
Co-precipitation of MnO and Cu in an ionic liquid as a first step toward self-formed barrier layers†
Paul-Henri
Haumesser
 *ab,
Catherine C.
Santini
c,
Laurence
Massin
d and
Jean-Luc
Rousset
d
*ab,
Catherine C.
Santini
c,
Laurence
Massin
d and
Jean-Luc
Rousset
d
aUniv. Grenoble Alpes, F-38000 Grenoble, France
bCEA, LETI, MINATEC Campus, F-38054 Grenoble, France. E-mail: paul.haumesser@cea.fr; Fax: +33 438783034; Tel: +33 438785759
cUniv. Lyon, CNRS-UMR 5265, 43 Bd du 11 Novembre 1918, F-69616, Villeurbanne Cedex, France
dUniv. Lyon, IRCELYON, UMR 5256, CNRS, Villeurbanne, France
First published on 27th November 2019
Abstract
The deposition and sintering of nanoparticles (NPs) containing both Cu and Mn could be an economically and technically interesting alternative to current processes for the metallisation of through Si vias (TSVs). We propose to use ionic liquids (ILs) to (i) synthesise suspensions of suitable NPs and (ii) spread and anneal those suspensions directly on technological substrates. Indeed, ILs readily stabilise NPs and possess good thermal stability. Suspensions of metallic Cu and MnO are formed upon decomposition of solutions of selected Cu and Mn precursors in an IL, C1C4ImNTf2. Upon decomposition of mixed solutions of both precursors, single populations of NPs are obtained, a good indication that both elements are incorporated in these NPs. Finally, upon spreading and annealing these suspensions on samples coated with SiO2, typical of TSV technology, a partial reduction of SiO2 is observed, in agreement with the formation of a MnSiO3 phase typical of self-formed barriers.
1 Introduction
Microelectronics devices have adopted Cu as a standard interconnecting material. However, Cu integration requires suitable barrier layers against diffusion, and a Cu seed layer to initiate electroplating.1 In the 3D chip-stacking approach, a major challenge is the deposition of a continuous barrier and seed layers in Through Silicon Vias (TSVs) at low cost.2 The TaN/Ta barrier and Cu seed layers are currently formed by physical vapour deposition (PVD) under vacuum. However, PVD-based processes inherently suffer from limited step coverage. There exist hardware solutions to improve it, but these sophisticated PVD processes are costly. Therefore, a more conformal deposition technique is needed, if possible at low cost to be attractive for TSV applications. A chemical “wet” process would be particularly well adapted in this situation.In recent years, the so-called self-formed barriers have attracted much attention.3 This approach uses Mn-doped Cu seed layers directly deposited onto the interline dielectric material. Under thermal treatment, Mn migrates towards the interface to form mixed oxides with silica, which act as a barrier against copper diffusion. Usually, the Cu(Mn) seed layer is deposited by the same PVD process used for the deposition of pure Cu. To our knowledge, processes in the liquid phase have never been described for this application.
Indeed, a new way to elaborate metallic films at low cost was proposed by annealing colloidal suspensions of metallic nanoparticles (NPs). This process has been shown to be capable of growing Cu films on solid surfaces4 or even Ag seed layers in TSVs.5 However, it has only been described for NPs stabilised in organic solvents, which are not readily usable for the targeted application, mostly due to surface contamination. A possible workaround would be to form and stabilise metallic NPs in ionic liquids (ILs).6 ILs are molten salts composed of organic cations and inorganic or organic anions. They are generally liquid at room temperature, and thermally and chemically stable, with extremely low volatility.7 In addition, ILs are ideal media to form and stabilise small and monodisperse NPs.8
Unlike traditional solvents, ionic liquids (ILs) can be used to generate a large variety of NPs mainly from Group VIII metals (Fe, Co, Ru, Rh, Pd, Os, Ir, and Pt) and from noble metals (Cu, Ag, and Au) by several physical and chemical routes, and stabilise them in the absence of further additives.8 In order to avoid surface contamination, the synthesis of NPs from organometallic (OM) precursors is largely reported. After decomposition under a H2 atmosphere, they precipitate the metal while forming volatile by-products that can be easily removed from the solution.8,9 Moreover, ILs are self-organised, as a result of additional interactions between their anionic and cationic constituents. For instance, if the ions contain an alkyl chain, hydrophobic interactions lead to the formation of non-polar nano-domains of fixed size, which may act as a template for the formation of the metallic particles, resulting in an exceptionally narrow size distribution of these nano-objects.10
Recently, we have demonstrated the successful synthesis of Cu11 and Ru–Cu NPs12 in ILs. There are surface reactions during the precipitation of these NPs that cause the association of the two metals, even if Ru and Cu are not miscible.12
The present study aims at applying this strategy to the co-precipitation of NPs containing both Cu and Mn and at investigating the possibility to use them as precursors for the deposition of Cu(Mn) seed layers compatible with the elaboration of self-formed barriers. In the first section, the precipitation of NPs is investigated. The two metals are studied first separately. In particular, a suitable Mn-containing OM precursor is selected, and its decomposition under H2 is investigated. Then, the co-decomposition of both precursors is studied, and the NPs produced are analysed. Finally, the formation of thin films starting with these various suspensions is examined.
2 Experimental
All experiments were conducted under an inert atmosphere using a glove box and schlenk lines. The synthesis of 1-butyl-3-methylimidazolium bis(trifluoromethylsulphonyl)imide, C1C4ImNTf2, was performed as reported in the literature13 and it was dried overnight under high vacuum (10−5 Pa, 12 h) before use. Its purity was checked by using NMR spectra, recorded on a Bruker Advance spectrometer at 300 MHz for 1H and at 75.43 MHz for 13C. After purification, the halide and water contents were found to be below 100 ppm (high resolution mass spectrometry) and ∼12 ppm (limit of Karl Fischer titration), respectively. Mesitylcopper (CuMes, purchased from Nanomeps and used without further purification) was used as an OM precursor for the Cu-NPs.11 Bis(neopentyl) manganese(II), Mn(tBuCH2)2, was selected as the Mn precursor (vide infra) and was synthesised as described in the literature.14 Each OM precursor was dissolved under inert conditions in C1C4ImNTf2 and these solutions, or mixtures of them, were decomposed under H2.All suspensions were analysed by using Transmission Electron Microscopy (TEM) using a Philips CM120 at 120 kV and by using High Resolution Transmission Electron Microscopy (HRTEM) using a Jeol JEM 2010FEF at 200 kV. The analysis was performed directly on the liquid samples, as our IL does not evaporate even under high vacuum. Size histograms were determined from a minimum of 200 particles per sample and fitted by a log-normal law.
These suspensions were used to coat samples prepared from a Si wafer coated with 500 nm of SiO2 by silane decomposition. For each sample, 0.05 mL of suspension was deposited onto a 2 × 2 cm Si coupon. The sample was purged for 5 min under primary vacuum, and flushed for 3 min at 500 mL min−1 of N2 (3%H2). Under a reduced flow of forming gas (100 mL min−1), the sample was heated at 250 °C within a few seconds and kept for 1 h at this temperature. The sample was then cooled down and washed 3 times with acetone, dried and finally stored under air.
The suspensions and coated Si samples were examined by using X-ray photoelectron spectroscopy (XPS). In a glove box, NP suspensions diluted with distilled and degassed acetonitrile were filtered 3 times. The paper samples containing NPs and IL traces were transferred to the spectrometer without air break via a shuttle. The analyses were carried out using a Kratos Ultra DLD spectrometer using monochromatic Al Kα radiation at 1486.6 eV (10 mA, 15 kV) as the photon source. All spectra were recorded in the hybrid (combined electrostatic and magnetic lens) mode at room temperature under ultra-high vacuum (10−9 Torr). Survey scans were conducted between 1200 eV and 0 binding energies using a 1 eV step size, a dwell time of 100 ms per step and 160 eV analyser pass energy. For all samples, high-resolution scans were carried out at 40 eV analyser pass energy. All photoelectron peaks were background-subtracted using a Shirley background and curve-fitted in the same manner, i.e. a Lorentzian-to-Gaussian ratio of 30%. All spectra were then calibrated using the adventitious C1s contaminant peak at 284.6 eV. Quantification was carried out with the VISION software.
3 Elaboration of Cu and MnO NPs
3.1 Elaboration of Cu-NPs
A previous study11 has shown that the decomposition of a solution of 5 × 10−2 mol L−1 CuMes in C1C4ImNTf2 affords Cu-NPs of 5.3 ± 1.1 nm (see also Fig. 4a). However, in the cited work, the Cu-NPs have only been analysed by HRTEM. Therefore, a suspension of Cu-NPs was characterised by XPS and XAES in the present study. Indeed, both techniques are needed to unambiguously determine the valence state of Cu, as the 2p3/2 lines of Cu(0) and Cu(I) are not resolved in XPS.15 The 2p3/2 line of Cu was measured at 932.2 eV by XPS, whereas the Cu LMM peak was detected at 918.9 eV by XAES (Fig. 1a and b). The graphical display (scatter plot) of the most intense photoelectron line binding energies (abscissa, oriented in the negative direction) versus the kinetic energy position of the sharpest core–core–core Auger line (ordinate) is known as a Wagner plot or chemical state plot.16 It has been demonstrated that such a plot increases the ability of XPS to identify chemical states of copper species.17Fig. 1c shows how the three chemical states of copper, namely, CuO, Cu2O and Cu, may be easily identified by using a Wagner plot. In the plot, the boxes corresponding to Cu, Cu2O and CuO are drawn to take into account the dispersion of the data obtained from the literature.18 Our measured values of binding energy of Cu 2p3/2 XPS and kinetic energy of Cu LMM Auger lines unambiguously fall in the region of the Wagner plot corresponding to metallic Cu.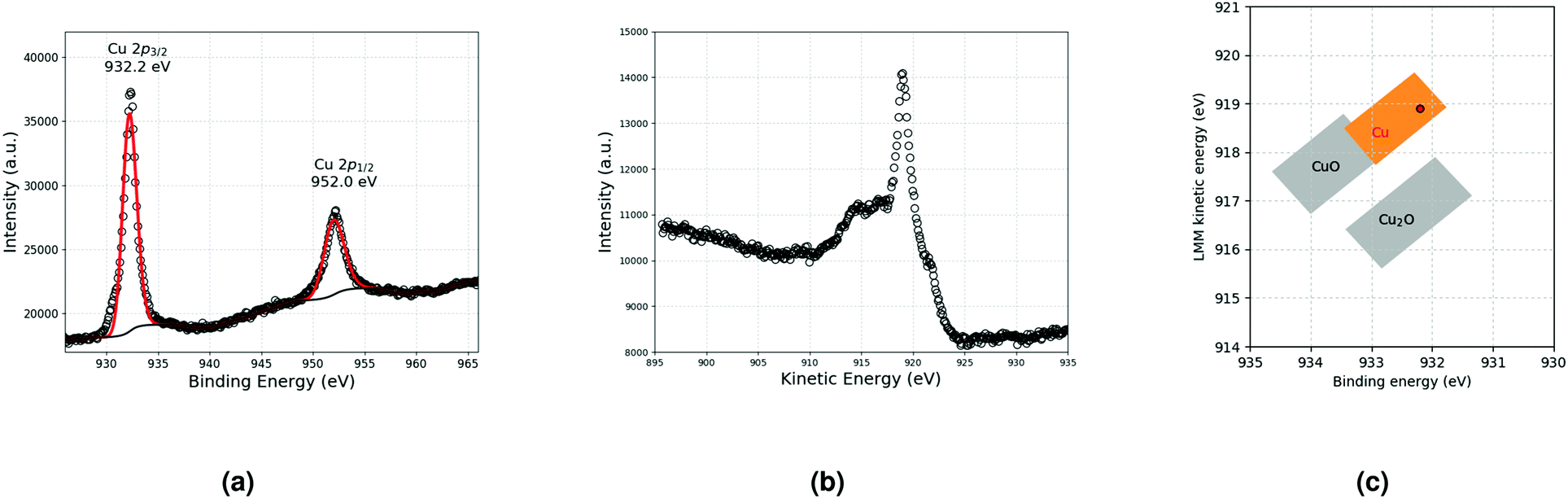 | ||
| Fig. 1 (a) Cu 2p lines and (b) Cu LMM Auger line measured by XPS and XAES, respectively. (c) Resulting Wagner plot.15 | ||
3.2 Elaboration of MnO-NPs
Mn-NPs in ionic liquids have been reported by Janiak and co-workers by decomposing carbonyl-based precursors using microwaves.19 In our case, no NP could be observed when a solution (5 × 10−2 mol L−1) of Mn2(CO)10 or (η5-MeCp)Mn(CO)3 in C1C4ImNTf2 was treated under 0.4 MPa H2 at 120 °C. By contrast, (η5-EtCp)2Mn (1.1 × 10−2mol L−1) in C1C4ImNTf2 afforded 5 nm NPs after 3 days under various experimental conditions. As this long reaction time is not compatible with the use of CuMes, the more reactive bis(neopentyl) manganese(II), Mn(tBuCH2)2, was selected instead.14The reactivity of Mn(tBuCH2)2 (5 × 10−2 mol L−1) in the solution of C1C4ImNTf2 (4 mL) was studied in a 100 mL autoclave connected to a continuous flow reactor under H2 (0.4 MPa) at 100 °C. The gas phase was continuously analysed by gas chromatography. Fig. S1 (ESI†) displays the instantaneous concentration of volatile products with respect to the concentration of metal in the solution. After 24 h, the equivalent of 8.5 C per Mn (i.e. 1.7 (tBuCH2) ligand out of 2 per Mn atom) evolved as alkanes (mainly methane C1, ethane C2, neopentane neoC5 and propane C3). The formation of C1, C2 and C3 could be explained by the alkane hydrogenolysis of neoC5 on putative Mn hydrides.20 Hence, the decomposition reaction is most probably completed after 24 h only at 100 °C.
In a first attempt to form Mn-based NPs, a solution of Mn(tBuCH2)2 (5 × 10−2 mol L−1) in C1C4ImNTf2 was treated at 50 °C under 0.9 MPa H2 for 48 h. The yellow solution turned brown after the reaction, which usually indicates the formation of NPs. This was verified by TEM inspection: 2.2 ± 0.5 nm NPs were observed in the IL. EDX analysis only detected Mn in areas where NPs were present (Fig. S2, ESI†). A typical HRTEM picture of one of these NPs is displayed in Fig. S3 (ESI†). From this image, the crystalline nature of the NP is clear. Accordingly, the Fourier transform of the HRTEM image exhibited well defined spots. This pattern could be successfully indexed using the cubic structures of β-Mn and α-Mn. The latter structure provides interplanar distances closer to the experimental values (Table S1, ESI†). However, it should be noted that MnO also possesses a cubic structure, with a lattice parameter half that of α-Mn. For this reason, this compound cannot be excluded in this analysis. In addition, the experimental pattern could also be indexed considering a tetragonal phase with lattice parameters a = 6.12 Å and c = 9.38 Å. Considering that Mn3O4 crystallises in a tetragonal phase with comparable lattice parameters (a = 5.76 Å and c = 9.47 Å),21 one cannot rule out this structure either. To really assess the nature of these NPs (metal or oxide), another technique was thus needed.
X-ray photoelectron spectroscopy (XPS) is the most established surface analysis technique to determine the oxidation state of Mn for which three dominant oxidation states (II, III, and IV) exist. Several studies have determined the oxidation of Mn in environmental oxide samples using the Mn 3p and Mn 3s regions. Indeed, each of those peaks displays several photolines (multiplet splitting), the energy differences of which depend on the oxidation state.22 However, the Scofield cross sections of the Mn 3s and Mn 3p peaks are low and both peaks are superimposed, in our case, respectively, to Mg 2s and Mg 2p lines. Nevertheless, we used the intense Mn 2p peak for a partial identification of Mn chemical states. Fig. 2 shows the Mn 2p XPS lines of a suspension of Mn-NPs. The binding energy of the Mn 2p3/2 peak at 640.9 eV can be unambiguously attributed to oxidised Mn species since the energy of the corresponding peak in metallic Mn is much lower.23
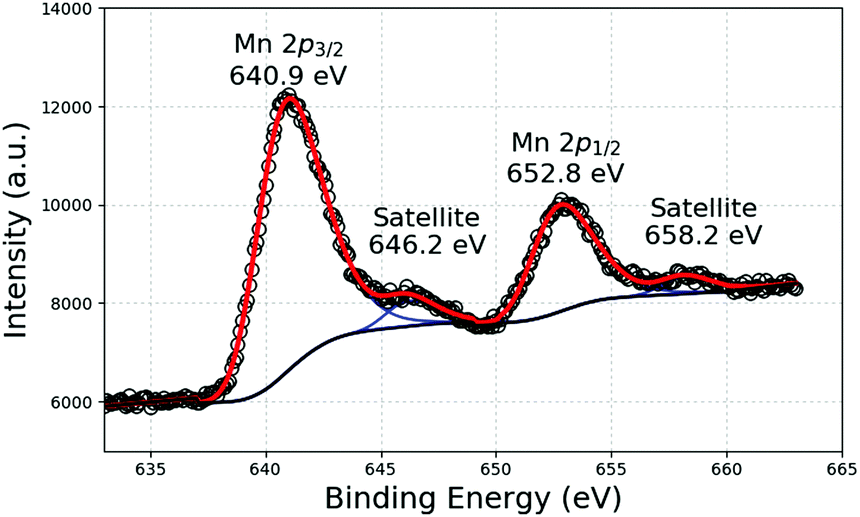 | ||
| Fig. 2 Mn 2p XPS response of a suspension of NPs formed after the decomposition of Mn(tBuCH2)2 in C1C4ImNTf2. | ||
In general, only the binding energy shifts are sufficient to identify chemical states, but the Mn 2p peaks are rather broad and the binding energies shifts of the different oxides are not large enough to clearly distinguish between them, especially when two or more species are simultaneously present. A possibility of further identification is given from analysis of the satellite structure, which is located at 5–6 eV higher binding energy than the Mn 2p peak for MnO oxide and at about 10 eV for Mn III and IV species. The latter satellite peak cannot be used since it obviously overlaps with the Mn 2p1/2 peak. In our case, a strong satellite structure is observed at 646.2 eV, showing a large presence of MnO even if we cannot rule out the presence of more oxidised species.
Finally, both HRTEM and XPS analyses demonstrate that the NPs formed by the decomposition of Mn(tBuCH2)2 under H2 in C1C4ImNTf2 are oxidised, and are mostly MnO-NPs. This is quite surprising, considering that all operations were conducted under inert or reducing atmospheres, in the strict absence of water and oxygen. In addition, the probable hydrogenolysis of neoC5 observed during synthesis (vide supra) suggests that metallic Mn was formed.20 If this hypothesis is correct, it would mean that metallic Mn is not stable in our IL. Similar results have been reported in the case of Fe-based NPs elaborated under very comparable conditions (decomposition under H2 in the same IL). A systematic oxidation of the NPs into FeO was observed, attributed to the presence of residual water in this hygroscopic IL.24 Indeed, both Fe and Mn are known to be readily oxidised by water.25 The formation of MnO instead of Mn-NPs contrasts with the PVD-based process, which produces a metallic Cu(Mn) deposit. The impact of this Mn oxide on the contact resistance at the bottom of TSVs will have to be assessed. Nevertheless, its presence on the TSV sidewalls is expected to have no negative consequences considering that (i) this oxidised layer will have a limited thickness compared to the typical TSV diameter (leading to no measurable increase in TSV resistance), and (ii) the formation of the barrier layer upon thermal treatment requires the oxidation of Mn anyway.26
In order to further investigate the kinetics of MnO-NP nucleation and growth, the same solution was treated at 100 °C under 0.4 MPa H2 for 1 h to 48 h. The corresponding size distribution profiles are shown in Fig. 3. Unexpectedly, smaller NPs with a narrower size dispersion were observed as the reaction duration increased.
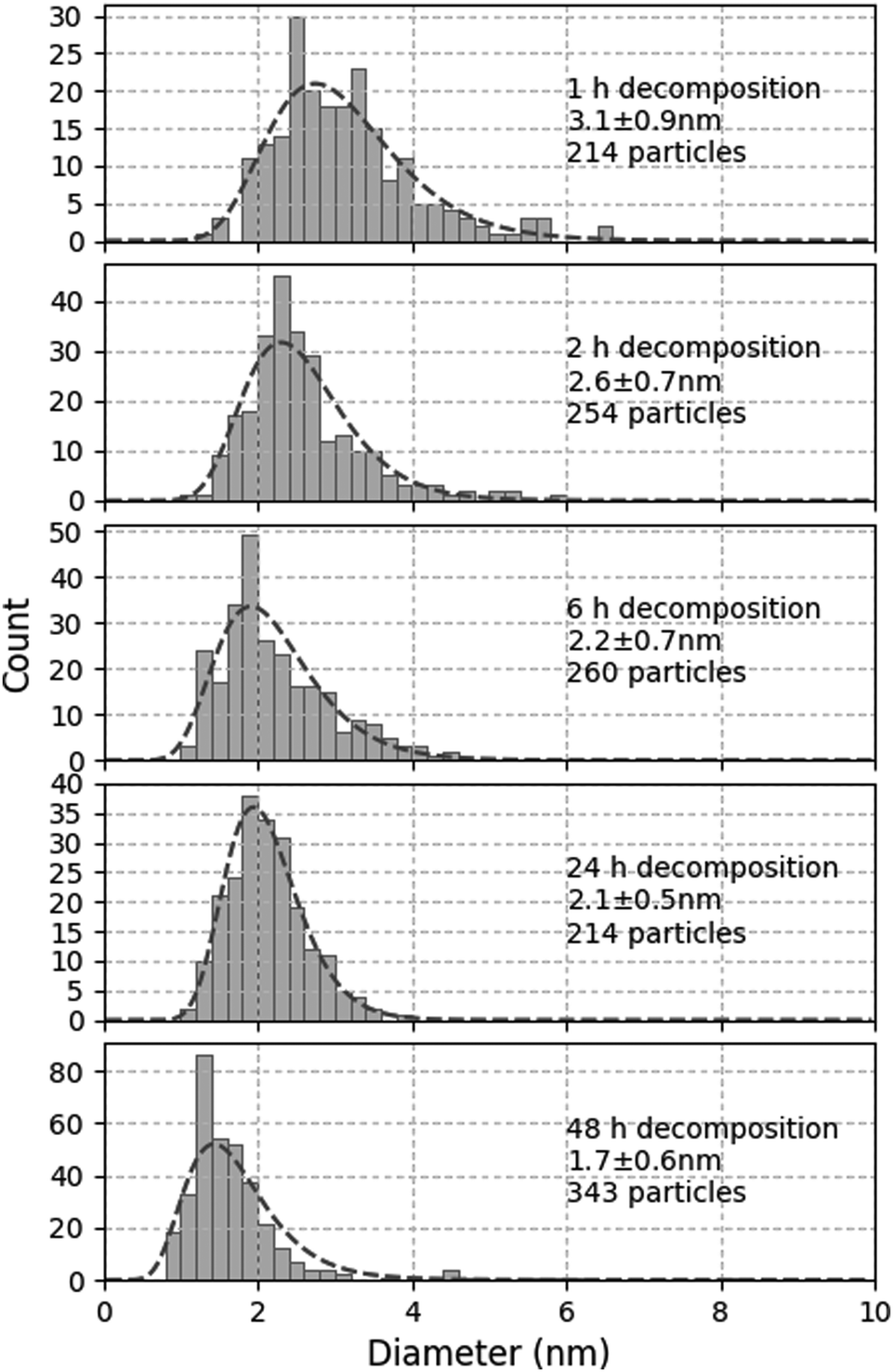 | ||
| Fig. 3 Evolution of size distribution of MnO-NPs generated from Mn(tBuCH2)2 in C1C4ImNTf2 under 0.4 MPa H2 at 100 °C for 1, 2, 6, 24 and 48 h. | ||
In the ideal case, this synthesis should produce monodisperse NPs by instantaneous nucleation followed by the growth of the metal, as described by LaMer.27 This mechanism supposes that the decomposition reaction is fast and provides atoms for the nucleation of the metal. Nucleation is only active for a short time in the early stages of the precipitation, when the concentration of atoms is high enough. When nuclei are formed, this concentration becomes too low to sustain further nucleation and all metallic nuclei continue to grow at the same pace, affording monodisperse NPs. Any deviation from this model is expected to deteriorate the size distribution.28 For instance, if the nucleation and growth steps are not well separated, a so-called progressive nucleation regime is established that leads to a wider size distribution. However, even in this situation, the average size of the NPs would increase, rather than decrease, with increasing synthesis duration. Similarly, any aggregative process during synthesis is expected to lead to larger NPs.
By contrast, any uncontrolled aggregation or growth mechanism occurring after the synthesis itself could explain this observation. To verify this hypothesis, the long term evolution of a suspension synthesised at 100 °C under 0.4 MPa H2 for 1 h only was monitored. After 1 month under Ar, the size distribution remained unchanged at 3.1 ± 0.8 nm (to be compared to the first graph in Fig. 3). Therefore, this suspension was stable upon storage under an inert atmosphere at room temperature. By contrast, when this suspension was exposed to 0.4 MPa H2 for 23 h at 100 °C, the size of the NPs increased to 4.2 ± 1.2 nm. Therefore, the suspensions resulting from incomplete synthesis are not stable under the conditions of synthesis, and the initially formed NPs continue to grow.
Considering that 24 h is required to completely decompose Mn(tBuCH2)2 (vide supra), unreacted precursor must remain after the 1 h reaction. This unreacted compound is stable under Ar (the NP size remains unchanged), but decomposes under H2, as expected. The fact that the NP size increased during the complement 23 h reaction suggests that this secondary decomposition preferentially caused the growth of existing MnO-NPs rather than a new nucleation. Such a behaviour would be consistent with the previously reported slow nucleation but fast NP growth by autocatalytic decomposition of metal precursors under H2.29 Such a reaction could in principle occur in the medium during cooling down the autoclave after incomplete decomposition of the OM precursor. This secondary reaction would occur at uncontrolled and decreasing temperatures, which would explain not only the overall increase in NP size, but also the wider size distribution.
To avoid this secondary reaction, it was thus necessary to consume all the Mn(tBuCH2)2 during the synthesis, which required 24 h under the conditions tested so far. In order to accelerate this synthesis and improve size distribution, further experiments were conducted under higher H2 pressure, namely 0.9 MPa. After 1 h at 100 °C, smaller NPs were obtained (2.4 ± 0.8 nm), comparable to those obtained after 24 h at lower H2 pressure. This suggests that increasing the H2 pressure indeed accelerates the reaction. In turn, this indicates that the mass transport of H2 within the liquid phase probably contributes to limiting the reaction rate. After 4 h (which corresponds to the typical duration for Cu-NP synthesis), the result was the same (2.4 ± 0.8 nm, see Fig. 4a). However, when the reaction was continued for 48 h, rod-shaped objects were formed, with a typical size of several tens of nm, likely due to the extended coalescence of the MnO-NPs under these conditions.
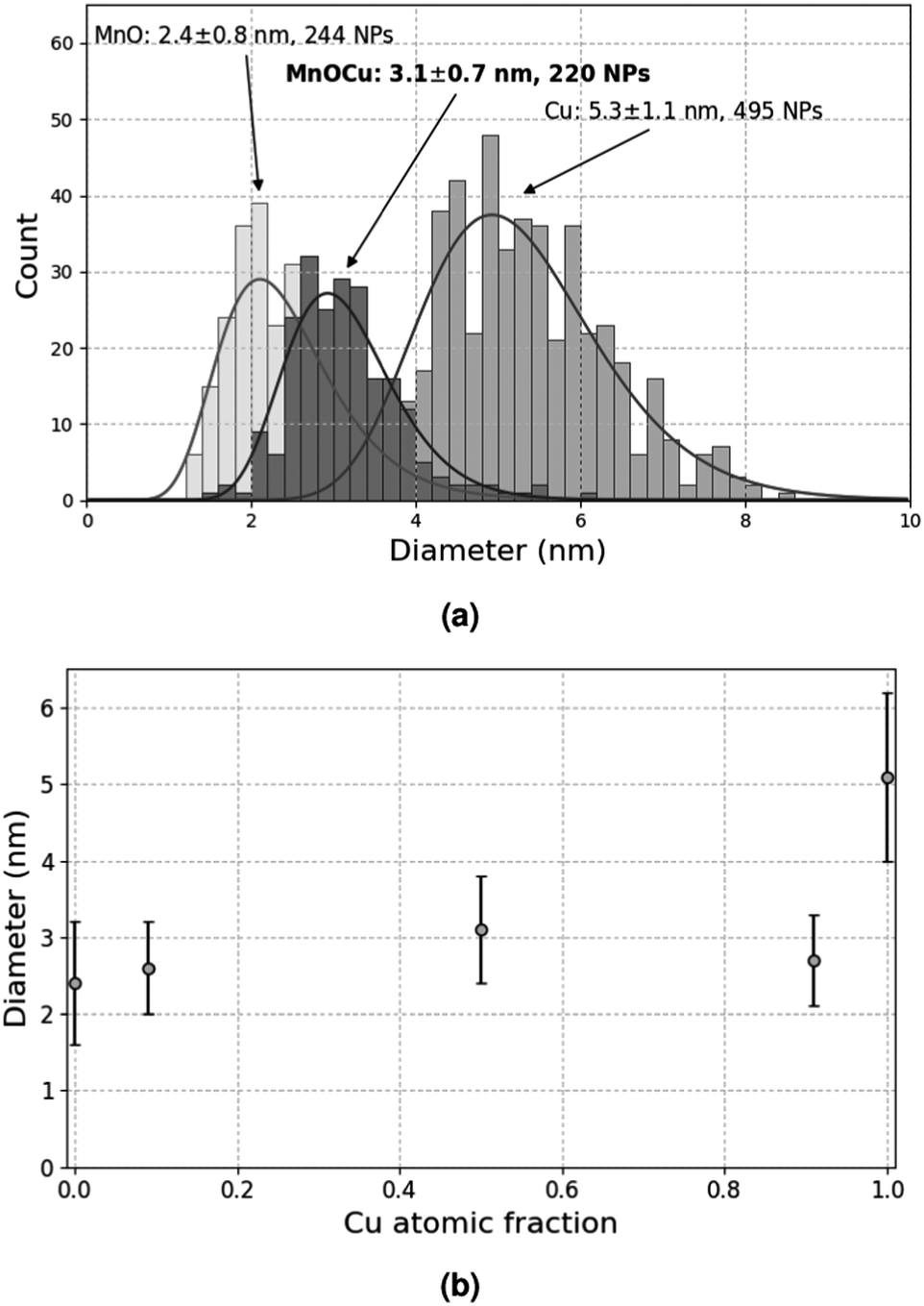 | ||
| Fig. 4 (a) Comparison of size distributions of Cu-, MnO- and MnO–Cu-NPs (Cu molar fraction = 0.5) formed in C1C4ImNTf2 under 0.9 MPa H2 at 100 °C for 4 h. (b) Size evolution of MnO–Cu-NPs with Cu molar fraction. | ||
4 Elaboration of MnO–Cu-NPs
In an attempt to co-precipitate MnO and Cu as mixed NPs, two solutions of 5 × 10−2 mol L−1 CuMes and 5 × 10−2 mol L−1 Mn(tBuCH2)2 were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio, and the mixture was decomposed under 0.9 MPa H2 at 100 °C for 4 h. This experiment afforded well dispersed NPs with only one population at 3.1 ± 0.7 nm, which is an intermediate value between pure Cu and pure MnO (Fig. 4a). Even more interestingly, this observation holds for various Cu:Mn volume ratios corresponding to Cu molar fractions ranging from 0.09 to 0.91 (Fig. 4b). These results strongly suggest that these NPs contain both elements: otherwise, two populations centered around 2.4 nm and 5.3 nm would be expected, corresponding to separate MnO- and Cu-NPs. Indeed, the same behaviour was observed and extensively studied in the Cu-Ru system, with the production of monodisperse NPs composed of a Ru core and a Cu shell.12,30
:1 volume ratio, and the mixture was decomposed under 0.9 MPa H2 at 100 °C for 4 h. This experiment afforded well dispersed NPs with only one population at 3.1 ± 0.7 nm, which is an intermediate value between pure Cu and pure MnO (Fig. 4a). Even more interestingly, this observation holds for various Cu:Mn volume ratios corresponding to Cu molar fractions ranging from 0.09 to 0.91 (Fig. 4b). These results strongly suggest that these NPs contain both elements: otherwise, two populations centered around 2.4 nm and 5.3 nm would be expected, corresponding to separate MnO- and Cu-NPs. Indeed, the same behaviour was observed and extensively studied in the Cu-Ru system, with the production of monodisperse NPs composed of a Ru core and a Cu shell.12,30
To further characterise these objects, a HRTEM analysis was undertaken. It indicates that the aggregates are crystalline (Fig. S4, ESI†). Surprisingly, the Fourier pattern corresponding to the NP could not be indexed considering the cubic structures of the metals. By contrast, a quite good agreement was found considering a tetragonal crystal structure with lattice parameters a = 5.95 Å and c = 9.40 Å (Table S2, ESI†). This structure is similar to either the Mn3O4 or Cu4O3 crystal structures.21,31 This could indicate that this particular NP is made of one of these two phases. It could also be a mixed phase between these two oxides. In all cases, if this interpretation is correct, it would mean that Cu is oxidised in these NPs, and/or Mn is more oxidised as well.
To verify this, the sample was also characterised by using XPS and XAES spectroscopies. Concerning copper, the 2p3/2 line was measured at 932.6 eV by XPS, whereas the Cu LMM peak was found to be very broad with a maximum detected in the low energy range around 914.9 eV by XAES (Fig. S6, ESI† and Fig. 5b). The large shift of the Cu LMM line to lower binding energy with respect to Cu metal indicates the presence of Cu2O species only,32 as also shown by the corresponding data in the Wagner plot in Fig. 5b. As far as the Mn XPS response is concerned, no satellite structure was observed, indicating the absence of MnO (Fig. 5a). Moreover, the binding energy of the Mn 2p3/2 peak at 641.2 eV was shifted to higher binding energies with respect to the pure metal. This can be attributed to oxidised Mn III and IV species. Hence, all these spectroscopic measurements confirm that Cu is oxidised, and that Mn also adopts a higher oxidation state in these NPs produced upon decomposing simultaneously both Cu and Mn precursors. Considering that Cu(0) and Mn(II) were observed for the individual metals, this result is surprising and certainly deserves further investigation, as it suggests that together, the two metals are more sensitive to oxidation than separately.
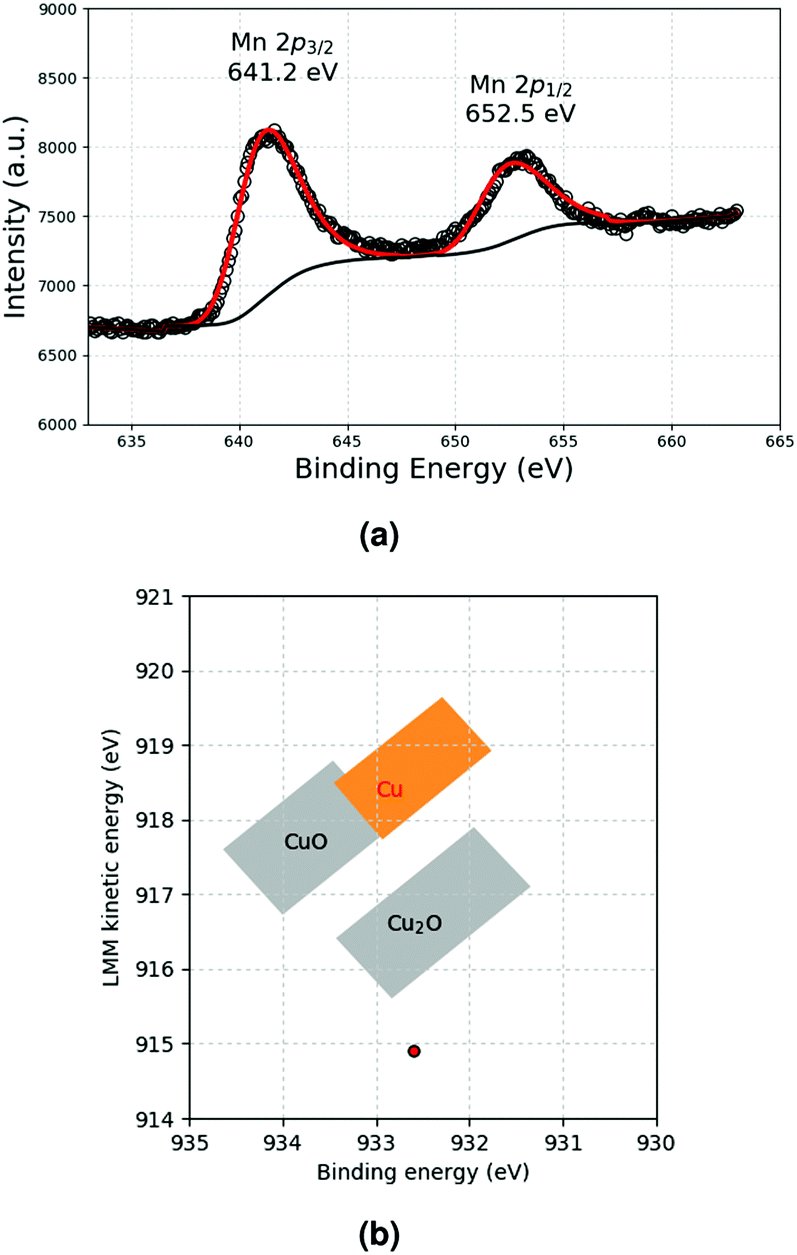 | ||
| Fig. 5 (a) Mn 2p XPS response and (b) Cu Wagner plot recorded for MnO–Cu-NPs. | ||
5 Formation of thin films
The conversion of these NPs into thin films was also attempted in this study. For this purpose, Si/SiO2 substrates were coated with fresh suspensions and subjected to a thermal treatment. As C1C4ImNTf2 is reputed to be thermally stable up to 350 °C,33 the experiments were conducted at 250 °C for 1 h. To mitigate the possible oxidation of the metals during sample preparation in air, these treatments were performed under a N2/H2 atmosphere. In all cases, solid deposits were formed, although with different morphologies (as observed by SEM, Fig. 6). Their elemental composition was analysed by using EDX (Fig. S8–S10, ESI†). In the case of Cu, the metal agglomerated on the surface into clusters that were significantly larger than the initial Cu-NPs (Fig. 6a). MnO afforded smoother but still discontinuous deposits in the form of flat, large islands (Fig. 6b). Finally, the MnO–Cu deposit exhibited an intermediate morphology, with the formation of smaller Cu agglomerates (Fig. 6c). Interestingly enough, the simultaneous presence of Cu and Mn was detected by EDX outside these Cu clusters (Fig. S10, ESI†). | ||
| Fig. 6 Top view SEM pictures of (a) Cu, (b) MnO and (c) MnO–Cu deposits on silica formed by in situ thermal treatment of corresponding suspensions in IL at 250 °C for 1 h under N2/H2. | ||
The chemical composition of the metallic deposits was examined by using XPS (Table 1). As expected and in agreement with the EDX results, Mn and Cu were only detected where expected (i.e. in the corresponding single deposit or in the mixture). In addition, F, N and S were detected, which stem from the remaining IL. Before going into the detailed discussion of the XPS peaks of the various elements, interesting observations can be made. First, the ratio between Cu (resp. Mn) and Si varies in the different samples. Upon comparing pure MnO and pure Cu deposits, the atomic ratio Mn/Si is equal to 0.11 while Cu/Si is equal to 0.03. This effect can be ascribed to the different morphologies of the two deposits: the Cu agglomerates cover less of the oxide surface than the flat MnO islands. For the MnO–Cu deposit, the Mn/Si ratio is essentially the same, but an even higher content of Cu is obtained (0.06) as compared to pure Cu. This suggests that Cu is better distributed over the sample surface, probably also incorporated into the flat areas. Hence, our results indicate that during thermal treatment, MnO–Cu-NPs are sintered into a discontinuous solid film. However, in the same time, partial segregation of both elements could occur, accounting for Cu agglomeration (which is observed in the case of pure Cu). For practical purposes, this agglomeration must be prevented. Suppressing this agglomeration would require a separate study that goes beyond the scope of the present article.
| Elements (at%) | Ratios | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ionic liquid | Deposit | Substrate | Cu/Si | Mn/Si | Si(III)/Si 2p | ||||||
| C 1s | O 1s | F 1s | N 1s | S 2p | Mn 2p3/2 | Cu 2p3/2 | Si 2p | ||||
| <dl: below detection limit. | |||||||||||
| Cu | 40.1 | 39.6 | 0.8 | 0.5 | 0.7 | <dl | 0.5 | 17.8 | 0.03 | 0.35 | |
| MnO | 48.8 | 30.4 | 2.0 | 1.0 | 0.5 | 1.7 | <dl | 15.6 | 0.11 | 0.70 | |
| MnO–Cu | 34.9 | 41.7 | 2.0 | 0.5 | 0.2 | 2.0 | 1.0 | 17.7 | 0.06 | 0.11 | 0.46 |
The XPS reponse of Mn and the Wagner plot of Cu in MnO and Cu-based films, respectively, are shown in Fig. 7a and c, respectively. For Cu, the Cu 2p3/2 peak (932.5 eV) and LMM band (914.2 eV) correspond to Cu2O (Fig. S5, ESI†). This oxidation is most probably related to the storage of the samples in air after film formation and before XPS analysis. For the MnO-based film, the structure of the Mn 2p peaks is preserved, and the satellite characteristics of Mn(II) are still present. This indicates that the MnO deposit is stable under air exposure. In the MnO–Cu deposit, MnO (Fig. 7b) and Cu2O (Cu 2p3/2 peak at 932.6 eV and LMM band at 915.5 eV, Fig. S7, ESI† and Fig. 7d) are detected. The observation of MnO is surprising, considering that a higher oxide was observed in the initial suspension. This could be ascribed to a reduction of this oxide under the forming gas atmosphere used during annealing.
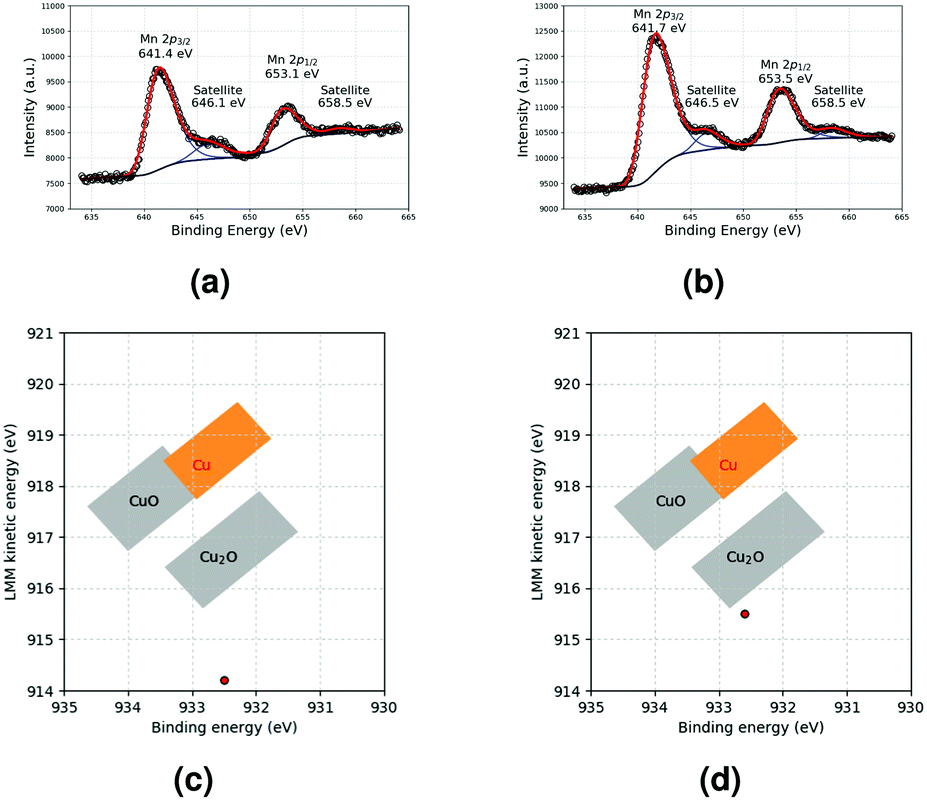 | ||
| Fig. 7 Mn 2p XPS response of (a) MnO and (b) MnO–Cu deposits and Wagner plots of Cu in (c) Cu and (d) MnO–Cu deposits. | ||
Finally, the Si 2p peak in the various samples provides interesting information. Indeed, two components could be resolved from this peak (Fig. S11, ESI†). For SiO2, the only expected contribution is Si(IV) (or Q4 species) between 103.4 and 103.7 eV.34,35 In our samples, an additional peak is detected between 102.1 and 102.3 eV. This contribution can be interpreted as Si(III)34 species in SiO2. The important observation is that the ratio between these peaks is different in the various samples. In the case of pure Cu, the ratio corresponds to about 65% Si(IV) and 35% Si(III). With the MnO–Cu deposit, the amount of Si(III) reaches 46%. With MnO only, an even higher contribution of Si(III) of 70% is recorded. This is strong evidence that some reaction occurred between MnO and the underlying SiO2 layer during thermal treatment at 250 °C. This result is in good agreement with the formation of a MnSiO3 phase already reported by others.26
6 Conclusions
The deposition and sintering of NPs containing both Cu and Mn could be an economically and technically interesting alternative to current processes of barrier and copper seed layer deposition in TSVs. Indeed, this approach would remove one process step (barrier deposition) and use less expensive equipment as it does not require high vacuum conditions. To achieve this goal, we propose to (i) synthesise suspensions of suitable NPs in ILs and (ii) spread and anneal those suspensions directly on technological substrates. This approach is made possible by two key properties of ILs: their ability to stabilise NPs (whose surface is not contaminated by ligands), and their good thermal stability.1-Butyl-3-methylimidazolium bistrifluoromethylsulphonylimide (C1C4ImNTf2) was the IL selected for this study. It is well suited for the controlled synthesis of Cu-NPs. In this work, it was demonstrated by combined XPS and Auger analyses that these NPs are constituted by metallic Cu. A major achievement of this study is the selection of an appropriate OM precursor for the synthesis of Mn-based NPs. The decomposition of solutions of Mn(tBuCH2)2 in C1C4ImNTf2 was extensively studied and suitable conditions were found to produce NPs in the desired size range (about 2.5 nm in diameter). However, XPS analysis showed that these NPs are not metallic, but rather MnO-NPs. It is probable that metallic Mn is formed after the decomposition of Mn(tBuCH2)2, but it is not stable and it is possibly oxidised by residual water in the IL.
An important observation is that upon decomposition of mixed solutions of CuMes and Mn(tBuCH2)2, single populations of NPs were obtained in a broad Cu/Mn composition range. This is a good indication that both elements were incorporated together in these NPs. Surprisingly, both metals were found to be oxidised (Cu(I) and Mn(III) or (IV)). Based on the observation of stable Cu and MnO-NPs in the separate suspensions, this oxidation can only be ascribed to unwanted exposure to air after synthesis and prior to analysis.
Finally, these suspensions were spread onto Si samples coated with SiO2, typical of TSV technology. MnO suspensions afforded flat deposits, whereas Cu preferentially formed agglomerates. An intermediate morphology was obtained for the MnO–Cu-NPs. Again, XPS analysis revealed that copper was oxidised in the films, certainly due to the storage of the samples in air. By contrast, MnO remained stable under the same conditions. Interestingly enough, a partial reduction of SiO2 was measured by XPS for MnO-containing films, in agreement with the formation of a MnSiO3 phase reported in other studies on self-formed barriers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed within the Plateforme Nanochimie between CEA Grenoble and CPE Lyon, in collaboration with IRCELYON.Notes and references
- P. H. Haumesser, S. Maitrejean, A. Roule and G. Passemard, Fabtech, 2006, 29, 108–114 Search PubMed.
- M. G. Farooq, ECS Trans., 2011, 35, 83–94 CrossRef CAS.
- J. Koike, M. Wada, S. Takahashi, N. Shimizu, H. Shibata, S. Nishikawa, T. Usui, H. Nasu and M. Yoshimaru, US7304384 B2, 2007.
- C. Barrière, G. Alcaraz, O. Margeat, P. Fau, J. Quoirin, C. Anceau and B. Chaudret, J. Mater. Chem., 2008, 18, 3084–3086 RSC.
- Y. Ham, D. Kim, K. Baek, K. Park, M. Kim, K. Kwon, K. Lee and L. Do, Electrochem. Solid-State Lett., 2012, 15, H145–H147 CrossRef CAS.
- W. Darwich, P.-H. Haumesser, C. C. Santini and F. Gaillard, Int. J. Mol. Sci., 2016, 17, 876 CrossRef PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, John Wiley & Sons, Weinheim, 2008 Search PubMed.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC.
- P. Campbell, M. Prechtl, C. C. Santini and P.-H. Haumesser, Curr. Org. Chem., 2013, 17, 414–429 CrossRef CAS.
- T. Gutel, C. C. Santini, K. Philippot, A. Padua, K. Pelzer, B. Chaudret, Y. Chauvin and J.-M. Basset, J. Mater. Chem., 2009, 19, 3624–3631 RSC.
- P. Arquillière, P.-H. Haumesser and C. C. Santini, Microelectron. Eng., 2012, 92, 149–151 CrossRef.
- I. S. Helgadottir, G. Freychet, P. Arquillière, M. Maret, P. Gergaud, P.-H. Haumesser and C. C. Santini, Nanoscale, 2014, 6, 14856–14862 RSC.
- L. Magna, Y. Chauvin, G. P. Niccolai and J.-M. Basset, Organometallics, 2003, 22, 4418–4425 CrossRef CAS.
- R. A. Andersen, E. Carmona-Guzman, J. F. Gibson and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1976, 2204–2211 RSC.
- C. D. Wagner and A. Joshi, J. Electron Spectrosc. Relat. Phenom., 1988, 47, 283–313 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- G. Moretti, Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, IM Publications, Chichester, 2003, p. 501 Search PubMed.
- G. Moretti, Handbook of Heterogeneous Catalysis, John Wiley & Sons, Ltd, 1997, p. 632 Search PubMed.
- D. Marquardt, Z. Xie, A. Taubert, R. Thomann and C. Janiak, Dalton Trans., 2011, 40, 8290–8293 RSC.
- A. Clearfield, J. Basset, B. Gates, J. Candy, A. Choplin, M. Leconte, F. Quignard and C. Santini, Surface Organometallic Chemistry: Molecular Approaches to Surface Catalysis, NATO ASI, Kluwer, Dordrecht, 1988, pp. 271–298 Search PubMed.
- C. R. Ross, D. C. Rubie and E. Paris, Am. Mineral., 1990, 75, 1249–1252 CAS.
- J. M. Cerrato, M. F. Hochella, W. R. Knocke, A. M. Dietrich and T. F. Cromer, Environ. Sci. Technol., 2010, 44, 5881–5886 CrossRef CAS PubMed.
- V. Di Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117–123 CrossRef CAS.
- B. C. Leal, J. D. Scholten, M. C. M. Alves, J. Morais, I. de Pedro, L. F. Barquin and J. Dupont, Inorg. Chem., 2016, 55, 865–870 CrossRef CAS.
- M. Pourbaix, Atlas d'équilibres Électrochimiques, Gauthier-Villars & Cie, Paris, 1963 Search PubMed.
- J. M. Ablett, J. C. Woicik, Z. Tökei, S. List and E. Dimasi, Appl. Phys. Lett., 2009, 94, 042112 CrossRef.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- P.-H. Haumesser, Nucleation and Growth of Metals, Elsevier, Oxford, 2016, pp. 59–70 Search PubMed.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- I. S. Helgadottir, P. P. Arquillière, P. Brea, C. C. Santini, P.-H. Haumesser, K. Richter, A.-V. Mudring and M. Aouine, Microelectron. Eng., 2013, 107, 229–232 CrossRef CAS.
- M. O'Keeffe and J. O. Bovin, Am. Mineral., 1978, 63, 180–185 Search PubMed.
- P. E. Larson, J. Electron Spectrosc. Relat. Phenom., 1974, 4, 213–218 CrossRef CAS.
- L. Chancelier, A. O. Diallo, C. C. Santini, G. Marlair, T. Gutel, S. Mailley and C. Len, Phys. Chem. Chem. Phys., 2014, 16, 1967–1976 RSC.
- F. J. Himpsel, F. R. McFeely, A. Taleb-Ibrahimi, J. A. Yarmoff and G. Hollinger, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 6084–6096 CrossRef CAS PubMed.
- R. Sawyer, H. W. Nesbitt and R. A. Secco, J. Non-Cryst. Solids, 2012, 358, 290–302 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Monitoring of the decomposition reaction, HRTEM analyses of the produced NPs and detailed XPS analysis of the Si 2p peak of the substrates. See DOI: 10.1039/c9nj03842f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |