
Highly active mesoionic chalcogenone zinc(II) derivatives for C–S cross-coupling reactions†
Moulali
Vaddamanu
a,
Kavitha
Velappan
b and
Ganesan
Prabusankar
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, TS 502285, India. E-mail: prabu@iith.ac.in; Fax: +914023016032; Tel: +914023016089
bDepartment of Chemistry, GITAM University, Hyderabad, TS 502329, India
First published on 19th November 2019
Abstract
The first mesoionic heavier chalcogenones, L1–L3 [L1 = 1-(2-mesitylene)-3-methyl-4-phenyltriazolin-5-selone; L2 = 1-(2-mesitylene)-3-methyl-4-phenyltriazolin-5-thione; L3 = 1-(benzyl)-2-3(methyl)-4-phenyltriazolin-5-selone], were isolated and characterised. Density functional theory was used to obtain insights into the σ donor and π accepting nature of mesoionic chalcogenones. Using these new ligands, a series of the first zinc(II) mesoionic chalcogenone complexes were isolated. Three mono nuclear zinc(II) chalcogenone complexes, [(L1)Zn(Cl)2(HOMe)] (1), [(L2)Zn(Cl)2(HOMe)] (3) and [(L2)Zn(Br)2(HOMe)] (4), and two dinuclear zinc complexes, [(L1)Zn(Br)(μ2-Br)]2 (2) and [(L3)Zn(Br)(μ2-Br)]2 (5), containing mesoionic thione and selone ligands were synthesized and characterised. These new complexes 1–5 represent the first structurally characterized mesoionic chalcogenone supported metal derivatives. Furthermore, all zinc complexes were characterized by thermogravimetric analysis and UV-vis spectroscopy. The solid-state structures of all zinc complexes were determined by single-crystal X-ray diffraction. The catalytic activities of the zinc(II) complexes in thioetherification reactions were investigated without scrubbing of oxygen. The scope of the catalytic reactions was explored with a wide range of thiophenols and aryl halides. The diaryl thioethers were obtained in very good yield under mild conditions. The present protocol furnishes a synthetic route for the C–S cross-coupling of thiophenols and aryl halides without scrubbing oxygen and moisture.
Introduction
The application of mesoionic carbene (MIC) or “abnormal” N-heterocyclic carbene (aNHC) supported metal derivatives has continued to expand at a rapid rate in catalysis compared to N-heterocyclic carbene (NHC) metal derivatives.1–4 The better σ donor and poor π accepting nature of MICs along with low steric hindrance compared to NHCs provide greater control on the metal centers for improved catalytic performance.7–9 The first MIC carbenes and their transition metal derivatives were reported by Crabtree and co-workers.10 Subsequently, several MIC–metal complexes have subsequently been reported with potential roles in synthetic and catalytic applications.1–9 Though the MICs and related chemistry have been known for a while, the triazolylidene type of MIC chemistry is relatively new. The triazolylidene family of MICs was discovered by Begtrup et al. in 1975.11 Begtrup isolated the first mesoionic oxide (I) through short-lived 1H-1,2,3-triazol-5-ylidene by deprotonation of the triazolium salt with NaH at 0 °C in the presence of oxygen (Scheme 1a).11 Later Enders used triazolylidenes as organocatalysts in 1995.12 Albrecht reported the synthesis of 1,2,3-triazolylidenes using a Huisgen cycloaddition between azides and alkynes in 2008.13 Thus the first 1,2,3-triazolylidene based MIC-Pd/Ag/Rh/Ir molecules were reported.13 Later, Bertrand succeeded in isolating and characterizing the first free triazolylidene.14 Notably, the triazole framework is versatile for introducing substituents and electronically unique as the carbenic carbon in triazolylidenes is flanked by only one heteroatom.15 The most recent review by Bertrand summarizes the advancement of triazolylidene chemistry.9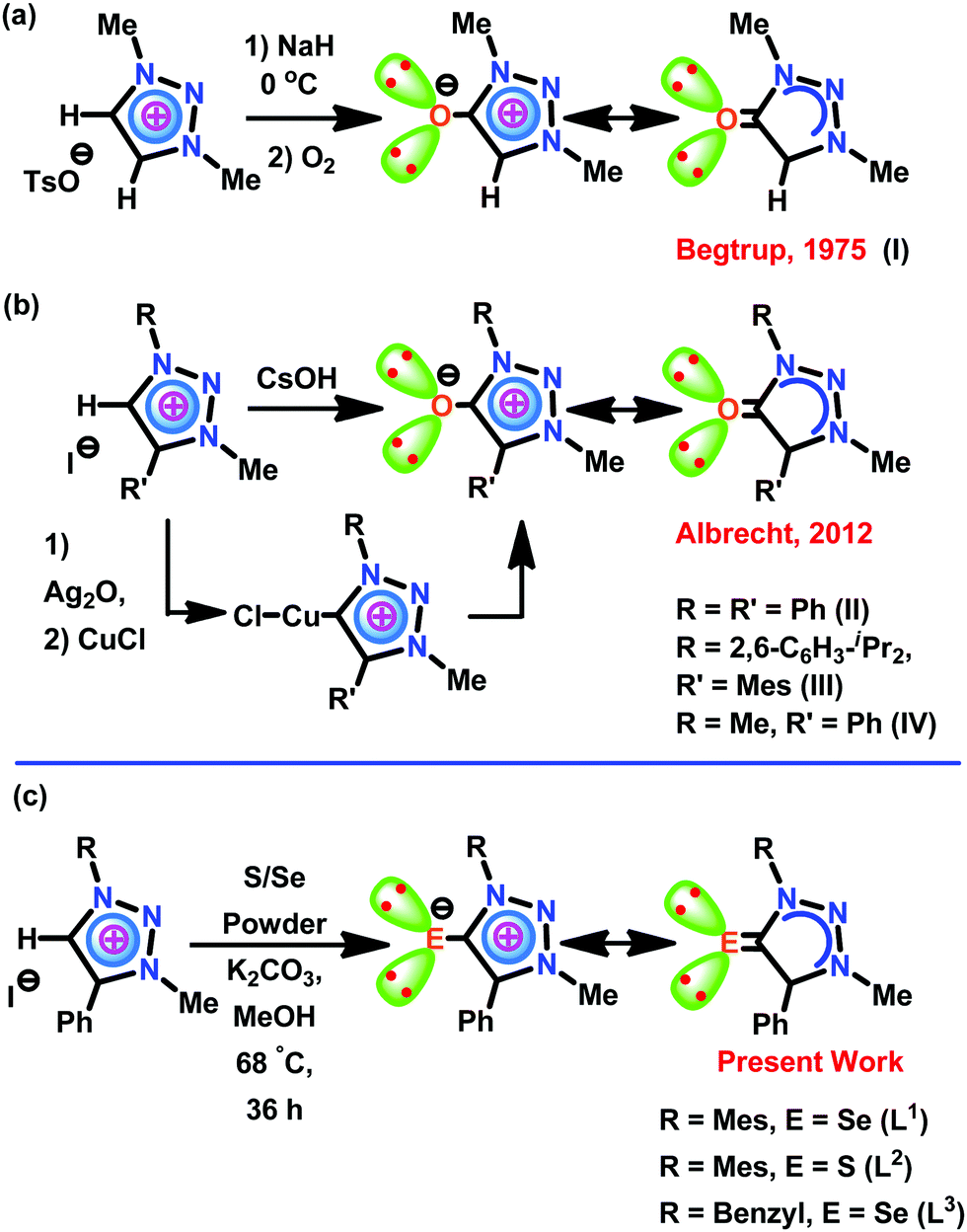 | ||
| Scheme 1 Synthesis of mesoionic chalcogenones. | ||
Though Begtrup accounted for the first mesoionic oxide (I) via short-lived 1H-1,2,3-triazol-5-ylidene, thirty-seven years later, Albrecht reported the first solid-state structure of mesoionic oxide II–IV (Scheme 1b).16aII–IV were derived via the reaction between Cu(I) triazolylidenes or triazolium salts and CsOH. Also, the in situ generation of II·CuCl was used as a catalyst in [3+2] dipolar cycloaddition of alkynes and azides (a ‘click’ reaction). Notably, the catalytic efficiency of II·CuCl was comparable with the corresponding copper(I) 1,2,3-triazol-5-ylidene complex.16b,c However, the heavier congeners of 1,2,3-triazole based mesoionic chalcogenone have not been isolated yet.
However, despite the growing number of examples documenting imidazole chalcogenone (IMC) chemical reactivity, over the last ten years, IMCs have proven to be pivotal supporting ligands for a variety of catalytically active metal complexes. We also noted the seminal contribution of Nolan, Son, Sing, and others, who have reported a broad range of imidazole chalcogenones (IMCs), such as imidazole thione (IMT) and imidazole selone (IMSe). The IMC Pd(II), Ir(III), Au(I), Au(III), Cu(I), Zn(II), Cd(II), Bi(III), Ni(II), Ru(II) and Rh(III) derivatives were used as catalysts in the Heck coupling reaction,17 Suzuki–Miyaura coupling,18 Sonogashira coupling,18b oxidative coupling of benzylamine to imine under visible-light,19 regioselective borylation of internal alkynes,20,21 hydroamination reactions,22,23 click chemistry,23,24 nitroarene reduction to aromatic amines,25 Barbier reactions in aqueous media,26o-acylative cleavage of cyclic ethers,27 triarylmethane synthesis,28 norbornene polymerization,29 oxidation of alcohols,30 catalytic transfer hydrogenation reactions,31–33N-alkylation of anilines,33 and cross-dehydrogenative coupling of alkynes with silanes.24 In most cases, the catalytic efficiency of IMC–metal complexes is comparable or even much more efficient compared to the corresponding metal–NHC derivatives.20,23,24
In contrast, the literature reveals a dearth of studies about the chemistry of mesoionic chalcogenone (MIC) metal complexes. Thus, we believe that the heavier mesoionic type of triazole chalcogenone (TAC) complexes are expected to have great potential for the evolution of new catalysts with unprecedented ligand-induced catalytic control. On the basis of this evolution, herein, we report the first examples of heavier mesoionic chalcogenones, L1–L3 [L1 = 1-(2-mesitylene)-3-methyl-4-phenyltriazolin-5-selone; L2 = 1-(2-mesitylene)-3-methyl-4-phenyltriazolin-5-thione; L3 = 1-(benzyl)-2-3(methyl)-4-phenyltriazolin-5-selone], through a one-pot synthesis. Besides, L1–L3 were used as ligands in isolating the mononuclear zinc(II) metal complexes [(L1)Zn(Cl)2(HOMe)] (1), [(L2)Zn(Cl)2(HOMe)] (3), and [(L2)Zn(Br)2(HOMe)] (4) and dinuclear zinc(II) metal complexes [(L1)Zn(Br)(μ2-Br)]2 (2), and [(L3)Zn(Br)(μ2-Br)]2 (5). Subsequently, 1–5 were used as catalysts for C–S cross-coupling reactions between thiophenols and aryl halides. These catalytic reactions can be carried out at 80 °C and less reaction time without scrubbing of oxygen, comprising a substantial advantage over known ZnEt2-mediated cross-coupling reactions.
Results and discussion
Recently we have performed a comparative study between Ph3P, NHC, and IMC supported copper(I) complexes for C–N and C–Si bond formation reactions.24 Based on the σ-donor and π-accepting strength, we assumed that [(IMes![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)CuI] [IMesS = 1,3-bis(2,4,6-trimethylphenyl)imidazoline-2-thone] was more active than [(IMes)CuCl] [IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-yilidine] for the synthesis of 1,2,3-triazoles and for the cross-dehydrogenative coupling of alkynes with silanes. However, the σ-donor and π-accepting nature of NHCs vs. IMCs remains unclear. In particular, the chemistry of the mesoionic type of heavier chalcogenones has never been investigated. Therefore Density Functional Theory (DFT) calculations were carried out at the B3LYP level using an all-electron basis set which includes polarization and diffuse functions (6-311++G**) for 1,3,4-trimethyl-1,2,3-triazolin-5-ylidene (I), 1,3,4-trimethyl-1,2,3-triazolin-5-thione (II) and 1,3,4-trimethyl-1,2,3-triazolin-5-selone (III) to understand the σ-donor and π-accepting nature (Fig. 1). Notably, the HOMO energy of II and III is considerably higher than that of I, while the LUMO energy of II and III is lower than that of I. Thus, mesoionic or abnormal NHC analogs II and III can become a good σ-donor and π-acceptor ligand compared to NHC. As a result, II or III can be considered as a potential option to compete with the catalytic properties of mesoionic or abnormal NHC metal derivatives.
S)CuI] [IMesS = 1,3-bis(2,4,6-trimethylphenyl)imidazoline-2-thone] was more active than [(IMes)CuCl] [IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-yilidine] for the synthesis of 1,2,3-triazoles and for the cross-dehydrogenative coupling of alkynes with silanes. However, the σ-donor and π-accepting nature of NHCs vs. IMCs remains unclear. In particular, the chemistry of the mesoionic type of heavier chalcogenones has never been investigated. Therefore Density Functional Theory (DFT) calculations were carried out at the B3LYP level using an all-electron basis set which includes polarization and diffuse functions (6-311++G**) for 1,3,4-trimethyl-1,2,3-triazolin-5-ylidene (I), 1,3,4-trimethyl-1,2,3-triazolin-5-thione (II) and 1,3,4-trimethyl-1,2,3-triazolin-5-selone (III) to understand the σ-donor and π-accepting nature (Fig. 1). Notably, the HOMO energy of II and III is considerably higher than that of I, while the LUMO energy of II and III is lower than that of I. Thus, mesoionic or abnormal NHC analogs II and III can become a good σ-donor and π-acceptor ligand compared to NHC. As a result, II or III can be considered as a potential option to compete with the catalytic properties of mesoionic or abnormal NHC metal derivatives.
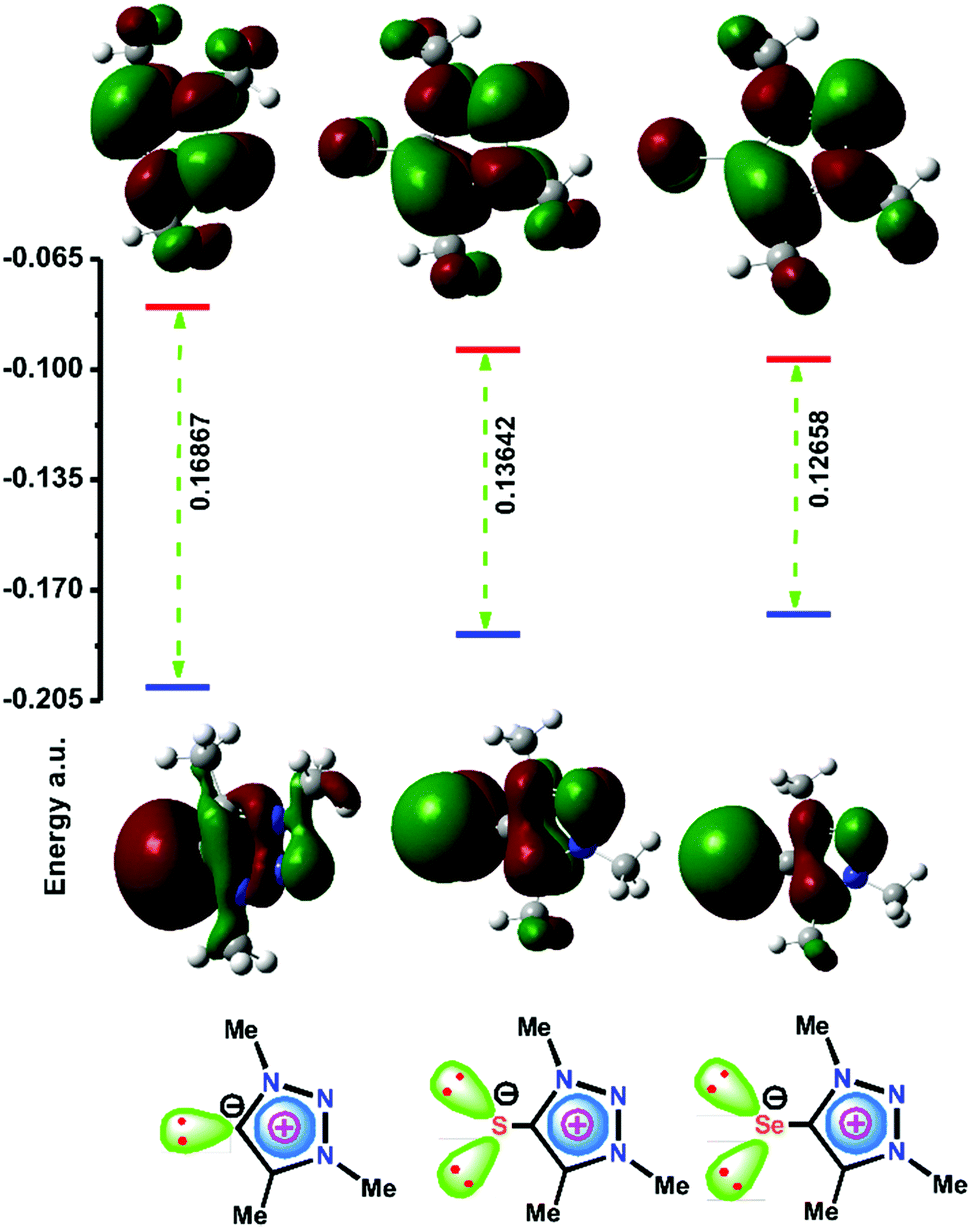 | ||
| Fig. 1 The comparison of the HOMO and LUMO energy differences between 1,3,4-trimethyl-1,2,3-triazolin-5-ylidene (I), 1,3,4-trimethyl-1,2,3-triazolin-5-thione (II) and 1,3,4-trimethyl-1,2,3-triazolin-5-selone (III). Density Functional Theory (DFT) calculations were carried out at the B3LYP level using an all electron basis set which includes polarization and diffuse functions (6-311++G**). | ||
The triazolium salts were prepared via a copper(I)-catalyzed azide–alkyne cycloaddition reaction followed by methylation of the triazole at the N3-position.16 The direct thionation or seleniation of N-alkylated-1,3 disubstituted 1,2,3 triazolium iodides with elemental sulfur or selenium powder in the presence of potassium carbonate gave corresponding mesoionic chalcogenone ligands L1, L2 and L3 in excellent yield.34,35L1, L2, and L3 are soluble in common nonpolar solvents and mid-polar solvents. These compounds were characterized by FT-IR, UV-vis, and multinuclear NMR techniques. In 1H NMR, the formation of the chalcogenone derivatives (L1, L2, and L3) was supported by the disappearance of an acidic triazolium proton (δH(range) 9.52 ppm).16 In 13C NMR, the chemical shift value of the carbon attached to the chalcogenone in L1 or L3 is about 10 ppm upfield shifted compared to L2. The 13C NMR chemical shift value of the carbon attached to the chalcogenone in L1 or L2 is comparable with that of IMesSe (IMesSe = 1,3-dimesitylimidazole-2-selone).28 The solid-state structures of L1, L2, and L3 were determined by single-crystal X-ray diffraction analysis. The molecular structures of L1, L2, and L3 are depicted in Fig. 2. L1, L2, and L3 are the first structurally characterized mesoionic NHC analogs of 1,2,3-triazolin-5-chalcogenones. The CS bond distance found in L2 (1.687(2) Å) is 0.017 Å longer than that of IPrS (1.670(3) Å) (IPrS = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-thione).28 The CSe bond distance found in L1 (1.840(4) Å) and L3 (1.841(5) Å) is marginally longer than that of IMesSe (1.827(6) Å) (IMesSe = 1,3-dimesitylimidazole-2-selone).28 The C(2)–C(1)–N(1) bond angles found in L1, L2, and L3 are comparable. The C(2)–C(1)–N(1) bond angles found in L1, L2 and L3 are comparable to that of IPrS (104.6(2)°) and IMesSe (104.8(5)°).28 Like 1,2,3-triazolin-5-ylidene,9 the N–N or C–N or C–C bond distances in L1, L2, and L3 are nearly comparable, which supports the aromatic nature of 1,2,3-triazolin-5-chalcogenones.
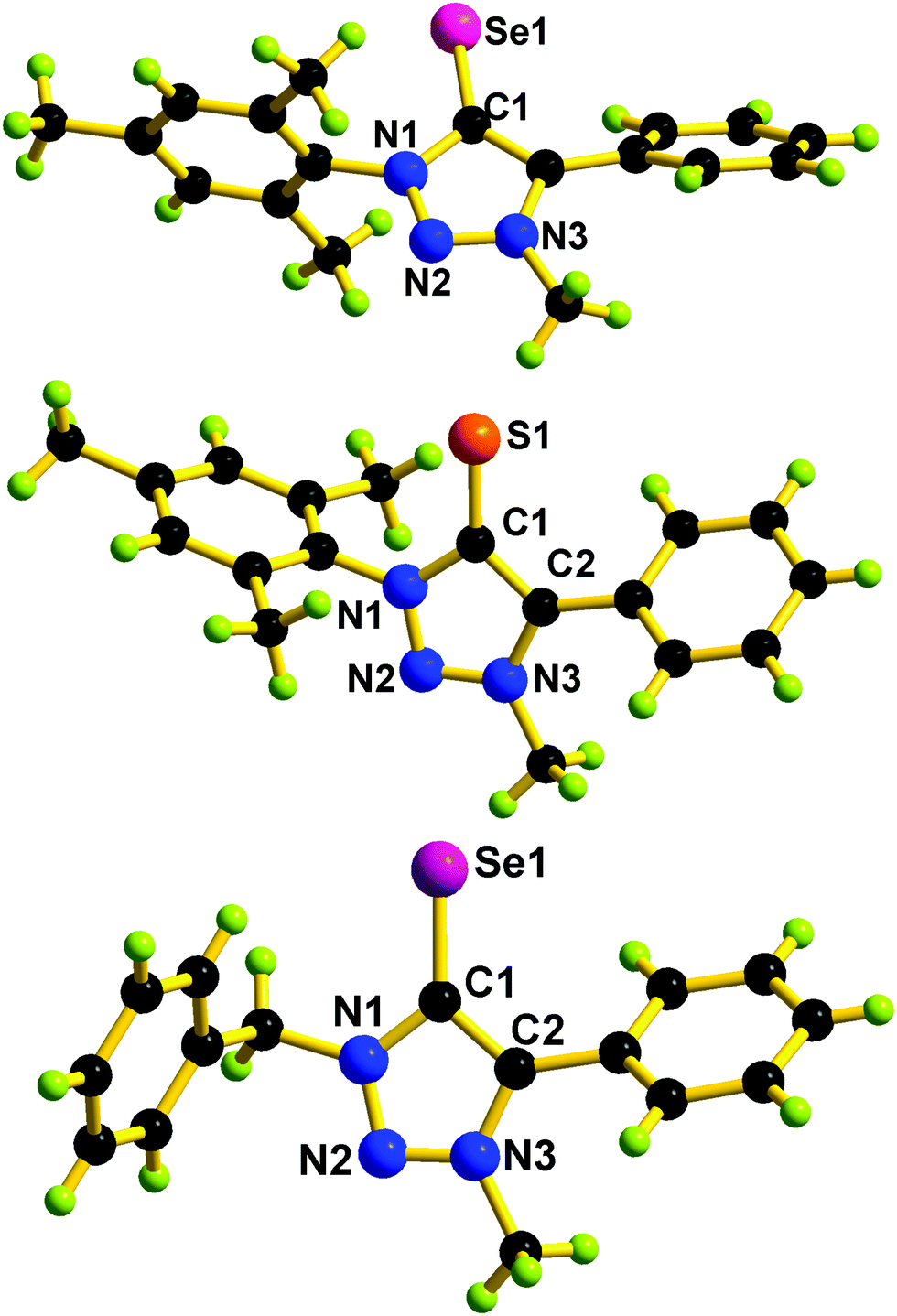 | ||
| Fig. 2 Top: Molecular structure of L1. Selected bond lengths (Å) and bond angles (°): C(1)–Se(1) 1.840(4), N(1)–C(1) 1.377(5), N(3)–C(2) 1.365(5), C(1)–C(2) 1.404(5), N(1)–N(2) 1.338(4), N(2)–N(3) 1.328(5), N(1)–N(2)–N(3) 103.20 (3), C(2)–C(1)–N(1) 103.30(3). Middle: Molecular structure of L2. Selected bond lengths (Å) and bond angles (°): C(1)–S(1) 1.687(2), N(1)–C(1) 1.385(3), N(3)–C(2) 1.359(3), C(1)–C(2) 1.400(3), N(1)–N(2) 1.339(2), N(2)–N(3) 1.324(3), N(1)–N(2)–N(3) 103.43 (17), C(2)–C(1)–N(1) 102.9(2). Bottom: Molecular structure of L3. Selected bond lengths (Å) and bond angles (°): C(1)–Se(1) 1.841(4), N(1)–C(1) 1.361(6), N(3)–C(2) 1.360(5), C(1)–C(2) 1.415(7), N(1)–N(2) 1.325(5), N(2)–N(3) 1.307(6), N(1)–N(2)–N(3) 103.50 (4), C(2)–C(1)–N(1) 105.8(4). | ||
The coordination capabilities of these new ligands were investigated with zinc halides in toluene (Schemes 2 and 3). L1 or L2 was treated with zinc(II) dihalides followed by recrystallization from a mixed solvent of methanol and chloroform to isolate the mono nuclear zinc(II) chalcogenone complexes [(L1)Zn(Cl)2(HOMe)] (1), [(L2)Zn(Cl)2(HOMe)] (3) and [(L2)Zn(Br)2(HOMe)] (4). Under similar reaction conditions, the reaction between ZnBr2 and L1 or L3 gave dinuclear zinc complexes [(L1)Zn(Br)(μ2-Br)]2 (2) and [(L3)Zn(Br)(μ2-Br)]2 (5), respectively.
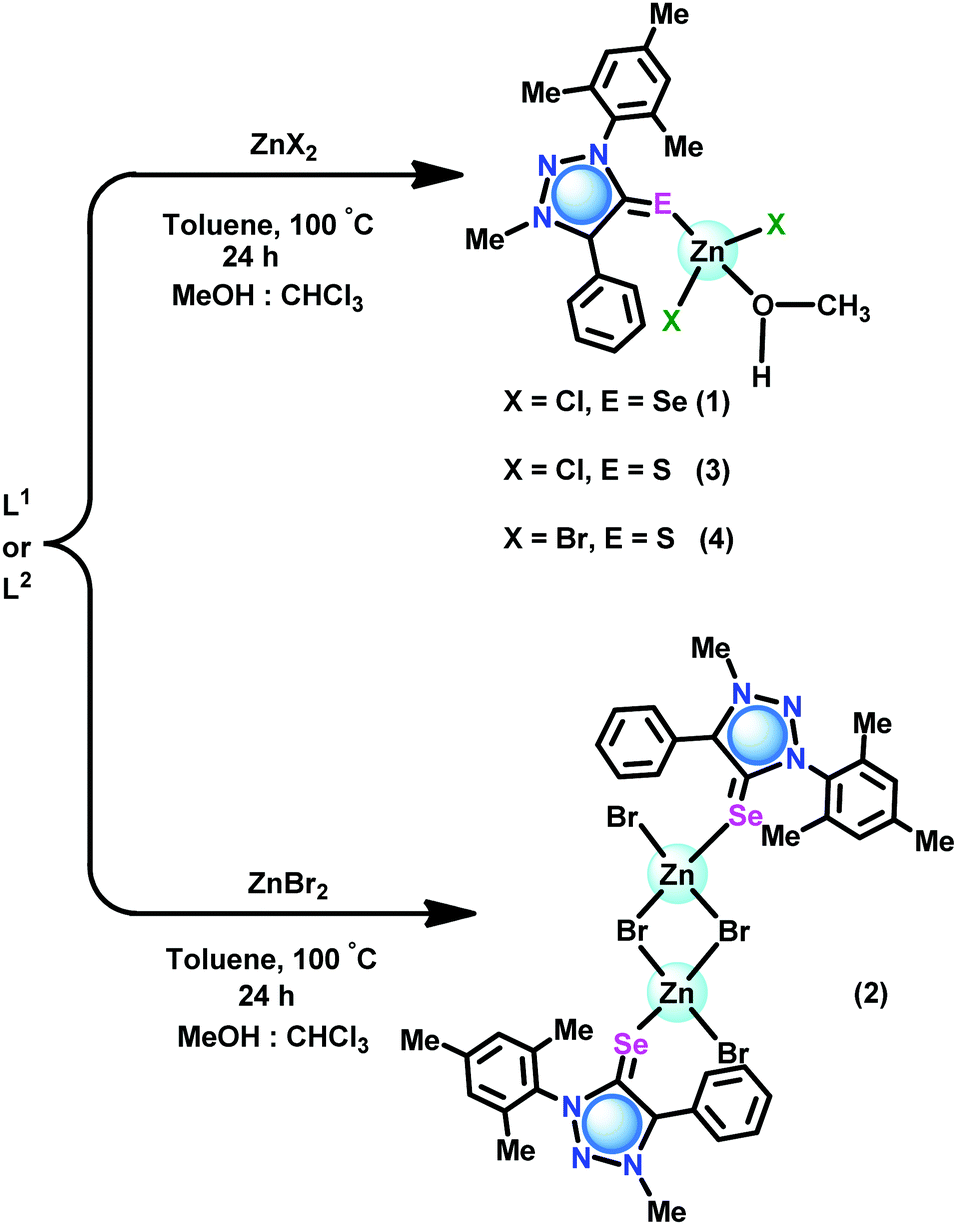 | ||
| Scheme 2 Synthesis of 1–4. | ||
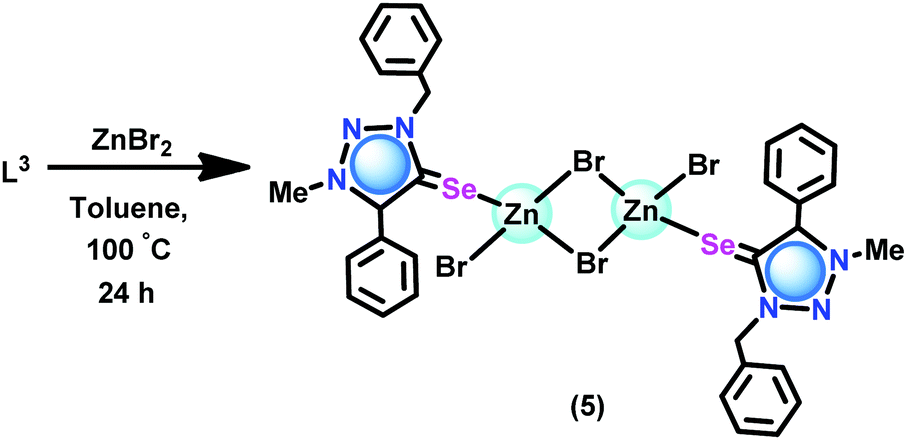 | ||
| Scheme 3 Synthesis of 5. | ||
The mesoionic chalcogenone supported zinc complexes (1–5) were characterized by FT-IR, UV-vis, TGA, and multinuclear NMR studies and X-ray diffraction techniques. In the FT-IR spectra, the ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) CE stretching frequency for 1–5 appeared at a 50 cm−1 lower wavenumber (1122–1126 cm−1) compared to the corresponding uncoordinated mesoionic thione ligand, L2 (1173 cm−1), or uncoordinated mesoionic selone ligand (L1 and L3) (1160–1170 cm−1).
CE stretching frequency for 1–5 appeared at a 50 cm−1 lower wavenumber (1122–1126 cm−1) compared to the corresponding uncoordinated mesoionic thione ligand, L2 (1173 cm−1), or uncoordinated mesoionic selone ligand (L1 and L3) (1160–1170 cm−1).
L1, L2, and L3 and 1–5 show an absorption band around 310 nm and 370 nm (Fig. 3, see the ESI†). The UV-vis absorption pattern of 1–5 is comparable with that of L1, L2, and L3. The absorption band observed around 310 nm could be attributed to the electron transition associated with π to π*. The absorption band at 370 nm can be attributed to the n to π* transitions. 1 shows both hyperchromic and bathochromic shifts compared to free ligand L1. 3 and 4 depict a hypochromic shift compared to free ligand L2. Besides, the 310 nm zone absorption bands of 3 and 4 show a bathochromic shift compared to free ligand L2. The dinuclear zinc(II) complex 2 shows a hypochromic shift compared to L1, while 5 exhibits a hyperchromic shift compared to L3. Based on the molar extinction coefficient, it is clear that the good σ donor capacity of the mesoionic chalcogenone was noticed with Zn(II)Cl2 compared to Zn(II)Br2.
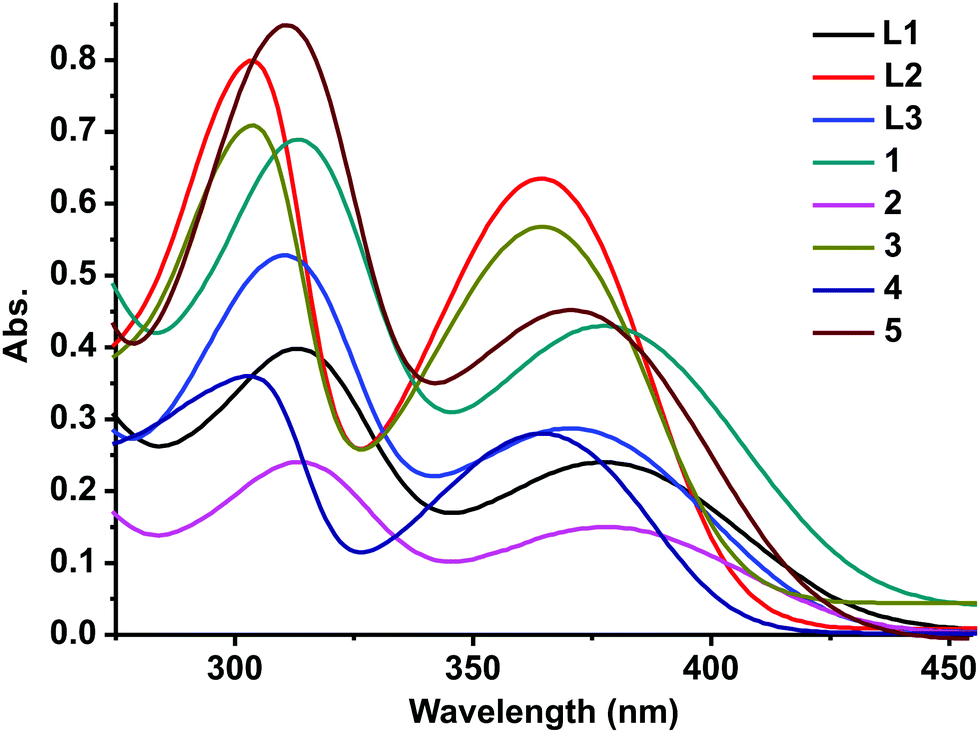 | ||
| Fig. 3 Solution-state UV-vis spectra of L1, L2, and L3 and 1–5 in DMSO at RT (1.04 × 10−4 M). | ||
The thermal stability of 1–5 was analyzed by TGA. Fig. 4 presents the amount and rate of change in the weight of the material as a function of temperature (10 °C per minute) under a nitrogen atmosphere. 1, 3 and 4 showed two steps of weight loss due to loss of methanol in step 1 till 100 °C then the organic moiety in step 2 till 500 °C. 5 shows gradual weight loss till 225 °C (8% weight loss) and then gradual weight loss till 560 °C (85% weight loss) due to the loss of organic moieties, while 2 shows three-step weight loss to yield the residue. The unchanged residue of 1–5 must be the corresponding zinc chalcogenides.
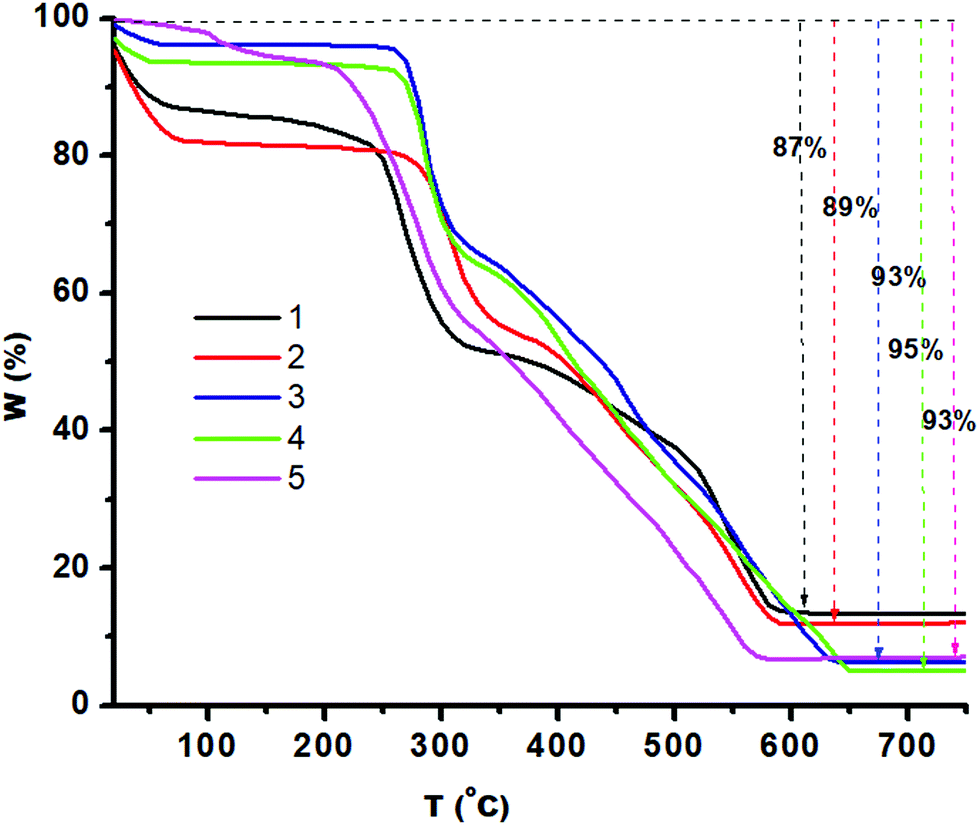 | ||
| Fig. 4 The thermal stability of compounds 1–5 (10 °C per minute) under a nitrogen atmosphere. | ||
The solid-state structures of 1–5 were confirmed by single-crystal X-ray diffraction studies. X-ray quality crystals were isolated from a chloroform and methanol mixture for 1–4, while from toluene for 5. The molecular structures of 1–5 are reported in Fig. 5–7. The crystallographic data for 1–5 are listed in Table S1 (see the ESI†). Molecules 1, 3, and 4 are mononuclear zinc(II) complexes, while 2 and 5 are dinuclear zinc(II) complexes. 1, 3, and 4 are isostructural molecules; thus, the structural features of 1, 3, and 4 are discussed collectively. The Zn(II) center in 1, 3 and 4 is tetra coordinated by one chalcogenone ligand and two halides and a methanol molecule. The geometry of the zinc(II) center in 1, 3, and 4 can be described as tetrahedral. The sums of angles around the zinc(II) center in 1, 3, and 4 are comparable. The Zn–Se bond distance found in 1 (2.436(6) Å) is 0.042 Å shorter than that of dichlorobis[1,3-dimethyl-2(3H)-imidazoleselone]zinc(II) (2.469 and 2.487 Å)36 and perchlorate tetra-1-(2,6-diisopropylphenyl)-3-(2-methoxy-2-oxoethyl)-imidazoline zinc(II) (2.463(6) Å).26 Similarly, the Zn–S bond distance found in 3 (2.326(9) Å) and 4 (2.326(18) Å) is ∼0.042 Å shorter then that of dichloridobis(1,3-diisopropyl-4,5-dimethyl-2H-imidazole-2-thione)zinc(II) (2.366(9) and 2.370(10) Å).37
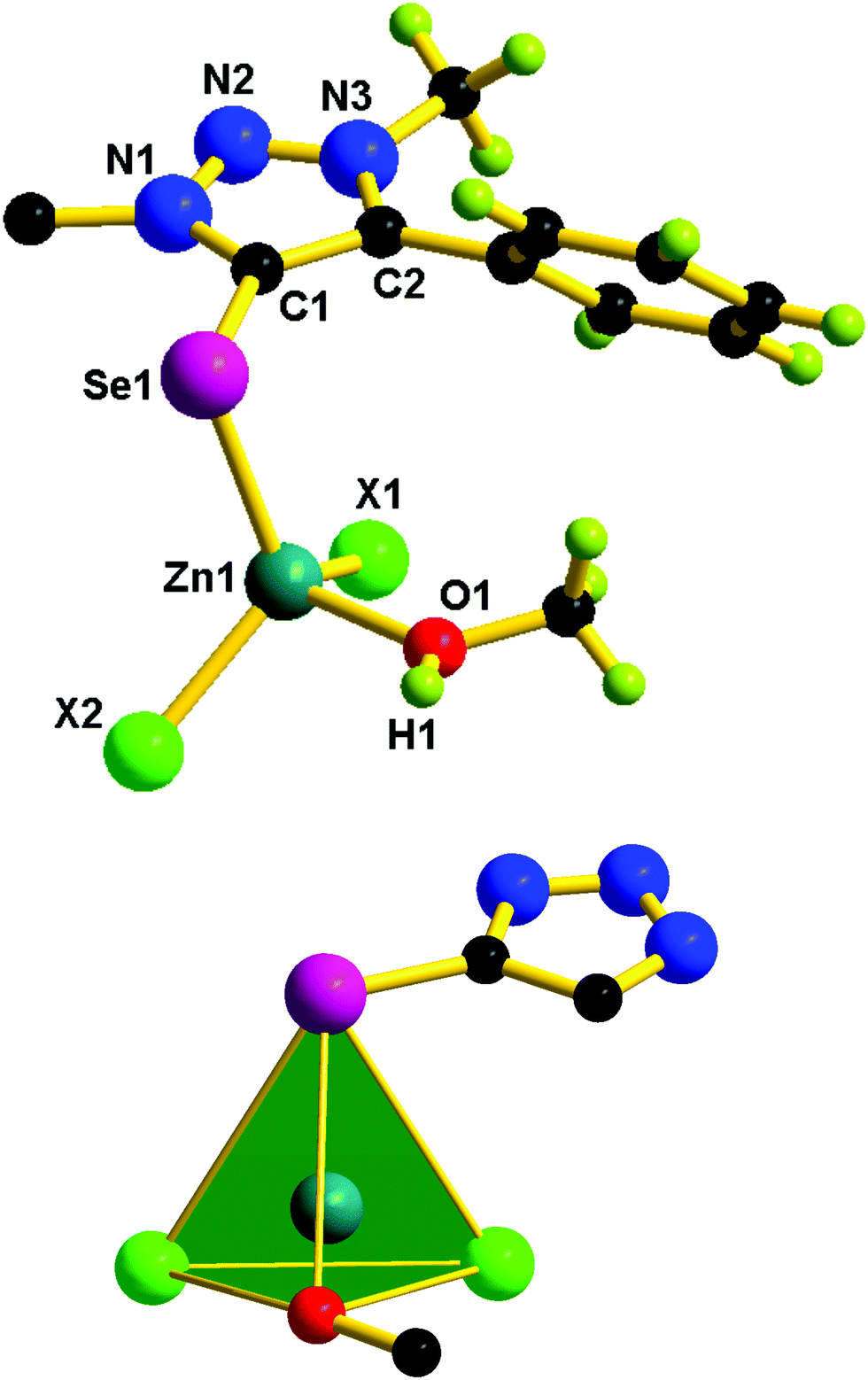 | ||
| Fig. 5 Molecular structures of 1, 3 and 4. The N(1) substituent is omitted for clarity. Selected bond lengths (Å) and bond angles (°) of 1: C(1)–Se(1) 1.879(4), Zn(1)–Se(1) 2.436(6), Zn(1)–Cl(1) 2.209(11), Zn(1)–Cl(2) 2.253(13), Zn(1)–O(1) 2.032(3), N(1)–N(2) 1.339(4), N(2)–N(3) 1.320(4), Se(1)–Zn(1)–Cl(1) 95.78(10), Se(1)–Zn(1)–Cl(2) 113.64(4), Se(1)–Zn(1)–O(1) 108.45(10), N(1)–N(2)–N(3) 103.00(3). Selected bond lengths (Å) and bond angles (°) of 3: C(1)–S(1) 1.719(3), Zn(1)–S(1) 2.326(9), Zn(1)–Cl(1) 2.209(10), Zn(1)–Cl(2) 2.250(9), Zn(1)–O(1) 2.029(3), N(1)–N(2) 1.334(3), N(2)–N(3) 1.318(4), S(1)–Zn(1)–Cl(1) 112.46(3), S(1)–Zn(1)–Cl(2) 113.44(4), S(1)–Zn(1)–O(1) 108.96(10), N(1)–N(2)–N(3) 103.40(2). Selected bond lengths (Å) and bond angles (°) for 4: C(1)–S(1) 1.717(6), Zn(1)–S(1) 2.326(18), Zn(1)–Br(1) 2.383(11), Zn(1)–Br(2) 2.341(9), Zn(1)–O(1) 2.029(4), N(1)–N(2) 1.342(6), N(2)–N(3) 1.328(6), S(1)–Zn(1)–Br(1) 113.94(5), S(1)–Zn(1)–Br(2) 112.85(5), S(1)–Zn(1)–O(1) 109.10(2), N(1)–N(2)–N(3) 102.50(4). | ||
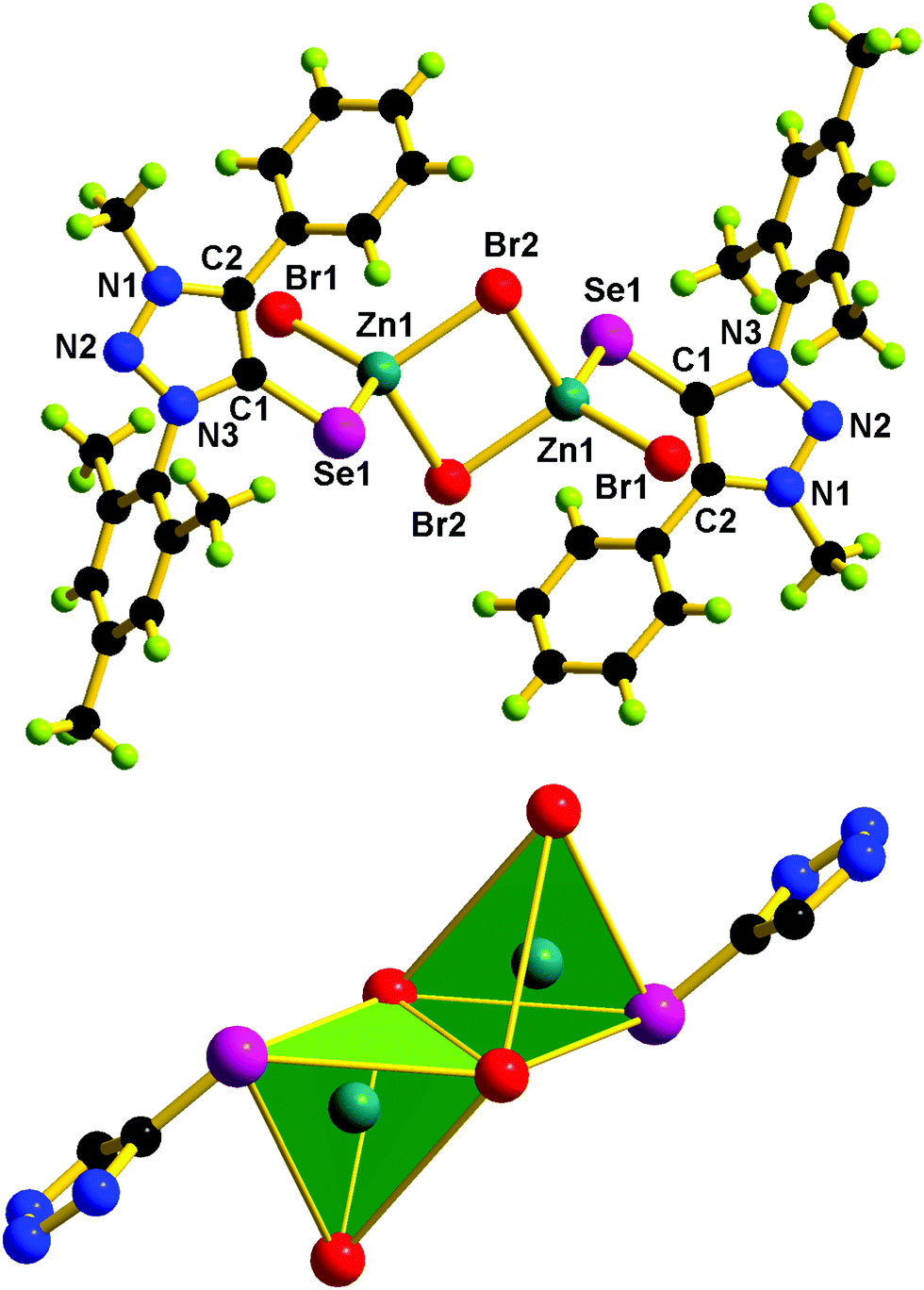 | ||
| Fig. 6 Molecular structure of 2. Selected bond lengths (Å) and bond angles (°): C(1)–Se(1) 1.884(5), Zn(1)–Se(1) 2.416(8), Zn(1)–Br(1) 2.509(9), Zn(1)–Br(2) 2.343(9), N(1)–N(2) 1.320(6), N(2)–N(3) 1.344(5), C(1)–Se(1)–Zn(1) 99.33(15), Br(2)–Zn(1)–Se(1) 103.31(3), Br(1)–Zn(1)–Br(2) 114.84(4), N(1)–N(2)–N(3) 103.20(4). | ||
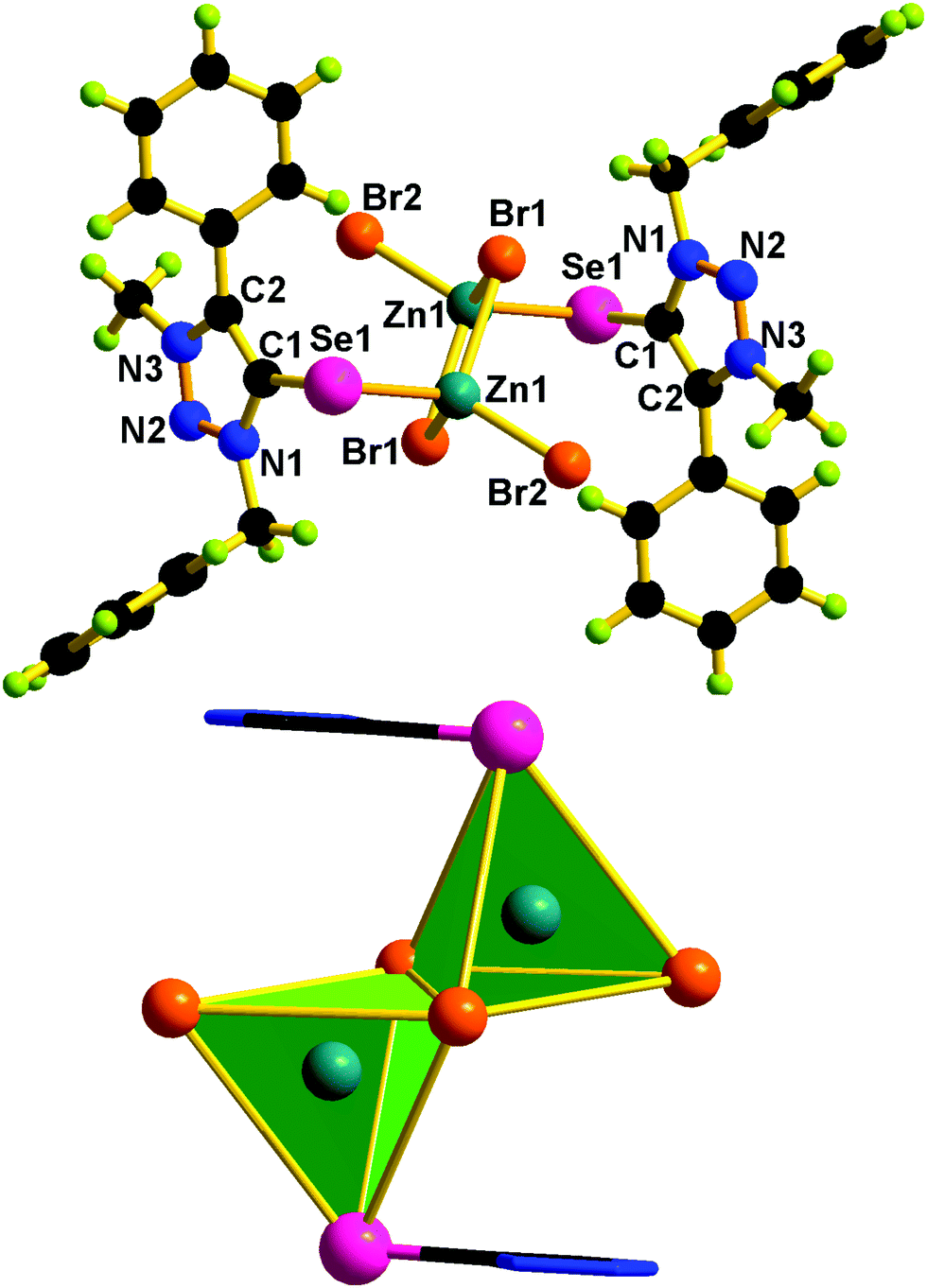 | ||
| Fig. 7 Molecular structure of 5. Selected bond lengths (Å) and bond angles (°): C(1)–Se(1) 1.881(5), Zn(1)–Se(1) 2.428(9), Zn(1)–Br(2) 2.498(8), Zn(1)′–Br(2) 2.500(9), Zn(1)–Br(1) 2.347(7), N(1)–N(2) 1.315(5), N(2)–N(3) 1.319(5), C(1)–Se(1)–Zn(1) 97.56(15), Br(2)–Zn(1)–Se(1) 108.74(3), Br(1)–Zn(1)–Br(2′) 111.20(3), N(1)–N(2)–N(3) 103.50(4). | ||
Molecules 2 and 5 consist of a dinuclear zinc(II) selone core. The core of 2 and 5 is composed of [Zn2(Br)2(μ2-Br)2(Se)2]. The coordination environment around the zinc center is satisfied by three bromide ions and one selone ligand, which leads to a tetrahedral geometry. Two tetrahedrons are fused through the Br–Br edge to result in the core [Zn2(Br)2(μ2-Br)2(Se)2].
The bond length and bond angle parameters of 2 and 5 are not comparable. The Zn–Se bond distance in 2 is 0.012 Å shorter than that of 5. The bridging Zn–Br bond distances in 2 are considerably shorter than the terminal Zn–Br bond distances, while in 5 the terminal Zn–Br bond distances are considerably shorter than the bridging Zn–Br bond distances. The C–Se–Zn bond angle in 2 is slightly wider than that of 5. The Br–Zn–Br angles in 2 are much wider than those of 5. The N–N, N–C and C–C bond distances within the five-membered heterocyclic ring are nearly comparable, which supports the existence of resonances within the ring system.
Catalyst 1–5 mediated C–S bond formation reactions
Thioethers are important motifs found in organic synthesis, biological chemistry, and materials science. The synthesis of thioethers can be achieved through cross-coupling of thiophenols and aryl halides using Pd(II), Cu(I), Cu(II), Ir(I), Ni(II), Fe(III), Mn(II), Co(II), In(III), Au(I), Rh(I), Ag(I), Bi(III) and La(III).38–46 Recently, the C–S coupling protocol was made attractive from an economic standpoint with the use of Et2Zn under anaerobic conditions as an alternative to expensive methods.39a,46 Notably Zn(II) based catalysts for cross-coupling of thiophenols and aryl halides under aerobic conditions are not known.39–46 Thus molecules 1–5 were used as catalysts to investigate the cross-coupling of thiophenols and aryl halides under aerobic conditions. For example, we have screened catalyst 1 with different bases and solvents at different temperatures to optimize the coupling reaction.The cross-coupling reaction between 1-bromo-4-nitro benzene and thiophenol was investigated with selected solvents and bases using catalyst 1 as a model system (Scheme 4). Polar to mid-polar solvents such as toluene, MeCN, and THF and bases such as K2CO3, Cs2CO3, and tBuONa were screened (Fig. 8). The catalytic reactions in solvents toluene and THF were not fruitful. Similarly, bases K2CO3 and Cs2CO3 were not suitable for the catalytic conversion. The control reactions using only ZnX2 or ZnX2·6H2O with NaOtBu (2 equiv.) in MeCN were unsuccessful, while ZnBr2 with NaOtBu (2 equiv.) in MeCN gave a poor yield (for more information see the ESI,† Tables S5 and S6).46 The best results were obtained in acetonitrile and NaOtBu at 80 °C within 15 h. Thus, the desired product was isolated and conveniently separated by column chromatography.
 | ||
| Scheme 4 Optimization of the solvent and base for the catalyst 1 mediated cross-coupling reaction between 1-bromo-4-nitro benzene and thiophenol. | ||
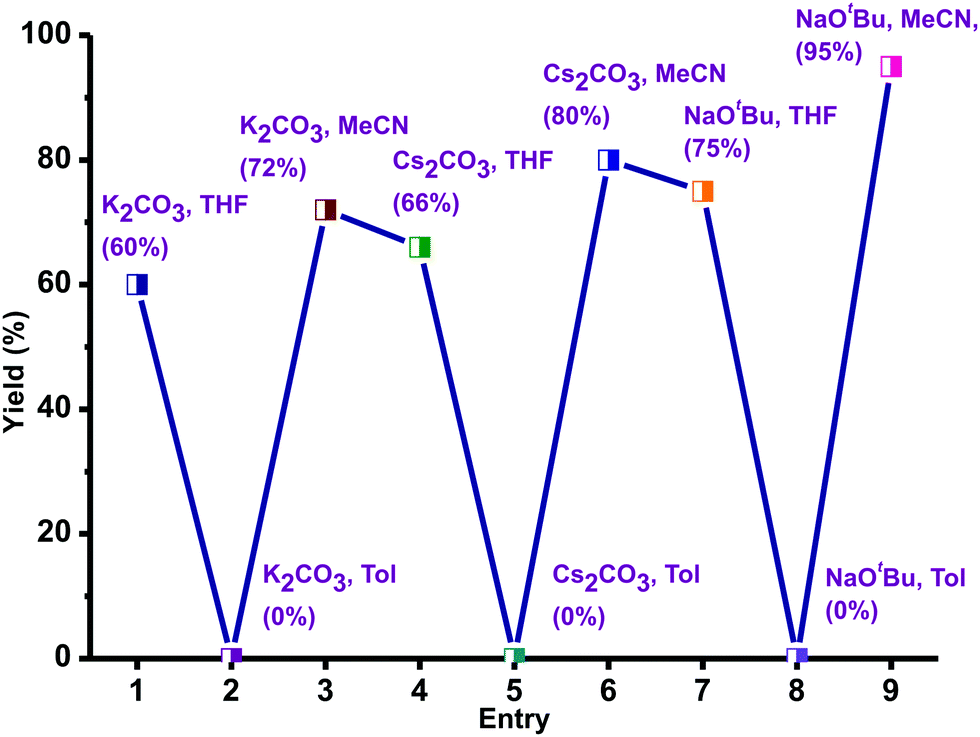 | ||
| Fig. 8 Optimization of the solvent and bases in catalyst 1 mediated thioetherification of aryl halides. Reaction conditions: 1-bromo-4-nitro benzene (1 mmol), thiophenol (1 mmol), base (2 equiv.), catalyst 1 (10 mol%), L-proline (10 mol%), 80 °C, 15 h. | ||
The cross-coupling reaction between 1-bromo-4-nitro benzene and thiophenol can also be catalyzed by ZnCl2 or ZnBr2 or strong bases.46 However, the isolated yield was very poor (5% to 25%) under very high catalyst loading with a high temperature in more than 24 h.
To compare the catalytic efficiency of 1–5, catalytic reactions were carried out between 1-bromo-4-nitrobenzene and thiophenol using the zinc catalyst (10 mol%) and L-proline (10 mol%) (Scheme 5). Catalysts 1–5 are very active and yielded the corresponding thioether in excellent yield within 15 h (Fig. 9). Among catalysts 1–5, catalyst 1 is found to be very active. As reported,39,46 more than 50% of known catalysts were used in 10% catalyst loading, while up to 20% catalyst loading has been utilized in most cases. Therefore, the catalyst loading in the present report is well within the reported range for C–S cross-coupling reactions. This is essential to decrease the reaction time (for more information see the ESI,† Table S6).
 | ||
| Scheme 5 Catalyst 1–5 mediated cross-coupling reaction between 1-bromo-4-nitro benzene and thiophenol. | ||
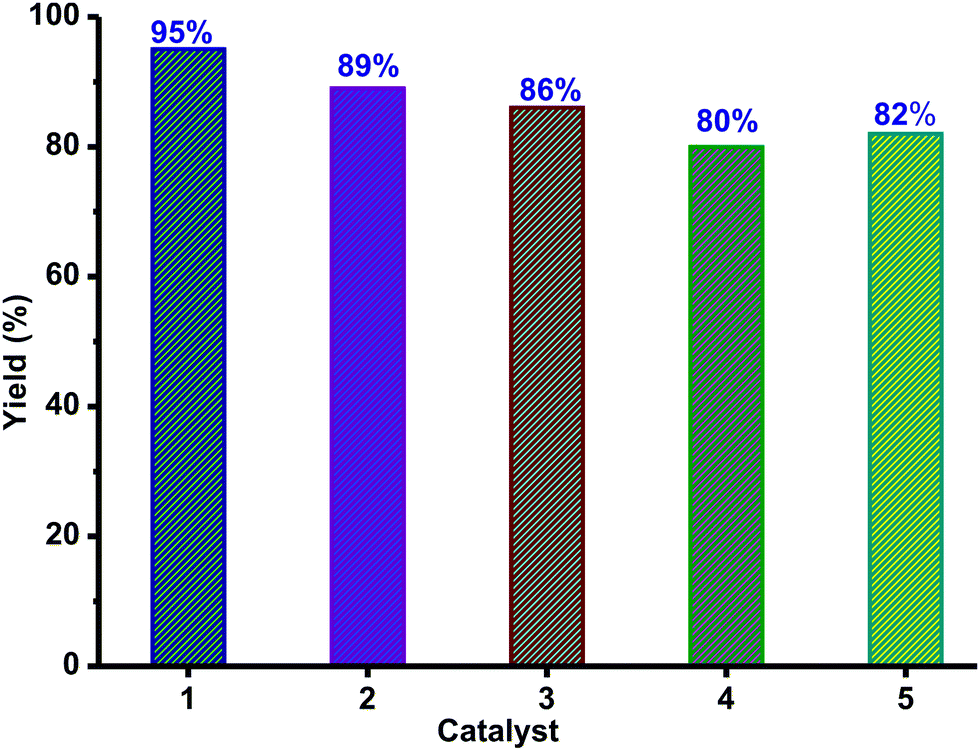 | ||
| Fig. 9 The catalyst 1–5 mediated cross-coupling reaction between 1-bromo-4-nitro benzene and thiophenol. Reaction conditions: 1-bromo-4-nitro benzene (1 mmol), thiophenol (1 mmol), NaOtBu (2 equiv.), catalyst (10 mol%), L-proline (10 mol%), 80 °C, 15 h. | ||
Thus, we have assessed the scope of catalyst 1 with a different combination of electron-deficient aryl halides or thiophenols (Scheme 6). The cross-coupling reactions between thiophenol and 1-halo-EWG-benzene (EWG = 4-NO2 (entry 1), 2-NO2 (entry 2), 4-CN (entry 3), 4-COMe (entry 4)) gave excellent yields. Similarly, excellent cross-coupling conversion was achieved between 4-chloro thiophenol and 4-nitro/2-nitro/4-cyano aryl halide (Scheme 6, entries 5–7)
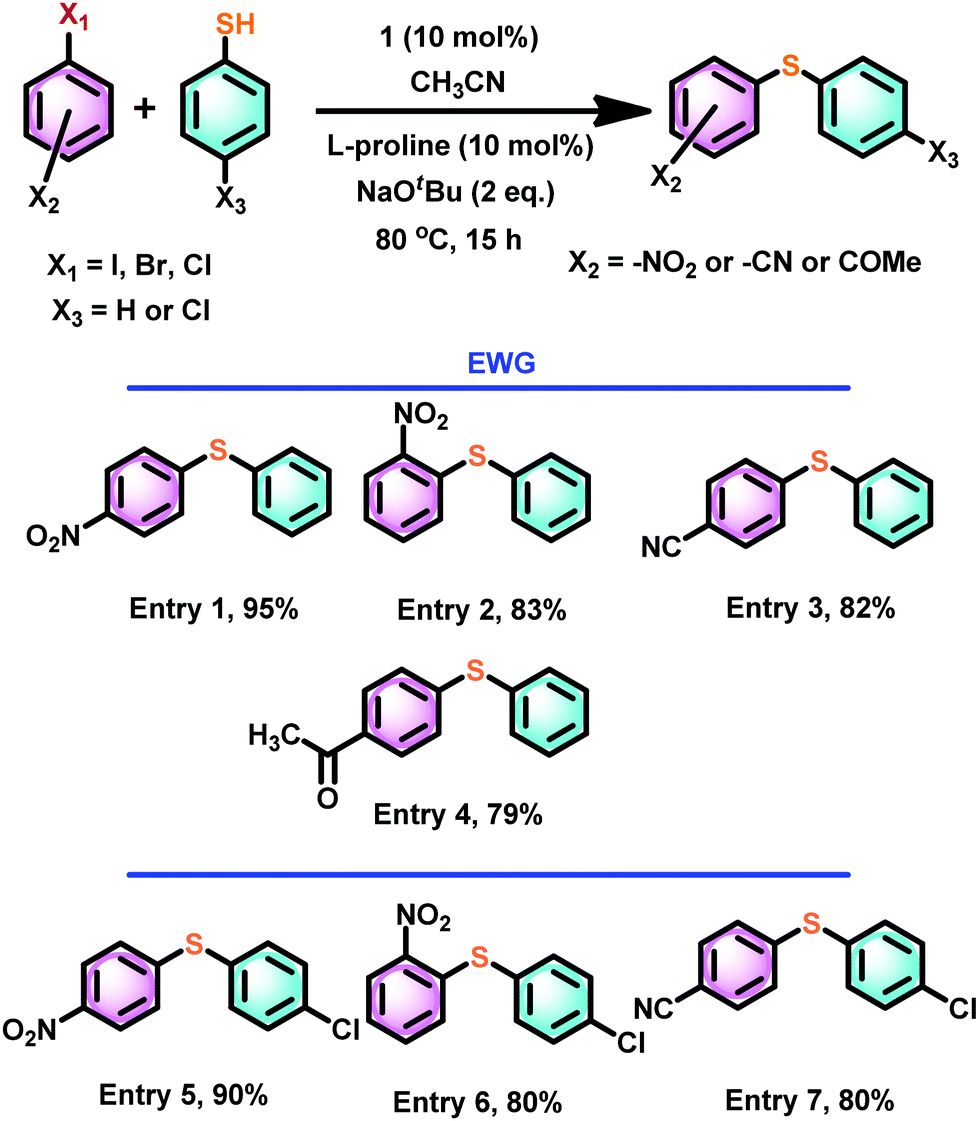 | ||
| Scheme 6 Scope of catalyst 1 mediated thioetherification of electron withdrawing group substituted aryl halides. | ||
The coupling reaction between an aryl halide such as 1-bromo-3-methoxy-benzene and thiophenol under the same conditions gave about 30% less yield compared to much more electron-deficient aryl halides (Scheme 7, entry 8). However, the combination of both 4-nitro-1-bromo benzene and 4-methyl and 4-methoxy aryl thiol gave a very good yield (Scheme 7, entries 9 and 10).
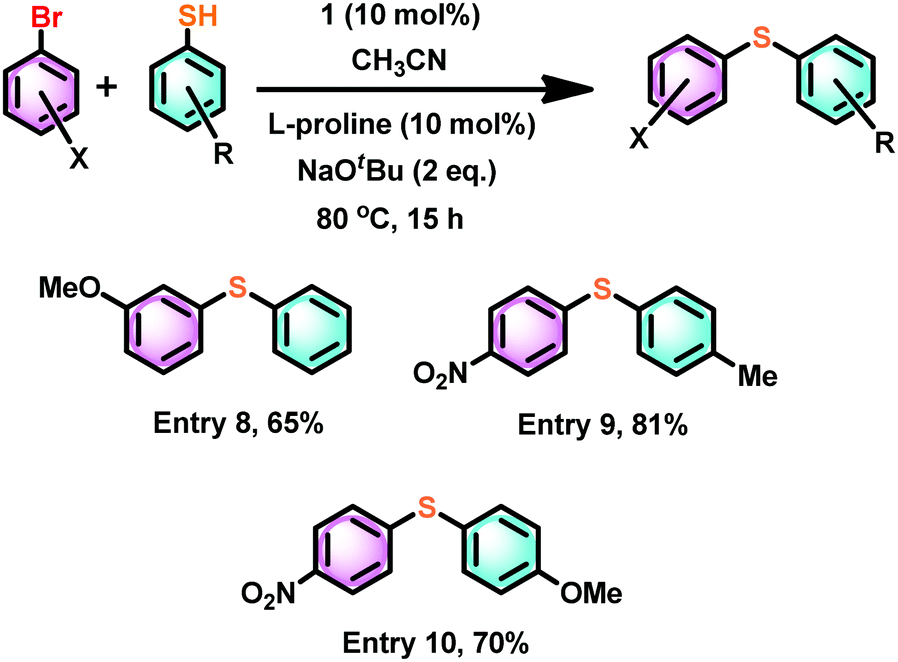 | ||
| Scheme 7 The influence of an electron-rich aryl halide or aryl thiol in catalyst 1 mediated thioetherification reactions. | ||
Similarly, the cross-coupling reaction between 2-bromo-3-nitropyridine and 4-methoxy thiophenol gave a cross-coupled product with good yield (Scheme 8). Hence, the electron-rich aryl halides are compatible with these conditions to yield cross-coupled products in excellent yield.
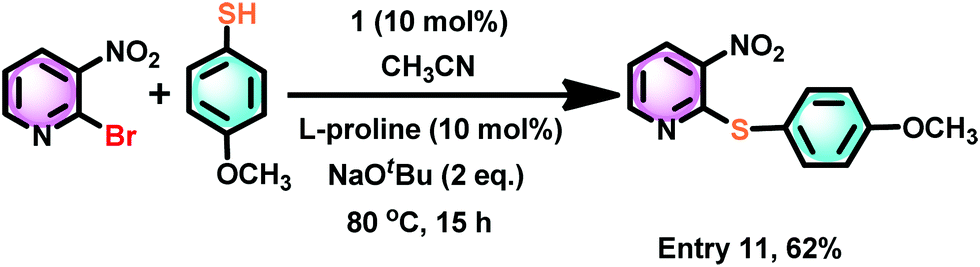 | ||
| Scheme 8 The catalyst 1 mediated thioetherification reaction between 2-bromo-3-nitropyridine and 4-methoxy thiophenol. | ||
2-(Arylthio)benzothiazoles are considered to be a potential class of bioactive ingredients in the pharmaceutical industry.47 Thus, the catalytic efficiency of 1 was investigated for the thioetherification between 2-mercaptobenzothiazole and 4-nitro thiophenol (Scheme 9). 2-(4-Nitrophenylthio)benzothiazole was isolated in excellent yield (86%). The catalyst 1 mediated synthesis of 2-(4-nitrophenylthio)benzothiazole appears to be more efficient than the CuBr mediated tandem one-pot synthesis at 80 °C for 15 h (70%),48 the FeCl3–N,N′-dimethylethylenediamine meditated reaction at 135 °C for 24 h (83%),49 enaminone ligand assisted copper(II)-catalyzed at 100 °C for 12 h (51%),50 and copper(I)-catalyzed at 100 °C for 24 h (83%).51
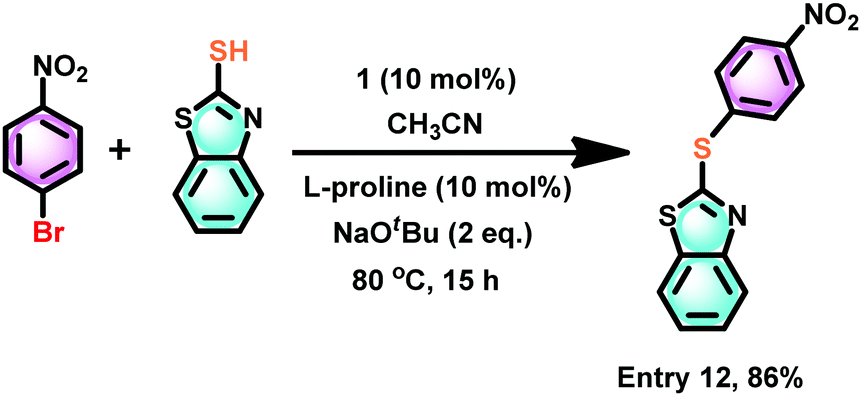 | ||
| Scheme 9 The catalyst 1 mediated thioetherification of 2-mercaptobenzothiazole with an electron withdrawing group substituted aryl halide, such as 4-nitro thiophenol. | ||
Also, we have attempted to isolate polyaromatic scaffolds, such as anthracene thioether derivatives. Similar molecules have shown promising features in anti-counterfeiting technology,52 medicinal chemistry,53 and the electronics industry as soft materials,54 to name a few. Under the above-optimized reaction conditions with catalyst 1, the C–S cross-coupling strategy was explored with 9-bromo anthracene. The C–S bond formation reaction between 9-bromo anthracene and 4-halo aryl thiols in the presence of catalyst 1 gave 9-(4-halo phenylthio)-anthracene in very good yield (74–70%) (Scheme 10, entries 13–15). It is noteworthy that the 9-(4-fluoro phenylthio)-anthracene, 9-(4-chloro phenylthio)-anthracene and 9-(4-bromo phenylthio)-anthracene derivatives have never been isolated. The present strategy results the formation of these rare molecules in pure form. The solid-state structure of 9-(4-chloro phenylthio)-anthracene has been characterised by the single-crystal X-ray diffraction technique (Fig. 10). The C(9)–S(1) and C(3)–S(1) bond distances are nearly comparable. The C(9)–S(1)–C(3) angle is 102.16(7)°. In the molecular packing of 9-(4-chloro phenylthio)-anthracene, pairs of 9-(4-chloro phenylthio)-anthracene molecules are arranged orthogonal to each other without any van der Waals interaction (see the ESI†). The present protocol seems to be competitive enough with the recently developed most efficient C–S cross-coupling reaction to isolate an unfunctionalized 9-(phenylthio)-anthracene using an air and moisture stable bimetallic catalyst [(tBu3P)Pd(I)]2, 89%).55
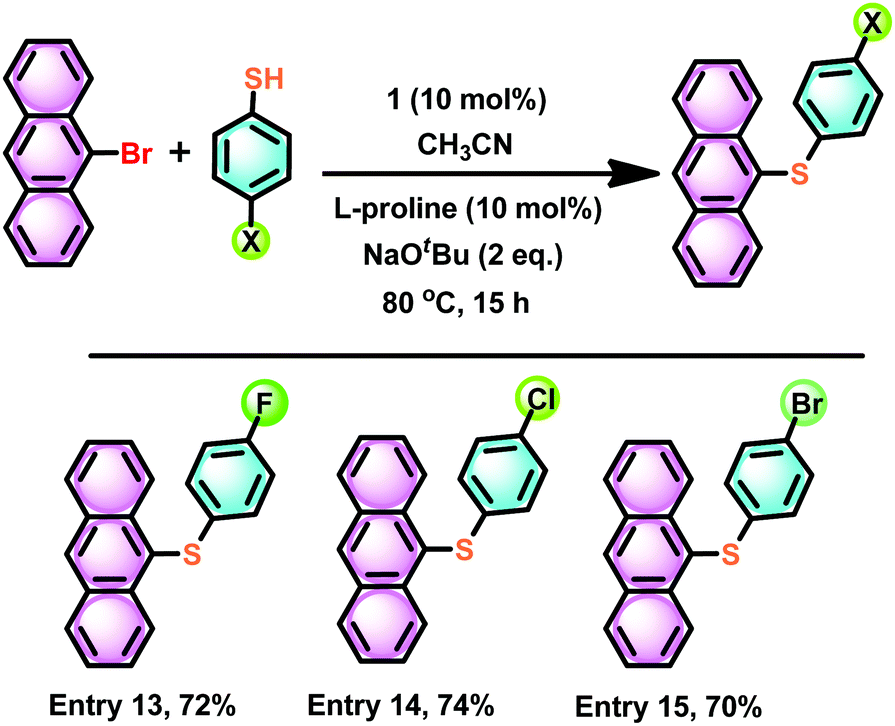 | ||
| Scheme 10 The catalyst 1 mediated thioetherification of 4-halo thiophenol with anthracene bromide. | ||
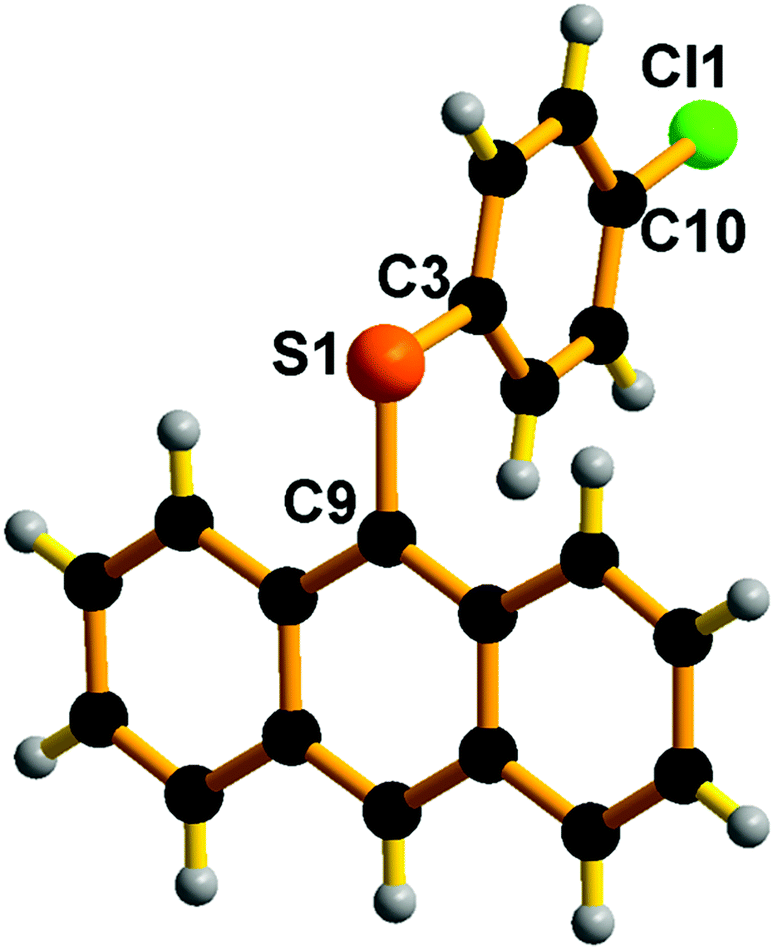 | ||
| Fig. 10 Molecular structure of E14. Selected bond lengths (Å) and bond angles (°): C(3)–S(1) 1.770(17), C(9)–S(1) 1.776(17), C(9)–S(1)–C(3) 102.16(7). | ||
Besides, the isolated yield under the above synthetic methodology is more commodious and economical under mild reaction conditions compared to the known methodologies.39
It is important to note that the addition of L-proline is very much essential to activate the catalyst. The catalyst is inactive in the absence of L-proline or the presence of less than 10 mol% L-proline. Furthermore, the blank reaction under optimized conditions gave zero progress in the catalytic conversion. Besides, the reaction mechanism and scope of the metal choice are yet to be explored further. Currently, we are working in this direction.
Experimental section
Materials and methods
All manipulations were carried out under an argon atmosphere using standard Schlenk techniques. The solvents were purchased from commercial sources and purified according to standard procedures and freshly distilled under an argon atmosphere before use. Phenyl acetylene, methyl iodide, sulfur powder, selenium powder, zinc chloride, and zinc bromide were purchased from commercial sources. Aromatic azides, CuBr(PPh3)3, 1-mesityl-3-methyl-4-phenyl-1H-1,2,3-triazol-3-ium iodide, and 1-benzyl-3-methyl-4-phenyl-1H-1,2,3-triazol-3-ium iodide, were prepared as described in the literature.13,16Caution! Organic azides and sodium azide are potentially-explosive substances. Azide salts tend to be highly heat and shock-sensitive explosives. Avoid strong acids, which can lead to the formation of hydrazoic acid, which is highly toxic, volatile, and explosive. Handle sodium azide powder or solutions >5% only in a properly functioning fume hood. Wear respiratory protection. Wear personal protective equipment. Avoid dust formation. Do not get in the eyes, or on the skin, or clothing. Use only under a chemical fume hood. Do not breathe vapors/dust. Do not ingest.
FT-IR measurements (neat) were carried out on a Bruker Alpha-P Fourier transform spectrometer. The UV-vis spectra were recorded on a T90+UV-vis spectrophotometer. The thermogravimetric analysis (TGA) was performed using a TASDT Q600. The NMR spectra were recorded on Bruker Ultrashield-400 spectrometers at 25 °C unless otherwise stated. Chemical shifts are given relative to TMS and were referenced to the solvent resonances as internal standards. Single crystals of complexes suitable for single-crystal X-ray analysis were obtained from their reaction mixture in a chloroform and methanol mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) at room temperature. Suitable crystals of L1–L3, 1–5 and E14 were selected and mounted on a SuperNova, Dual, Cu at zero, Eos diffractometer. The crystal was kept at 298 K during data collection. Using Olex2,56 the structure was solved with the olex2.solve57 structure solution program using direct methods and refined with the olex2.refine58 refinement package using Gauss–Newton minimization. Absorption corrections were performed based on multiple scans. Non-hydrogen atoms were anisotropically refined. Hydrogen atoms were included in the refinement in the calculated positions riding on their carrier atoms. L1 and L3 gave one “A” level alert due to a negligible difference in the measured fraction theta full value (0.861 for L1 and 0.925 for L3). Attempts to rectify these issues were not fruitful. Molecule 5 gave one “B” level alert due to the disordered carbon atoms of the benzene/toluene molecule. Though the reaction was carried out and crystallized in toluene, the lattice solvent molecule in 5 was fixed as benzene due to the heavily disordered nature of the lattice solvent molecule. Our efforts to locate the toluene methyl group were not successful. However the disordered lattice solvent molecule in 5 has no implication for the structural properties of metal complex 5.
:1 ratio) at room temperature. Suitable crystals of L1–L3, 1–5 and E14 were selected and mounted on a SuperNova, Dual, Cu at zero, Eos diffractometer. The crystal was kept at 298 K during data collection. Using Olex2,56 the structure was solved with the olex2.solve57 structure solution program using direct methods and refined with the olex2.refine58 refinement package using Gauss–Newton minimization. Absorption corrections were performed based on multiple scans. Non-hydrogen atoms were anisotropically refined. Hydrogen atoms were included in the refinement in the calculated positions riding on their carrier atoms. L1 and L3 gave one “A” level alert due to a negligible difference in the measured fraction theta full value (0.861 for L1 and 0.925 for L3). Attempts to rectify these issues were not fruitful. Molecule 5 gave one “B” level alert due to the disordered carbon atoms of the benzene/toluene molecule. Though the reaction was carried out and crystallized in toluene, the lattice solvent molecule in 5 was fixed as benzene due to the heavily disordered nature of the lattice solvent molecule. Our efforts to locate the toluene methyl group were not successful. However the disordered lattice solvent molecule in 5 has no implication for the structural properties of metal complex 5.
Synthesis of 1-mesityl-3-methyl-4-phenyl-1,2,3-triazole selone (L1)
To dry potassium carbonate (1.36 g, 9.87 mmol), 1-mesityl-3-methyl-4-phenyl-1H-1,2,3-triazol-3-ium iodide (1.6 g, 3.95 mmol), Se powder (0.4 g, 5.19 mmol) and methanol (20 mL) were added under an argon atmosphere. The reaction mixture was stirred for 36 h at 68 °C. The progress of the reaction was monitored by TLC. After completion of the reaction, water was added to the reaction mixture and extracted with dichloromethane (3 × 15 mL). The organic extract was washed with brine solution, and dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure to isolate analytically pure L1. Yield: 77% (based on the triazolium salt). M.p.: 190–195 °C. 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, J = 7.4 Hz, 2H, HAr), 7.52 (m, 3H, HAr), 7.03 (s, 2H, HMes), 4.16 (s, 3H, NCH3), 2.36 (s, 3H, Ar-CH3), 2.09 (s, 6H, Ar-CH3). 13C NMR (CDCl3, 100 MHz): δ 148.95 (CSe), 141.42 (Ctrz–Mes), 140.67 (Ctrz–Ph), 135.28 (CAr), 132.81 (CAr), 130.13 (CAr), 129.25 (CAr), 128.92 (CAr), 126.06 (CAr), 38.62 (N-CH3), 21.32 (ArCH3), 18.09 (ArCH3). FT-IR (neat, ): 2920(m), 2855(w), 1600(m), 1523(w), 1445(s), 1319(w), 1285(w), 1170(s) (CSe), 1113(m), 1069(s), 1020(m), 855(s), 763(m), 693(s), 539(s) cm−1.
Synthesis of 1-mesityl-3-methyl-4-phenyl-1,2,3-triazole thione (L2)
L2 was prepared by a procedure analogous to that of L1 starting from 1-mesityl-3-methyl-4-phenyl-1H-1,2,3-triazol-3-ium iodide (1.6 g, 3.95 mmol) and sulfur powder (0.25 g, 7.36 mmol). Yield: 80% (based on the triazolium salt). M.p.: 170–175 °C. 1H NMR (CDCl3, 400 MHz): δ 7.81 (d, J = 8.12 Hz, 2H, HAr) 7.49 (m, 3H, HAr), 7.02 (s, 2H, HMes), 4.11 (s, 3H, N-CH3), 2.34 (s, 3H, Ar-CH3), 2.11 (s, 6H, Ar-CH3). 13C NMR (CDCl3, 100 MHz): δ 158.76 (CS), 140.55 (Ctrz–Mes), 137.10 (Ctrz–Ph), 135.55 (CAr), 131.94 (CAr), 129.76 (CAr), 129.26 (CAr), 128.91 (CAr), 126.12 (CAr), 38.80 (N-CH3), 21.30 (Ar-CH3), 17.96 (Ar-CH3). FT-IR (neat, ): 2915(w), 2853(w), 1602(m), 1523(w), 1461(s), 1400(w), 1336(s), 1286(m), 1173(s) (CSe), 1116(m), 1063(m), 1020(s), 861(s), 797(m), 759(s), 697(s), 546(s) cm−1.
Synthesis of 1-benzyl-3-methyl-4-phenyl-1,2,3-triazole selone (L3)
L3 was prepared by a procedure analogous to that of L1 starting from 1-benzyl-3-methyl-4-phenyl-1H-1,2,3-triazol-3-ium iodide (1.5 g, 3.97 mmol) and selenium powder (0.31 g, 3.97 mmol). Yield: 84% (based on the triazolium salt). M.p.: 152–155 °C. 1H NMR (DMSO-d6, 400 MHz): δ 7.64 (m, 4H, HAr), 7.48 (m, 3H, HAr), 7.35 (m, 3H, HAr), 5.84 (s, 2H, N-CH2), 3.97 (s, 3H, N-CH3). 13C NMR (DMSO-d6, 100 MHz): δ 147.04 (CSe), 141.12 (Ctrz–Ar), 134.28 (CAr), 130.16 (CAr), 130.02 (CAr), 129.40 (CAr), 128.92 (CAr), 128.83 (CAr), 128.70 (CAr), 126.15 (CAr), 53.42 (N-CH2) 38.21 (N-CH3). FT-IR (neat, ): 3034(w), 2931(w), 1600(w), 1524(w), 1470(w), 1416(m), 1367(m), 1310(s), 1246(m), 1213(w), 1160(s) (CSe), 1069(s), 1018(m), 838(m), 761(m), 729(m), 690(s), 576(s) cm−1.
Synthesis of [(L1)Zn(Cl)2(HOMe)] (1)
L1 (0.12 g, 0.34 mmol) was added to a solution of ZnCl2 (0.05 g, 0.33 mmol) in toluene under an argon atmosphere at ambient temperature. Subsequently, the reaction mixture was stirred at 100 °C for 24 h to give a colourless precipitate. The solvent was decanted, and the solid residue was washed with hexane (2 × 5 mL) and dried under a high vacuum. The resulting powder was dissolved in a chloroform and methanol mixture (1:1 ratio). Colourless crystals of 1 were obtained at room temperature. Yield: 80% (based on ZnCl2). M.p.: 200–205 °C (decomposed). 1H NMR (DMSO-d6, 400 MHz): δ 7.91 (d, J = 7.28 Hz, 2H, HAr), 7.56 (m, 3H, HAr), 7.09 (s, 2H, HMes), 4.19 (s, 3H, N-CH3), 3.17 (d, 3H, OCH3), 2.34 (s, 3H, Ar-CH3), 1.98 (s, 6H, Ar-CH3). 13C NMR (DMSO-d6, 100 MHz): δ 146.65 (CSe), 140.38 (Ctrz–Mes), 139.85 (Ctrz–Ph), 135.07 (CAr), 132.75 (CAr), 130.11 (CAr), 129.70 (CAr), 128.68 (CAr), 128.37 (CAr), 126.19 (CAr), 48.59 (N-CH3), 38.91 (OCH3), 20.71 (Ar-CH3), 17.41 (Ar-CH3). FT-IR (neat, ): 3300(m), 2920(s), 2853(m), 2512(w), 1608(s), 1541(w), 1446(s), 1377(m), 1320(w), 1285(m), 1196(s) (CSe), 1122(w), 1072(m), 1001(s), 857(s), 778(s), 694(s), 546(m) cm−1.
Synthesis of [(L1)Zn(Br)(μ2-Br)]2 (2)
2 was prepared by a procedure analogous to that of 1, starting from L1 (0.11 g, 0.30 mmol) and ZnBr2 (0.07 g, 0.29 mmol). Yield: 74% (based on ZnBr2). M.p.: 200–205 °C (decomposed). 1H NMR (DMSO-d6, 400 MHz): δ 7.90 (d, J = 6.32 Hz, 2H, HAr), 7.57 (m, 3H, HAr), 7.09 (s, 2H, HMes), 4.20 (s, 3H, N-CH3), 2.34 (s, 3H, Ar-CH3), 1.98 (s, 6H, Ar-CH3). 13C NMR (DMSO-d6, 100 MHz): δ 147.34 (CSe), 140.17 (Ctrz–Mes), 139.77 (Ctrz–Ph), 135.07 (CAr), 132.85 (CAr), 130.09 (CAr), 129.61 (CAr), 128.63 (CAr), 128.32 (CAr), 126.35 (CAr), 48.56 (N-CH3), 20.66 (Ar-CH3), 17.39 (Ar-CH3). FT-IR (neat, ): 3374(s), 2319(w), 1611(s), 1472(m), 1378(w), 1320(w), 1284(m), 1194(m) (CSe), 1122(w), 1071(w), 1016(w), 851(m), 686(s) cm−1.
Synthesis of [(L2)Zn(Cl)2(HOMe)] (3)
3 was prepared by a procedure analogous to that of 1, starting from L2 (0.11 g, 0.35 mmol) and ZnCl2 (0.05 g, 0.35 mmol). Yield: 70% (based on ZnCl2). M.p.: 210–215 °C (dec.). 1H NMR (DMSO-d6, 400 MHz): δ 7.90 (d, J = 7.04 Hz, 2H, HAr), 7.56 (m, 3H, HAr), 7.07 (s, 2H, HMes), 4.15 (s, 3H, NCH3), 2.32 (s, 3H, ArCH3), 1.99 (s, 6H, ArCH3). 13C NMR (DMSO-d6, 100 MHz): δ 157.00 (CS), 139.76 (Ctrz–Mes), 136.02 (Ctrz–Ph), 135.32 (CAr), 131.97 (CAr), 129.71 (CAr), 129.36 (CAr), 128.66 (CAr), 128.37 (CAr), 126.26 (CAr), 48.59 (NCH3), 20.68 (ArCH3), 17.31 (ArCH3). FT-IR (neat, ): 3319(s), 1615(s), 1541(w), 1442(s), 1368(m), 1326(s), 1289(s), 1203(s), 1126(m) (CS), 1071(m), 1001(s), 860(s), 790(w), 753(w), 692(s), 545(s) cm−1.
Synthesis of [(L2)Zn(Br)2(HOMe)] (4)
4 was prepared by a procedure analogous to that of 1, starting from L2 (0.11 g, 0.34 mmol) and ZnBr2 (0.08 g, 0.33 mmol). Yield: 73% (based on ZnBr2). M.p.: 215–220 °C (decomposed). 1H NMR (DMSO-d6, 400 MHz): δ 7.96 (d, J = 7.02 Hz, 2H, HAr) 7.60 (m, 3H, HAr), 7.13 (s, 2H, HMes), 4.21 (s, 3H, N-CH3), 3.21 (d, 3H, OCH3), 2.38 (s, 3H, Ar-CH3), 2.05 (s, 6H, Ar-CH3). 13C NMR (DMSO-d6, 100 MHz): δ 157.11 (CS), 139.74 (Ctrz–Mes), 135.97 (Ctrz–Ph), 135.32(CAr), 131.99 (CAr), 129.70(CAr), 129.34 (CAr), 128.66 (CAr), 128.36 (CAr), 126.29 (CAr), 48.58 (N-CH3), 39.02 (OCH3), 20.69 (Ar-CH3), 17.32 (Ar-CH3). FT-IR (neat, ): 3363(s), 2195(w), 2120(w), 1610(s), 1473(m), 1365(w), 1328(w), 1289(s), 1194(s) (CS), 1122(m), 1069(m), 1017(s), 851(s), 688(s), 549(m) cm−1.
Synthesis of [(L3)Zn(Br)(μ2-Br)]2 (5)
L3 (0.10 g, 0.31 mmol) was added to a solution of ZnBr2 (0.07 g, 0.31 mmol) in toluene under an argon atmosphere at ambient temperature. Subsequently, the reaction mixture was stirred at 100 °C for 24 h to give a colourless clear solution. Colourless crystals of 5 were obtained at room temperature. Yield: 77% (based on ZnBr2). M.p.: 110–115 °C (decomposed). 1H NMR (CD3CN, 400 MHz): δ 7.61 (m, 5H, HAr), 7.51 (d, J = 5.82 Hz, 2H, HAr), 7.44 (m, 3H, HAr), 7.23 (d, J = 6.40 Hz, 2H, HAr), 5.86 (s, 2H, N-CH2) 4.15 (s, 3H, N-CH3). 13C NMR (CD3CN, 100 MHz): δ 145.47 (CSe), 138.90 (Ctrz–Ar), 136.47 (CAr), 134.43 (CAr), 131.88 (CAr), 131.66 (CAr), 131.50 (CAr), 130.01(CAr), 129.92 (CAr), 129.83(CAr), 129.22 (CAr), 126.26 (CAr), 125.78 (CAr), 118.35 (CAr), 56.12 (N-CH2), 39.55 (N-CH3). FT-IR (neat, ): 3354(s), 2332(w), 2198(w), 2102(w), 2008(w), 1611(s), 1486(m), 1444(m), 1314(s), 1250(m), 1160(m) (CSe), 1081(s), 1018(s), 807(m), 759(m), 694(s), 639(m), 568(s), 528(m) cm−1.
1–5 catalysed C–S cross-coupling reactions
The catalytic reactions were performed using a standard Schleck technique using zinc complexes 1–5 as catalysts for the C–S bond coupling reactions of aryl halides and thiols as reported.39,46 A dry Schlenk tube was charged with 1-bromo-4-nitro benzene (0.20 g, 1 mmol) and thiophenol (0.1 g, 1 mmol), L-proline (10 mol%, 0.005 g), and NaOtBu (0.09 g, 0.10 mmol) in CH3CN (3 mL). The Schlenk was heated in an oil bath at 80 °C for 15 h. The reaction mixture was then cooled and extracted with ethyl acetate (3 × 15 mL), and the ethyl acetate layer was washed with a saturated aqueous NaCl solution. The organic layer was dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure in a rotary evaporator. The crude residue was purified by column chromatography using EtOAc–hexane as the eluent to obtain the product as a yellow solid.Conclusion
In summary, the first mesoionic heavier chalcogenones, L1–L3, were synthesized and characterized. The σ donor and π accepting nature of the mesoionic chalcogenones were investigated using density functional theory. Compared to mesoionic triazolin-5-ylidene, mesoionic heavier chalcogenones are likely to become a good σ-donor and π-acceptor ligand. Utilizing these ligands, five monomeric and dimeric zinc complexes were prepared in good yields. Compounds 1–5 were characterized in solution by NMR and UV-vis spectroscopy as well as in the solid-state by TGA, FTIR, and single-crystal X-ray diffraction. The UV-vis absorption, thermal, and structural properties of the zinc complexes were significantly influenced by varying the N-substituents of the mesoionic ligands. Single crystal X-ray studies show that the zinc(II) centers in 1–5 are four-coordinated and exhibit distorted tetrahedral geometry. Subsequently, the catalytic application of 1–5 in cross-coupling reactions between thiophenols and aryl halides was demonstrated. 1–5 were active towards C–S bond formation reactions under mild reaction conditions, and this reveals the potential of easily accessible mesoionic chalcogenone Zn(II) compounds in catalysis. Notably, mononuclear zinc(II) complex 1 was more active than 2–5. The introduction of the heavier chalcogenone ligand in the zinc(II) salt led to increasing catalytic activity, presumably due to its optimized steric and electronic support with the metal center. This strategy contrasts with the previous use of highly reactive diethyl zinc under an inert atmosphere. The present catalysts may thus render a cheap yet effective alternative for expensive late transition metal catalysts in addition to a broad substrate scope.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the DST-SERB (EMR/2017/001211) for financial support. VM thanks UGC for the fellowship. VK thanks DST-WOS-A (WOS-A/CS-65/2016) for the fellowship.References
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, B. P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 236, 556–559 CrossRef PubMed.
- P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596–609 CrossRef CAS.
- M. Albrecht, Chem. Commun., 2008, 3601–3610 RSC.
- (a) D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 554–584 CrossRef CAS; (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- J. M. Aizpurua, M. Sagartzazu-Aizpurua and Z. Monasterio, Top. Heterocycl. Chem., Springer-Verlag, Berlin Heidelberg, 2015, vol. 40, pp. 211–268 Search PubMed.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- K. O. Marichev, S. A. Patil and A. Bugarin, Tetrahedron, 2018, 74, 2523–2546 CrossRef CAS.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- G. G. Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef.
- S. Gründemann, A. Kovacevic, M. Albrecht, J. F. Faller and R. H. Crabtree, Chem. Commun., 2001, 2274–2275 RSC.
- M. Begtrup, J. Chem. Soc., Chem. Commun., 1975, 334–335 RSC.
- D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J. P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed. Engl., 1995, 34, 1021–1023 CrossRef CAS.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762 CrossRef CAS PubMed.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- (a) A. Petronilho, H. Müller-Bunz and M. Albrecht, Chem. Commun., 2012, 48, 6499–6501 RSC; (b) T. Nakamura, T. Terashima, K. Ogata and S.-I. Fukuzawa, Org. Lett., 2011, 134, 620–623 CrossRef PubMed; (c) S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067–3075 CrossRef CAS.
- (a) W.-G. Jia, Y.-B. Huang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2008, 5612–5620 RSC; (b) N. Ghavale, S. T. Manjare, H. B. Singh and R. J. Butcher, Dalton Trans., 2015, 44, 11893–11900 RSC.
- (a) G. K. Rao, A. Kumar, J. Ahmed and A. K. Singh, Chem. Commun., 2010, 46, 5954–5956 RSC; (b) L.-M. Zhang, H.-Y. Li, H.-X. Li, D. J. Young, Y. Wang and J.-P. Lang, Inorg. Chem., 2017, 56, 11230–11243 CrossRef CAS PubMed.
- J. Jin, H.-W. Shin, J. H. Park, J. H. Park, E. Kim, T. K. Ahn, D. H. Ryu and S. U. Son, Organometallics, 2013, 32, 3954–3959 CrossRef CAS.
- H. R. Kim, I. G. Jung, K. Yoo, K. Jang, E. S. Lee, J. Yun and S. U. Son, Chem. Commun., 2010, 46, 758–760 RSC.
- K. Srinivas, C. N. Babu and G. Prabusankar, Dalton Trans., 2015, 44, 15636–15644 RSC.
- E. Alvarado, A. C. Badaj, T. G. Larocque and G. G. Lavoie, Chem. – Eur. J., 2012, 18, 12112–12121 CrossRef CAS PubMed.
- K. Srinivas and G. Prabusankar, Dalton Trans., 2017, 46, 16615–16622 RSC.
- K. Srinivas and G. Prabusankar, RSC Adv., 2018, 8, 32269–32282 RSC.
- H.-N. Zhang, W.-G. Jia, Q.-T. Xu and C.-C. Ji, Inorg. Chim. Acta, 2016, 450, 315–320 CrossRef CAS.
- C. N. Babu, K. Srinivas and G. Prabusankar, Dalton Trans., 2016, 45, 6456–6465 RSC.
- K. Srinivas, P. Suresh, C. N. Babu, A. Sathyanarayana and G. Prabusankar, RSC Adv., 2015, 5, 15579–15590 RSC.
- K. Srinivas, A. Sathyanarayana, C. N. Babu and G. Prabusankar, Dalton Trans., 2016, 45, 5196–5209 RSC.
- Y.-B. Huang, W.-G. Jia and G.-X. Jin, J. Organomet. Chem., 2009, 694, 86–90 CrossRef CAS.
- A. K. Sharma, H. Joshi, R. Bhaskar and A. K. Singh, Dalton Trans., 2017, 46, 2228–2237 RSC.
- O. Prakash, K. N. Sharma, H. Joshi, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 2535–2543 CrossRef CAS.
- A. K. Sharma, H. Joshi, K. N. Sharma, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 3629–3639 CrossRef CAS.
- P. Dubey, S. Gupta and A. K. Singh, Dalton Trans., 2018, 47, 3764–3774 RSC.
- M. Vaddamanu, R. Karupnaswamy, K. Srinivas and G. Prabusankar, ChemistrySelect, 2016, 1, 4668–4671 CrossRef CAS.
- A. Sathyanarayana, K. Srinivas, A. Mandal, S. Gharami and G. Prabusankar, J. Chem. Sci., 2014, 126, 1589–1595 CrossRef CAS.
- D. J. Williams, K. M. White, D. VanDerveer and A. P. Wilkinson, Inorg. Chem. Commun., 2002, 5, 124–126 CrossRef CAS.
- U. Flörke, A. Ahmida, H. Egold and G. Henkel, Acta Crystallogr., 2014, E70, m384 Search PubMed.
- (a) A. Ghaderi, Tetrahedron, 2016, 72, 4758–4782 CrossRef CAS; (b) L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185–1195 CrossRef CAS PubMed.
- For selected examples: (a) C.-F. Lee, Y.-C. Liu and S. S. Badsara, Chem. – Asian J., 2014, 9, 706–722 CrossRef CAS PubMed and references there in; (b) P. Shaw, A. R. Kennedy and D. J. Nelson, Dalton Trans., 2016, 45, 11772–11780 RSC; (c) L.-H. Zou, C. Zhao, P.-G. Li, Y. Wang and J. Li, J. Org. Chem., 2017, 82, 12892–12898 CrossRef CAS PubMed; (d) V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef CAS; (e) K. D. Jones, D. J. Power, D. Bierer, K. M. Gericke and S. G. Stewart, Org. Lett., 2018, 20, 208–211 CrossRef CAS PubMed; (f) K. Choudhuri, S. Maiti and P. Mal, Adv. Synth. Catal., 2019, 361, 1092–1101 CrossRef CAS; (g) P. H. Gehrtz, V. Geiger, T. Schmidt, L. Sršan and I. Fleischer, Org. Lett., 2018, 21, 50–55 CrossRef PubMed; (h) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef CAS PubMed; (i) C.-W. Chen, Y.-L. Chen, D. M. Reddy, K. Du, C.-E. Li, B.-H. Shih, Y.-J. Xue and C.-F. Lee, Chem. – Eur. J., 2017, 23, 10087–10091 CrossRef CAS PubMed; (j) J. L. Farmer, M. Pompeo, A. J. Lough and M. G. Organ, Chem. – Eur. J., 2014, 20, 15790–15798 CrossRef CAS PubMed; (k) C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590–608 CrossRef CAS.
- M. Kosugi, T. Shimizu and T. Migita, Chem. Lett., 1978, 13–14 CrossRef CAS.
- G. T. Venkanna, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2014, 4, 2941–2950 CrossRef CAS.
- X. Liu, S.-B. Zhang, H. Zhu and Z.-B. Dong, J. Org. Chem., 2018, 83, 11703–11711 CrossRef CAS PubMed.
- S.-C. Lee, H.-H. Liao, A. Chatupheeraphat and M. Rueping, Chem. – Eur. J., 2018, 24, 3608–3612 CrossRef CAS PubMed.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760–1763 CrossRef CAS.
- A. P. Thankachan, K. S. Sindhu, K. K. Krishnan and G. Anilkumar, RSC Adv., 2015, 5, 32675–32678 RSC.
- M. A. B. Azam and B. Suresh, Sci. Pharm., 2012, 80, 789–823 CrossRef CAS PubMed.
- X. Liu, S.-B. Zhang, H. Zhu and Z.-B. Dong, J. Org. Chem., 2018, 83, 11703–11711 CrossRef CAS PubMed.
- A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880–2883 CrossRef CAS.
- Y. Liu, B. Huang, X. Cao, D. Wu and J.-P. Wan, RSC Adv., 2014, 4, 37733–37737 RSC.
- G. He, Y. Huang, Y. Tong, J. Zhang, D. Zhao, S. Zhou and S. Han, Tetrahedron Lett., 2013, 54, 5318–5321 CrossRef CAS.
- P. R. Christensen and M. O. Wolf, Adv. Funct. Mater., 2016, 26, 8471–8477 CrossRef CAS.
- (a) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS; (b) D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed; (c) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (d) M. Feng, B. Tang, S. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS; (e) G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS.
- For example: (a) X. Gao, J. L. Hodgson, D. Jiang, S. B. Zhang, S. Nagase, G. P. Miller and Z. Chen, Org. Lett., 2011, 13, 3316–3319 CrossRef CAS PubMed; (b) P. R. Christensen, B. O. Patrick, É. Caron and M. O. Wolf, Angew. Chem., Int. Ed., 2013, 52, 12946–12950 CrossRef CAS PubMed.
- T. Scattolin, E. Senol, G. Yin, Q. Guo and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 12425–12429 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., 2015, A71, 59–75 CrossRef.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., 2015, A71, 59–75 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of the synthesized complexes. Solid state packing. Computational data. CCDC 1893235 (L1), 1893237 (L2), 1893236 (L3), 1893242 (1), 1893241 (2), 1893240 (3), 1893238 (4), 1893239 (5) and 1895992 (E14). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj04440j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |