DOI:
10.1039/C9NJ03663F
(Paper)
New J. Chem., 2020,
44, 239-257
Design, synthesis, characterization and evaluation of the anticancer activity of water-soluble half-sandwich ruthenium(II) arene halido complexes†
Received
15th July 2019
, Accepted 18th November 2019
First published on 18th November 2019
Abstract
The synthesis, detailed characterization, and investigation of the DNA- and BSA-binding affinity and cytotoxicity of three half-sandwich Ru(II) arene chlorido complexes (1–3) are reported. Specifically, organometallic complexes [(η6-p-cymene)(L1)RuCl]Cl (1), [(η6-p-cymene)(L2)RuCl]Cl (2) and [(η6-p-cymene)(L3)RuCl]Cl (3) [L1 = 2-(1H-benzo[d]imidazol-2-yl)quinolone, L2 = 2-(quinolin-2-yl)benzo[d]oxazole, and L3 = 2-(quinolin-2-yl)benzo[d]thiazole] were synthesized using [((η6-p-cymene)Ru(μ-Cl)Cl)2] and L1–L3. All the complexes were characterized by various spectroscopic and analytical methods. Complex 1 was also characterized by single crystal X-ray diffraction analysis, the results of which are in-line with the piano-stool structure elucidated from the spectroscopic methods. Cyclic voltammetry of 1–3 showed good stability of the Ru(II) state. The interactions of 1–3 with CT-DNA were investigated by UV-Vis studies and competitive binding with ethidium bromide (EthBr) using emission spectroscopy. Steady-state fluorescence quenching and synchronous fluorescence studies were performed to understand the interactions between 1–3 and bovine serum albumin (BSA). The geometry optimization of 1–3 was performed using the density functional theory (DFT). Complexes 1 and 2 were investigated for DNA binding using theoretical calculations. Molecule complexes 1-DNA and 2-DNA were huge (482–483 atoms, 2442 electrons) for any level of quantum chemical analysis. The semi-empirical PM6 method, which is more accurate than the force-field methods, was used, and the result suggested that several strong and weak electrostatic and intermolecular hydrogen bonding interactions exist between the outer edge of DNA and complexes 1 and 2. In addition, the in vitro cytotoxicity of L1–L3 and complexes 1–3 against a breast cancer cell line (MCF-7) was investigated by MTT cytotoxicity analyses. The studies performed here show that these complexes exhibit a good binding behavior with DNA and BSA and show good anticancer potency towards MCF-7 cells.
1. Introduction
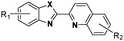
Increased cellular proliferation due to mutations at the genetic level causes cancer. Since most of the patients are diagnosed at an advanced stage and with distant metastasis, chemotherapy is one of the major accessible and effective tools for treatment.1–5 Currently, metal-based chemotherapeutics are an engrossing option with effective and emerging possibilities.6–9 The most conspicuously used chemotherapeutic agent for antiblastic chemotherapy is cisplatin, the biological potential of which was identified by Rosenberg et al. in 1964.10 Since the time cisplatin was discovered, both in vitro and in vivo studies on metal-based complexes have increased drastically.2,6,11–17 The outcome of this was observed in the form of new platinum-based complexes, viz., carboplatin,18 oxaliplatin19 and nedaplatin.20 Cisplatin has its limitations, such as nephrotoxicity, neurotoxicity and problems due to the resistance (natural or acquired) developed by many tumours.21,22
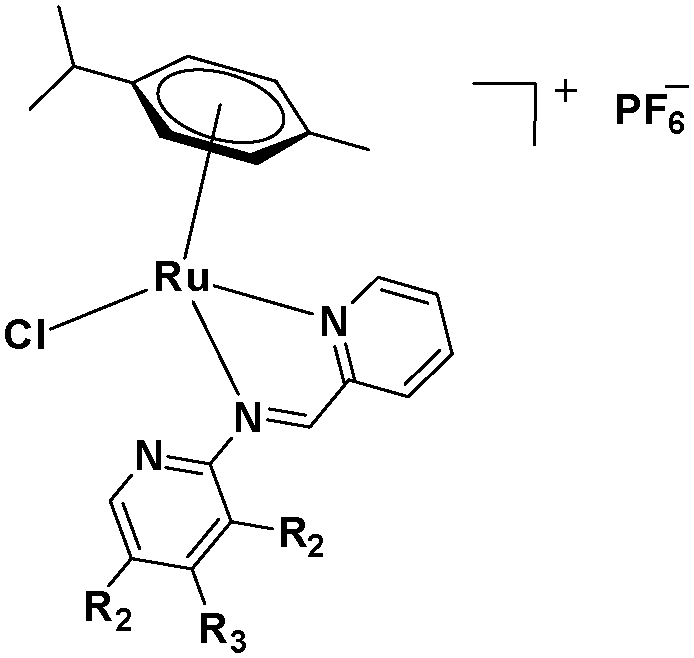
The challenge ahead was, therefore, to develop new and more effective metal-based anticancer agents. A wide range of small molecules and transition metal-based therapeutic agents were developed subsequently.13–16,23–25 Among these potential therapeutic agents, Ru-based complexes are emerging as attractive drugs with the most promising anticancer activities. Ru-based complexes are very special due to their variable oxidation states, optimum ligand exchange rates, low general toxicity and high selective cytotoxicity towards proliferative cells, water solubility and octahedral geometry that enables the tuning of their electronic and steric properties.4,5,16,22,26–28 Some of the Ru-based complexes like [imiH]trans-[Ru(N-imi)(S-DMSO)Cl4] (NAMI-A), [indH]trans-[Ru(N-ind)2Cl4] (KP1019) and [Na]trans-[Ru(N-ind)2Cl4] (NKP-1339) have reached the clinical trial stage (Scheme 1).1,29–31
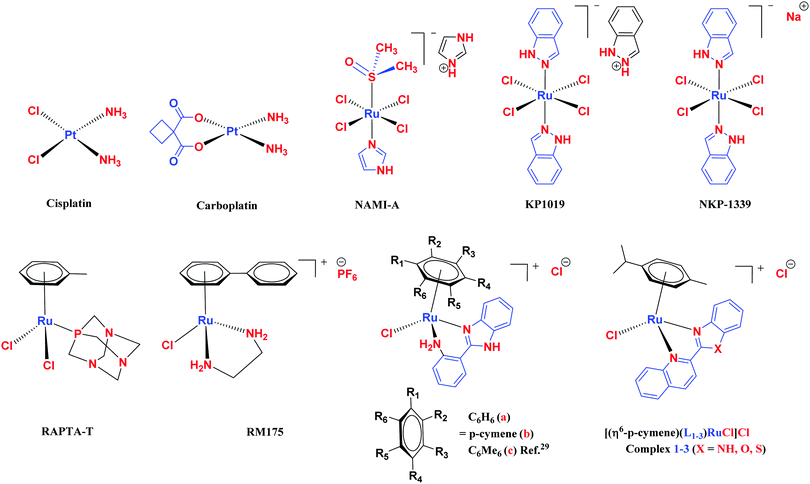 |
| | Scheme 1 Chemical structures of reported metal-based chemotherapeutic agents and the complexes used in this work 1–3. | |
Ruthenium arene complexes, also known as piano-stool or half-sandwich complexes, have been shown to be very promising drugs towards a wide spectrum of cancer types (breast, colon, lung, and pancreatic cancer cells).5,32–37 The design of these complexes holds amphiphilic properties imparted by the hydrophilic metal center and the hydrophobic arene moiety.3–5
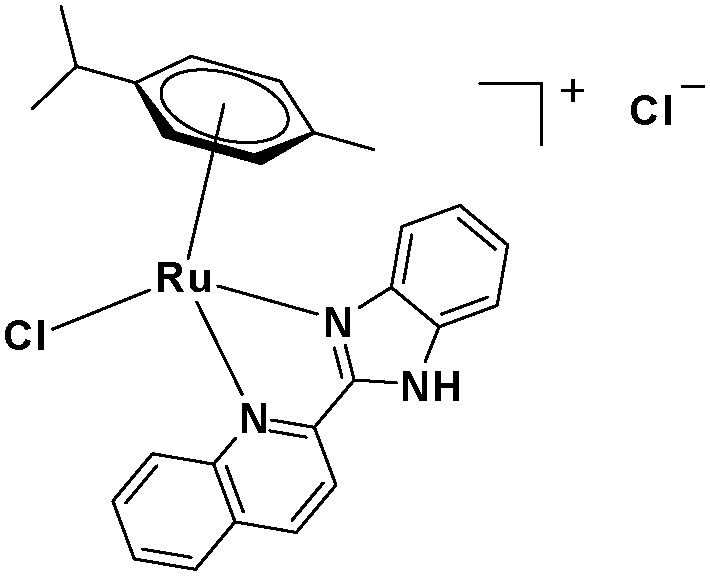
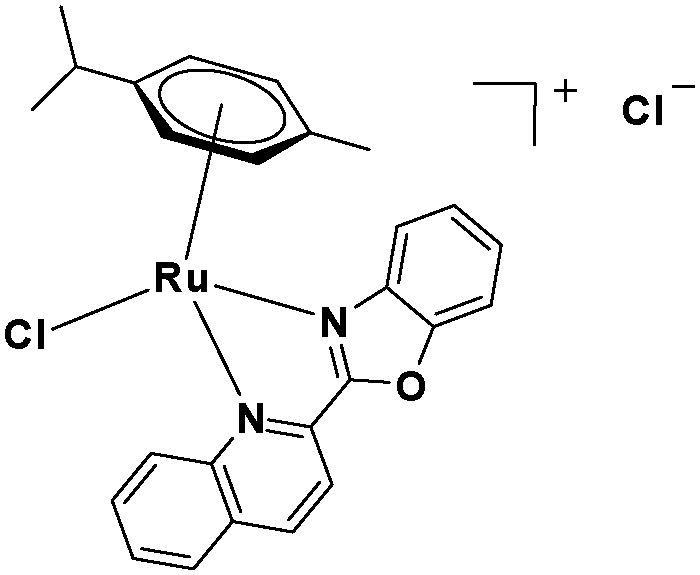
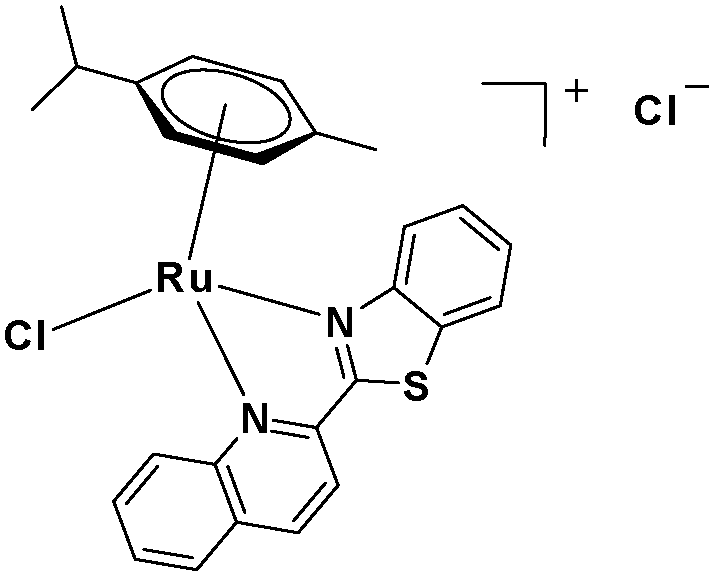
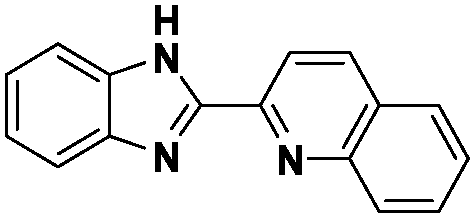
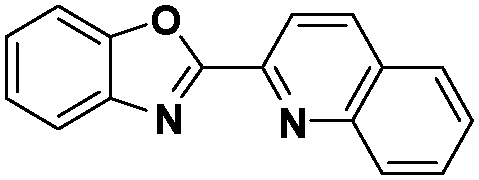
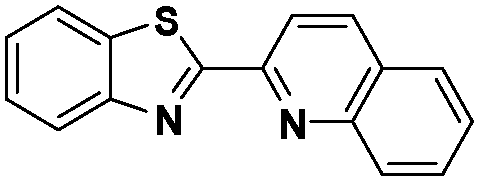
In the design of Ru-arene anticancer drugs, the nature of the arene part,38–41 the coordinating halide group38,42 and the bidentate chelating ligand39,40,43 plays a pivotal role in the observed biological implications.23,38,44,45 The properties of such complexes can be altered by a modification in any of these three parts, especially by varying the nature of the bidentate chelating ligand. In this context, several studies have shown the effect of quinoline27,45,46 and benzimidazole/benzoxazole/benzothiazole47–50 moieties, which are known to possess various pharmacological effects. To the best of our knowledge, there is no report on the design and use of adducts of these moieties and the biological applications of such complexes. In order to study the combined effect of the adduct of quinoline and benzazole, three bidentate ligands, namely 2-(1H-benzo[d]imidazol-2-yl)quinoline (L1), 2-(quinolin-2-yl)benzo[d]oxazole (L2) and 2-(quinolin-2-yl)benzo[d]thiazole (L3), were chosen for the preparation of the Ru-arene complexes [(η6-p-cymene)(L1)RuCl]Cl (1), [(η6-p-cymene)(L2)RuCl]Cl (2) and [(η6-p-cymene)(L3)RuCl]Cl (3), respectively (Scheme 1). The characterization of these complexes was accomplished by various spectroscopic and analytical methods. The 1H–1H COSY NMR spectra helped in assigning the chemical shifts and the structure elucidation of 1–3. The crystal structure of 1 confirmed the elucidated structure obtained by spectroscopic and analytical methods. The electrochemical behavior and stability of the Ru(II) state in these complexes was established by cyclic voltammetry. For a complete understanding of the complexes and their interactions, state-of-the-art theoretical studies have been performed. The development of new ab initio methods in the formative years along with the increasing computing power has progressively turned quantum chemistry into an accurate predictive tool for molecular systems involving light elements. However, the situation is more challenging for systems containing transition metals, where inappropriate methods and basis sets may lead to deceptive results. To this end, theoretical calculations have been reported based on the well-established DFT for the understanding of the structural features of 1–3, as well as their interactions with the model DNA. The binding affinity of 1–3 towards CT-DNA and BSA, along with the cytotoxic studies, has also been demonstrated here. The in vitro cytotoxic studies of 1–3 against breast cancer (MCF-7) cells by the MTT assay demonstrated the good cytotoxic activity of 1–3. Complex 1 was found to have the best anti-cancer properties among the three complexes reported here, and a rationale for the observed trend is discussed.
2. Experimental section
2.1 Materials used
All common chemicals and solvents were purchased from Sigma Aldrich, Thermo-Fisher Scientific, Molychem and CDH, India and used without further purification. Solvents used for spectroscopic analyses were purified and dried by standard methods.51,52 Metal chloride (RuCl3·3H2O), α-terpinene, quinaldine, 2-amino phenol, 2-amino thiophenol, o-phenylene diamine, ethidium bromide (EthBr), Tris–HCl buffer, phosphate buffer saline (PBS), calf thymus-deoxyribonucleic acid (CT-DNA) and bovine serum albumin (BSA) were procured from Sigma Aldrich and TCI, India. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma-Aldrich, MO, USA.
2.2 Physical measurements
The melting point was measured using the Büchi Melting Point B-540 apparatus. Infrared spectra were recorded at room temperature using a PerkinElmer FTIR spectrometer in the 4000–400 cm−1 range. Conductivity measurements were carried out using the Systronics Conductivity Meter 304. Elemental analysis was performed on a Thermo Scientific Flash 2000 elemental analyzer. The 1H, 13C and COSY NMR spectra were recorded in DMSO-d6 and CDCl3 solvents on a Bruker Avance III-500 MHz spectrometer at 298 K. UV-Vis spectrophotometric studies were performed at room temperature on an Agilent Technologies Cary 60 UV-Vis spectrophotometer. ESI-mass spectra were recorded on a Thermo Scientific Exactive Plus mass spectrometer. Fluorescence studies were conducted using an Agilent Cary Eclipse fluorescence spectrophotometer at 298 K. Crystallographic data was collected on a Bruker kappa APEX-II CCD diffractometer at 293(2) K using graphite monochromated Mo Kα radiation (λ = 0.71073 Å).
2.3 Syntheses and characterization
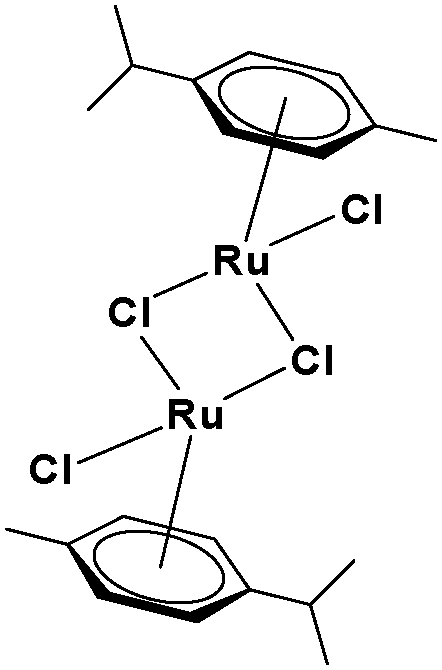
The ligands 2-(1H-benzo[d]imidazol-2-yl)quinoline (L1), 2-(quinolin-2-yl)benzo[d]oxazole (L2) and 2-(quinolin-2-yl)benzo[d]thiazole (L3) were synthesized using methods described in the literature.53,54 Ru(p-cymene)dimer [((η6-p-cymene)Ru(μ-Cl)Cl)2] was synthesized and purified according to a previously published method.55,56 Complex 1 was synthesized by a method different from the one reported in the literature.57
2.3.1 Synthesis and characterization of [(η6-p-cymene)(L1)RuCl]Cl·0.33CH2Cl2 (1).
To a solution of 2-(1H-benzo[d]imidazol-2-yl)quinoline (L1) (0.2 g, 0.816 mmol) in dry dichloromethane (30 mL), the Ru(p-cymene) dimer [((η6-p-cymene)Ru(μ-Cl)Cl)2] (0.25 g, 0.408 mmol) was added and stirred. This mixture was refluxed under nitrogen for 12 h. The reaction mixture was cooled to room temperature, and the solvent was removed under vacuum. The product was purified by silica gel (60–120 mesh) column chromatography (MeOH-DCM eluent system). Yellowish brown solid; yield = 87%. m.p. = 213 °C decomp. 1H NMR (500 MHz, DMSO-d6, δ ppm): 0.685 (3H, d, J = 6.0 Hz, –Ho1); 0.725 (3H, d, J = 6.5 Hz, –Ho2); 2.163–2.218 (1H, m, –Hn); 2.287 (3H, s, –Hk); 6.113 (1H, d, J = 5.5 Hz, –Hm1); 6.25 (1H, d, J = 6.0 Hz, –Hm2); 6.328 (2H, dd, J = 6.5 and 6.0 Hz, –Hl1+l2); 7.617–7.670 (2H, m, –H13,14); 7.897 (1H, d, J = 7.0 Hz, –H15); 7.969 (1H, t, J = 7.5 Hz, –H5); 8.166–8.198 (2H, m, –H4,12); 8.292 (1H, d, J = 7.5 Hz, –H6); 8.806 (2H, dd, J = 8.5 and 8.0 Hz, –H3,10); 8.992 (1H, d, J = 8.5 Hz, –H9). 13C NMR (125 MHz, DMSO-d6, δ ppm): 18.98 (–Ck), 21.62 (–Co), 22.12 (–Co), 30.76 (–Cn), 79.00, 84.09, 84.84, 85.72, 103.01, 105.19, 114.77, 118.52, 119.30, 125.82, 126.98, 129.33, 129.73, 129.85, 129.97, 133.74, 134.90, 141.95, 142.15, 148.17, 148.98, 150.49. Anal. calcd (%) for C26H25Cl2N3Ru·0.33CH2Cl2: C, 54.55; H, 4.46; N, 7.25. Found (%): C, 54.52; H, 4.71; N, 7.25. ESI-MS(+) in CH3OH (calcd, found, m/z) 516.0780, 516.0780, [M+ = 1 − Cl]. FTIR (KBr, cm−1) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (N–H) 3391.88, (C–H aromatic + aliphatic) 3047.88, 2963.68, 2909.89, 2860.51, (C
(N–H) 3391.88, (C–H aromatic + aliphatic) 3047.88, 2963.68, 2909.89, 2860.51, (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) 1595.95, (CC) 1476.34. UV-Vis: (in PBS (pH = 7.4)), λmax (nm) (λmax, dm3 mol−1 cm−1): 250 (3.12 × 104), 301 (1.96 × 104), 370 (2.27 × 104); (in DMSO
N) 1595.95, (CC) 1476.34. UV-Vis: (in PBS (pH = 7.4)), λmax (nm) (λmax, dm3 mol−1 cm−1): 250 (3.12 × 104), 301 (1.96 × 104), 370 (2.27 × 104); (in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBS (pH = 7.4) (1:10 v/v)) λmax (nm) (λmax, dm3 mol−1 cm−1): 258 (2.10 × 104), 305 (1.61 × 104), 371 (2.20 × 104).
:PBS (pH = 7.4) (1:10 v/v)) λmax (nm) (λmax, dm3 mol−1 cm−1): 258 (2.10 × 104), 305 (1.61 × 104), 371 (2.20 × 104).
2.3.2 Synthesis and characterization of [(η6-p-cymene)(L2)RuCl]Cl·0.5CH2Cl2·0.5CH3OH (2).
To a solution of 2-(quinolin-2-yl)benzo[d]oxazole (L2) (0.2 g, 0.816 mmol) in dry dichloromethane (30 mL), the Ru(p-cymene) dimer [((η6-p-cymene)Ru(μ-Cl)Cl)2] (0.25 g, 0.408 mmol) was added and stirred. This mixture was refluxed under nitrogen for 12 h. The reaction mixture was cooled to room temperature, and the solvent was removed under vacuum. The product was purified by silica gel (60–120 mesh) column chromatography (MeOH-DCM eluent system). Reddish brown solid; yield = 82%. m.p. = 110 °C decomp. 1H NMR (500 MHz, CDCl3, δ ppm): 0.971 (3H, d, J = 7.0 Hz, –Ho1); 1.019 (3H, d, J = 7 Hz, –Ho2); 2.247 (3H, s, –Hk); 2.562–2.617 (1H, m, –Hn); 6.135 (1H, d, J = 6.0 Hz, –Hm1); 6.263 (1H, d, J = 6.0 Hz, –Hm2); 6.314 (1H, d, J = 6.0 Hz, –Hl2); 6.397 (1H, d, J = 6.0 Hz, –Hl1); 7.724 (1H, t, J = 7.5 Hz, –H12); 7.794–7.855 (2H, m, –H13,14); 7.893 (1H, t, J = 7.5 Hz, –H4); 8.176 (1H, d, J = 8.5 Hz, –H3); 8.242 (1H, t, J = 8.0 Hz, –H5); 8.288 (1H, d, J = 8.0 Hz, –H15); 8.398 (1H, d, J = 8.0 Hz, –H10); 8.817 (2H, t, J = 9.0 Hz, –H6,9). 13C NMR (125 MHz, CDCl3, δ ppm): 19.05 (–Ck), 21.60 (–Co), 22.80 (–Co), 31.35 (–Cn), 81.88, 83.32, 84.35, 85.25, 104.21, 105.96, 113.07, 119.31, 119.90, 128.49, 128.80, 129.68, 129.75, 130.20, 130.82, 134.29, 138.79, 141.99, 144.08, 149.40, 150.97, 161.12. Anal. calcd (%) for C26H24Cl2N2ORu·0.5CH2Cl2·0.5CH3OH: C, 53.08; H, 4.45; N, 4.59. Found (%): C, 53.61; H, 4.96; N, 4.30. ESI-MS(+) in CH3OH (calcd, found, m/z) 517.0620, 517.0623, [M+ = 2 − Cl]. FTIR (KBr, cm−1) (C–H aromatic + aliphatic) 3049.10, 2963.37, 2923.94, 2869.10, (CN) 1615.97, (CC) 1587.94. UV-Vis: (in PBS (pH = 7.4)), λmax (nm) (λmax, dm3 mol−1 cm−1): 251 (1.91 × 104), 299 (1.48 × 104), 358 (1.62 × 104); (in DMSO:PBS (pH = 7.4) (1:10 v/v)) λmax (nm) (λmax, dm3 mol−1 cm−1): 252 (2.24 × 104), 301 (1.72 × 104), 360 (1.94 × 104).
2.3.3 Synthesis and characterization of [(η6-p-cymene)(L3)RuCl]Cl·0.5CH2Cl2 (3).
To a solution of 2-(quinolin-2-yl)benzo[d]thiazole (L3) (0.2 g, 0.816 mmol) in dry dichloromethane (30 mL), the Ru(p-cymene) dimer [((η6-p-cymene)Ru(μ-Cl)Cl)2] (0.25 g, 0.408 mmol) was added and stirred. This mixture was refluxed under nitrogen for 12 h. The reaction mixture was cooled to room temperature, and the solvent was removed under vacuum. The product was purified by silica gel (60–120 mesh) column chromatography (MeOH-DCM eluent system). Brown solid; yield = 78%. m.p. = 147 °C decomp. 1H NMR (500 MHz, CDCl3, δ ppm): 0.780–0.833 (6H, m, –Ho); 2.159–2.220 (1H, m, –Hn); 2.375 (3H, s, –Hk); 5.791 (1H, d, J = 5.0 Hz, –Hm1); 5.907 (1H, d, J = 5.0 Hz, –Hm2); 6.105 (1H, d, J = 6.0 Hz, –Hl2); 6.182 (1H, d, J = 5.5 Hz, –Hl1); 7.665 (1H, t, J = 7.5 Hz, –H13); 7.804 (1H, t, J = 7.5 Hz, –H4); 7.894 (1H, t, J = 8.0 Hz, –H14); 8.099–8.160 (2H, m, –H3,5); 8.285 (1H, d, J = 8.0 Hz, –H12); 8.413–8.462 (2H, m, –H10,15); 8.798 (2H, d, J = 8.0 Hz, –H6,9). 13C NMR (125 MHz, CDCl3, δ ppm): 19.02 (–Ck), 21.84 (–Co), 22.15 (–Co), 30.98 (–Cn), 82.71, 84.95, 85.02, 85.10, 105.05, 105.36, 120.88, 122.66, 124.71, 128.75, 129.07, 129.44, 129.74, 130.10, 130.39, 133.48, 134.06, 142.16, 149.39, 150.68, 151.57, 165.48. Anal. calcd (%) for C26H24Cl2N2RuS·0.5CH2Cl2: C, 52.09; H, 4.12; N, 4.59; S, 5.25. Found (%): C, 52.16; H, 4.61; N, 3.90; S, 5.06. ESI-MS(+) in CH3OH (calcd, found, m/z) 533.0392, 533.0394, [M+ = 3 − Cl]. FTIR (KBr, cm−1) (C–H aromatic + aliphatic) 3037.36, 2958.08, 2918.68, (CN) 1603.65, (CC) 1449.86. UV-Vis: (in PBS (pH = 7.4)), λmax (nm) (λmax, dm3 mol−1 cm−1): 260 (2.15 × 104), 304 (1.56 × 104), 375 (2.36 × 104); (in DMSO:PBS (pH = 7.4) (1:10 v/v)) λmax (nm) (λmax, dm3 mol−1 cm−1): 263 (2.16 × 104), 311 (1.45 × 104), 363 (2.06 × 104), 381 (2.40 × 104).
2.4 UV-Vis spectral studies
All the UV-Vis spectral studies were carried out on an Agilent Technologies Cary 60 UV-Vis spectrophotometer at 298 K. For aqueous stability studies, fixed concentration samples (26.66 μM) of 1–3 were prepared in phosphate buffered saline (PBS, pH = 7.4). A blank sample of only PBS (pH = 7.4) was also prepared as the control. Similarly, the stability of complexes 1–3 in DMSO was also studied using the same concentration (26.66 μM). These measurements were repeated after 12 h, 24 h and 48 h. These studies were also carried out for ligands L1–L3 and the Ru(p-cymene) dimer [((η6-p-cymene)Ru(μ-Cl)Cl)2].
2.5 Partition coefficient
The lipophilicity of complexes 1–3 was determined by the shake flask method between octanol and water phases.58,59 An Octanol and water (1:1 v/v) mixture was used for these studies. 25 μM solutions of complexes 1–3 were prepared in octanol saturated water (OSW), and then, these solutions were added to an equal amount of the water and octanol mixture. The mixtures were mechanically mixed for 24 hours. After the separation of two layers, they were subjected to UV-Vis spectroscopic analysis. To determine the relative concentrations, the absorption bands at 370 nm (for 1), 358 nm (for 2), and 375 nm (for 3) were considered. The partition coefficient (logP) values were calculated using the equation logP = log[(1–3)oct./(1–3)aq].59
2.6 X-ray crystallography
Crystallographic data of complex 1 was collected on a Bruker kappa APEX-II CCD diffractometer at 293(2) K using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). For unit cell determination, the single crystal was exposed to X-rays for 10 s in three sets of frames. The detector frames were integrated using the program SAINT,60 and the multi-scan absorption corrections were performed using the SADABS program.61 The structures were solved by direct methods and refined by the full-matrix least-squares method based on F2 using SHELXS-9762 and SHELXL-97.62 All non-hydrogen atoms were refined using anisotropic displacement parameters (except the disordered atoms), whereas the hydrogen atoms were placed in the calculated positions when possible and given the isotropic U values 1.2 times that of the atom, to which they are bonded. Some carbon atoms of the isopropyl groups in both cationic units were disordered over two positions with the fractional occupancies of 0.42/0.58 for C8/C8A, 0.4/0.6 for C9/C9A, 0.43/0.57 for C10/C10A, 0.66/0.34 for C34/C34A and 0.6/0.4 for C35B/C35A. One carbon atom (fractional occupancy 0.8/0.2 for C57/C57A) of the hexane solvent and the chlorine atom (fractional occupancy 0.6/0.4 for Cl9/Cl9A) of the dichloromethane solvent were also disordered over two positions. Materials for publication were prepared using the SHELXTL,62 PLATON,63 Olex2,64 and Mercury65 programs.
2.7 Cyclic voltammetry
Cyclic voltammetry experiments were carried out on an AUTOLAB PGSTAT 302N (Metrohm Autolab B.V., Netherlands) electrochemical analyzer. A three-electrode system, consisting of glassy carbon as the working electrode (GCE), a platinum wire as the auxiliary electrode and a silver wire as the pseudo-reference electrode, was used for cyclic voltammetric measurements. The electrochemical properties of all three Ru complexes were studied in CH3CN with 0.1 M tetrabutylammoniumperchlorate (TBAP) as the supporting electrolyte. The GCE, 3 mm diameter, was manually polished with a 0.3 μm Al2O3 slurry on a polishing cloth, rinsed with double distilled water followed by CH3CN and dried with argon. All solutions were purged with pure argon prior to measurement to remove oxygen. All electrochemical measurements were carried out by maintaining a blanket of argon over the solutions at room temperature.
2.8 Theoretical studies
A detailed investigation of the proposed complexes 1–3 was carried out using first-principles-based quantum mechanical methods. All stationary points were freely optimized and the corresponding geometries were obtained using the density functional theory (DFT)-based hybrid B3LYP66,67 functional with the 6-31G(d,p) basis set68 for all atoms except Ru, for which the LANL2DZ basis set69–71 with an effective core potential (ECP) was used. Harmonic vibrational frequencies were calculated at the same level of the equilibrium geometries to validate each calculated structure as a true minimum. All energy values in the main text are the electronic energies computed at the level of the theory, as mentioned above. All the theoretical calculations were performed using the GAUSSIAN 16 program package.72 For the complex-DNA structure and interactions, the semi-empirical PM6 method73 was used over the force-field methods for enhanced protein docking, satisfactory complex geometry and binding energy estimation, and free optimization of the DNA with the cationic parts of complexes 1 and 2.
2.9 UV-Vis spectrophotometric studies for DNA interaction
The UV-Vis absorption spectrophotometric titrations were carried out with a fixed concentration of 1–3 (10 μM) prepared in Milli-Q water. The UV-Vis absorption titration spectra were recorded with the addition of varying concentrations of CT-DNA (0–15 μM; T10E1 buffer, pH ∼ 7.6) at 298 K. These titrations were also performed with a fixed concentration of L1–L3 (25 μM; DMSO:water (5:100 v/v)) and varying concentrations of CT-DNA (0–4.3 × 10−7 M; T10E1 buffer, pH ∼ 7.6) at 298 K. The concentration of CT-DNA was calculated using the molar absorption coefficient of 6600 M−1 cm−1 at 260 nm. The equilibrium binding constants (Kb) and binding site size (s) were calculated using Bard's equation.
2.10 Emission spectral studies for DNA interaction
EthBr displacement fluorescence experiments were carried out on an Agilent Cary Eclipse fluorescence spectrophotometer. Fixed concentration solutions of CT-DNA (35 μM) and EthBr (35 μM) were prepared in T10E1 buffer (pH ∼ 7.6). After intercalation of EthBr with DNA, the EthBr–DNA complex showed good fluorescence intensity. The addition of increasing concentrations (0–360 μM; Milli-Q water) of 1–3 showed significant quenching in fluorescence intensity at 298 K. These studies were also carried out with increasing concentrations of the Ru(p-cymene) dimer (0–6.03 × 10−4 M; Milli-Q water), L1 (0–7.24 × 10−4 M; DMSO:water (5:100 v/v)) and L3 (0–4.42 × 10−4 M; DMSO:water (5:100 v/v)). The fluorescence spectral changes were observed at the excitation wavelength (λex.) and the emission wavelength (λem.), 480 nm and 597 nm, respectively.
2.11 Protein interaction studies
The steady-state fluorescence quenching experiments were carried out on an Agilent Cary Eclipse fluorescence spectrophotometer at 298 K. A fixed concentration solution of BSA (0.15 μM; pH ∼ 7.4) was prepared in PBS. The steady-state fluorescence quenching experiments were carried out with the addition of increasing concentrations (0–16 μM; PBS, pH ∼ 7.4) of 1–3 and Ru(p-cymene) dimer at the excitation wavelength (λex. = 280 nm) and the emission wavelength (λem. = 344 nm). Synchronous fluorescence emission titration of BSA (0.15 μM; PBS, pH ∼ 7.4) with varying concentrations of 1–3 and Ru(p-cymene) dimer (0–16 μM; PBS, pH ∼ 7.4) was performed at the wavelength difference of Δλ = 60 nm and Δλ = 15 nm.
The UV-Vis absorption spectrophotometric studies were done on an Agilent Technologies Cary 100 UV-Vis spectrophotometer. The stock solution of BSA (10 μM; pH ∼ 7.4) was prepared in PBS and stored in a dark place at 4 °C. UV-Vis spectra of BSA were recorded alone and in the presence of 1–3 and Ru(p-cymene) dimer (10 μM; PBS, pH ∼ 7.4) at 298 K. UV-Vis spectra of ligands L1–L3 (10 μM; DMSO:water (5:100 v/v)) were also recorded in the presence of BSA.
2.12 Cell viability assay
MCF-7 breast cancer cells were procured from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained at the in-house cell repository of the National Centre for Cell Science, Pune, India. Cells were routinely cultured in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, UT, USA or Gibco, NY, USA), penicillin (100 U mL−1) and streptomycin (100 mg mL−1) (Invitrogen Life Technologies, CA, USA) and maintained at 37 °C in a 5% CO2 humidified incubator (Thermo Fisher Scientific, OH, USA). The MCF-7 breast cancer cells were plated at a density of 5 × 103 cells per well in a 96-well plate and allowed to adhere. After 24 h, the cells were treated with the vehicle (DMSO) and different concentrations of complexes 1–3, ligands L1–L3 and Ru(p-cymene) dimer (0.1, 1, 10, 100 μM) as per the experimental requirements. The stock solutions of complexes 1–3, ligands L1–L3 and Ru(p-cymene) dimer were prepared in DMSO. After the treatment duration (48 h), the medium was removed and 50 μL of MTT (methylthiazole tetrazolium, 1 mg mL−1 in DMEM without phenol red) (Sigma-Aldrich, MO, USA) was added in each well and further incubated for 4 h at 37 °C. The formazan crystals were solubilized in 100 μL of isopropanol, and the absorbance was measured at 570 nm. Calculation of IC50 values was done by plotting the percentage viability v/s concentration on a logarithmic graph and fitting it with nonlinear regression using the dose response-inhibition equation in GraphPad Prism. It gave the concentration, at which 50% of the cells remained viable relative to the control. All experimental data were obtained from at least three independent experiments and are expressed as mean ± standard error.
3. Results and discussion
3.1 Synthesis and characterization
The synthesis of mononuclear arene-Ru(II) complexes [(η6-p-cymene)(L1–3)RuCl]Cl (1–3) was carried out by the single-step reaction of the Ru(p-cymene) dimer and ligands L1–L3 with excellent yields (Scheme 2). All the complexes were characterized by elemental analysis, FT-IR, 1H, 13C-NMR, UV-Vis and ESI mass spectroscopic studies. The structure determination of 1 was done by single crystal X-ray diffraction analysis. Complexes 1–3 were yellowish brown to brown crystalline solids, air-stable, non-hygroscopic and stable in the solid state for a long period of time. All the complexes were easily soluble in water, methanol, ethanol, chloroform, dichloromethane, acetone, acetonitrile, dimethylformamide, and dimethylsulfoxide, while insoluble in hexane, diethyl ether and petroleum ether. The solubility of the complexes in aqueous media were found to be 140 g L−1 for 1, 112 g L−1 for 2, and 29 g L−1 for 3.41 The conductivity values were measured to be 66 S cm2 mole−1 for 1, 128 S cm2 mole−1 for 2, and 122 S cm2 mole−1 for 3 in acetonitrile at 298 K, which are comparable to the molar conductivity values of the 1:1 electrolytes. The values of molar conductivity indicated the mono-cationic nature of complexes 1–3.74 The elemental analysis of 1–3 showed the purity of these complexes along with the presence of a few solvent molecules. Data from the elemental analysis of 1–3 was collected using powder samples dried under high vacuum after column chromatography (Fig. S1–S3, ESI†). In FT-IR studies, the (N–H) stretching vibrations of complex 1 were observed at 3391.88 cm−1, which had shifted compared to the 3485.55 cm−1 peak observed for ligand L1. The vibrations corresponding to (C–H, aliphatic and aromatic) were observed in the range 3100–2800 cm−1 for complexes 1–3. The vibrations in the range 1650–1450 cm−1 corresponded to the CN and CC aromatic stretching bands. The most relevant IR frequencies are given in the experimental section (Fig. S4–S6, ESI†).
 |
| | Scheme 2 Scheme for syntheses of complex 1–3. | |
The formation of 1–3 was confirmed by the 1H, 13C and 1H–1H COSY NMR spectral results. The 1H, 13C and 1H–1H COSY NMR spectra of 1 were recorded in DMSO-d6, while for 2 and 3, CDCl3 was used as the solvent. In the 1H-NMR spectra of 1–3 (Fig. S7–S9, ESI†), the signals observed in the range δ 0.685–1.019 ppm corresponded to two methyl groups of the isopropyl substituent placed at the p-cymene moiety. One multiplet observed in the range δ 2.159–2.617 ppm was related to the single proton of the isopropyl group. Three protons of the methyl substituent showed up as a singlet in the range δ 2.247–2.375 for all the complexes. Two aromatic protons of p-cymene could be seen as two doublets in the range δ 5.791–6.263 ppm, while the other two aromatic protons showed a signal in the range δ 6.105–6.397 ppm. The other aromatic protons of the quinoline-based ligand part were found in the range δ 7.617–8.992 ppm. Peaks observed in the 1H-NMR spectra were in good agreement with the chemical structures of 1–3. 1H-NMR peak assignment and structure elucidation for all the complexes were performed by 1H–1H COSY NMR spectra (Fig. 1 and Fig. S10–S17, ESI†). The protons corresponding to the two methyl groups (Ho1 and Ho2) of the isopropyl substituent showed a correlation with the middle proton (Hn) of the isopropyl group placed at the p-cymene moiety for all the complexes (Fig. S10, S12 and S15, ESI†). The aromatic protons of the p-cymene moiety (Hm) correlated with the other aromatic protons (Hl) in complexes 1–3. For complex 1, proton H4 correlated with H3 and H5, while H5 also correlated with H6. Moreover, protons H9, H12 and H14 correlated with H10, H13 and H15, respectively, as shown in Fig. 1. Similarly, in complex 2, proton H4 correlated with H3 and H5, while proton H5 also correlated with H6 simultaneously. In addition to that, protons H9, H12 and H14 correlated with H10, H13 and H15, respectively, as shown in Fig. S13 (ESI†). In the aromatic region of complex 3, protons H3, H5 and H9 were in correlation with protons H4, H6 and H10, respectively. In addition to this, proton H13 showed a correlation with H12 and H14. Proton H14 also correlated with proton H15, as shown in Fig. S16 (ESI†). 13C-NMR data (Fig. S18–S20, ESI†) associated with the methyl group of p-cymene was seen between δ 18.98–19.05 ppm, while the two methyl groups of the isopropyl substituent were observed separately in the range δ 21.60–22.80 ppm. The methanetriyl carbon of the isopropyl group appeared between δ 30.76–31.35 ppm. Carbons associated with C6H4 of the p-cymene moiety exhibited signals in the range δ 79.00–105.96 ppm. Other carbons related to the ligand parts of all the complexes were observed in the range δ 113.07–165.48 ppm.
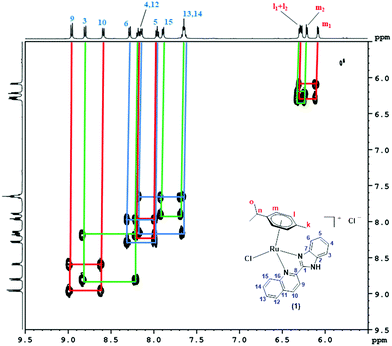 |
| | Fig. 1 Aromatic region of the 1H–1H-COSY NMR spectrum of 1 recorded in DMSO-d6. | |
To further support the identity of complexes 1–3, their mass spectra were recorded using ESI-MS (positive mode). The ESI mass spectra of 1–3 show the molecular ion peak corresponding to the [(η6-p-cymene)(L1–3)RuCl]+ fragment as the base peak (Fig. S21–S23, ESI†). The m/z values for the complexes 1–3 are detailed in the experimental section. The solution phase studies, such as mass spectrometry, conductivity measurements and NMR spectroscopy, revealed the solution phase structure to be mono-cationic for the complexes 1–3.
3.2 Electronic spectral studies
For all the three complexes, three absorption bands were observed in their respective electronic spectra. Two strong HE (high energy) bands were observed at ∼250 nm (250 nm for 1; 251 nm for 2; and 260 nm for 3) and ∼300 nm (301 nm for 1; 299 nm for 2; and 304 nm for 3), which could be assigned to the intra-ligand charge transfer (ILCT) transitions. The LE (low energy) strong absorption band observed at ∼370 nm (370 nm for 1; 358 nm for 2; and 375 nm for 3; Fig. 2) would be for the metal to ligand charge transfer (MLCT).
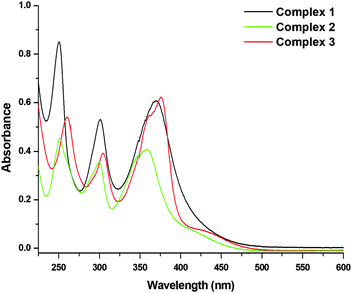 |
| | Fig. 2 UV-Vis absorption spectra of 1, 2 and 3 [26.66 μM] recorded in PBS (pH = 7.4) at 298 K. | |
3.3 Stability in aqueous and DMSO media
The aqueous stability studies of 1–3 were carried out using UV-Vis and 1H-NMR spectroscopy in the aqueous medium (PBS, pH = 7.4) and DMSO. The UV-Vis spectra in PBS and DMSO were recorded after 0 h, 12 h, 24 h and 48 h of incubation (Fig. S24–S26, ESI†). Complexes 1–3 showed characteristic absorption bands in the range of 250 to 375 nm. UV-Vis spectra in PBS at various time intervals suggested that complexes 1 and 3 exhibited good aqueous stability, although a slight change in UV-Vis spectra was observed, which is likely due to hydrogen bonding in the aqueous PBS buffer. However, complex 2 in PBS showed a decrease in the intensity of absorption bands over the period of study. It is clear that complex 2 was less stable compared to 1 and 3 in the aqueous medium.
The UV-Vis spectra of complexes 1–3 were also recorded in DMSO to further assess the stability of these complexes (Fig. S27–S30, ESI†). UV-Vis spectra in DMSO suggested that complexes 1 and 3 were quite stable in DMSO. A slight shift in the UV-Vis absorption maxima of 1 was observed probably due to interaction with DMSO. Complex 2 showed changes in not only intensity but also the position of the absorption bands over the period of study in DMSO.75
In order to obtain further insights into the stability of complexes 1–3, the stability was also studied using 1H NMR spectroscopy in DMSO-d6. The spectra were recorded at 0 h, 0.5 h, 2 h, 6 h, 24 h and 48 h time intervals (Fig. S31–S33, ESI†). The NMR spectra showed that complexes 1 and 3 were quite stable even after 48 h in the DMSO medium. Complex 2, as anticipated from the UV-Vis studies, showed degradation over the period of study. The NMR spectra of complex 2 clearly showed increasing peaks of free p-cymene with time. However, there was no evidence for the dissociation of ligand L2 from complex 2. All the ligands are also stable in DMSO, as shown in Fig. S34–S37 (ESI†). The Ru(p-cymene) dimer was stable in PBS but showed small changes in the absorption bands in DMSO over the period of study (Fig. S38 and S39, ESI†).75
3.4 Partition coefficient
The behavior of drug-like compounds can be understood by the partition coefficient.58,76 Compounds with logP values in the range −5 to 0 can cross both cellular and nuclear membranes, while logP values falling in the range 0 to +5 indicate mitochondrial and endoplasmic reticulum accumulation.77,78 The logP value obtained by the shake flask method for 1 was −0.700 ± 0.006, 2 was −1.429 ± 0.084 and 3 was −1.002 ± 0.021. The values of logP < 0 indicated the lipophilic behavior of these complexes. The lipophilicity calculated from the experiment follows the order: 1 > 3 > 2.
3.5 X-ray crystallographic studies
X-ray diffraction analysis was performed to confirm the spectrometric structure elucidation of complex 1. The structure of the cation [(η6-p-cymene)(L1)RuCl]+ (1) is shown in Fig. 3. The crystallographic data are listed in Table 1, and selected bond lengths and angles are given in Table S1 (ESI†). Data collection and refinement details are given in the experimental section. The structure was solved in a triclinic system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. Two independent molecules were present in the asymmetric unit (the other two complexes were symmetric). Each cationic unit had a characteristic “piano-stool” geometry of the Ru(II) arene complexes, with a η6 π-bonded p-cymene ring forming the seat and three other donor atoms (two nitrogen atoms of quinoline and benzimidazole units and one chloride ion) forming the three legs of the stool. The Ru–N bond lengths were 2.072(5) and 2.171(5) Å in one molecule and 2.075(5) and 2.170(5) Å in the other molecule. The Ru–Cl bond lengths were 2.3901(16) and 2.3966(18) Å, respectively. The Ru–C (p-cymene) bond lengths were in the range of 2.167(7)–2.250(7) and 2.170(8)–2.235(7) Å. The aforementioned data is comparable with those of similar systems reported earlier.41,57,79,80 Intermolecular π–π stacking was observed among the quinoline rings of the chelating ligand (Fig. S40 and Table S2, ESI†). The CH/NH protons of the chelating ligand (L1), bound chloride, counter ions (chloride), and solvent molecules in the unit cell were involved in the network of H-bonding interactions, as shown in Fig. S41 (ESI†), and the related parameters are listed in Table S3 (ESI†). Suitable crystals for 2 and 3 could not be obtained for structure determination. Since the spectroscopic data of 2 and 3 showed similar behavior to that of 1, we expect structural similarity between all three complexes. Therefore, the structure of 1 is considered the representative structure for complexes 1–3, which was further supported by structural optimization through DFT calculations.
space group. Two independent molecules were present in the asymmetric unit (the other two complexes were symmetric). Each cationic unit had a characteristic “piano-stool” geometry of the Ru(II) arene complexes, with a η6 π-bonded p-cymene ring forming the seat and three other donor atoms (two nitrogen atoms of quinoline and benzimidazole units and one chloride ion) forming the three legs of the stool. The Ru–N bond lengths were 2.072(5) and 2.171(5) Å in one molecule and 2.075(5) and 2.170(5) Å in the other molecule. The Ru–Cl bond lengths were 2.3901(16) and 2.3966(18) Å, respectively. The Ru–C (p-cymene) bond lengths were in the range of 2.167(7)–2.250(7) and 2.170(8)–2.235(7) Å. The aforementioned data is comparable with those of similar systems reported earlier.41,57,79,80 Intermolecular π–π stacking was observed among the quinoline rings of the chelating ligand (Fig. S40 and Table S2, ESI†). The CH/NH protons of the chelating ligand (L1), bound chloride, counter ions (chloride), and solvent molecules in the unit cell were involved in the network of H-bonding interactions, as shown in Fig. S41 (ESI†), and the related parameters are listed in Table S3 (ESI†). Suitable crystals for 2 and 3 could not be obtained for structure determination. Since the spectroscopic data of 2 and 3 showed similar behavior to that of 1, we expect structural similarity between all three complexes. Therefore, the structure of 1 is considered the representative structure for complexes 1–3, which was further supported by structural optimization through DFT calculations.
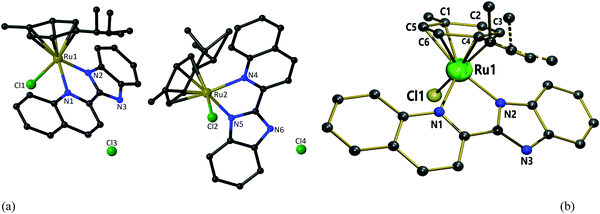 |
| | Fig. 3 (a) Two independent molecules in the asymmetric unit of 1 with partial atomic numbering. (b) The “piano-stool” geometry of 1 [Hydrogen atoms, solvent molecules and counter anion omitted for clarity]. | |
Table 1 Crystallographic parameters associated with the crystal structure of 1
| Crystal parameters |
Complex 1 |
| Sum formula |
C115H124Cl18N12Ru4 |
| Formula weight |
2716.27 |
| Crystal system |
Triclinic |
| Space group |
P |
|
a (Å) |
11.511(5) |
|
b (Å) |
12.353(5) |
|
c (Å) |
22.155(5) |
|
α (deg) |
78.665(5) |
|
β (deg) |
75.648(5) |
|
γ (deg) |
68.751(5) |
|
V (Å3) |
2824.6(18) |
|
Z
|
1 |
|
λ (Å) |
0.71073 |
| Colour and habit |
Red, needle |
| Size (mm3) |
0.25 × 0.20 × 0.13 |
|
T (K) |
293(2) |
|
D
calcd (g cm−3) |
1.605 |
|
μ (mm−1) |
1.006 |
|
F(000) |
1394 |
| 2θ (°) |
1.99 to 50.00 |
| Index ranges |
−13 ≤ h ≤ 13; −14 ≤ k ≤ 14; −26 ≤ l ≤ 26 |
| Reflections collected |
30633 |
| Independent reflections (Rint) |
9825 (0.0431) |
| Data/restraints/parameters |
9825/0/673 |
| GOF on F2 |
1.144 |
| Final R indices I > 2σ(I) |
R
1 = 0.0604, wR2 = 0.1814 |
|
R indices (All data) |
R
1 = 0.0778, wR2 = 0.2239 |
| Largest peak and hole (e Å−3) |
1.879 and −2.261 |
3.6 Electrochemical properties
In order to get some insights into the stability of the Ru(II) state in 1–3, the electrochemical behavior of complexes 1–3 were studied by cyclic voltammetry at various scan rates. The representative cyclic voltammograms of complex 1 are shown in Fig. 4 and Fig. S42 (ESI†). The anodic peak observed at 1.381 V (Ia) for complex 1 was related to the oxidation of Ru2+ to Ru3+, but no cathodic peak was observed corresponding to Ia, which indicated the irreversible nature of this process. The other anodic peak at 1.765 V (IIa) and cathodic peak at 1.672 V (IIc) corresponded to the Ru3+ ↔ Ru4+ redox responses. The peak to peak separation (ΔEpII = 0.093) of the above peaks suggested that this redox process was quasi-reversible. The oxidation (Epa) and reduction (Epc) potentials along with the peak to peak separation (ΔEp) values of all the investigated complexes 1–3 are listed in Table 2. Complexes 2 and 3 showed irreversible oxidation peaks (Epa) for Ru2+ to Ru3+ at 1.296 V and 1.296 V, respectively (Fig. S43 and S44, ESI†). The oxidation peaks (Epa) for Ru3+ to Ru4+ and the reduction peaks (Epc) for Ru4+ to Ru3+ were seen in the ranges 1.865–1.872 V and 1.748–1.774 V, respectively. The peak to peak separation (ΔEp) values for complexes 2 and 3 were 0.098 V and 0.117 V, respectively, which showed the quasi-reversible and irreversible nature of their redox processes, respectively. The data presented here are comparable with those of similar systems reported previously.81–83 Overall, the oxidative responses occurred at very high potentials and the Ru(II) state in the complexes 1–3 was quite stable to be oxidized by common biological oxidants.
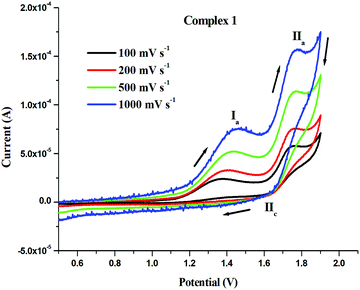 |
| | Fig. 4 Cyclic voltammograms of 1 (1 mM) solution in CH3CN with 0.1 M [nBu4N][ClO4] at scan rates: 100 mV s−1, 200 mV s−1, 500 mV s−1, 1000 mV s−1 at room temperature. | |
Table 2 Potential values of anodic peaks (Epa), cathodic peaks (Epc) and peak-to-peak separation (ΔEp) of 1–3 at the scan rate of 100 mV s−1
| Complexes |
E
pIa (V) |
E
pIIa (V) |
E
pIIc (V) |
ΔEpII (V) |
|
1
|
1.381 |
1.765 |
1.672 |
0.093 |
|
2
|
1.269 |
1.872 |
1.774 |
0.098 |
|
3
|
1.296 |
1.865 |
1.748 |
0.117 |
3.7A DFT studies for geometry optimization of 1–3 and their DNA binding
Full geometry optimization of all the three Ru complexes (1–3) was carried out using the DFT-based hybrid B3LYP functional with the 6-31G(d,p) basis set for all atoms except Ru, for which the LanL2DZ basis set with ECP was used. In the quest for lowest energy conformers for each case, a systematic conformational analysis was performed. From an initial pool of structures, the lowest energy conformers for the cationic parts of the Ru complexes 1–3 were determined. For each case, Mulliken charge analysis was performed to find the most electro-positive hydrogen, which ought to be the probable site of interaction with the counter chloride anion. Then Cl− was inserted in those places and re-optimization was performed. Finally, following the energy hierarchy, the global minima structures were determined for 1–3 (Fig. S45A and B, ESI†). In addition, harmonic frequency calculations were performed at the same level of the theory to validate the final optimized structures. In all the cases, the cationic fragment of the optimized structure showed a typical three-legged piano stool geometry. The Ru center was coordinated with two nitrogens of the ligand and one Cl−, whereas the p-cymene ring was coordinated in a  -manner. The geometrical parameters of the optimized structures are in good agreement with those of previously obtained crystal structures of related systems.41 We also assessed the structures of the complexes with DCM as the solvent (using polarization continuum mode) following the same experimental conditions.
-manner. The geometrical parameters of the optimized structures are in good agreement with those of previously obtained crystal structures of related systems.41 We also assessed the structures of the complexes with DCM as the solvent (using polarization continuum mode) following the same experimental conditions.
The structural analysis showed some interesting facts about the geometry of the complexes, particularly about the position of the counter chloride anion when a solvent is used. In 1, the most electro-positive hydrogen is the NH of the ligand, as expected, which interacted with the counter Cl− to form the most stable conformer in the gas phase, as well as the solvent phase. The corresponding bond lengths of H–N (1.107 Å and 1.052 Å without and with solvent, respectively) and H⋯Cl− (1.812 Å and 2.020 Å without and with solvent, respectively) showed that, due to the effect of solvent, the H⋯Cl− bond length increased noticeably, leading to decoupling between H and Cl− (Table S4, ESI†). The effect of solvent was more prominent for the structures of 2 and 3, where no such highly electro-positive hydrogen atoms were available (Table S5, ESI†). In such cases, Cl− interacted weakly with the different aliphatic and aromatic hydrogen atoms and behaved like a roaming atom, giving rise to several low energy conformers. In this case, the solvent affects the structures to different extents by modifying the position of Cl− extensively. The effect of solvent also changes the sequence of the lowest energy conformation pattern. In Table 3, the relative energies of the four lowest energy conformers are given for 2 with and without the solvent. To validate these observations, we further carried out these calculations using higher basis sets (6-31+G(d,p) for all atoms except Ru, for which Def2TZVP with ECP basis was used). As can be seen from Table 3, conformer A was the lowest energy structure in the gas phase for both the basis sets, and conformer B possessed the minimum energy structures while using a solvent. This showed that the counter Cl− ion was quite labile, and depending on external perturbation, it changed its position to attain the most favorable structure. The same pattern was also found for complex 3.
Table 3 Relative energies (in kcal mol−1) for the different conformers of 2 with and without solvent using different basis sets
| Conformer |
Basis1 |
Basis2 |
| Gas phase |
Solvent phase |
Gas phase |
Solvent phase |
| Basis1: B3LYP with 6-31G(d,p) for light atoms and LanL2DZ with ECP for Ru. Basis2: B3LYP with 6-31G(d,p) for light atoms and Def2TZVP with ECP for Ru. |
|
A
|
0.0 |
1.56 |
0.0 |
1.09 |
|
B
|
4.5 |
0.0 |
4.1 |
0.0 |
|
C
|
2.6 |
1.12 |
5.7 |
0.83 |
|
D
|
4.2 |
1.10 |
4.3 |
0.73 |
The frontier molecular orbital diagrams also showed a rich variety of complexes with and without solvent similar to the conformational structures. For 1, the electron density of LUMO was localized throughout the ligand (L1) and the metal center and was found to be nearly the same with and without a solvent due to non-significant structural changes. On the other hand, the HOMO for the gas and solvent phases were quite different due to noticeable structural changes, mainly due to the positional change of the counter chloride anion. While the HOMO in the gas phase was located in the counter anion, as well as on the entire complex, the contour of the HOMO in the solvent phase was mainly located on the counter chloride anion and very slightly dispersed on the nearby nitrogen atoms of the ligand. This observation is in harmony with the structural features found, where the counter anion is largely decoupled with the rest of the molecule in the solvent phase. It can be seen from Table S4 (ESI†) that the Cl−⋯H–N bond length increased considerably in the solvent phase from 1.812 Å to 2.020 Å, which validates this observation.
3.7B DNA binding interaction studies of 1–3 by the semi-empirical PM6 method
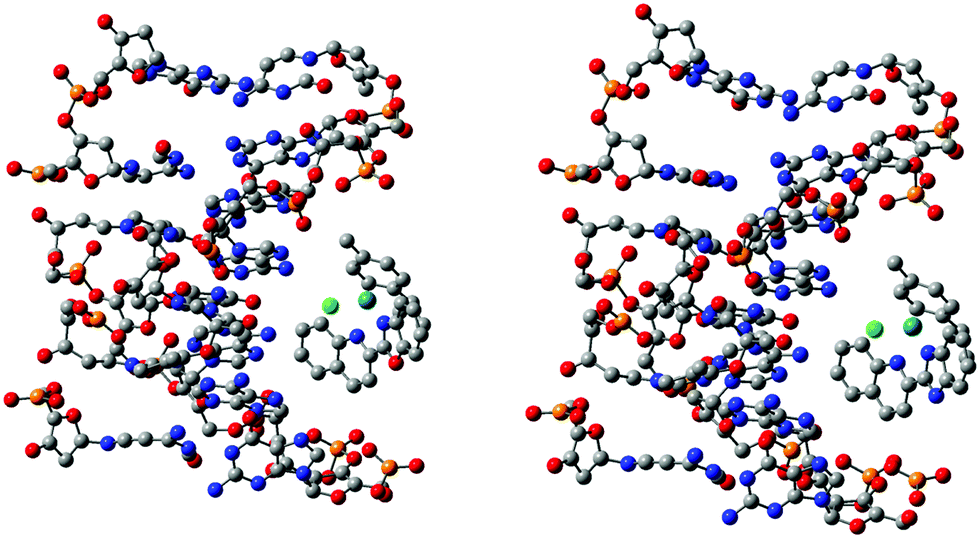
The interactions of complexes 1 and 2 with a B-DNA dodecamer d(CGCGAATTCGCG) were investigated. The coordinates of the B-DNA, which is a right-handed Watson–Crick family model, were taken from the Protein Data Bank, of which seven DNA strands were retained for simplicity in this study. Since, the investigated molecules, complex 1-DNA and complex 2-DNA, are huge (482–483 atoms, 2442 electrons) for any level of quantum chemical methods, it is unrealistic to execute calculations using the present state-of-the-art computers. To date, empirical or force-field methods have been routinely used to elucidate such huge structures besides docking studies, which is an obvious choice due to much faster calculations but with a compromise on the accuracy. In this study, the challenge was to investigate such a huge system using the semi-empirical PM6 method, which should be more accurate compared to the force-field methods, for a better understanding of the complex-DNA structure and interactions. This method was used for a fair balance between accuracy and computational time. The optimized structure suggested that several strong and weak electrostatic and intermolecular hydrogen bonding interactions existed between the outer edge of the DNA and the complex (Fig. 5 and Fig. S46, ESI†). Actually, the complexes aligned at the outer surface of four adjacent base pairs and also with the backbone of the DNA. In the preferred conformational structure, van der Waals interactions and hydrogen bonding probably play the major role. The Cl attached to Ru interacts with the shortest bond length, 2.57 Å and 1.51 Å, in 1 and 2, respectively. The multiple aromatic and aliphatic hydrogens from the C–H bonds of the complex interact with the oxygen of the phosphate group of the DNA. Interestingly, due to the interaction of the π-electrons in the ligand with N–H of DNA, the strands take a curved shape to fit with the DNA nicely. The calculated binding energies for the 1 and 2 systems were very similar (25.6 and 21.8 kcal mol−1 for 1 and 2, respectively) which establishes a strong interaction between them. The structural features and binding energies described above indicate the competency of the characteristic binding of the complexes with DNA.
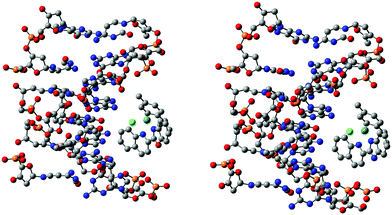 |
| | Fig. 5 Molecular docked models of 1 (right) and 2 (left) with B-DNA optimized by the PM6 method. Hydrogen atoms are omitted for clarity. | |
3.8 UV-Vis spectrophotometric studies for DNA interaction
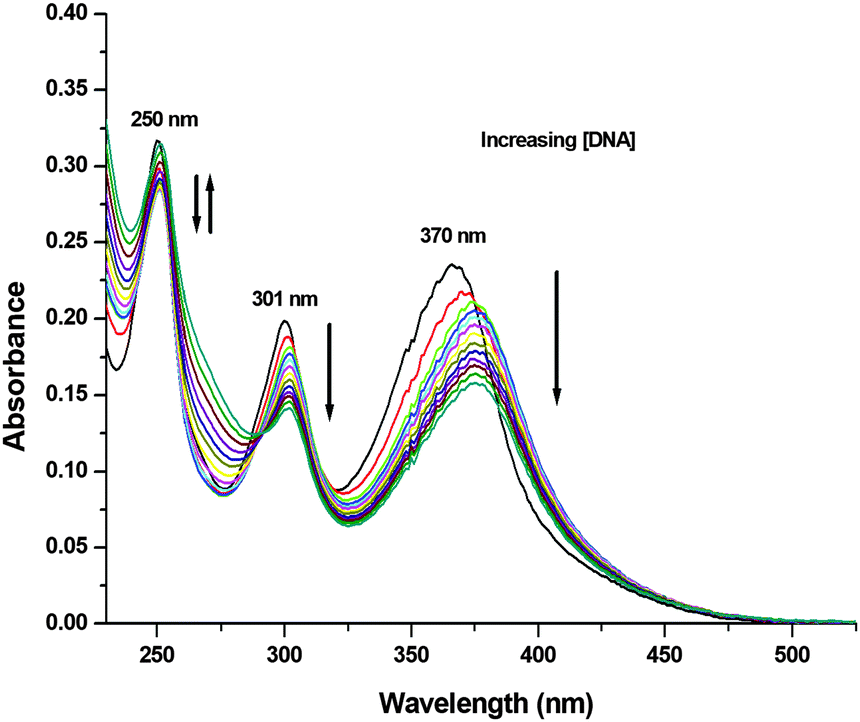
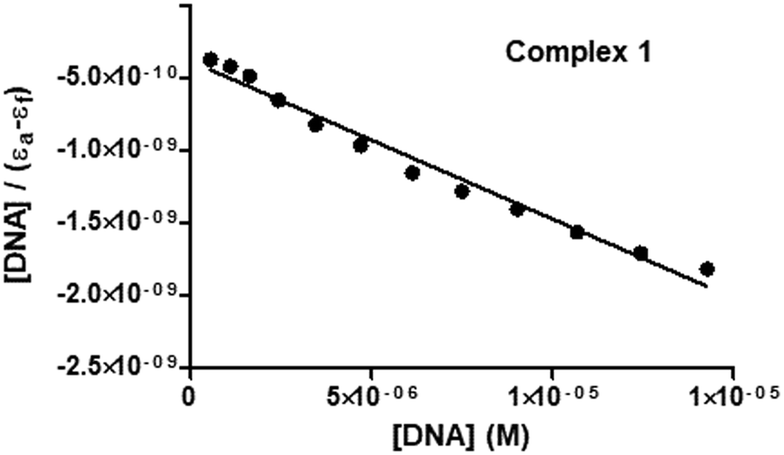
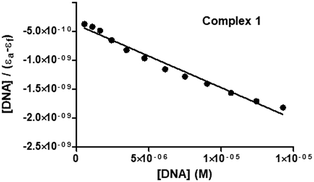
To experimentally investigate the binding affinity of 1–3 with DNA, the reliable UV-Vis absorption spectroscopy tool was employed.84–86 Metal complexes can bind to the DNA through covalent bonds, as well as non-covalent interactions, such as intercalation, groove binding and electrostatic interactions.86 The nature and pattern of the UV-Vis absorption titration curve can provide valuable information regarding the interaction mode of the compounds with DNA. In order to evaluate the equilibrium binding constant (Kb) and binding site size (s), complexes 1–3 (10 μM; Milli-Q water) were titrated against increasing concentrations of CT-DNA (0–15 μM; T10E1 buffer, pH ∼ 7.6; Fig. 6 and Fig. S47, S48, ESI†). As discussed above, complexes 1–3 showed three absorption bands in their electronic spectra. With an incremental addition of CT-DNA (0–15 μM) to the solution of 1, the HE band at 250 nm showed hypochromism followed by hyperchromism with a small redshift (Δε = 3.47 × 103 M−1 cm−1, Δε = 2.90 × 103 M−1 cm−1, Δλ = 2 nm; Table S6, ESI†). The first LE band at 301 nm showed hypochromism with a small redshift (Δε = 5.64 × 103 M−1 cm−1, Δλ = 1 nm). The second LE band around 370 nm for complex 1 also showed hypochromism with a significant redshift (Δε = 7.84 × 103 M−1 cm−1, Δλ = 6 nm). Similar results were observed for complexes 2 and 3 (Table S6, ESI†), but with less redshift compared to those observed for complex 1. Hypochromism followed by hyperchromism was observed for the HE band at 250 nm, indicating binding through intercalation, as well as electrostatic interaction.87 The significant hypochromic shifts at 301 nm and 370 nm in the UV-Vis spectra suggested a strong interaction between the complexes and CT-DNA through intercalation.84 The redshift observed for the bands indicated the participation of the aromatic groups of DNA in the interaction. In addition, the existence of the -NH group in complex 1 increases the possibility of DNA interaction taking place via hydrogen bonding. The equilibrium binding constant (Kb) was calculated (Fig. 7 and Fig. S49, S50, ESI†) according to the following equation:41,88–91
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) |
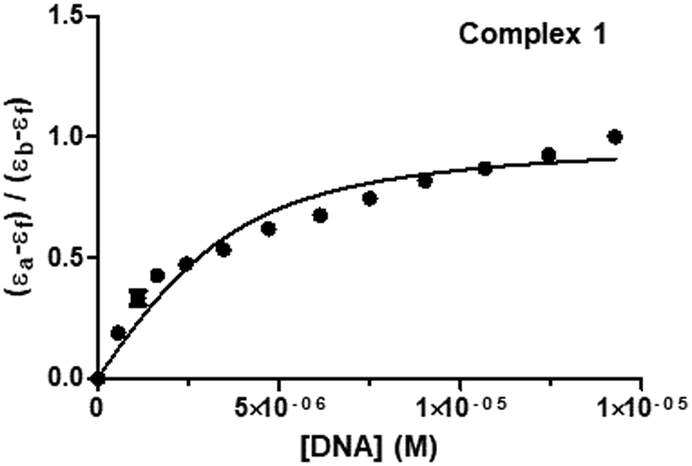
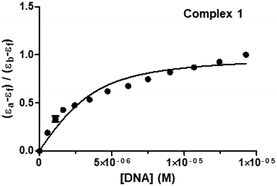
where [DNA] is the concentration of DNA in M (in base pairs), εa is the molar absorptivity at the given DNA concentration, εf is the molar absorptivity of the free complex, and εb is the molar absorptivity of the complex in the fully bound form. In order to calculate the “s” (binding site size) (Fig. 8 and Fig. S51, S52, ESI†), Bard's equation was employed based on the McGhee–von Hippel (MvH) model.85,92,93
| (εa − εf)/(εb − εf) = (b − (b2 − 2Kb2ct[DNA]/s)1/2)/2Kbct |
| b = 1 + Kbct + Kb[DNA]/2s |
where ct is the total metal complex concentration, and b is a constant. The equilibrium binding constants (Kb) and binding site sizes (s, per base pair) for complexes 1–3 were calculated (2.83 ± 0.23 × 105, 0.162 for 1; 3.25 ± 0.23 × 104, 0.071 for 2; 5.23 ± 1.01 × 104, 0.100 for 3; Table 4). The values of Kb for 1–3 suggested a strong binding interaction with DNA, and they fall in a range comparable to those of similar systems reported in the literature.41,90 The binding affinity of the complexes toward CT-DNA followed the order 1 > 3 > 2. Binding site size (s) is the number of base pairs interacting per bound protein molecule. The value of “s” suggests the nature of interaction taking place between the complex and DNA. Since these complexes showed moderately strong binding and smaller “s” values in which indicate the existence of electrostatic interactions between 1–3 and DNA by groove and/or surface binding.41,85
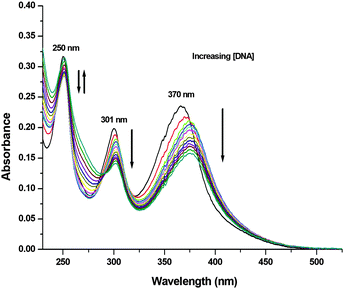 |
| | Fig. 6 UV-Vis absorption titration spectra of 1 (10 μM; Milli-Q water) with the addition of varying concentrations of CT-DNA (0–15 μM; T10E1 buffer, pH ∼ 7.6) at 298 K. UV-Vis absorption spectra of 1 (black). | |
 |
| | Fig. 7 Plot for the calculation of equilibrium binding constant (Kb) associated with the titration of 1 and CT-DNA at 298 K. | |
 |
| | Fig. 8 Plot associated with the titration of 1 and CT-DNA at 298 K to fit the model of Bard and Thorp. | |
Table 4 Data from the UV-Vis absorption studies on the DNA binding ability of complexes 1–3
| Complexes |
Equilibrium binding constant (Kb) (in M−1) |
Binding site size (s) (per base pair) |
|
1
|
2.83 ± 0.23 × 105 |
0.162 |
|
2
|
3.25 ± 0.23 × 104 |
0.071 |
|
3
|
5.23 ± 1.01 × 104 |
0.100 |
In addition to complexes 1–3, the interaction of ligands L1–L3 with DNA was also studied. Graphs are shown in ESI† (Fig. S53–S61). The Ru(p-cymene) dimer could not be studied due to the presence of overlapping absorption bands with that of DNA. However, the values of equilibrium binding constants (Kb) and binding site sizes (s, per base pair) for ligands L1–L3 (1.73 ± 0.03 × 106, 0.0061 for L1; 2.04 ± 0.03 × 106, 0.0058 for L2; 1.16 ± 0.02 × 106, 0.0065 for L3) could be determined (Table S7, ESI†).
3.9 Emission spectral studies for DNA interaction
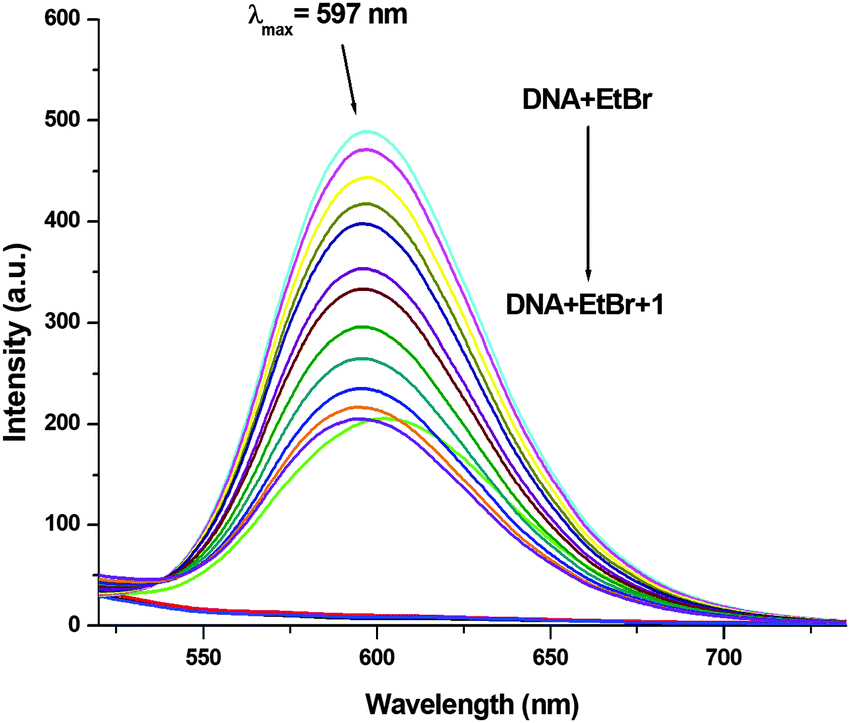
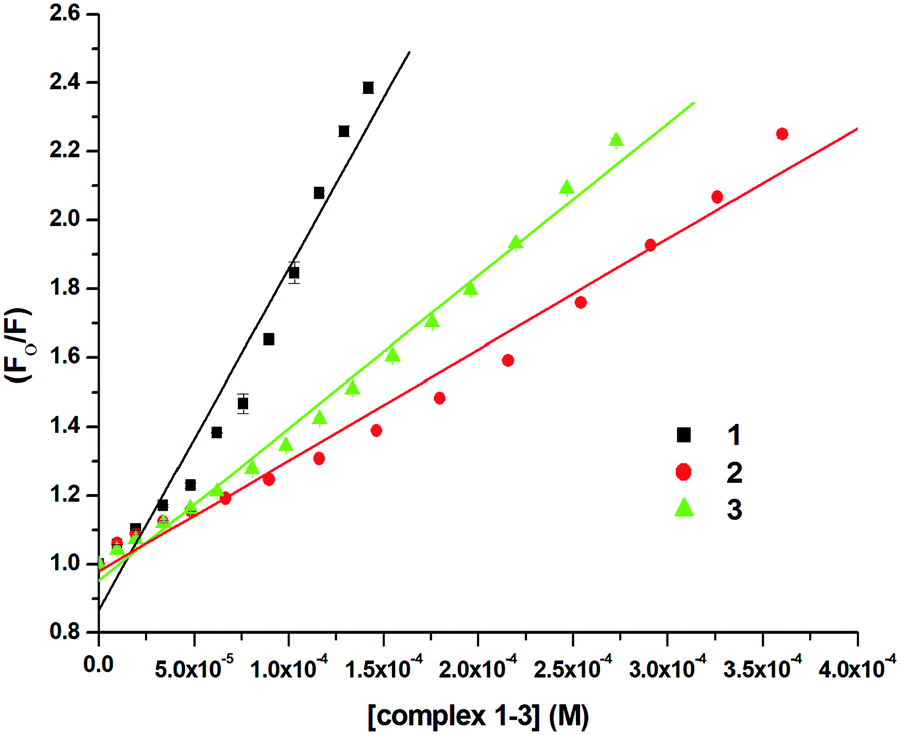
In order to examine the interaction of 1–3 with CT-DNA, ethidium bromide (EthBr) was employed as an intercalator of CT-DNA. EthBr is a well-known DNA intercalator, which does not show fluorescence in its free-state. The ability of EthBr to intercalate into the DNA helix allows it to show strong fluorescence in the bound state with CT-DNA.94,95 Our experiment showed significant quenching in the fluorescence intensity of the DNA–EthBr complex with increasing concentrations of 1–3 (0–140 μM). This was due to the displacement of EthBr from the EthBr–DNA complex. The EthBr–DNA complex was excited at 480 nm, and the emission intensity was monitored at 597 nm. The spectra (Fig. 9) obtained from the above titrations exhibited a noticeable blueshift (2.5 nm) along with a significant decrease in the fluorescence intensity of complex 1. No shift in the peak maxima was observed for complexes 2 and 3 (Fig. S62 and S63, ESI†) though there was significant quenching of the fluorescence intensity (58.08%, 1; 55.15%, 2; 55.56%, 3; Fig. 9 and Fig. S62, S63, ESI†). These results suggested that 1–3 were able to interact with the DNA binding sites strongly and displace EthBr from the DNA–EthBr complex.84,95,96 The DNA–EthBr fluorescence quenching studies for 1–3 were performed following the Stern–Volmer equation:41,88–91
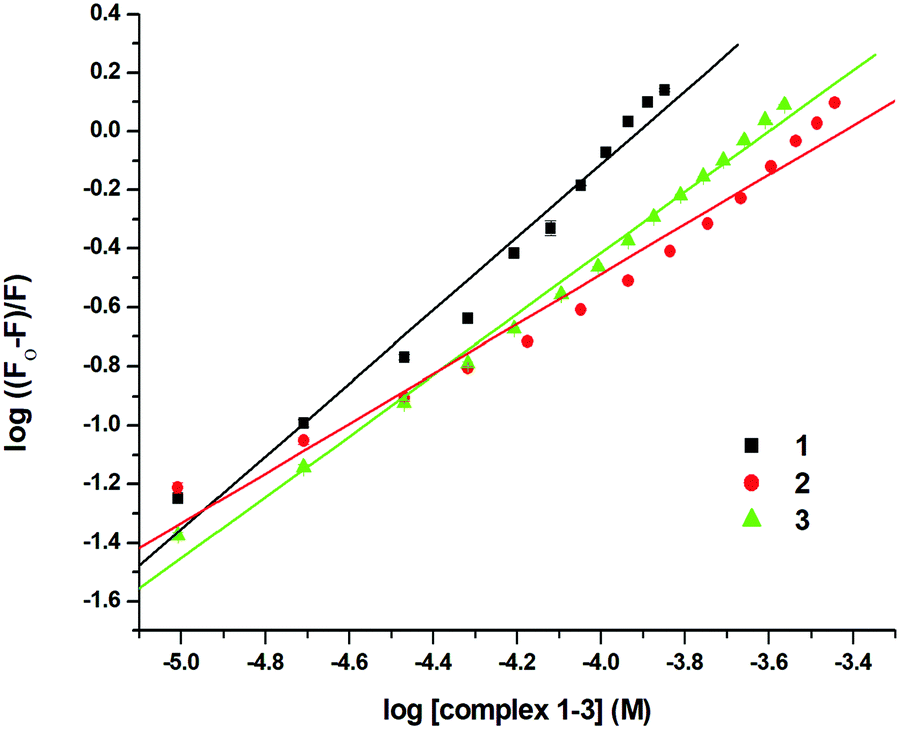
where F0 and F are the emission intensities of EthBr–DNA in the absence and presence of 1–3, respectively, Ksv is the quenching constant, and [Q] is the concentration of 1–3. The nature of the Stern–Volmer plots for 1–3, suggested that the quenching was not only static but also dynamic. In order to verify this, data was also plotted using the modified Stern–Volmer or Scatchard equation:91,97
| log((F0 − F)/F) = logKa + nlog[Q] |
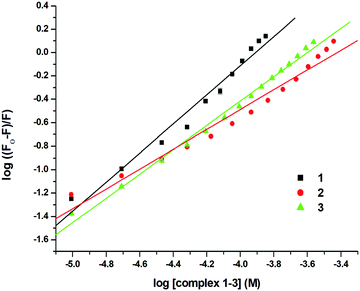
where Ka is the association constant. The values of Ksv, Ka and n for 1–3 were calculated using the above equations and are shown in Fig. 10, 11 and Fig. S64–S69 (ESI†) and Table 5. The values of Ksv and Ka suggested the interaction of 1–3 with DNA in the following order: 1 > 3 > 2.
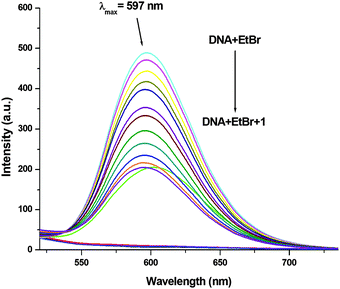 |
| | Fig. 9 Molecular fluorescence emission profile associated with the titration of EthBr–DNA (35 μM; T10E1 buffer, pH ∼ 7.6; λex. 480 nm; λem. 597 nm) with varying concentrations of 1 (0–140 μM; Milli-Q water) at 298 K. Fluorescence spectra of only buffer (black), only DNA (red), only EthBr (green), only 1 (blue) and EthBr–DNA(cyan). | |
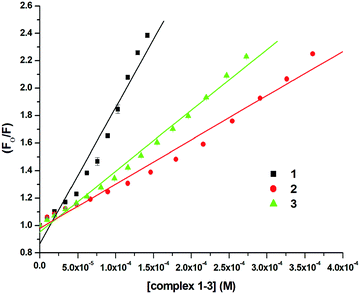 |
| | Fig. 10 Stern–Volmer plot for the calculation of quenching constant (Ksv) associated with the titration of EthBr–DNA and 1–3 at 298 K. | |
 |
| | Fig. 11 Scatchard plot for the calculation of association constant (Ka) for the interaction of EthBr–DNA and 1–3 at 298 K. | |
Table 5 Data from the emission spectroscopic studies on the DNA binding ability of complexes 1–3
| Complexes |
Quenching constant (Ksv) (M−1) |
Association Constant (Ka) (M−1) |
No. of binding site (n) |
Apparent DNA binding constant (Kapp) (M−1) |
|
1
|
9.92 ± 0.055 × 103 |
7.28 ± 0.60 × 104 |
1.24 |
5.13 ± 0.036 × 106 |
|
2
|
3.22 ± 0.016 × 103 |
7.88 ± 0.54 × 102 |
0.84 |
2.24 ± 0.008 × 106 |
|
3
|
4.43 ± 0.009 × 103 |
5.45 ± 0.27 × 103 |
1.03 |
2.96 ± 0.004 × 106 |
Further, the apparent DNA binding constants (Kapp) were calculated using the equation:41,90,97
| KEthBr[EthBr] = Kapp[complexes] |
where
KEthBr is the binding constant of EthBr with DNA, [EthBr] is the concentration of EthBr, and [complexes] is the concentration of
1–3, at which the fluorescence intensity of EthBr–DNA reduces to 50%. The calculated apparent DNA binding constants (
Kapp) are listed in
Table 5, and they follow the order:
1 >
3 >
2. The results obtained from the above studies suggested that
1 was more capable of displacing EthBr from the DNA–EthBr complex, and the complexes followed the trend:
1 >
3 >
2. The values of the calculated parameters also confirm that all three complexes strongly interact with DNA, and other spectroscopic results are in good agreement with those of the EthBr displacement studies by fluorescence quenching.
These studies were also performed for ligands L1, L3 and the Ru(p-cymene) dimer to check the binding affinity (Fig. S70–S78, ESI†). Due to the interference of the intrinsic fluorescence of ligand L2, these studies could not be performed for ligand L2. The calculated quenching constants (Ksv) (1.51 ± 0.023 × 103 for L1; 2.34 ± 0.033 × 103, for L3; 1.35 ± 0.027 × 103, for Ru(p-cymene) dimer) and association constants (Ka) (1.10 ± 0.32 × 103 for L1, 1.62 ± 0.06 × 103, for L3; 4.01 ± 0.67 × 102, for Ru(p-cymene) dimer) are shown in Table S8 (ESI†). The quenching constants (Ksv) and association constants (Ka) of ligands L1, L3 and Ru(p-cymene) dimer were less compared to the respective complexes 1 and 3. This showed that ligands L1, L3 and Ru(p-cymene) dimer had poorer ability to displace EthBr form the DNA–EthBr complex compared to their respective complexes.
Overall, it can be understood that complexes 1–3 could interact with DNA with good binding affinities, as determined by the UV-Vis absorption studies and EthBr displacement assays. The binding of complexes 1–3 to DNA is through non-covalent interactions, as understood from the binding energy obtained from the theoretical calculations, as well as the binding constants obtained from the experimental data. Theoretical studies suggest that the complexes aligned with the outer surface of four adjacent base pairs and also with the backbone of the DNA. Thus, groove, as well as surface binding is indicated, and in the preferred conformational structure, van der Waals interactions and hydrogen bonding probably play the major role. EthBr displacement studies suggest the intercalation of the complexes, inducing direct displacement, with the complexes 1–3 replacing EthBr from the EthBr–DNA complex. However, indirect displacement is possible if complexes 1–3 induce conformational changes in order to fit in the structure (groove and/or surface) of DNA, which can also contribute to breaking the EthBr–DNA complex. Ligands L1–L3 showed stronger binding with DNA than their corresponding Ru complexes 1–3, probably through electrostatic interaction and hydrogen bonding interactions. Since ligands L1–L3 are planar in nature, capable of intercalation and smaller in size, they can easily approach the binding sites available on the DNA. Therefore, the stronger binding interaction of ligands L1–L3 with DNA compared to their complexes 1–3 is understandable.
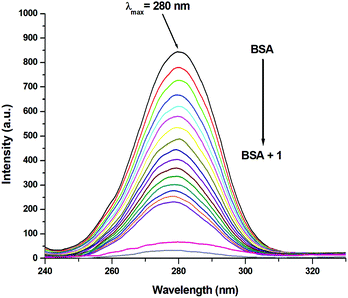
3.10 Protein interaction studies
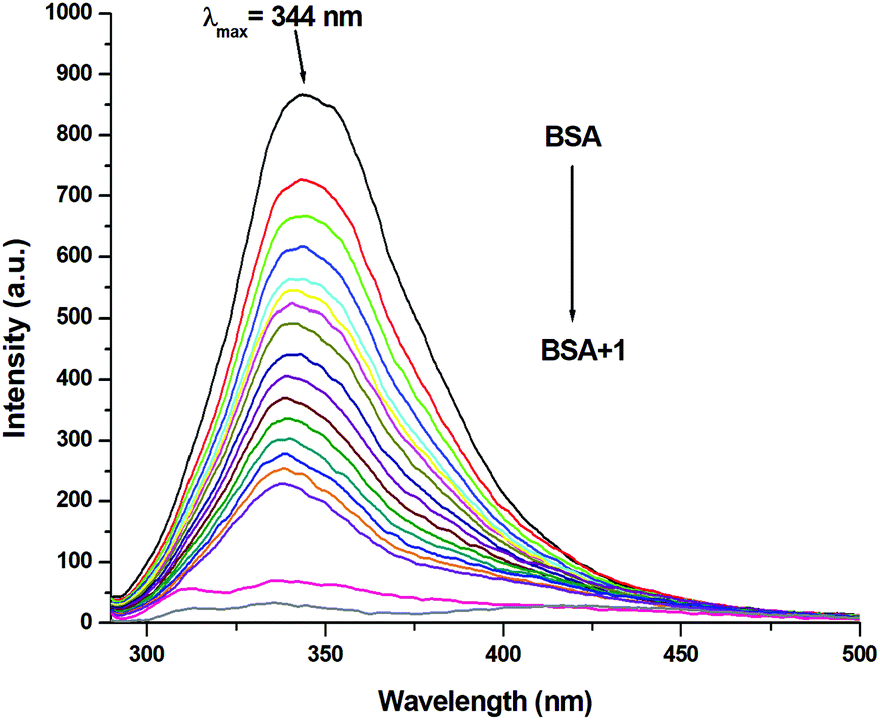
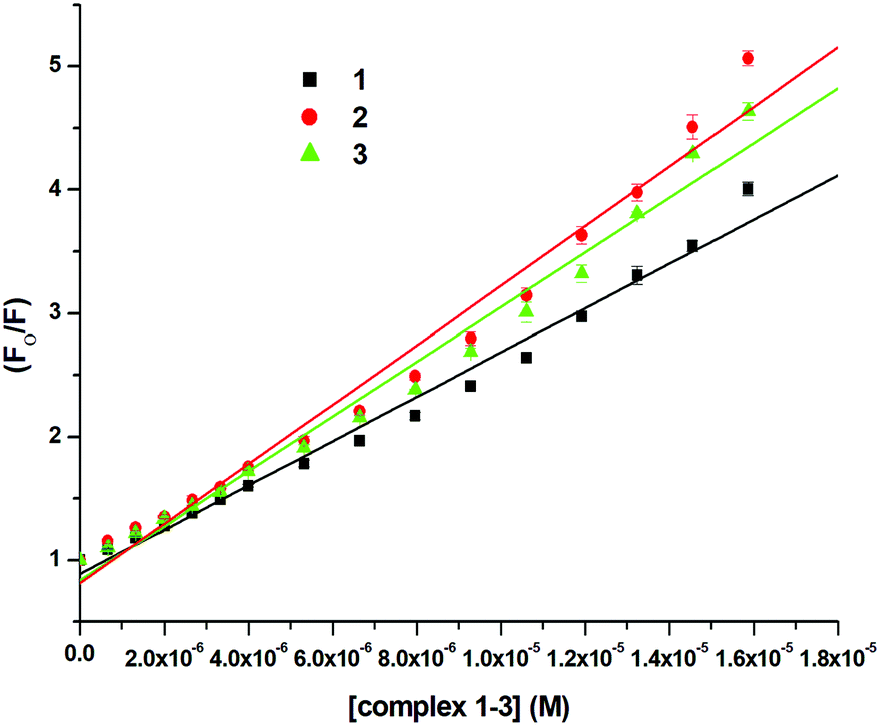
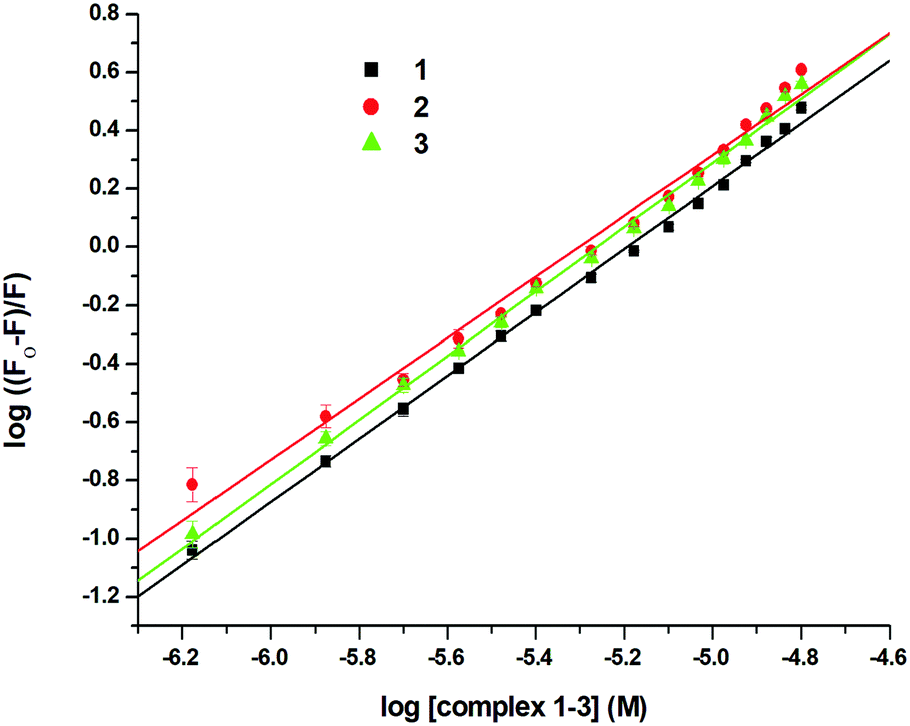
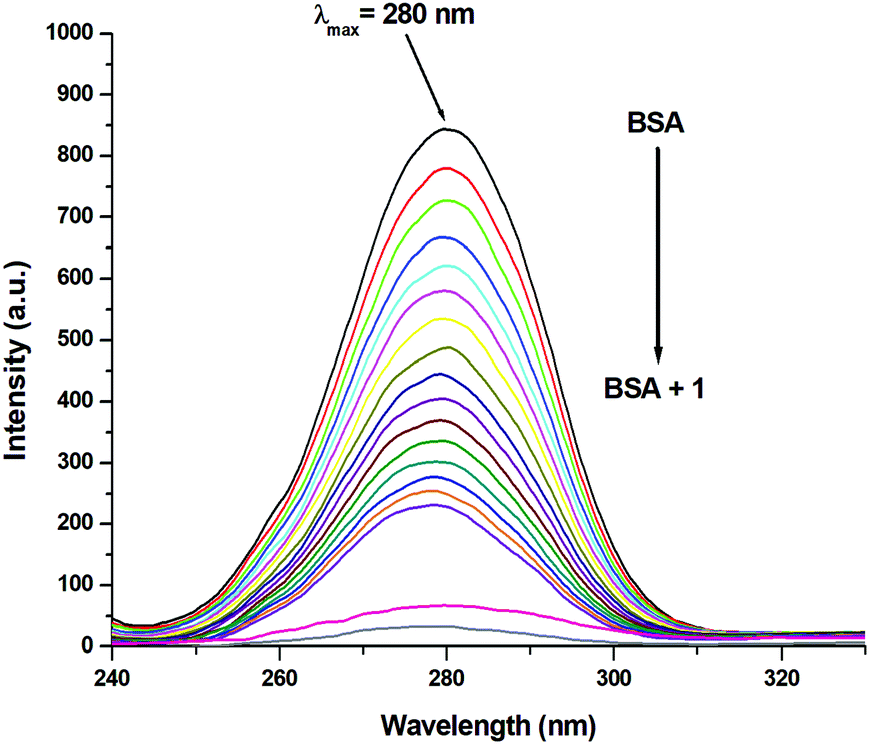
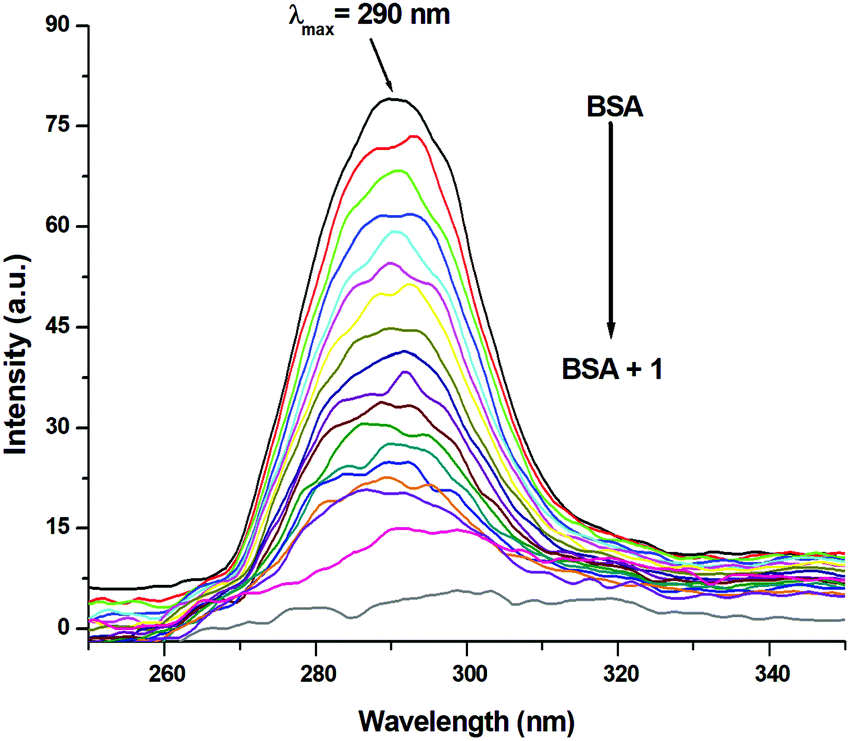
Since the structure of bovine serum albumin (BSA) is comparable to that of human serum albumin (HSA), it has been extensively studied to reveal its roles in drug transport and metabolism.98,99 The emission property of BSA originates from the tryptophan, tyrosine and phenylalanine residues present in the protein.100,101 Interaction of compounds with BSA can lead to significant quenching of the fluorescence intensity. This quenching can be attributed to various phenomena, such as protein conformational changes, substrate binding, subunit association and denaturation of the protein.102,103 Fluorescence quenching studies were carried out to understand the protein binding profile of 1–3 using bovine serum albumin (BSA). A representative fluorescence titration profile accompanying the interaction of BSA (c, 0.15 μM, in PBS) with increasing concentrations of 1 (c, 0–16 μM, in PBS) is shown in Fig. 12. These fluorescence titrations were also performed for 2 and 3 under similar experimental conditions. The excitation of BSA at 280 nm led to a strong emission peak at 344 nm in the fluorescence spectra. With an increasing concentration of 1–3, the band at 344 nm was quenched remarkably, showing a blueshift (75.04%, 6 nm for 1, 80.26%, 7 nm for 2 and 78.42%, 6 nm for 3; Fig. 12 and Fig. S79, S80, ESI†). These results suggested that complexes 1–3 strongly bound to the interaction sites on BSA.98–103 An additional fluorescence band was observed around 387 nm, which increased with an increasing concentration of 2. This was due to the degradation of complex 2 in the medium. The rate of this process was observed to be very slow and we could easily accomplish the experiment with no significant effect on the results. The quenching constant or Stern–Volmer constant (Ksv) was calculated from the F0/F vs. [Q] plot using the Stern–Volmer equation given below:41,88–91
The equilibrium binding constant (Kb) for this bio-molecular binding event was evaluated using Scatchard equation:91,97
| log((F0 − F)/F) = logKb + nlog[Q] |
where F0 and F are the emission intensities of BSA in the absence and presence of 1–3, respectively, Ksv is the quenching constant, Kb is the equilibrium binding constant, and [Q] is the concentration of 1–3. The values of Ksv, Kb and n for 1–3 are listed in Table 6, and all graphs associated with the calculations are shown in Fig. 13, 14 and Fig. S82–S88 (ESI†). The values of Ksv and Kb suggested the interaction of 1–3 with BSA and followed the order 1 > 3 > 2. The value of n obtained from calculations was ∼1, suggesting a single binding site for the interaction of 1–3 with BSA.
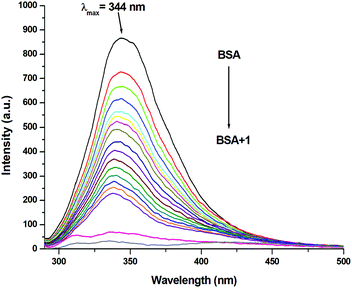 |
| | Fig. 12 Molecular fluorescence emission profile associated with titration of BSA (0.15 μM; PBS, pH ∼ 7.4; λex. 280 nm; λem. 344 nm) with varying concentrations of 1 (0–16 μM; PBS, pH ∼ 7.4) at 298 K. Fluorescence spectra of BSA (black), only buffer (pink) and only complex (grey). | |
Table 6 Data from the emission spectroscopic studies on the interaction of BSA with complexes 1–3
| Complexes |
Quenching constant (Ksv) (M−1) |
Binding constant (Kb) (M−1) |
No. of binding site (n) |
|
1
|
1.79 ± 0.026 × 105 |
4.21 ± 0.88 × 105 |
1.08 |
|
2
|
2.41 ± 0.040 × 105 |
3.63 ± 1.29 × 105 |
1.04 |
|
3
|
2.21 ± 0.019 × 105 |
6.52 ± 1.66 × 105 |
1.10 |
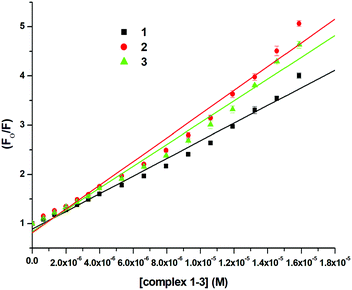 |
| | Fig. 13 Stern–Volmer plot for the calculation of quenching constant (Ksv) associated with the interaction of BSA and 1–3 at 298 K. | |
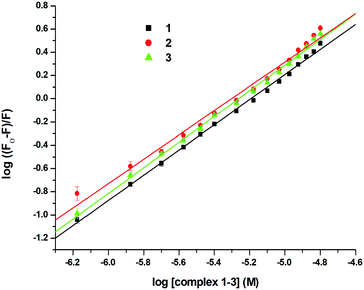 |
| | Fig. 14 Scatchard plot for the calculation of binding constant (Kb) associated with the interaction of BSA and 1–3 at 298 K. | |
In addition to the Ru complexes 1–3, the protein interactions of ligands L1–L3 and Ru(p-cymene) dimer were also investigated for direct comparison with the complexes. Ligands L1–L3 could not be studied since they exhibited interference due to their intrinsic fluorescence overlapping with BSA emission. However, the protein interaction of Ru(p-cymene) dimer was studied (Fig. S81, ESI†), and the binding parameters could be calculated (Ksv = 3.32 ± 0.099 × 105; Kb = 4.73 ± 0.84 × 107 and n = 1.42, Fig. S85, S89 and Table S9, ESI†). The Ksv and Kb values obtained for the Ru(p-cymene) dimer are higher compared to the values obtained for the complexes 1–3. This is likely due to its smaller size, the participation of the two p-cymenes and the two chlorides in non-covalent interactions, and the presence of two metal centers in the Ru(p-cymene) dimer. The higher value for binding sites (n) also supports this theory.
Additionally, the UV-Vis absorption spectra of BSA in the presence of 1–3, L1–L3 and Ru(p-cymene) dimer were also recorded. UV-Vis absorption spectra are given in ESI† (Fig. S90 and S91). The results support the observations from the emission spectroscopic studies. The increase in the absorption band at 280 nm was assigned to the formation of BSA-compound adducts, which indicates static quenching.41 In dynamic quenching, short-lived excited states may be formed due to collisions, which do not change the nature of the absorption bands.41
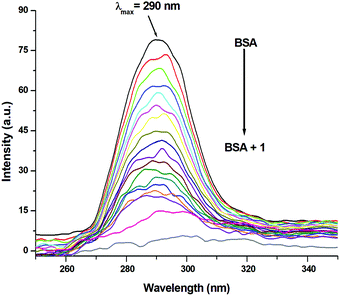
3.11 Involvement of tryptophan and tyrosine residues in the binding
Synchronous emission spectra were employed to investigate the changes in the microenvironment and conformation of BSA during its interaction with 1–3. Synchronous fluorescence spectrometry is a powerful investigative tool to understand the microenvironment of multicomponent systems.104,105 It provides information regarding the sites, where conformational changes occur. At small Δλ (15 nm) values, tyrosine residues contribute to the characteristic synchronous spectrum of BSA protein, while at large Δλ (60 nm) values, the characteristic synchronous spectrum of BSA is due to the tryptophan residues.105,106 Fluorescence titration profiles resulting from the interaction of BSA (c, 0.15 μM, in PBS) with increasing concentrations of 1 (c, 0–16 μM, in PBS) are shown in Fig. 15 and 16. Identical studies were carried out for 2, 3 and Ru(p-cymene) dimer under the same experimental conditions (Fig. S92–S97, ESI†). Synchronous emission spectra could not be recorded for ligands L1–L3 due to the interference of their intrinsic fluorescence. Using a large Δλ (60 nm) value for the synchronous fluorescence spectroscopy of BSA led to a strong emission at 280 nm. This band intensity decreased continuously (72.94% for 1; 81.44% for 2; 77.81% for 3 and 82.32% for Ru(p-cymene)dimer) with the addition of incremental amounts of 1–3, as shown in Fig. 15 and Fig. S92–S94 (ESI†). These results strongly suggested the participation of the tryptophan moiety in the binding event. Similarly, the fluorescence intensity of spectra having a small Δλ (15 nm) value at 290 nm also diminished with the addition of increasing concentrations of 1–3 (74.52% for 1; 73.27% for 2; 74.45% for 3 and 66.27% for Ru(p-cymene) dimer), as shown in Fig. 16 and Fig. S95–S97 (ESI†). It also indicated that the tyrosine moiety was involved in binding. Since the spectra show that the quenching of fluorescence occurs due to both residues, this is indicative of strong binding occurring along with conformational changes, hydrophobicity enhancement and reduced polarity of the BSA molecule.41,105,106 At both large and small Δλ values, complex 2 showed an additional synchronous fluorescence band around 330 nm and 350 nm, respectively, due to the displacement of the ligand by water present in the media. However, we easily managed to carry out the experiment with no significant effect on the results as the rate of this process was very slow.
 |
| | Fig. 15 Synchronous fluorescence emission profile associated with the titration of BSA (0.15 μM; PBS, pH ∼ 7.4) with varying concentrations of 1 (0–16 μM; PBS, pH ∼ 7.4) at the wavelength difference of Δλ = 60 nm. Fluorescence spectra of BSA (black), only buffer (pink) and only complex (grey). | |
 |
| | Fig. 16 Synchronous fluorescence emission profile associated with titration of BSA (0.15 μM; PBS, pH ∼ 7.4) with varying concentrations of 1 (0–16 μM; PBS, pH ∼ 7.4) at the wavelength difference of Δλ = 15 nm. Fluorescence spectra of BSA (black), only buffer (pink) and only complex (grey). | |
3.12 Anticancer activity
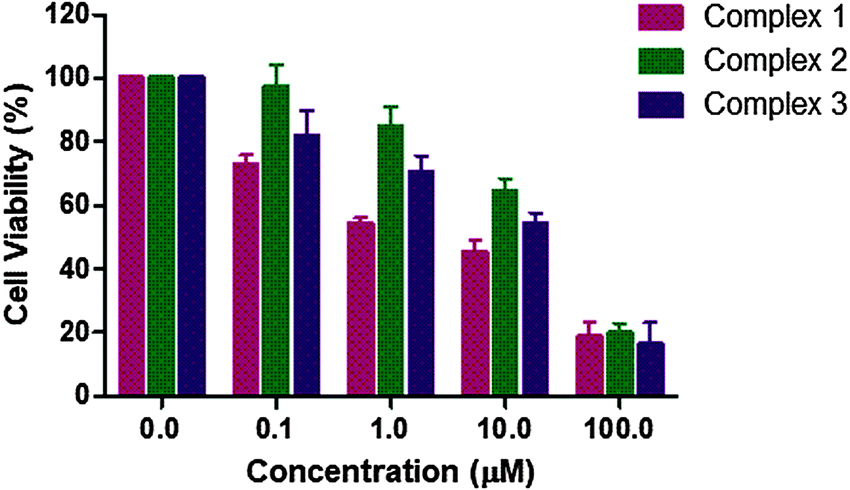
In order to check the antiproliferative activity of the Ru complexes 1–3 along with L1–L3, cell toxicity studies were performed against the MCF-7 breast cancer cell line using the MTT assay. The results are shown in Fig. 17 and Table 7. All experiments included untreated cells as the control. The anticancer activity was determined using a concentration range of 0–100 μM for the complexes. The cell viability graphs of the complexes 1–3, ligands L1–L3 and Ru(p-cymene) dimer, along with the graphs corresponding to the calculation of IC50 values, are demonstrated in Fig. S98 and S99 (ESI†). The IC50 values of the two stable complexes 1 and 3 were found to be 2.642(2) μM and 7.359(2) μM, respectively. The IC50 values of both these complexes were better than their corresponding ligands L1 [6.087(5) μM] and L3 [9.019(4) μM]. Complex 1 was found to exhibit a lower IC50 value compared to that of Ru(p-cymene) dimer [5.271(2) μM], while complex 3 showed a slightly higher IC50 value. For the Ru complex 2, which is unstable in PBS and DMSO, the IC50 value of 18.211(2) μM was obtained. It is surprisingly higher than that of its ligand L2, which has a very low IC50 value of 2.606(6) μM. It is worth mentioning that the IC50 values of such ligands vary from 1.62–124 μM, as reported previously against another cancer cell line.111 The decreased activity of complex 2 may be due to low stability in DMSO-containing media, in which the stock solutions were prepared. The NMR spectrum of complex 2 in DMSO-d6 showed that it was unstable, and upon degradation, the p-cymene peaks increased with time, whereas those of free L2 did not. This may be the reason for the decreased cytotoxicity of complex 2. Interestingly, all the three ligands L1–L3 and their Ru complexes 1–3 were active against the MCF-7 cell line. It should be noticed that although ligands L1–L3, as well as the Ru(p-cymene) dimer, were found to have good IC50 values, the design and synthesis of the complexes are still important, especially for selectivity, stability and good aqueous solubility. As observed in these studies, ligands (L1–L3) are not water-soluble, while the Ru complexes (1–3) are. It is also worth mentioning that the observed trend of their binding affinities towards DNA and BSA is in-line with the cell toxicity data. Overall, the complexes, especially 1 and 3, ligands L1–L3 and Ru(p-cymene) dimer exhibit good IC50 values, comparable to similar compounds previously reported.107–111 A comparison of the anticancer activity of the compounds reported in this study against the MCF-7 cell line and other similar complexes reported previously is given below for comprehensive understanding (Table 7).
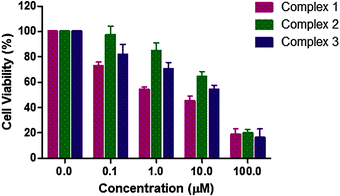 |
| | Fig. 17
In vitro cytotoxicity of complexes 1–3 against MCF-7 cells measured by the MTT assay after 48 h. | |
Table 7 Comparison of the IC50 values (in μM) of ligands (L1–L3), Ru(p-cymene)dimer, complexes 1–3 and previously reported Ru complexes against cancer cell lines
| S. no. |
Compounds |
Chemical structure |
IC50 value (μM) |
Ref. |
|
MCF-7-breast cancer.
MDA MB231-breast cancer.
A549-lung cancer.
|
| 1 |
Complex 1 |
|
2.64a |
This Work |
| 2 |
Complex 2 |
|
18.21a |
This Work |
| 3 |
Complex 3 |
|
7.36a |
This Work |
| 4 |
Ru(p-cymene)dimer |
|
5.27a |
This Work |
| 5 |
L1
|
|
6.09a |
This Work |
| 6 |
L2
|
|
2.61a |
This Work |
| 7 |
L3
|
|
9.02a |
This Work |
| 8 |
Cisplatin |
|
21.62a |
112
|
| 9 |
Ruthenium pyridinylmethylene scaffolds |
|
7.76–25.42a |
107 and 109
|
| 10 |
Ru p-cymene NHC |
|
2.4–500a |
113
|
| 11 |
Aryldiazole |
|
26–300a |
114
|
| 12 |
Carbosilane |
|
2.5–54a |
115
|
| 13 |
Aroylhydrazone |
|
10.9–15.8a |
116
|
| 14 |
Heteroarenyl-benzimidazoles |
|
2.04–79.43b |
111
|
| 1.62–124.90c |
4. Conclusion
In conclusion, three half-sandwich Ru(II) arene chlorido complexes 1–3 are presented along with a detailed investigation of their spectroscopic, analytical and biophysical properties. Electrochemical studies established that the higher oxidation states of these complexes are achievable only at very high redox potentials and the Ru(II) state is quite stable. The structure and geometry optimization of 1–3 was carried out using the density functional theory (DFT), the results of which are in-line with the experimental data. Theoretical calculations using the PM6 method revealed structural features and binding energies, which suggest that complexes 1 and 2 bind with DNA through groove and surface interactions. Since the investigated complexes 1-DNA and 2-DNA are extremely large for any level of quantum chemical methods, the semi-empirical PM6 method, which is more accurate than the force-field methods, is used for a better understanding of the complex-DNA structure and interactions and for a fair balance between accuracy and computational time. In addition, the binding interactions of 1–3 with CT-DNA and BSA protein were studied in detail using experimental methods, which reveal a strong affinity of these complexes towards CT-DNA and BSA. Interestingly, the in vitro cell viability studies of 1–3 against breast cancer (MCF-7) cells revealed their good anticancer potency. The anticancer activities of complexes 1–3, ligands L1–L3, and Ru(p-cymene) dimer against the MCF-7 cells were in good correlation with their binding behavior and stability. The order of activity of 1–3 in the cytotoxicity studies, as well as the DNA and BSA binding studies, was observed to be 1 > 3 > 2. Further studies to evaluate the anticancer potential of these molecules on other cell lines and in animal models will be useful.
Conflicts of interest
The authors declare that there are no competing financial interests.
Acknowledgements
AKS acknowledges University Grants commission for UGC-BSR Research Start-up-grant (Ref. No. F.30.414/2018(BSR)); Science and Engineering Research Board for financial support (grant ref.: EMR/2016/001452) and DST-INSPIRE research grant (IFA-13, CH-97). TAK is grateful to the UGC for NET-SRF; KB is grateful to SERB for NPDF (PDF/2017/000929) fellowships. Authors acknowledge the instrumental facilities at Department of Chemistry, Central University of Rajasthan supported by DST-FIST (ref. no. SR/FST/CSI-257/2014(C)). We are thankful to Department of Chemistry, IIT, Jodhpur for X-ray crystallographic facilities. We are also thankful to Mr Abhijeet Singh and Prof. Manoj Kumar Bhat, NCCS, Pune, India and Dr Subbaiah Edupalli, Central University of Rajasthan, India for helping with the cell toxicity studies. Authors dedicate the manuscript to Late Dr Sunil G. Naik for his support in the initiation of this work.
References
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- J. Furrer and G. Süss-Fink, Coord. Chem. Rev., 2016, 309, 36–50 CrossRef CAS.
- A. K. Singh, D. S. Pandey, Q. Xu and P. Braunstein, Coord. Chem. Rev., 2014, 270, 31–56 CrossRef.
- A. F. Peacock and P. J. Sadler, Chem. – Asian J., 2008, 3, 1890–1899 CrossRef CAS PubMed.
- S. Banerjee and A. R. Chakravarty, Acc. Chem. Res., 2015, 48, 2075–2083 CrossRef CAS PubMed.
- A. Gatti, A. Habtemariam, I. Romero-Canelón, J.-I. Song, B. Heer, G. J. Clarkson, D. Rogolino, P. J. Sadler and M. Carcelli, Organometallics, 2018, 37, 891–899 CrossRef CAS.
- P. J. Jarman, F. Noakes, S. Fairbanks, K. Smitten, I. K. Griffiths, H. K. Saeed, J. A. Thomas and C. Smythe, J. Am. Chem. Soc., 2019, 141(7), 2925–2937 CrossRef CAS PubMed.
- A. P. Basto, N. Anghel, R. Rubbiani, J. Müller, D. Stibal, F. Giannini, G. Süss-Fink, V. Balmer, G. Gasser and J. Furrer, Metallomics, 2019, 11, 462–474 RSC.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS PubMed.
- V. Ramu, S. Gautam, A. Garai, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2018, 57, 1717–1726 CrossRef CAS PubMed.
- H. Shi, I. Romero-Canelón, M. Hreusova, O. Novakova, V. Venkatesh, A. Habtemariam, G. J. Clarkson, J.-i. Song, V. Brabec and P. J. Sadler, Inorg. Chem., 2018, 57, 14409–14420 CrossRef CAS PubMed.
- K. Suntharalingam, W. Lin, T. C. Johnstone, P. M. Bruno, Y.-R. Zheng, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 14413–14416 CrossRef CAS PubMed.
- K. Suntharalingam, S. G. Awuah, P. M. Bruno, T. C. Johnstone, F. Wang, W. Lin, Y.-R. Zheng, J. E. Page, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 2967–2974 CrossRef CAS PubMed.
- J. P. Coverdale, H. E. Bridgewater, J.-I. Song, N. A. Smith, N. P. Barry, I. Bagley, P. J. Sadler and I. Romero-Canelón, J. Med. Chem., 2018, 61, 9246–9255 CrossRef CAS PubMed.
- F. Chen, I. Romero-Canelón, J. J. Soldevila-Barreda, J.-I. Song, J. P. Coverdale, G. J. Clarkson, J. Kasparkova, A. Habtemariam, M. Wills, V. Brabec and P. J. Sadler, Organometallics, 2018, 37, 1555–1566 CrossRef CAS PubMed.
- M. K. Raza, S. Gautam, P. Howlader, A. Bhattacharyya, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2018, 57, 14374–14385 CrossRef CAS PubMed.
- A. J. Wagstaff, A. Ward, P. Benfield and R. C. Heel, Drugs, 1989, 37, 162–190 CrossRef CAS PubMed.
- E. Raymond, S. Chaney, A. Taamma and E. Cvitkovic, Ann. Oncol., 1998, 9, 1053–1071 CrossRef CAS PubMed.
- M. Shimada, H. Itamochi and J. Kigawa, Cancer Manage. Res., 2013, 5, 67 CrossRef CAS PubMed.
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed.
- K. Irena, Curr. Med. Chem., 2006, 13, 1085–1107 CrossRef PubMed.
- J.-W. Chen and C.-C. Chang, ACS Appl. Mater. Interfaces, 2016, 8, 29883–29892 CrossRef CAS PubMed.
- S. Mukhopadhyay, R. K. Gupta, R. P. Paitandi, N. K. Rana, G. Sharma, B. Koch, L. K. Rana, M. S. Hundal and D. S. Pandey, Organometallics, 2015, 34, 4491–4506 CrossRef CAS.
- R. Aird, J. Cummings, A. Ritchie, M. Muir, R. Morris, H. Chen, P. Sadler and D. Jodrell, Br. J. Cancer, 2002, 86, 1652 CrossRef CAS PubMed.
- H. Mishra and R. Mukherjee, J. Organomet. Chem., 2010, 695, 1753–1760 CrossRef CAS.
- R. P. Paitandi, S. Mukhopadhyay, R. S. Singh, V. Sharma, S. M. Mobin and D. S. Pandey, Inorg. Chem., 2017, 56, 12232–12247 CrossRef CAS PubMed.
- Y.-J. Liu, C.-H. Zeng, Z.-H. Liang, J.-H. Yao, H.-L. Huang, Z.-Z. Li and F.-H. Wu, Eur. J. Med. Chem., 2010, 45, 3087–3095 CrossRef CAS PubMed.
- A. Bergamo, C. Gaiddon, J. Schellens, J. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- P.-S. Kuhn, V. Pichler, A. Roller, M. Hejl, M. Jakupec, W. Kandioller and B. Keppler, Dalton Trans., 2015, 44, 659–668 RSC.
- S. W. Chang, A. R. Lewis, K. E. Prosser, J. R. Thompson, M. Gladkikh, M. B. Bally, J. J. Warren and C. J. Walsby, Inorg. Chem., 2016, 55, 4850–4863 CrossRef CAS PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- F. Wang, A. Habtemariam, E. P. van der Geer, R. Fernández, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. Fabbiani and P. Lozano-Casal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- S. W. Magennis, A. Habtemariam, O. Novakova, J. B. Henry, S. Meier, S. Parsons, I. D. Oswald, V. Brabec and P. J. Sadler, Inorg. Chem., 2007, 46, 5059–5068 CrossRef CAS PubMed.
- T. Bugarcic, O. Nováková, L. Zerzánková, O. i. Vrána, J. Kašpárková, A. Habtemariam, S. Parsons, P. J. Sadler and V. Brabec, J. Med. Chem., 2008, 51, 5310–5319 CrossRef CAS PubMed.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734–17743 CrossRef CAS PubMed.
- F. J. Ballester, E. Ortega, V. Porto, H. Kostrhunova, N. Davila-Ferreira, D. Bautista, V. Brabec, F. Domínguez, M. D. Santana and J. Ruiz, Chem. Commun., 2019, 55, 1140–1143 RSC.
- Q. Du, Y. Yang, L. Guo, M. Tian, X. Ge, Z. Tian, L. Zhao, Z. Xu, J. Li and Z. Liu, Dyes Pigm., 2019, 162, 821–830 CrossRef CAS.
- P. R. Florindo, D. M. Pereira, P. M. Borralho, C. M. Rodrigues, M. Piedade and A. C. Fernandes, J. Med. Chem., 2015, 58, 4339–4347 CrossRef CAS PubMed.
- A. Kumar, A. Kumar, R. K. Gupta, R. P. Paitandi, K. B. Singh, S. K. Trigun, M. S. Hundal and D. S. Pandey, J. Organomet. Chem., 2016, 801, 68–79 CrossRef CAS.
- S. Mokesch, D. Schwarz, M. Hejl, M. H. Klose, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Molecules, 2019, 24, 2373 CrossRef CAS PubMed.
- M. Ramesh and G. Venkatachalam, J. Organomet. Chem., 2019, 880, 47–55 CrossRef CAS.
- R. Dayam, F. Aiello, J. Deng, Y. Wu, A. Garofalo, X. Chen and N. Neamati, J. Med. Chem., 2006, 49, 4526–4534 CrossRef CAS PubMed.
- K. D. Upadhyay, N. M. Dodia, R. C. Khunt, R. S. Chaniara and A. K. Shah, ACS Med. Chem. Lett., 2018, 9, 283–288 CrossRef CAS PubMed.
- O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871–910 CrossRef CAS PubMed.
- J. Camacho, A. Barazarte, N. Gamboa, J. Rodrigues, R. Rojas, A. Vaisberg, R. Gilman and J. Charris, Bioorg. Med. Chem., 2011, 19, 2023–2029 CrossRef CAS PubMed.
- M. L. Richards, S. C. Lio, A. Sinha, K. K. Tieu and J. C. Sircar, J. Med. Chem., 2004, 47, 6451–6454 CrossRef CAS PubMed.
- A. Sharma, V. Luxami and K. Paul, Bioorg. Med. Chem. Lett., 2013, 23, 3288–3294 CrossRef CAS PubMed.
- Y. Tong, J. J. Bouska, P. A. Ellis, E. F. Johnson, J. Leverson, X. Liu, P. A. Marcotte, A. M. Olson, D. J. Osterling and M. Przytulinska, J. Med. Chem., 2009, 52, 6803–6813 CrossRef CAS PubMed.
-
D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, New York, 1st edn, 1988 Search PubMed.
-
B. S. Furniss, Vogel's textbook of practical organic chemistry, Pearson Education India, 1989 Search PubMed.
- J. Hu, J. Li, J. Qi and J. Chen, New J. Chem., 2015, 39, 843–848 RSC.
- T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, J. Am. Chem. Soc., 2012, 135, 118–121 CrossRef PubMed.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Eur. J. Inorg. Chem., 2013, 4558–4562 CrossRef.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC.
- O. Dayan, M. Tercan and N. Özdemir, J. Mol. Struct., 2016, 1123, 35–43 CrossRef CAS.
- A. Gilewska, B. Barszcz, J. Masternak, K. Kazimierczuk, J. Sitkowski, J. Wietrzyk and E. Turlej, JBIC, J. Biol. Inorg. Chem., 2019, 1–16 Search PubMed.
- R. K. Gupta, R. Pandey, G. Sharma, R. Prasad, B. Koch, S. Srikrishna, P.-Z. Li, Q. Xu and D. S. Pandey, Inorg. Chem., 2013, 52, 3687–3698 CrossRef CAS PubMed.
- SAINT: SAX Area-Detector Integration Program, Siemens Industrial Automation, Inc., 1995.
- SADABS: Siemens Area Detector Absorption Correction Program, Bruker AXS Inc., 1998.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. v. d. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji and X. Li, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- Z. Bikadi and E. Hazai, J. Cheminf., 2009, 1, 15 Search PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, Chem. – Eur. J., 2013, 19, 14768–14772 CrossRef CAS PubMed.
- F. Aman, M. Hanif, W. A. Siddiqui, A. Ashraf, L. K. Filak, J. h. Reynisson, T. Söhnel, S. M. Jamieson and C. G. Hartinger, Organometallics, 2014, 33, 5546–5553 CrossRef CAS.
- O. Mazuryk, K. Magiera, B. Rys, F. Suzenet, C. Kieda and M. Brindell, JBIC, J. Biol. Inorg. Chem., 2014, 19, 1305–1316 CrossRef CAS PubMed.
- R. W. Horobin, J. C. Stockert and F. Rashid-Doubell, Histochem. Cell Biol., 2006, 126, 165–175 CrossRef CAS PubMed.
- A. Skoczynska, M. Małecka, M. Cieslak, J. Kazmierczak-Baranska, K. Krolewska-Golinska, A. Leniart and E. Budzisz, Polyhedron, 2017, 127, 307–314 CrossRef CAS.
- W. Ginzinger, G. Mühlgassner, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, M. Reithofer, W. Berger and B. K. Keppler, J. Med. Chem., 2012, 55, 3398–3413 CrossRef CAS PubMed.
- M. Auzias, B. Therrien, G. Süss-Fink, P. Štěpnička, W. H. Ang and P. J. Dyson, Inorg. Chem., 2008, 47, 578–583 CrossRef CAS PubMed.
- M. Yadav, A. K. Singh, B. Maiti and D. S. Pandey, Inorg. Chem., 2009, 48, 7593–7603 CrossRef CAS PubMed.
- F. Marchetti, C. Pettinari, R. Pettinari, A. Cerquetella, L. M. Martins, M. F. C. Guedes da Silva, T. F. Silva and A. J. Pombeiro, Organometallics, 2011, 30, 6180–6188 CrossRef CAS.
- Z.-C. Liu, B.-D. Wang, Z.-Y. Yang, Y. Li, D.-D. Qin and T.-R. Li, Eur. J. Med. Chem., 2009, 44, 4477–4484 CrossRef CAS PubMed.
- D. Herebian and W. S. Sheldrick, J. Chem. Soc., Dalton Trans., 2002, 966–974 RSC.
- N. Grover, T. W. Welch, T. A. Fairley, M. Cory and H. H. Thorp, Inorg. Chem., 1994, 33, 3544–3548 CrossRef CAS.
- A. Paul, R. K. Gupta, M. Dubey, G. Sharma, B. Koch, G. Hundal, M. S. Hundal and D. S. Pandey, RSC Adv., 2014, 4, 41228–41236 RSC.
- K. Ghosh, P. Kumar, N. Tyagi and U. P. Singh, Inorg. Chem., 2010, 49, 7614–7616 CrossRef CAS PubMed.
- S. Baskaran, M. Murali Krishnan and M. N. Arumugham, J. Coord. Chem., 2015, 68, 4395–4407 CrossRef CAS.
- R. Pettinari, F. Marchetti, A. Petrini, C. Pettinari, G. Lupidi, B. Fernández, A. R. Diéguez, G. Santoni and M. Nabissi, Inorg. Chim. Acta, 2017, 454, 139–148 CrossRef CAS.
- S. Jain, T. A. Khan, Y. P. Patil, D. Pagariya, N. Kishore, S. Tapryal, A. D. Naik and S. G. Naik, J. Photochem. Photobiol., B, 2017, 174, 35–43 CrossRef CAS.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS PubMed.
- M. T. Carter, M. Rodriguez and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 8901–8911 CrossRef CAS.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- F. M. Almes, Biochemistry, 1993, 32, 4246–4253 CrossRef PubMed.
- M. Howe-Grant, K. C. Wu, W. R. Bauer and S. J. Lippard, Biochemistry, 1976, 15, 4339–4346 CrossRef CAS PubMed.
- X.-B. Fu, D.-D. Liu, Y. Lin, W. Hu, Z.-W. Mao and X.-Y. Le, Dalton Trans., 2014, 43, 8721–8737 RSC.
- E. Ramachandran, D. S. Raja, N. S. Bhuvanesh and K. Natarajan, Dalton Trans., 2012, 41, 13308–13323 RSC.
- D. S. Raja, N. S. Bhuvanesh and K. Natarajan, Dalton Trans., 2012, 41, 4365–4377 RSC.
- C. Dang, R. F. Ebert and W. R. Bell, J. Biol. Chem., 1985, 260, 9713–9719 CAS.
- C.-X. Wang, F.-F. Yan, Y.-X. Zhang and L. Ye, J. Photochem. Photobiol., A, 2007, 192, 23–28 CrossRef CAS.
- M. Eftink and C. Ghiron, J. Phys. Chem., 1976, 80, 486–493 CrossRef CAS.
- D. S. Raja, N. S. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2011, 46, 4584–4594 CrossRef CAS.
- J. Miller, J. Pharm. Biomed. Anal., 1983, 1, 525–535 CrossRef CAS.
- M. E. Pacheco and L. Bruzzone, J. Lumin., 2013, 137, 138–142 CrossRef CAS.
- J. Tang, F. Luan and X. Chen, Bioorg. Med. Chem., 2006, 14, 3210–3217 CrossRef CAS PubMed.
- G. R. Jadhav, S. Sinha, M. Chhabra and P. Paira, Bioorg. Med. Chem. Lett., 2016, 26, 2695–2700 CrossRef CAS.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599 CrossRef CAS PubMed.
- P. Malvi, B. Chaube, V. Pandey, M. V. Vijayakumar, P. R. Boreddy, N. Mohammad, S. V. Singh and M. K. Bhat, Mol. Oncol., 2015, 9, 689–703 CrossRef CAS PubMed.
- M. F. Baig, S. P. Shaik, V. L. Nayak, A. Alarifi and A. Kamal, Bioorg. Med. Chem. Lett., 2017, 27, 4039–4043 CrossRef CAS PubMed.
- P. Liu, B.-Y. Wu, J. Liu, Y.-C. Dai, Y.-J. Wang and K.-Z. Wang, Inorg. Chem., 2016, 55, 1412–1422 CrossRef CAS PubMed.
- F. Hackenberg, H. Müller-Bunz, R. Smith, W. Streciwilk, X. Zhu and M. Tacke, Organometallics, 2013, 32, 5551–5560 CrossRef CAS.
- M. Martínez-Alonso, N. Busto, F. A. Jalón, B. R. Manzano, J. M. Leal, A. M. Rodríguez, B. García and G. Espino, Inorg. Chem., 2014, 53, 11274–11288 CrossRef PubMed.
- M. Maroto-Díaz, B. T. Elie, P. Gómez-Sal, J. Pérez-Serrano, R. Gómez, M. Contel and F. J. de la Mata, Dalton Trans., 2016, 45, 7049–7066 RSC.
- N. Mohan, S. Muthumari and R. Ramesh, J. Organomet. Chem., 2016, 807, 45–51 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data and graphs of elemental analysis, IR spectra, NMR spectra, HRMS, UV-Vis spectra, crystallography figures and parameters, cyclic voltammetry, theoretical calculation graphs and tables, DNA binding data and graphs, BSA binding studies and MTT assay. CCDC 1920293 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj03663f |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b and
Anuj K.
Sharma
*b and
Anuj K.
Sharma
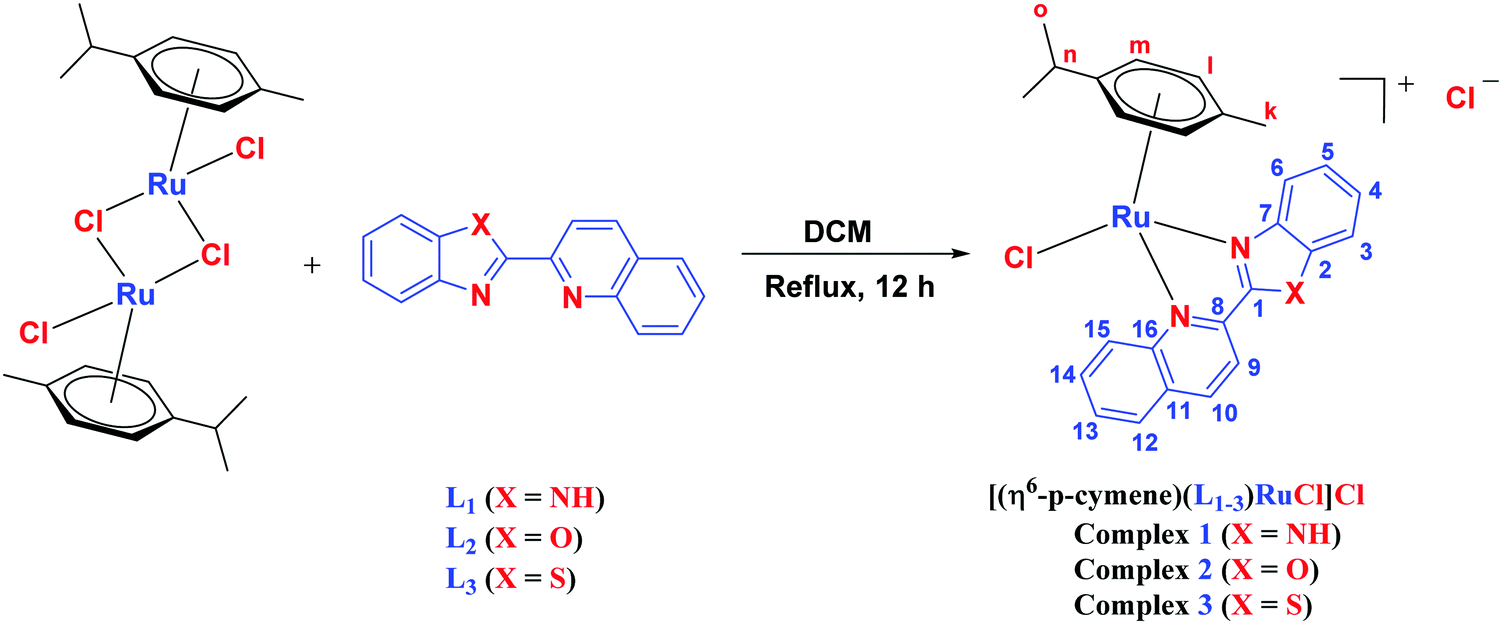
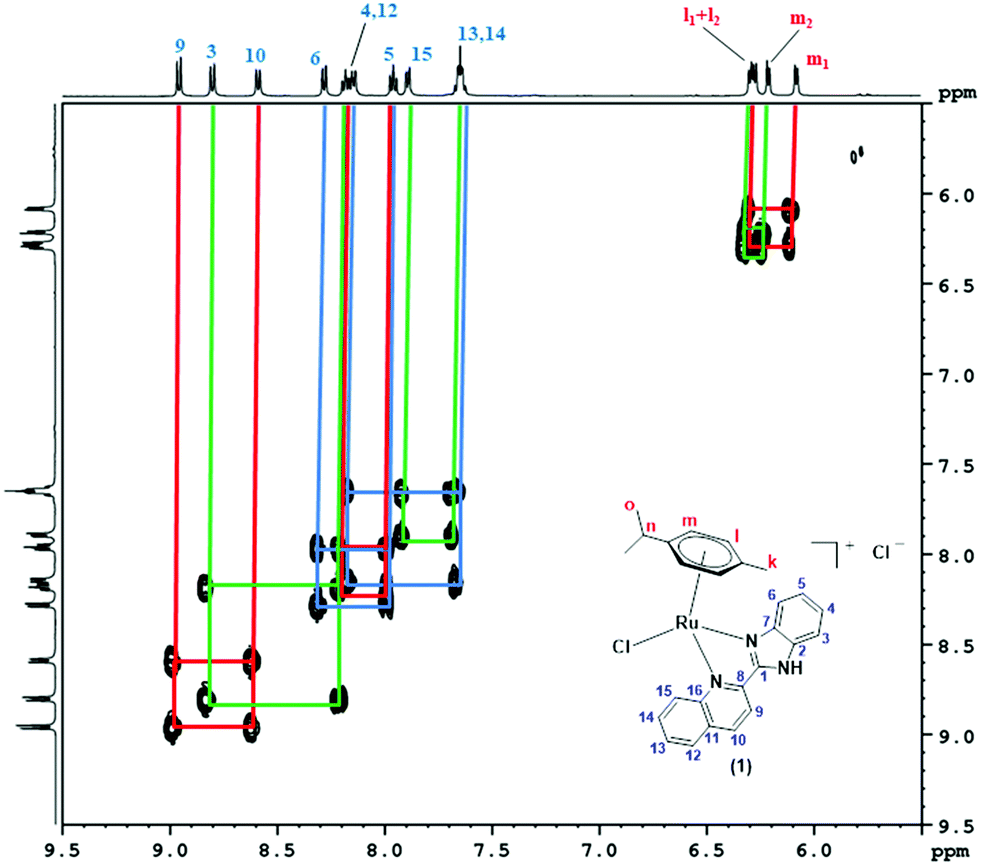
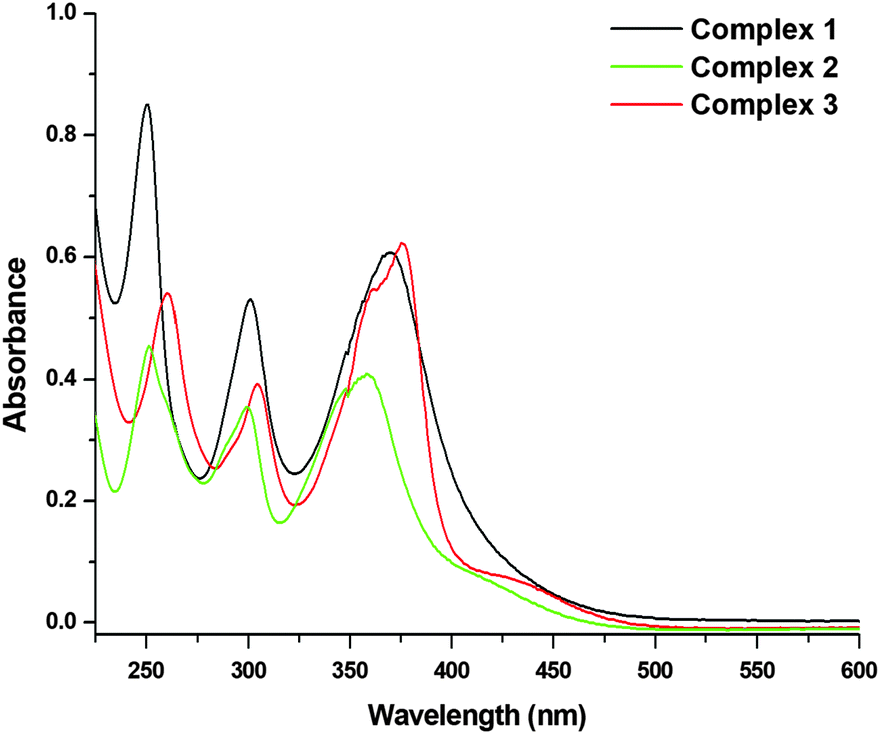
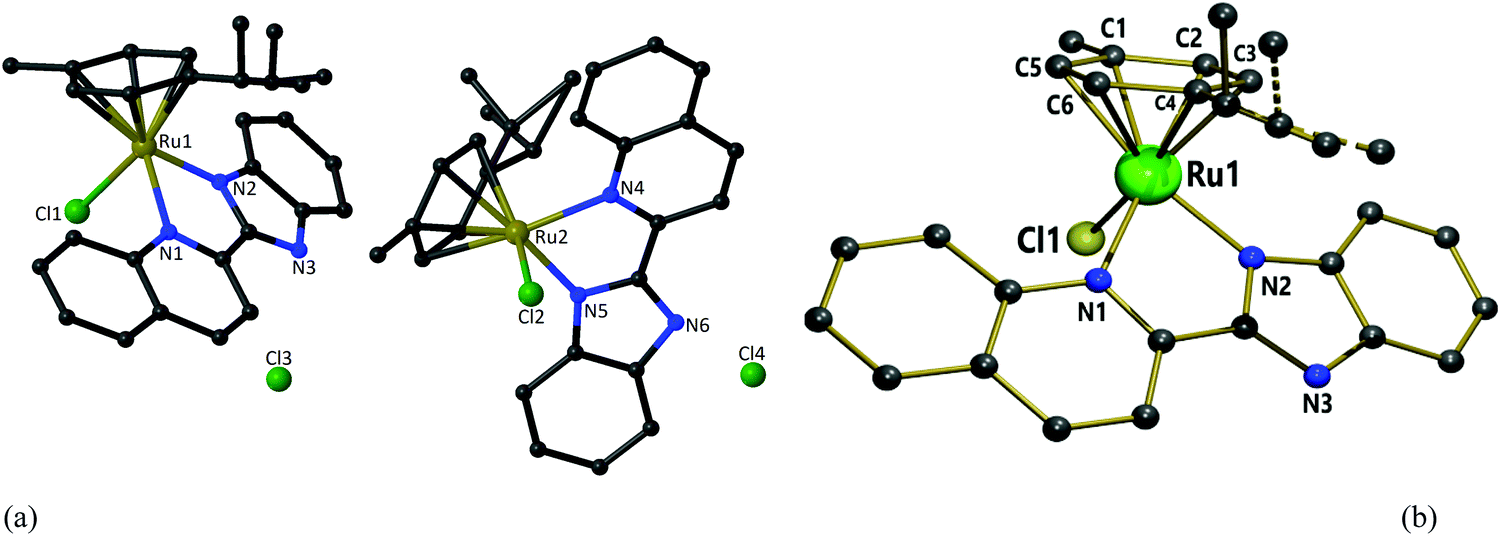
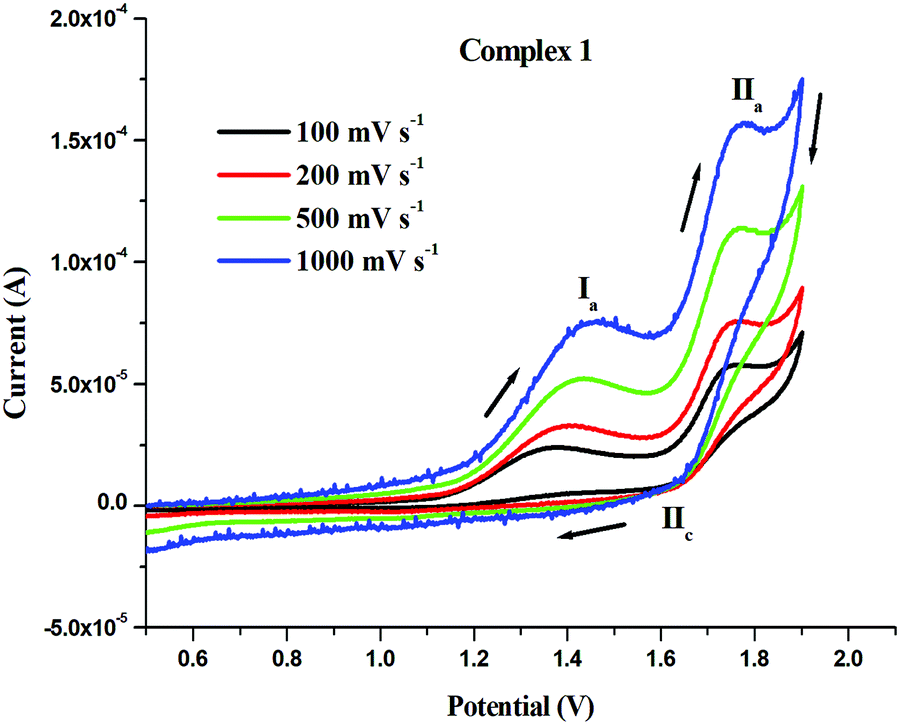
 -manner. The geometrical parameters of the optimized structures are in good agreement with those of previously obtained crystal structures of related systems.41 We also assessed the structures of the complexes with DCM as the solvent (using polarization continuum mode) following the same experimental conditions.
-manner. The geometrical parameters of the optimized structures are in good agreement with those of previously obtained crystal structures of related systems.41 We also assessed the structures of the complexes with DCM as the solvent (using polarization continuum mode) following the same experimental conditions.