zhu and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2019, 58, 1178–1182 CrossRef CAS PubMed.
- Y. Pan, T. Xu, Y.-S. Ge and X.-B. Lu, Organometallics, 2011, 30, 5687–5694 CrossRef CAS.
- Y. Pan, A. Zhao, Y. Li, W. Li, Y.-M. So, X. Yan and G. He, Dalton Trans., 2018, 47, 13815–13823 RSC.
- Y. Pan, T. Xu, S. Fu, G.-W. Yang and X.-B. Lu, Macromolecules, 2013, 46, 8790–8796 CrossRef CAS.
- Y. Pan, T. Xu, G.-W. Yang, K. Jin and X.-B. Lu, Inorg. Chem., 2013, 52, 2802–2808 CrossRef CAS PubMed.
- Y. Pan, W. Li, N.-N. Wei, Y.-M. So, Y. Li, K. Jiang and G. He, Dalton Trans., 2019, 48, 3583–3592 RSC.
- Y. Pan, W. Li, N.-N. Wei, Y.-M. So, X. Lai, Y. Li, K. Jiang and G. He, Dalton Trans., 2019, 48, 9079–9088 RSC.
- A. O. Tolpygin, T. A. Glukhova, A. V. Cherkasov, G. K. Fukin, D. V. Aleksanyan, D. Cui and A. A. Trifonov, Dalton Trans., 2015, 44, 16465–16474 RSC.
- P. Zhang, H. Liao, H. Wang, X. Li, F. Yang and S. Zhang, Organometallics, 2017, 36, 2446–2451 CrossRef CAS.
- J. A. Dean, Lange's handbook of chemistry, McGraw-Hill, New York, 15th edn, 1999, Section 4: 4.5 Search PubMed.
- J. Raynaud, J. Y. Wu and T. Ritter, Angew. Chem., Int. Ed., 2012, 51, 11805–11808 CrossRef CAS PubMed.
- B. Liu, L. Li, G. Sun, D. Liu, S. Li and D. Cui, Chem. Commun., 2015, 51, 1039–1041 RSC.
- S. Loughmari, A. Hafid, A. Bouazza, A. El Bouadili, P. Zinck and M. Visseaux, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2898–2905 CrossRef CAS.
- S. Georges, A. Q. Touré, M. Visseaux and P. Zinck, Macromolecules, 2014, 47, 4538–4547 CrossRef CAS.
- E. Laur, A. Welle, A. Vantomme, J.-M. Brusson, J.-F. Carpentier and E. Kirillov, Catalysts, 2017, 7, 361–372 CrossRef.
- S. Georges, O. H. Hashmi, M. Bria, P. Zinck, Y. Champouret and M. Visseaux, Macromolecules, 2019, 52, 1210–1219 CrossRef.
- X. Yu, M. Li, J. Hong, X. Zhou and L. Zhang, Chem. – Eur. J., 2019, 25, 2569–2576 CAS.
- W. Li, J. Zhao, X. Zhang and D. Gong, Ind. Eng. Chem. Res., 2019, 58, 2792–2800 CrossRef CAS.
- S. Zhang, L. Han, H. Ma, P. Liu, H. Shen, L. Lei, C. Li, L. Yang and Y. Li, Ind. Eng. Chem. Res., 2019, 58, 12845–12853 CrossRef CAS.
- Y. Ren, J. T. Miller, S. T. Polderman, T. D. Vo, A. C. M. Wallace, J. M. O. Cue, S. T. Tran, M. C. Biewer and M. C. Stefan, RSC Adv., 2019, 9, 3345–3350 RSC.
- S. Georges, M. Bria, P. Zinck and M. Visseaux, Polymer, 2014, 55, 3869–3878 CrossRef CAS.
- X. Kang, Y. Luo, G. Zhou, X. Wang, X. Yu, Z. Hou and J. Qu, Macromolecules, 2014, 47, 4596–4606 CrossRef CAS.
- G. Zhou, X. Kang, X. Wang, Z. Hou and Y. Luo, Organometallics, 2018, 37, 551–558 CrossRef CAS.
- L. F. Sanchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2005, 24, 3792–3799 CrossRef CAS.
- L. Li, S. Li and D. Cui, Macromolecules, 2016, 49, 1242–1251 CrossRef CAS.
- M. F. Lappert and R. Pearce, J. Chem. Soc., Chem. Commun., 1973, 4, 126 RSC.
- P. I. Binda, S. Abbina and G. Du, Synthesis, 2011, 2609–2618 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1956486 (1a) and 1956487 (1b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj05081g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |