
Light-triggered dual-modality drug release of self-assembled prodrug-nanoparticles for synergistic photodynamic and hypoxia-activated therapy†
Dongyang
Zhao
a,
Wenhui
Tao
a,
Songhao
Li
a,
Lingxiao
Li
a,
Yixin
Sun
a,
Guanting
Li
a,
Gang
Wang
b,
Yang
Wang
b,
Bin
Lin
c,
Cong
Luo
a,
Yongjun
Wang
a,
Maosheng
Cheng
c,
Zhonggui
He
*a and
Jin
Sun
 *a
*a
aDepartment of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China. E-mail: hezhonggui@vip.163.com; sunjin@syphu.edu.cn
bCollege of Pharmacy, Guangxi University of Chinese Medicine, Nanning 530200, China
cKey Laboratory of Structure-Based Drug Design and Discovery, Shenyang Pharmaceutical University, Shenyang 110016, China
First published on 17th March 2020
Abstract
Photodynamic therapy (PDT) leads to tumor hypoxia which could be utilized for the activation of hypoxia-activated prodrugs (HAPs). However, conventional photosensitizer-loaded nanoformulations suffer from an aggregation-caused quenching (ACQ) effect, which limits the efficiency of PDT and synergistic therapy. Herein, prodrug-nanoparticles (NPs) are prepared by the self-assembly of heterodimeric prodrugs composed of pyropheophorbide a (PPa), hypoxia-activated prodrug PR104A, and a thioether or thioketal linkage. In addition, a novel dual-modality drug release pattern is proposed on the basis of the structural states of prodrug-NPs. Under light irradiation, PR104A is released via photoinduced electron transfer (PET) due to the aggregation state of prodrugs. With the disassembly of prodrug-NPs, the ACQ effect is relieved, and PPa produces singlet oxygen which further promotes the reactive oxygen species (ROS)-sensitive release of PR104A. Such prodrug-NPs turn the disadvantage of the ACQ effect to facilitate drug release, demonstrating high-efficiency synergy in combination with PDT and hypoxia-activated therapy.
New conceptsFor conventional photosensitizer-loaded nano-drug delivery systems (nano-DDSs), the therapeutic outcome of photodynamic therapy (PDT) or PDT-participant synergistic therapy is severely limited by an aggregation-caused quenching (ACQ) effect. Based on this, multiple strategies have been exploited to disintegrate nano-DDSs via responding to tumor stimuli. However, we proposed a concept that the disadvantage of photosensitizer aggregation could be used as the new power for drug release. In the aggregation state, due to the ACQ effect, singlet oxygen generation is limited whereas photoinduced electron transfer (PET) arises. The PET-induced drug release is a tailor-made release pattern for nano-DDSs in the aggregation state. With the disassembly of nano-DDSs, the ACQ effect is relieved, and singlet oxygen could be generated to promote reactive oxygen species-sensitive drug release. Our findings provide new insights into photosensitizer-aided drug release and possibilities for the development of drug co-loading and co-delivery dual-synergistic nano-DDSs. |
1. Introduction
Photodynamic therapy (PDT) is an important clinically approved treatment modality for various malignant diseases such as cancers. Under light irradiation, PDT exploits reactive oxygen species (ROS) produced via the reaction between photosensitizers and oxygen to achieve a curative effect. However, oxygen consumption during PDT usually aggravates tumor hypoxia,1,2 which is one of the main reasons for the poor response to PDT as well as conventional chemotherapy and radiotherapy.3,4 Recently, it has been reported that the treatment effect of PDT could be significantly improved by using different methods to relieve tumor hypoxia.5–8 As an opposite strategy, several reports have found that a PDT-induced tumor hypoxic microenvironment could be utilized for the activation of hypoxia-activated prodrugs (HAPs), contributing to an effective synergistic combination cancer therapy.9–11Multiple nano-drug delivery systems (nano-DDSs) have been exploited to simultaneously encapsulate two drugs into one carrier for combination therapy.12,13 However, the use of conventional nanocarriers for noncovalent co-loading of drugs has long been criticized due to several limitations, including low co-loading efficiency (typically <10%), poor stability, and high risk of premature drug leakage in blood circulation. A self-assembled prodrug-nanosystem is a novel self-delivering nanoplatform formed by drug conjugates themselves, which has demonstrated multiple integrated advantages of a prodrug strategy and a nano-DDS.14–16 Therefore, self-assembled prodrug-nanoparticles (NPs) have great potential in combination therapy.
Light-triggered drug release of nano-DDSs has attracted special attention due to its noninvasive nature, high controllability, and precise temporal and spatial release. Different mechanisms have been exploited for light-activated nano-DDSs, especially photoinduced cleavage and photooxidation.17,18 In photochemical studies, sulfur radical cations are generally produced by photoinduced electron transfer (PET) between excited sensitizers and low oxidation potential organic sulfides. C–S fragmentation to form an alkyl cation and a sulfur radical is one of the most studied decomposition pathways of sulfur radical cations, which might be exploited to trigger drug release.19–21 The rate of PET is related to the distance between electron donors and acceptors. The smaller the distance, the faster the PET reaction rate.22,23 Since prodrug-NPs exhibit a small intermolecular distance, drug release via PET might be employed as a tailor-made release pattern for carrier-free prodrug-NPs. To the best of our knowledge, a drug release nano-DDS relying on photoinduced C–S fragmentation and closely packed prodrug-NPs has not been reported to date. In addition, the PDT efficiency of a photosensitizer-loaded nano-DDS is usually limited due to an aggregation-caused quenching (ACQ) effect,24,25 whereas here, the aggregation of photosensitizer-loaded prodrug-NPs may turn the disadvantage of the ACQ effect into an advantage to promote drug release.
Herein, we designed novel heterodimeric prodrugs (PR104A-S-PPa and PR104A-TK-PPa) by conjugating hypoxia-activated prodrug PR104A to pyropheophorbide a (PPa) via a thioether or thioketal linkage, and a nonsensitive conjugate (PR104A-PPa) was also synthesized. Thioether and thioketal were selected as S-containing linkages due to the different responses to PET and ROS given the subtle structural difference, therefore resulting in different PR104A release rates and combination treatment effects. The self-assembled prodrug-NPs possess a dense intermolecular-packing structure. Upon laser irradiation, the excited PPa preferentially reacts directly with the neighboring S-containing linkages to form free radical cations via PET, which leads to the cleavage of C–S bonds and the release of PR104A. With the disassembly of prodrug-NPs, the ACQ effect is relieved, and the excited PPa could also convert the surrounding oxygen into singlet oxygen (1O2) via energy transfer, which promotes the ROS-sensitive release of PR104A as well. Meanwhile, PDT depletes oxygen and generates severe hypoxia, which can facilitate the activation of the released PR104A (Fig. 1). Such a unique nanoplatform integrates PPa and PR104A into one system with high-efficiency co-delivery and on-demand dual-modality drug release properties and is promising to overcome a series of obstacles in the combination of PDT and hypoxia-activated therapy.
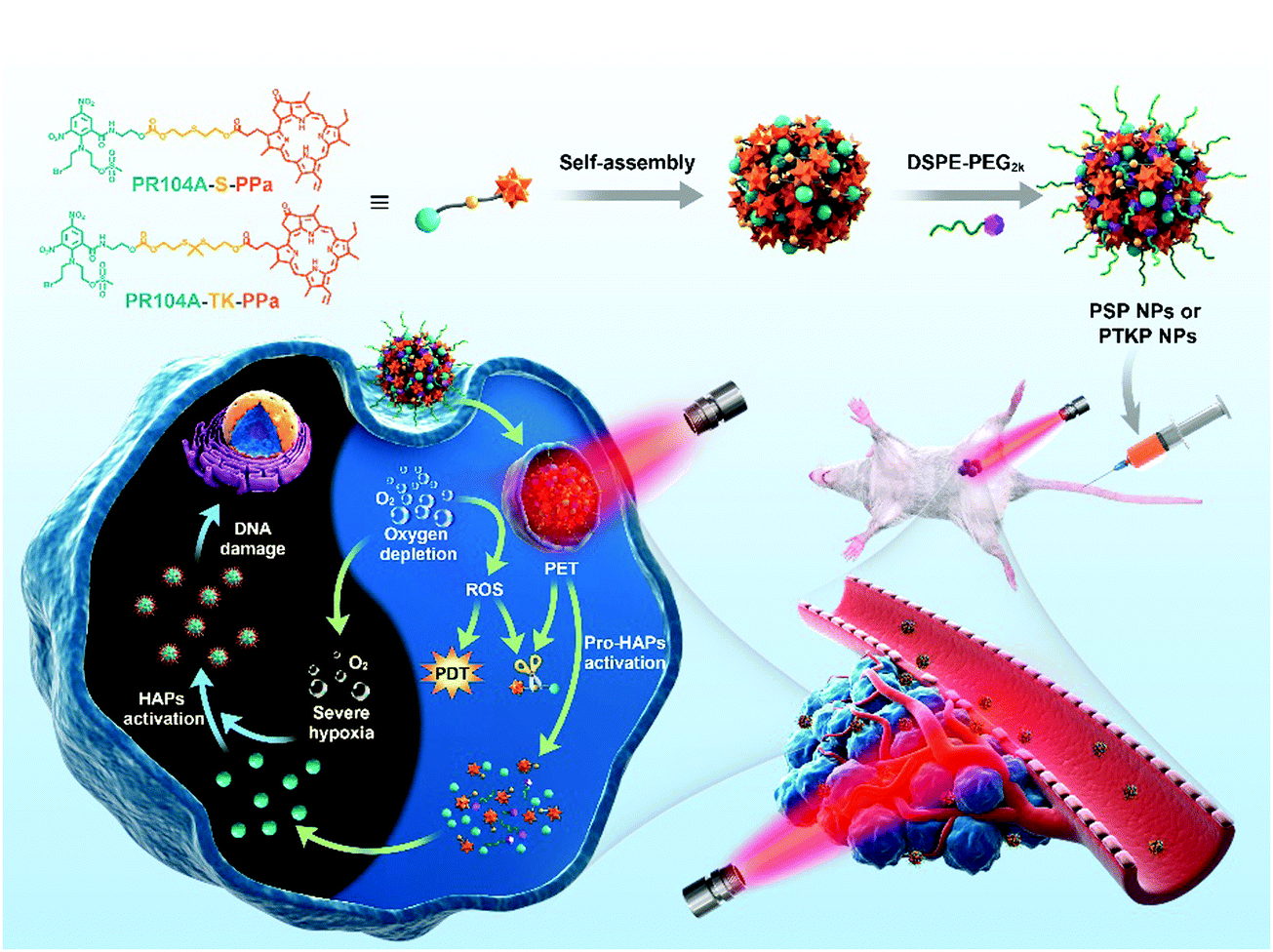 | ||
| Fig. 1 Schematic illustration of the preparation of self-assembled heterodimeric prodrug-NPs, the light-triggered dual-modality activation of prodrug-NPs, and the PDT-induced activation of HAPs. | ||
2. Results and discussion
2.1 Design and synthesis of heterodimeric prodrugs
First, TKOH, the thioketal linkage, was obtained through a simple two-step reaction (Fig. S1A and S2, ESI†).26 PR104A was synthesized according to a previous method (Fig. S3, ESI†).27,28 Then, three novel heterodimeric prodrugs were synthesized by conjugating PR104A with PPa using thioether, thioketal, or carbon chain linkages (Fig. S1B, ESI†). PR104A was reacted with 2,2′-thiodiethanol, TKOH, and 1,6-hexandiol to produce PR104A-S-OH, PR104A-TK-OH, and PR104A-OH, respectively. The intermediate products were confirmed by ESI-MS and 1H NMR spectroscopy (Fig. S4–S6, ESI†). PR104A-S-OH, PR104A-TK-OH, and PR104A-OH were further reacted with PPa via ester formation to synthesize PR104A-S-PPa, PR104A-TK-PPa, and PR104A-PPa, respectively. The end products were also confirmed by ESI-MS and 1H NMR spectroscopy (Fig. S7–S9, ESI†).2.2 Preparation and characterization of prodrug-NPs
Prodrug-NPs were prepared using a one-step nanoprecipitation method according to our previous reports (Fig. 2A).14–16 Both PPa and PR104A are lipophilic organic molecules, and the hydrophobic heterodimeric prodrugs (PR104A-S-PPa, PR104A-TK-PPa, and PR104A-PPa) could spontaneously self-assemble into uniform NPs (PSP NPs, PTKP NPs, and PP NPs) after being dispersed in water. DSPE-PEG2k (20%, w/w) was added to improve the colloidal stability and prolong the systemic circulation time of prodrug-NPs. In our previous studies, we found that multiple mechanisms were involved in the self-assembly process, including chemical linkage, structural flexibility, and intermolecular π–π stacking. The prepared prodrug-NPs exhibited regularly spherical-shaped structures, with a mean diameter of approximately 90–100 nm and a zeta potential of approximately −30 mV (Fig. 2B and Fig. S10A, Table S1, ESI†). Notably, given that the prodrugs themselves act as both the carriers and the cargos, the prepared NPs exhibited a high drug loading capacity (33.6–35.8% for PR104A, 36.0–38.4% for PPa, w/w) (Table S1, ESI†), compared to the conventional encapsulated nanoformulations (typically <10%). High drug loading is one of the prominent advantages of self-assembled prodrug-NPs, resulting in significantly improved drug delivery efficiency and reduced excipient-related toxicity. Moreover, the prodrug-NPs showed good long-term store stability at 4 °C for 2 weeks and excellent colloidal stability in pH 7.4 PBS supplemented with 10% FBS at 37 °C for 24 h (Fig. S10B and C, ESI†).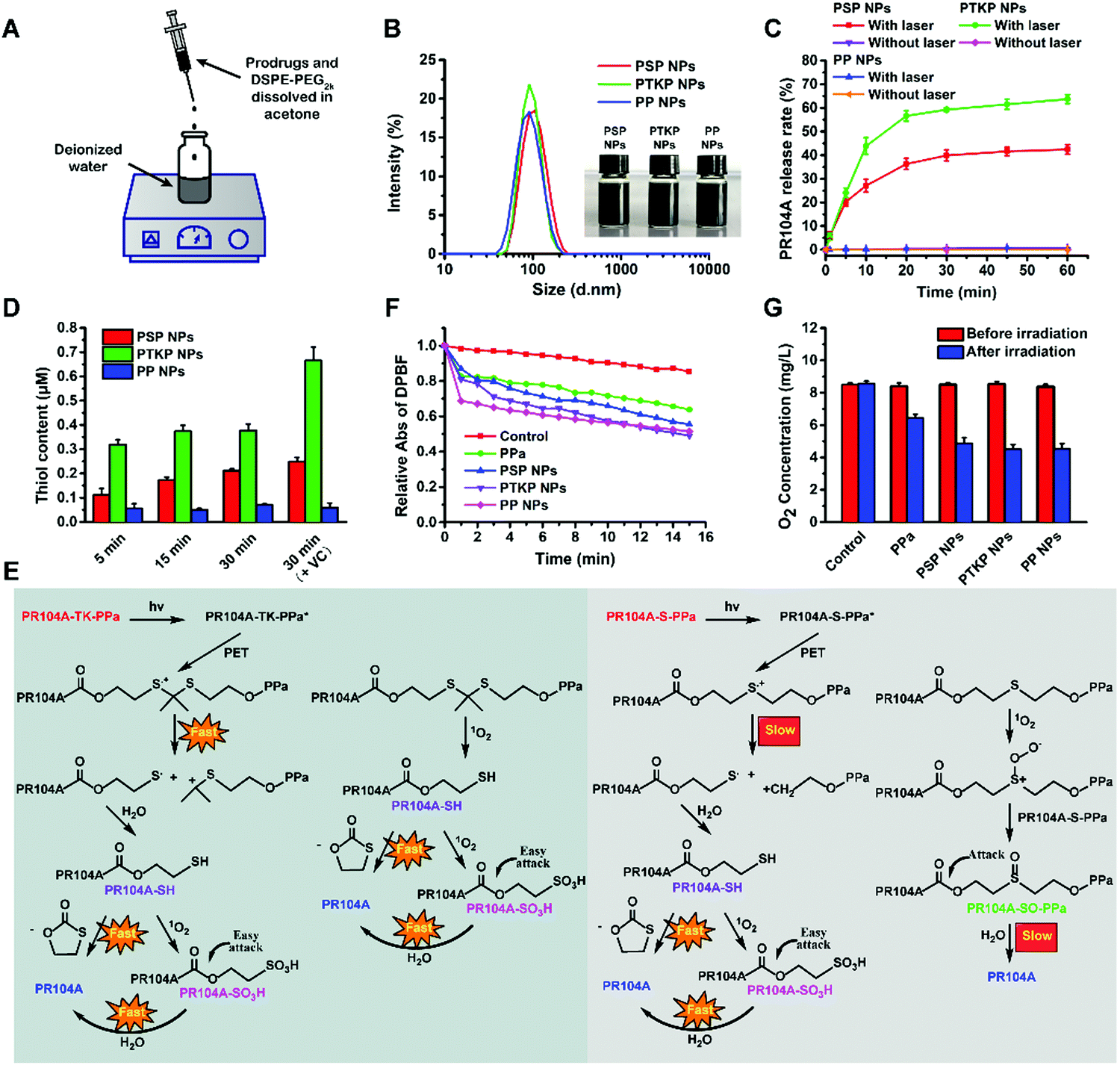 | ||
| Fig. 2 Characterization of prodrug-NPs. (A) Schematic representation of the one-step nanoprecipitation method. (B) DLS size distributions and photos of prodrug-NPs. (C) In vitro PR104A release of prodrug-NPs under laser irradiation (100 mW cm−2) (n = 3). (D) Thiol determination after prodrug-NPs receive laser irradiation (100 mW cm−2) in the presence of vitamin C or not (n = 3). (E) Light-triggered dual-modality drug release mechanisms of PR104A-TK-PPa and PR104A-S-PPa. (F) In vitro1O2 generation. Comparison of the decay rates of DPBF alone and in the presence of free PPa solution or prodrug-NPs under laser irradiation (100 mW cm−2). (G) O2 concentration determination of control, free PPa solution, and prodrug-NPs after laser irradiation (100 mW cm−2) for 15 min (n = 3). | ||
2.3 In vitro drug release from prodrug-NPs
PR104A, one of the HAPs, could be activated to hydroxylamine (PR104H) and amine (PR104M) under hypoxic conditions to induce DNA interstrand crosslink.29 To ensure the safety and effectiveness of treatment, selective PR104A tumor release is of great importance. Hence, we tested the PR104A release behavior of prodrug-NPs in 10 mM H2O2 release medium or under 660 nm laser irradiation. As shown in Fig. S11 (ESI†), minimal drug was released upon 10 mM H2O2 treatment. Two reasons accounted for this result. First, the compact structure of prodrug-NPs prevented H2O2 from penetrating into the inner part to promote oxidation. Second, the oxidizability of H2O2 was relatively weak compared with other ROS, such as singlet oxygen, superoxide anion radicals, or hydroxyl radicals. These two factors caused the prodrug-NPs to exhibit good stability against H2O2 oxidation (Fig. S12, ESI†). By contrast, 42.4% and 63.7% of PR104A were released when PSP NPs and PTKP NPs were irradiated with a 660 nm laser for 1 h, respectively. However, negligible drug release (<1%) was found in nonsensitive PP NPs under the same laser irradiation (Fig. 2C). This unique release behavior ensures the excellent safety of PSP NPs and PTKP NPs to avoid the off-target release triggered by oxide stimulation of hyperoxia in blood or ROS in normal inflamed tissues. Therefore, more PSP NPs and PTKP NPs could reach the tumor site to realize fast and precise light-triggered PR104A release.2.4 Drug release mechanism
To further investigate the mechanism of light-triggered PR104A release, the precise change in molecular weight was monitored by high-resolution mass spectroscopy (MS) after PSP NPs or PTKP NPs were exposed to 660 nm laser irradiation for 5 min (Fig. S13 and S14, ESI†). The MS revealed that the molecular weight of PR104A-SH appeared in both PSP NPs and PTKP NPs. To our best knowledge, thioketal could be oxidized into thiol in the presence of ROS, while thioether could only be oxidized to sulfoxide and sulfone.30 Therefore, the generation of thiol in PSP NPs and PTKP NPs was attributed to a novel mechanism induced by the PET reaction. The intermolecular distance in self-assembled prodrug-NPs was close enough to enable electron transfer between excited PPa and sulfur atoms to form sulfur radical cations. Sulfur radical cations could be decomposed via C–S fragmentation to form an alkyl cation and a sulfur radical, and this process represented an unusual type of SN1 reaction with the radical acting as the leaving group.19–21 The cleavage rate of C–S bonds via PET in PTKP NPs was faster than that in PSP NPs because the formed alkyl cation in PTKP NPs was more stable due to the electron-donating effect of the dimethyl group. Density functional theory (DFT) calculations were utilized to study the strength of the C–S bonds at the B3LYP/6-311G(d,p) level, demonstrating that the bond dissociation energy for the C–S bond in PR104A-S-PPa (61.5 kcal mol−1) was 9-fold higher than that of the C–S bond in PR104A-TK-PPa (6.7 kcal mol−1). As shown in Fig. 2D, the thiol content of PSP NPs and PTKP NPs increased with increasing irradiation time, and PTKP NPs generated more thiol than PSP NPs, indicating the occurrence of the PET reaction. By contrast, PP NPs exhibited no thiol formation. In addition, excess vitamin C was added to PSP NPs and PTKP NPs as a ROS scavenger; after irradiation for 30 min, the thiol production did not decrease but increased compared to the vitamin C-free groups. These results showed that the PET reaction has nothing to do with the existence of ROS.The generated PR104A-SH in both PSP NPs and PTKP NPs could undergo fast intramolecular nucleophilic substitution to produce a five-membered ring thiolactone to release PR104A. In addition, 1O2 could also oxidize PR104A-SH to PR104A-SO3H, which could also effectively promote the release of PR104A due to the extremely hydrophilic sulfonic group facilitating hydrolysis of the adjacent carbonate ester bond.31
In addition to PET-induced drug release, with the disassembly of prodrug-NPs, the generated 1O2 could also oxidize the ROS-sensitive thioketal to thiol, followed by a fast PR104A release. However, the ROS-sensitive thioether could only be oxidized to sulfoxide PR104A-SO-PPa (Fig. S13, ESI†), which demonstrated a slower PR104A release rate due to the β-position of the carbonate ester bond, according to our previous study.32 In general, PTKP NPs could release PR104A faster than PSP NPs due to both faster PET-induced and subsequently ROS-sensitive drug release (Fig. 2E).
2.5 In vitro 1O2 generation and O2 consumption
The ability of prodrug-NPs to produce ROS under laser irradiation was investigated using DPBF, an indicator whose UV absorption is irreversibly decreased under the action of 1O2 (Fig. 2F). Here, 1O2 generation in PPa aqueous solution was less than that of the prodrug-NPs due to its poor aqueous solubility. Three prodrug-NPs presented different 1O2 generation profiles. As expected, when PSP NPs or PTKP NPs were irradiated, the excited PPa preferentially transferred electrons with sulfur atoms to form sulfur radical cations, while nonsensitive PP NPs only utilized the surface prodrug molecules to produce 1O2via transferring energy to O2 at a slow rate. Therefore, PP NPs could generate more 1O2 in the beginning stage. However, with the increased cleavage of thioether or thioketal linkages, the 1O2 generation of PSP NPs or PTKP NPs could exceed that of PP NPs due to the conquering of the ACQ effect. Notably, PTKP NPs exhibited superior ability in addressing the challenge of the ACQ effect compared with PSP NPs, attributing to the faster PET reaction. Meanwhile, the oxygen concentrations were also measured after laser irradiation for 15 min using a portable dissolved oxygen meter. As illustrated in Fig. 2G, the higher the 1O2 generated, the lower the O2 concentration. This outcome strongly demonstrated that the photodynamic prodrug-NPs could generate further anabatic hypoxia for HAP activation.2.6 Cellular uptake
Next, the cellular uptake of prodrug-NPs was evaluated using confocal laser scanning microscopy (CLSM) and flow cytometry. As shown in Fig. 3A and B, free PPa treated cells showed significantly higher intracellular PPa fluorescence intensity compared with prodrug-NP treated cells. Meanwhile, the rank order of PPa fluorescence intensity for the three prodrug-NPs in 4T1 cells was PSP NPs > PTKP NPs > PP NPs. The intracellular fluorescence intensity did not accurately represent the cellular uptake amount because the PPa fluorescence in prodrug-NPs was quenched, whereas in free PPa it was not (Fig. S15, ESI†). Intracellular ROS oxidized a small portion of the external prodrug molecules of PSP NPs or PTKP NPs, which were then dissociated from NPs in a free state to increase the fluorescence intensity. PSP NPs had stronger fluorescence than PTKP NPs probably due to poor stability under oxidizing conditions (Fig. S12, ESI†). Nonsensitive PP NPs could not be oxidized, therefore maintaining the lowest fluorescence. To verify our hypothesis, the HPLC method was used to determine the intracellular contents of PPa, PR104A-S-PPa, PR104A-TK-PPa, or PR104A-PPa after free PPa solution, PSP NPs, PTKP NPs, or PP NPs were incubated with 4T1 cells for 4 h, respectively. An approximately 2-fold increase in cellular internalization was observed when comparing the prodrug-NPs with the control free PPa (Fig. 3C). PSP NPs were present at a lower concentration than the other two prodrug-NPs due to poor stability under intracellular oxidation as mentioned above. Our results clearly showed that the fluorescence intensity of NPs is typically affected by the particle aggregation state, and the HPLC direct determination method accurately reflects the concentration of prodrug-NPs. | ||
| Fig. 3 In vitro cell experiments. (A) CLSM images and (B) flow cytometry measurement of 4T1 cells incubated with free PPa or prodrug-NPs for 4 h. Scale bar represents 10 μm. (C) Cellular uptake in 4T1 cells determined by HPLC (n = 3). (D) CLSM images and flow cytometry analysis of 4T1 cells with ROS/hypoxia detection probes in different treatments: negative control, positive controls (hypoxia-induced and ROS-induced), and prodrug-NPs with laser irradiation (100 mW cm−2) for 1 min. Scale bar represents 100 μm. (E) PR104A released from prodrug-NPs under laser irradiation (100 mW cm−2) for 5 min after cellular uptake in 4T1 cells for 4 h (n = 3). (F) Cytotoxicity of PR104A to 4T1 cells under normoxia and hypoxia conditions after incubation for 24 h (n = 3). (G) Cytotoxicity of different treatments to 4T1 cells in the dark or under laser irradiation (100 mW cm−2) for 5 min after incubation for 24 h (n = 3). (+): laser irradiation. The levels of significance were set at probabilities of **p < 0.01 and ***p < 0.001. | ||
2.7 Intracellular ROS generation and aggravated hypoxia
We expected that the prodrug-NPs would convert oxygen into ROS to generate a hypoxic microenvironment in tumor cells; therefore, the released PR104A would be sequentially activated to selectively kill tumor cells. We treated 4T1 cells with prodrug-NPs, and ROS and hypoxia levels were analyzed after laser irradiation. As shown in Fig. 3D, cells treated with prodrug-NPs exhibited stronger fluorescence of both the ROS production (green) and the hypoxia generation (red) upon laser irradiation than the control. To guarantee a high cell survival rate, the irradiation time was set as 1 min. As explained above, the excited PPa of PSP NPs or PTKP NPs preferentially transferred electrons with sulfur atoms to form sulfur radical cations via the PET reaction. However, the PET reaction of PSP NPs was slower than that of PTKP NPs. Therefore, a small portion of excited PPa of PSP NPs also reacted with the surrounding O2 to form 1O2, resulting in stronger ROS and hypoxia fluorescence than PTKP NPs. The nonsensitive PP NPs could only utilize the surface prodrug molecules to produce 1O2via transferring energy to O2, leading to the highest ROS and hypoxia fluorescence after irradiation for 1 min. If the irradiation time was extended, PSP NPs or PTKP NPs would be disintegrated, especially PTKP NPs, and the ROS production and hypoxia generation would exceed those of PP NPs as a result of conquering of the ACQ effect as shown in Fig. 2F.2.8 Intracellular drug release from prodrug-NPs
We then investigated the drug release behavior of prodrug-NPs after incubation with 4T1 cells with or without laser irradiation. As shown in Fig. 3E, PTKP NPs exhibited approximately 1.9-fold higher PR104A release compared with PSP NPs under the same laser irradiation. Higher intracellular PR104A release was achieved with PTKP NPs given their both faster PET-induced and ROS-sensitive activation, as well as better stability to avoid premature elimination. By contrast, nonsensitive PP NPs, with very little PR104A released, showed no significant difference with or without laser treatment. The intracellular release result was consistent with the above in vitro PR104A release study.2.9 Cytotoxicity assay
Next, cytotoxicity against 4T1 cells was tested. It was found that PR104A exhibited stronger cytotoxicity toward 4T1 cells under hypoxia than normoxia, indicating the hypoxia-triggered activity of PR104A (Fig. 3F). In addition, we found that cells incubated with prodrug-NPs could be efficiently killed after 660 nm laser irradiation, while prodrug-NPs showed very low cytotoxicity without laser irradiation. The cell killing activity of PR104A & PPa (+) was greatly enhanced compared to PR104A or PPa (+) alone, demonstrating synergistic cytotoxicity (the calculated combination index (CI) < 1). Meanwhile, given that most intracellular PR104A was released under laser irradiation, PTKP NPs exhibited higher synergistic cell killing (IC50: 238.2 nM) compared with the other two prodrug-NPs (IC50: 324.5 nM for PSP NPs, IC50: 856.4 nM for PP NPs) (Fig. 3G and Table S2, ESI†).2.10 In vivo pharmacokinetic study
Sprague Dawley rats were used to investigate the pharmacokinetic behavior of prodrug-NPs after intravenous injection. The molar concentration–time curves of the prodrugs and the released PR104A are illustrated in Fig. 4A and B, and the pharmacokinetic parameters are summarized in Table S3 (ESI†). As shown in Fig. 4A, free PR104A was rapidly eliminated from blood resulting in a short half-life (t1/2, 0.3 h) as reported in a previous study.33 In comparison, the prodrug-NPs exhibited significantly prolonged circulation time compared with free PR104A. The area under the curve (AUC0–24h) of prodrugs in PSP NPs, PTKP NPs, and PP NPs was 321.5-fold, 374.8-fold, and 410.4-fold higher than that of free PR104A, respectively. It is well known that long blood circulation could contribute to high tumor accumulation of prodrug-NPs by exploiting the enhanced permeability and retention (EPR) effect. Furthermore, PSP NPs showed a slightly faster clearance in blood compared with the other two prodrug-NPs due to poor stability in the presence of blood oxidizing substances. Similar experimental results were also obtained in an in vitro plasma stability study (Fig. S16, ESI†). Meanwhile, nearly over 90% of the three prodrug-NPs were present in the prodrug form in the bloodstream, avoiding side effects on normal tissues (Fig. 4B). | ||
| Fig. 4 In vivo pharmacokinetic behavior and biodistribution of prodrug-NPs. Molar plasma concentration–time curves of (A) the prodrugs and (B) the released PR104A after i.v. administration to rats with PR104A or prodrug-NPs at a PR104A equivalent dose of 2.8 mg kg−1 (n = 3). (C) In vivo fluorescence images of the orthotopic 4T1 tumor-bearing mice at 4, 12, 24, and 36 h after i.v. administration to mice with free PPa solution or prodrug-NPs at a PPa equivalent dose of 3 mg kg−1. (D) Ex vivo fluorescence imaging of the normal organs and tumor collected from the mice at 36 h post i.v. injection. (E) Quantitative analysis of fluorescent signals of the tumor and normal organs at 36 h post i.v. injection (n = 3). (F) HPLC determination of drug content in the tumor at 36 h after i.v. administration (n = 3). (G) Ex vivo immunofluorescence staining and the quantitative results of tumor slices collected from prodrug-NP injected mice with laser irradiation (200 mW cm−2) for 5 min after 24 h tumor accumulation. Scale bar represents 100 μm. The levels of significance were set at probabilities of *p < 0.05 and **p < 0.01. | ||
2.11 In vivo biodistribution
Then, the biodistribution of prodrug-NPs was evaluated in female BALB/c mice bearing orthotopic 4T1 tumors. As expected, free PPa solution was distributed throughout the body and was rapidly eliminated after administration. In contrast, strong fluorescence signals in tumors were observed after treatment with PSP NPs, PTKP NPs, and PP NPs. In addition, the tumor accumulation of prodrug-NPs peaked at 24 h after injection and still retained strong fluorescence at 36 h (Fig. 4C). The increased tumor accumulation of prodrug-NPs was attributed to the long blood circulation and EPR effect. Meanwhile, the PEGylation and the small size of prodrug-NPs could reduce the phagocytosis of the reticuloendothelial system, avoiding high distribution in the liver and spleen. At 36 h after administration, the tumor and main organs were collected for fluorescence quantitative analysis. As shown in Fig. 4D and E, the tumor exhibited the highest fluorescence intensity compared with other organs. The tumor fluorescence intensities of three prodrug-NPs were different: PSP NPs > PTKP NPs > PP NPs. The fluorescence intensity probably did not represent the actual tumor accumulation, as emphasized above. PSP NPs were more unstable than PTKP NPs and PP NPs, thus exhibiting higher fluorescence intensity. To verify our hypothesis, the tumors were collected and homogenized after mice were injected with free PPa solution, PSP NPs, PTKP NPs, or PP NPs for 36 h. HPLC was used to determine the drug content of PPa, PR104A-S-PPa, PR104A-TK-PPa, or PR104A-PPa in tumor homogenates, respectively. As shown in Fig. 4F, although the strongest fluorescence was observed, the actual content of PSP NPs was lower compared with those of PTKP NPs and PP NPs. In contrast, nonsensitive PP NPs exhibited the lowest fluorescence but had the highest tumor accumulation due to their inert property. Meanwhile, the actual tumor accumulation of PSP NPs, PTKP NPs, and PP NPs was 6-fold, 9.2-fold, and 17.8-fold higher than that of free PPa solution, respectively, indicating the superior drug delivery of prodrug-NPs to the tumor. Similar to cellular uptake assay, HPLC determination could accurately reflect the content of prodrug-NPs in an aggregation state, compared to quantitative calculation based on fluorescence intensity.Furthermore, after tumor accumulation for 24 h, the tumor-bearing mice were irradiated, and we evaluated the tumor hypoxia status by utilizing ex vivo immunofluorescence staining. As shown in Fig. 4G, prodrug-NPs showed obviously increased tumor hypoxia compared to the control.
2.12 In vivo antitumor efficacy
To validate the advantages of our prodrug-NPs for combination therapy, we evaluated the antitumor efficiency in heterotopic and metastatic orthotopic 4T1 tumor-bearing mice. Since biodistribution imaging showed the highest tumor accumulation of prodrug-NPs at 24 h post-injection, tumor-bearing mice were intravenously administered four doses every two days and the tumors were irradiated after one day following each injection (Fig. 5A). As shown in Fig. 5B–F, tumor growth was the fastest in the saline-treated control group, while moderately limited tumor growth was observed using PP NPs, PSP NPs, and PTKP NPs. Mice treated with PR104A & PPa (+) exhibited no significant difference in tumor inhibition compared to PPa (+) alone. The poor combination therapy was ascribed to the poor co-delivery efficiency of the two free drug solutions, and intravenous co-administration did not ensure synchronous drug delivery in the body. By contrast, the laser-treated PTKP NP group exhibited unique superiority over other groups given its excellent stability, high tumor accumulation, and optimal light-triggered dual-modality PR104A release. PSP NPs showed poor blood circulation and tumor accumulation, and the PR104A release rate under irradiation was also slower than that of PTKP NPs, which led to an inferior antitumor effect compared to PTKP NPs. Notably, PP NPs exhibited the best ability to avoid premature oxidation and the highest tumor accumulation but the most inefficient tumor inhibition, demonstrating the necessity for sensitive thioketal or thioether linkages for PR104A release. There was no obvious change in body weight except for the PR104A & PPa (+) group, which exhibited a slight decrease during the period of drug administration. Meanwhile, the solution groups of PR104A, PPa (+), and PR104A & PPa (+) exhibited slight hepatic injury due to the use of organic solvents such as ethanol and Cremophor EL (Fig. S17, ESI†).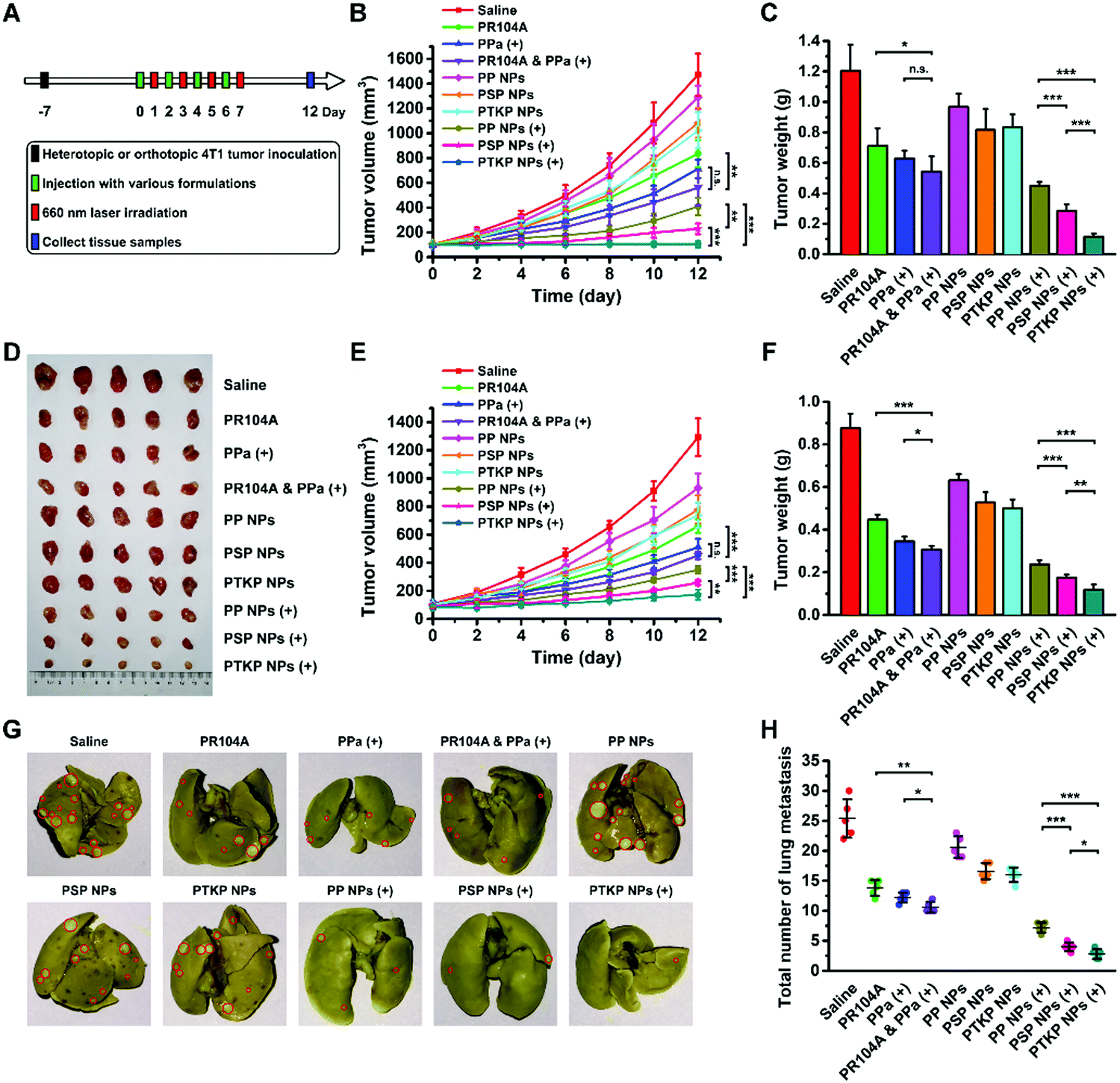 | ||
| Fig. 5 In vivo therapeutic efficacy and antimetastatic activity of prodrug-NPs. (A) Pharmacodynamics study protocol. (B) Tumor growth curves, (C) weights and (D) pictures of the collected heterotopic 4T1 tumors after different treatments at a PR104A equivalent dose of 2.8 mg kg−1 and a PPa equivalent dose of 3 mg kg−1 with laser irradiation (200 mW cm−2) for 5 min or without irradiation (n = 5). (E) Tumor growth curves and (F) weights of the collected orthotopic 4T1 tumors after different treatments. (G) Representative photos of the whole lungs from mice treated with different formulations. Red circles denote surface lung metastases. (H) Average number of surface lung metastases (n = 5). (+): laser irradiation. The levels of significance were set at probabilities of *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
The orthotopic 4T1 model could also spontaneously form lung metastases. Therefore, we also evaluated the antimetastatic activity by counting the number of metastatic pulmonary nodules and analyzing the hematoxylin & eosin (H&E) stained tissue sections of the lungs and liver. As shown in Fig. 5G and H, the treatment of prodrug-NPs, especially PTKP NPs, led to a significantly decreased number of metastatic pulmonary nodules upon laser irradiation compared to other groups. The results were further confirmed by analysis of the lung and liver tissue sections, which also showed a significant reduction in metastasis (Fig. S18, ESI†). The tumor metastasis inhibition of PTKP NPs might benefit from the rapid release and activation of PR104A.
3. Conclusions
In summary, we prepared prodrug-NPs (PSP NPs and PTKP NPs) by the self-assembly of heterodimeric prodrugs (PR104A-S-PPa and PR104A-TK-PPa) with high drug co-loading capacity for PR104A and PPa. The carrier-free prodrug-NPs possess a compact structure. Upon laser irradiation, PPa preferentially transfers electrons to the neighboring sulfur atoms of thioether or thioketal linkages to form sulfur radical cations via PET. The sulfur radical cations lead to the cleavage of C–S bonds, the release of PR104A, and the subsequent disintegration of prodrug-NPs. Then, ROS could be generated through the reaction between excited PPa and surrounding O2. The generated ROS could kill tumor cells as well as facilitate ROS-sensitive PR104A release. Meanwhile, hypoxia caused by PDT could activate PR104A for synergistic antitumor therapy. Such prodrug-NPs turn the disadvantage of the ACQ effect into an advantage to promote light-triggered dual-modality drug release. PTKP NPs exhibited better blood circulation, higher tumor accumulation, and faster light-triggered PR104A release than PSP NPs, therefore demonstrating the best combination therapy efficiency. Our findings provide new insights into light-triggered photosensitizer-aided drug release and possibilities for the development of drug co-loading and co-delivery dual-synergistic prodrug nano-DDSs.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 81872816 and U1608283), Liaoning Revitalization Talents Program (No. XLYC1808017), Key Projects of Technology Bureau in Shenyang (No. 18400408), and Key Projects of Liaoning Province Department of Education (No. 2017LZD03).References
- R. B. Moore, J. D. Chapman, J. R. Mercer, R. H. Mannan, L. I. Wiebe, A. J. Mcewan and M. S. Mcphee, J. Nucl. Med., 1993, 34, 405–411 CAS.
- Z. J. Zhou, J. B. Song, L. M. Nie and X. Y. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- B. W. Henderson and V. H. Fingar, Cancer Res., 1987, 47, 3110–3114 CAS.
- J. E. Moulder and S. Rockwell, Cancer Metastasis Rev., 1987, 5, 313–341 CrossRef CAS PubMed.
- D. L. Tao, L. Z. Feng, Y. Chao, C. Liang, X. J. Song, H. R. Wang, K. Yang and Z. Liu, Adv. Funct. Mater., 2018, 28, 1804901 CrossRef.
- Z. Y. Luo, H. Tian, L. L. Liu, Z. K. Chen, R. J. Liang, Z. Chen, Z. H. Wu, A. Q. Ma, M. B. Zheng and L. T. Cai, Theranostics, 2018, 8, 3584–3596 CrossRef CAS PubMed.
- P. Zhu, Y. Chen and J. L. Shi, ACS Nano, 2018, 12, 3780–3795 CrossRef CAS PubMed.
- D. W. Zheng, B. Li, L. Xu, Q. L. Zhang, J. X. Fan, C. X. Li and X. Z. Zhang, ACS Nano, 2018, 12, 6218–6227 CrossRef CAS PubMed.
- D. Cui, J. G. Huang, X. Zhen, J. C. Li, Y. Y. Jiang and K. Y. Pu, Angew. Chem., Int. Ed., 2019, 58, 5920–5924 CrossRef CAS PubMed.
- Y. Z. Wang, Y. Xie, J. Li, Z. H. Peng, Y. Sheinin, J. P. Zhou and D. Oupický, ACS Nano, 2017, 11, 2227–2238 CrossRef CAS PubMed.
- L. Z. Feng, L. Cheng, Z. L. Dong, D. L. Tao, T. E. Barnhart, W. B. Cai, M. W. Chen and Z. Liu, ACS Nano, 2017, 11, 927–937 CrossRef CAS PubMed.
- L. Ma, M. Kohli and A. Smith, ACS Nano, 2013, 7, 9518–9525 CrossRef CAS PubMed.
- C. B. He, J. Q. Lu and W. B. Lin, J. Controlled Release, 2015, 219, 224–236 CrossRef CAS PubMed.
- C. Luo, J. Sun, B. J. Sun and Z. G. He, Trends Pharmacol. Sci., 2014, 35, 556–566 CrossRef CAS PubMed.
- B. J. Sun, C. Luo, X. B. Zhang, M. R. Guo, M. C. Sun, H. Yu, Q. Chen, W. Q. Yang, M. L. Wang, S. Y. Zuo, P. Y. Chen, Q. M. Kan, H. T. Zhang, Y. J. Wang, Z. G. He and J. Sun, Nat. Commun., 2019, 10, 3211 CrossRef PubMed.
- C. Luo, B. J. Sun, C. Wang, X. B. Zhang, Y. Chen, Q. Chen, H. Yu, H. Q. Zhao, M. C. Sun, Z. B. Li, H. T. Zhang, Q. M. Kan, Y. J. Wang, Z. G. He and J. Sun, J. Controlled Release, 2019, 302, 79–89 CrossRef CAS PubMed.
- N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv. Drug Delivery Rev., 2012, 64, 1005–1020 CrossRef CAS PubMed.
- M. Karimi, Z. P. Sahandi, S. Baghaee-Ravari, M. Ghazadeh, H. Mirshekari and M. R. Hamblin, J. Am. Chem. Soc., 2017, 139, 4584–4610 CrossRef CAS PubMed.
- O. Lanzalunga and A. Lapi, J. Sulfur Chem., 2012, 33, 101–129 CrossRef CAS.
- M. Kamata, Y. Murakami, Y. Tamagawa, M. Kato and E. Hasegawa, Tetrahedron, 1994, 50, 12821–12828 CrossRef CAS.
- S. M. Bonesi, E. Carbonell, H. Garcia, M. Fagnoni and A. Albini, Appl. Catal., B, 2008, 79, 368–375 CrossRef CAS.
- S. Lu, S. Wang, J. Zhao, J. Sun and X. Yang, Anal. Chem., 2017, 89, 8429–8436 CrossRef CAS PubMed.
- N. Mirco, C. Sebastiano and S. Franco, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC.
- C. H. Ren, H. M. Wang, D. Mao, X. L. Zhang, Q. Q. Fengzhao, Y. Shi, D. Ding, D. L. Kong, L. Wang and Z. M. Yang, Angew. Chem., Int. Ed., 2015, 54, 4823–4827 CrossRef CAS PubMed.
- N. Zheng, Z. Zhang, J. Kuang, C. Wang, Y. Zheng, Q. Lu, Y. Bai, Y. Li, A. Wang and W. Song, ACS Appl. Mater. Interfaces, 2019, 11, 18224–18232 CrossRef CAS PubMed.
- L. H. Liu, W. X. Qiu, B. Li, C. Zhang, L. F. Sun, S. S. Wan, L. Rong and X. Z. Zhang, Adv. Funct. Mater., 2016, 26, 6257–6269 CrossRef CAS.
- G. J. Atwell and W. A. Denny, J. Labelled Compd. Radiopharm., 2007, 50, 7–12 CrossRef CAS.
- S. J. Yang, G. J. Atwell and W. A. Denny, Tetrahedron, 2007, 63, 5470–5476 CrossRef CAS.
- M. J. McKeage, M. B. Jameson, R. K. Ramanathan, J. Rajendran, Y. C. Gu, W. R. Wilson, T. J. Melink and N. S. Tchekmedyian, BMC Cancer, 2012, 12, 496 CrossRef CAS PubMed.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553–5585 CrossRef CAS PubMed.
- Q. Pei, X. Hu, X. Zheng, S. Liu, Y. Li, X. Jing and Z. Xie, ACS Nano, 2018, 12, 1630–1641 CrossRef CAS PubMed.
- B. J. Sun, C. Luo, H. Yu, X. B. Zhang, Q. Chen, W. Q. Yang, M. L. Wang, Q. M. Kan, H. T. Zhang, Y. J. Wang, Z. G. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS PubMed.
- K. Patel, S. S. Choy, K. O. Hicks, T. J. Melink, N. H. Holford and W. R. Wilson, Cancer Chemother. Pharmacol., 2011, 67, 1145–1155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00034e |
| This journal is © The Royal Society of Chemistry 2020 |