
Nano-size porous carbon spheres as a high-capacity anode with high initial coulombic efficiency for potassium-ion batteries†
Hehe
Zhang
ab,
Chong
Luo
a,
Hanna
He
b,
Hong-Hui
Wu
c,
Li
Zhang
 d,
Qiaobao
Zhang
*a,
Haiyan
Wang
*b and
Ming-Sheng
Wang
*a
d,
Qiaobao
Zhang
*a,
Haiyan
Wang
*b and
Ming-Sheng
Wang
*a
aDepartment of Materials Science and Engineering, College of Materials, Xiamen University, Xiamen, Fujian 361005, China. E-mail: zhangqiaobao@xmu.edu.cn; mswang@xmu.edu.cn
bHunan Provincial Key Laboratory of Chemical Power Sources, College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, P. R. China. E-mail: wanghy419@126.com
cBeijing Advanced Innovation Center for Materials Genome Engineering, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China
dSoochow Institute for Energy and Materials Innovations (SIEMIS), Key Laboratory of Advanced Carbon Materials and Wearable Energy Technologies of Jiangsu Province, Soochow University, Suzhou, Jiangsu 215006, China
First published on 10th March 2020
Abstract
Hard carbon materials have been recognized as a promising family of anode materials for potassium ion batteries (PIBs), but their practical application is severely hindered due to the inferior initial coulombic efficiency (ICE) and low capacity. Herein, we report our findings in simultaneously improved potassium storage capacity and ICE through the design of nano-size and porous structure and the appropriate selection of electrolytes. Benefiting from the high specific surface area, stable electrode|electrolyte interface, and fast potassium ion and electron transfer, the optimized electrode exhibits a high ICE of up to 68.2% and an outstanding reversible capacity of 232.6 mA h g−1 at 200 mA g−1. In particular, superior cycling stability of 165.2 mA h g−1 at 1000 mA g−1 and 129.7 mA h g−1 at 2000 mA g−1 can be retained after 1500 cycles, respectively. Quantitative analysis reveals that this optimized structure leads to an enhanced surface-controlled contribution, resulting in fast potassiation kinetics and electronic|ionic conductivities, which are regarded as essential features for potassium storage. Our findings in this work provide an efficient strategy to significantly improve potassium storage capacity while maintaining a high ICE for hard carbon electrodes.
New conceptsPotassium-ion batteries (PIBs) are regarded as ideal alternatives to lithium-ion batteries (LIBs) as potassium has abundant natural resources. Hard carbon with a disordered structure and large interlayer space is a promising anode with high capacity and long-cycle stability for PIBs owing to its ability to accommodate the insertion/extraction of the big potassium ion (2.72 Å). However, the low initial coulombic efficiency (ICE) and low capacity of previously reported porous carbon materials are unacceptable for practical application. Developing new strategies to improve the potassium storage performance while maintaining a high ICE is still challenging but highly desirable. In this work, we report a facile one-step method for the design of unique carbon spheres with nano-size and porous structure to boost the potassium storage performance. Further, a more suitable electrolyte for hard carbon electrodes is explored. Kinetics analysis reveals that this unique nanostructure can effectively enhance surface-controlled contribution and improve the potassium diffusion coefficient during potassiation/depotassiation. By virtue of structural advantages and higher diffusion kinetics, the optimized electrode not only achieves an ultrahigh reversible capacity while maintaining outstanding cycling stability but also attains a relatively high ICE, which offers a promising approach toward developing practical hard carbon electrodes for advanced PIBs. |
Introduction
With the spotlight on renewable electricity production, energy storage systems, especially lithium ion batteries (LIBs), have been rapidly developed due to their high energy density and long cycling life.1–5 Nevertheless, because of the rapid increase in the demand for LIBs, the high cost of lithium limits further large-scale energy storage applications of LIBs. As an alternative, potassium-ion batteries (PIBs) have received increasing attention owing to the natural abundance and low redox potential of potassium, and the potassium ion (K+) exhibits similar chemical characteristics to the lithium ion (Li+) in most electrode materials.6–8 Additionally, the energy density of PIBs is not as low as the common perception based on theoretical calculations (218 W h kg−1 for PIBs compared to 279 W h kg−1 for LIBs),9 which allows PIBs not only to meet the demand for large-scale energy storage applications but also to replace LIBs in portable electronic devices and electric vehicles. However, given the large atomic radius of K+ (1.38 Å), which can lead to a huge structural change during cycling, the choice of electrode materials is still limited.10,11 For instance, a volume change of 61% occurs when K+ intercalates into graphite, a commercial anode material in LIBs.12 Therefore, it is highly desirable, yet challenging, to develop suitable anode materials for PIBs.As one of the analogs of graphite, hard carbon, consisting of graphite-like microcrystallites and amorphous regions, possesses a disordered and rigid structure.13 Recent evidence suggests that a limited structural change occurs after K+ inserts into hard carbon,14 and the electrochemical properties including energy density and cyclic stability can be further improved through heteroatom doping (N, P, S, and O)15–20 and pore formation,21,22 thereby making hard carbon one of the most promising anodes for PIBs. However, the low initial coulombic efficiency (ICE) of carbon-based materials is actually unacceptable in practical applications, but most previous studies have not dealt with this issue. For example, Yan et al.23 reported N doped carbon nanosheets which delivered outstanding reversible capacities (367 mA h g−1 at 50 mA g−1 and 168 mA h g−1 at 2000 mA g−1) in PIBs, but the low ICE (20%) of this electrode means 80% of the intercalated K+ would be irrevocably trapped in the anode in a full battery. A considerable amount of literature ascribed the initial capacity loss to the undesirable side reactions and the formed SEI films on the electrode surface.16,17,23–25 Therefore, it is a common route in lithium-ion and sodium-ion batteries to increase the ICE by decreasing the specific surface area and the number of defects of hard carbon.26,27 Nevertheless, due to the larger atomic radius of K+, slow diffusion kinetics of K+ intercalation into hard carbon would result in poor rate performance.14 In contrast, much faster chemical kinetics is demonstrated when the potassium storage process occurs on the surface, but this kind of process is highly influenced by the surface defects and functional groups.15,23 Thus, this causes a contradiction, and the major issue is how to balance the potassium storage capacity and ICE in a hard carbon electrode.
Herein, to mitigate these obstacles, we report our findings in fabricating a hard carbon anode with both excellent capacity and a high ICE via a simple one-step sol–gel method. Through the design of nano-size and porous structure and the appropriate selection of electrolytes, the electrode finally delivers a high reversible capacity of 232.6 mA h g−1 at 200 mA g−1 while guaranteeing a satisfactory ICE (68.2%). A high rate capacity (138.4 mA h g−1 at 2000 mA g−1) and stable cycle performance (165.2 mA h g−1 at 1000 mA g−1 and 129.7 mA h g−1 at 2000 mA g−1 after 1500 cycles) are also obtained. Kinetics analysis reveals that the surface-controlled process plays a dominant role in the carbon spheres with nano-size and porous structure (SPCS), resulting in fast reaction kinetics for potassium storage.
Experimental
Material synthesis
Preparation of carbon spheres with nano-size and porous structure (SPCS): generally, 0.4 g resorcinol is firstly mixed in a solution containing 0.4 mL of ammonia solution (NH4OH), 40 mL of deionized water (DI) and 16 mL of absolute ethanol (EtOH). After stirring for 0.5 h, 0.4 g of sodium dodecyl sulfate (SDS) is added to form a homogeneous solution. 0.56 mL of formaldehyde solution (37%) is dispersed into the mixture, and 2 mL of tetraethyl orthosilicate (TEOS) is added quickly. The reaction is carried out for 24 h at room temperature, and then heated at 80 °C in a Teflon-lined autoclave for another 24 h. The solid powder is collected through filtration and air-dried at 80 °C overnight. After being carbonized at 700 °C for 3 h under an Ar atmosphere, the black powder (100 mg) is treated with 40 mL of hydrofluoric acid (5%) to dissolve the silica. Finally, SPCS can be obtained after centrifugal cleaning and vacuum drying. SPCS-800 and SPCS-900 are carbonized at 800 °C and 900 °C for 3 h, respectively.In contrast, CS is synthesized by a similar method except for adding SDS and TEOS, and SCS is synthesized without adding TEOS.
Material characterization
Morphologies of the prepared materials are observed using a scanning electron microscope (NovaNano SEM 230) and a transmission electron microscope (Tecnai G2 F20 S-TWIX). X-ray powder diffraction (XRD) is collected with a Cu-Kα1 source (λ = 1.5418 Å) on a Bruker D8 X-ray diffractometer. X-ray photoelectron spectroscopy (XPS) is conducted on an ESCALAB 250Xi spectrometer. The BET surface areas and pore size distribution are recorded on a V-Sorb 2800P. Raman spectra are recorded on a RAM HR-800.Electrochemical characterization
To prepare the working electrodes, active material, carboxymethyl cellulose (CMC) and acetylene black are mixed in DI with a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15 and rapidly pasted onto Cu foil. After drying in a vacuum overnight, the Cu foil is cut into circular electrodes. CR2016-type coin cells are assembled in an Ar-filled glovebox. Potassium metal is used as the counter electrode and glass fiber is applied as the separator. 0.8 M KPF6 or 1 M KFSI in EC/DEC solution (EC/DEC = 1:1, v/v), 0.8 M KPF6 in EC/PC solution (EC/PC = 1:1, v/v) and 1 M KFSI in DME solution are used as comparative electrolytes. Galvanostatic charge–discharge profiles are studied on a NEWARE battery testing system (CT-3008W) with a potential range of 0.01–3.0 V (vs. K+/K). Cyclic voltammetry (CV) measurements are characterized on an electrochemical workstation (Shanghai Chenhua). Galvanostatic intermittent titration (GITT) is performed by galvanostatic charging/discharging for 30 min at 15 μA g−1 and relaxing for 2 h.
:15:15 and rapidly pasted onto Cu foil. After drying in a vacuum overnight, the Cu foil is cut into circular electrodes. CR2016-type coin cells are assembled in an Ar-filled glovebox. Potassium metal is used as the counter electrode and glass fiber is applied as the separator. 0.8 M KPF6 or 1 M KFSI in EC/DEC solution (EC/DEC = 1:1, v/v), 0.8 M KPF6 in EC/PC solution (EC/PC = 1:1, v/v) and 1 M KFSI in DME solution are used as comparative electrolytes. Galvanostatic charge–discharge profiles are studied on a NEWARE battery testing system (CT-3008W) with a potential range of 0.01–3.0 V (vs. K+/K). Cyclic voltammetry (CV) measurements are characterized on an electrochemical workstation (Shanghai Chenhua). Galvanostatic intermittent titration (GITT) is performed by galvanostatic charging/discharging for 30 min at 15 μA g−1 and relaxing for 2 h.
Results and discussion
The preparation process of SPCS is schematically illustrated in Fig. 1a. Firstly, emulsion droplets are formed after the negatively charged RF precursor dispersed in the ternary solution (ammonia–ethanol–water).28,29 Then, the introduced sodium dodecyl sulfate (SDS) would be positioned on the surface of the emulsion droplets owing to the electrostatic interaction with ammonium ions (NH4+). In this process, SDS can supply the negative charges surrounding the surface of the emulsion droplets and further prevent their subsequent growth.30 Moreover, due to the presence of the anionic surfactant SDS, the last added TEOS cannot uniformly nucleate, but it rather formed a cage-like template around the droplets.31 In this case, TEOS is used not only to form pores, but also to further reduce the particle size. After removing SiO2, SPCS with nano-size and porous structure is finally obtained. | ||
| Fig. 1 (a) Schematic illustration of synthesizing SPCS. SEM images of (b) SiO2@SPCS consisting of a coated SiO2 template and embedded carbon spheres and (c) SPCS. (d) Size distributions of CS, SCS, and SPCS. (e) TEM images, (f) HRTEM image and (g–j) EDS elemental mappings of SPCS. | ||
From SEM and TEM images, it can be clearly observed that CS and SCS have smooth spherical morphologies with diameters of 681 nm and 284 nm (Fig. S1 and S2, ESI†), respectively. Particularly, after adding TEOS, a 3D core–shell structure consisting of a coated SiO2 template and embedded carbon spheres can be obtained (Fig. 1b and Fig. S3, ESI†). After removing the template, SPCS exhibits a rough spherical morphology with a porous structure and a small diameter of 152 nm (Fig. 1c and e). The detailed particle size distribution for CS, SCS, and SPCS is shown in Fig. 1d. Apparently, the particle size can be effectively reduced from 681 nm to 284 nm by adding SDS, demonstrating that the anionic surfactant has a significant effect on particle size regulation. It is well known that the energy storage performance of a device relies heavily on its nanostructure.32,33 In our case, the nano-size porous structure of SPCS will contribute to reduce the ion diffusion distance and increase active sites accommodating K+ adsorption, which is favorable for improving potassium storage capacity.34 The HRTEM image of SPCS exhibits an amorphous structure with an expanded interlayer distance of about 0.39 nm and EDS elemental mapping images confirm the homogeneous distributions of N, C, and O. As reported, compared with the limited interlayer spacing of graphite (0.335 nm), the expanded interlayer distance of SPCS (0.39 nm) enables more reversible intercalation of large K+.14
The nitrogen adsorption–desorption isotherms are verified for analyzing porosities of CS, SCS, and SPCS (Fig. 2a and Fig. S4, ESI†). The Brunauer–Emmett–Teller (BET) measured surface area of SPCS is 398.85 m2 g−1, which is much higher than those of CS (76.02 m2 g−1) and SCS (81.97 m2 g−1). In this connection, a low ICE (50.4%) of the SPCS electrode in the common KPF6-EC/DEC electrolyte also suggests the abundant porous structure of SPCS. Although a higher specific surface area is beneficial for reducing the diffusion pathways and contacting the electrolyte, the resulting low ICE should also be addressed. The X-ray diffraction (XRD) patterns show similar diffraction features for the three samples with two main broad diffractions at 22.5° and 43.5° (Fig. 2b). Furthermore, the average interlayer distance can be estimated to be 0.39 nm from the diffraction peak (2θ = 22.5°),35 which is in good agreement with the HRTEM result. This expanded interlayer distance facilitates the insertion of large-size K+. Raman spectra could further elucidate the extent of the disordered structure of the three samples (Fig. 2c). For carbon materials, the calculated intensity ratio of the D band (1336 cm−1) and G band (1565 cm−1) is often regarded as the degree of graphite disorder.36,37 SPCS has a higher intensity ratio (ID/IG) of 1.09 than those of CS (1.04) and SCS (1.04), demonstrating that more defects can be induced through the addition of TEOS. Besides, the increased defects would provide more active sites on the surface of SPCS, benefiting the reversible adsorption of K+ and further improving the potassium storage capacity. X-ray photoelectron spectroscopy (XPS) spectra are recorded to obtain further structural information of the three materials. As seen in Fig. S5 and Table S1 (ESI†), SPCS has higher N and O doping contents, which may be attributed to the addition of TEOS during the synthesis of SPCS. As reported, a higher heteroatom-doping content is conducive to the reversible adsorption of K+ and the wettability of the carbon electrodes, resulting in higher capacity and greater rate performance.38,39 The XPS survey spectra demonstrate the existence of N (399.4 eV), O (523.1 eV) and C (284.5 eV). The high-resolution N1s spectra can be deconvoluted into three typical nitrogen peaks, including pyridinic N (398.1 eV), pyrrolic N (400.2 eV) and graphitic N (401.1 eV), respectively.40 In recent studies, pyrrolic N and pyridinic N are proven to have higher K+-adsorption ability than graphitic N. For SPCS, the high content of this active N (63.5%, Table S3, ESI†) would provide more active sites, resulting in an enhanced surface-driven process in electrochemical reactions, and facilitate K+ adsorption.41
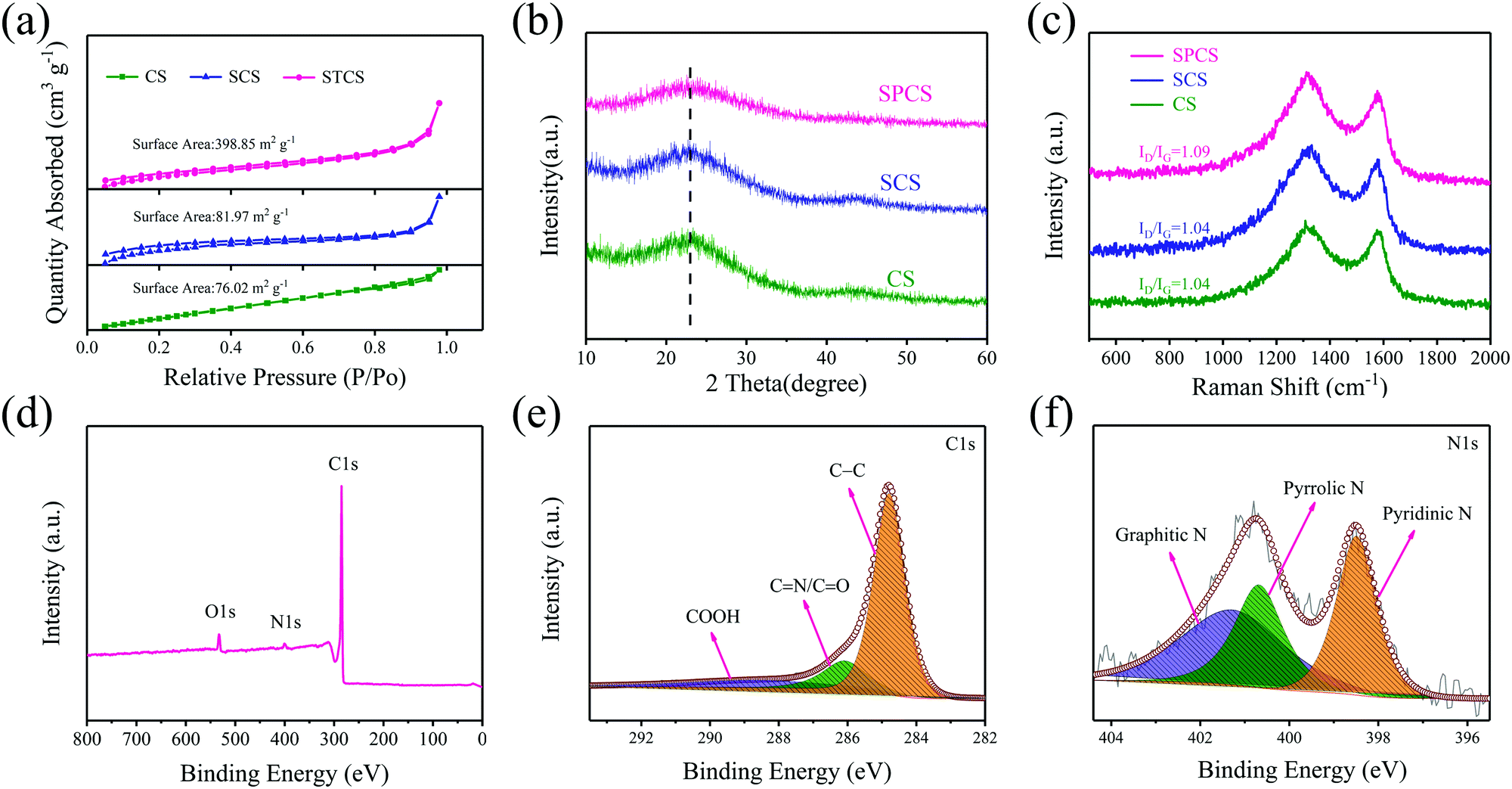 | ||
| Fig. 2 (a) Nitrogen adsorption–desorption isotherms of CS, SCS and SPCS. (b) X-ray diffraction (XRD) patterns and (c) Raman spectrum of all three samples. (d) XPS survey spectra, (e) C 1s spectra and (f) N 1s spectra of SPCS. | ||
The potassium storage performance of SPCS electrodes in different common electrolytes is firstly delivered in Fig. 3a and b. Although KPF6 electrolytes all show great cycling stability, the low ICE of 50.4% and 51.6% for them are unacceptable for practical application. In a recent study, it was confirmed that the KFSI-DME electrolyte facilitated the formation of a uniform and stable solid-electrolyte interphase (SEI) and exhibited excellent electrochemical stability in potassium metal batteries.42 As seen in Fig. 3a and b, the SPCS electrode with the KFSI-DME electrolyte delivers a relatively high ICE of 68.2%, and an ultrahigh capacity of 232.6 mA h g−1 even after 100 cycles at 200 mA g−1, which achieves the goal of improving the potassium storage capacity while maintaining a high ICE. Therefore, the following results are collected using the KFSI-DME electrolyte.
 | ||
| Fig. 3 Potassium storage behaviors of all electrodes: (a) cycle performance and (b) initial charge–discharge curves of SPCS with different electrolytes at 200 mA g−1. (c) The cyclic voltammetry (CV) curves of SPCS at 0.1 mV s−1. (d) Cycle performance and (e) initial charge–discharge profiles of CS, SCS, and SPCS at 200 mA g−1. (f) The rate capability of the three electrodes. (g) Long-term cycle stability of SPCS at high current densities of 1000 and 2000 mA g−1. | ||
As shown in Fig. 3c, the cyclic voltammetry (CV) curves of the SPCS electrode are tested at 0.1 mV s−1 for three cycles. The irreversible cathodic peak appears in the first cycle and disappears in subsequent cycles, which can be attributed to the formation of SEI layers and irreversible reactions.38,39 Besides, the subsequent cycles overlap well, suggesting excellent reversibility of the SPCS electrode.12 It is worth noting that the open circuit potentials (OCPs) of CS and SCS are 2.53 and 2.49 V (Fig. S6, ESI†), respectively, which are higher than that of SPCS (1.65 V). According to previous literature,43,44 a lower OCP reflects facile insertion of K+ into the surface of the electrode in the activation process, contributing to a high ICE. The cycling performance of CS, SCS, and SPCS is measured for 100 cycles at a current density of 200 mA g−1 (Fig. 3d). SPCS exhibits a high reversible capacity of 232.6 mA h g−1 after 100 cycles, which is higher than those of CS (121.5 mA h g−1) and SCS (179.5 mA h g−1). In addition, the initial charge–discharge curves for the three electrodes are shown in Fig. 3e. SCS delivers a much higher ICE of 74.5% than CS (60.3%). More importantly, SPCS with a high specific surface area still maintains a high ICE of 68.2%, which does not seem to match our cognition. Actually, no straightforward correlation between the specific surface area (and porosity) and irreversible capacity has been established.27 For example, Cao et al.45 increased the pyrolysis temperature of cellulose from 1300 to 1500 °C and the specific surface area was reduced from 117 to 43 m2 g−1. However, the ICE decreased from 63.4% to 55%, demonstrating several complex factors involved in the ICE. Some researchers have realized that sometimes the structure of carbon materials cannot be responsible for the low ICE, which must arise from the thicker SEI composed of the severely decomposed electrolyte and alkali metal ions. Zhang et al.46 demonstrated that a carbon electrode cycled in an ether-based solvent can deliver a higher ICE than in ester-based electrolytes. Compared with the thick SEI precipitated by large amounts of decomposed organic products in EC/DEC, the stable, thin and uniform SEI consumes fewer alkali metal ions and leads to a higher ICE in Diglyme. The superiority of ether-based electrolytes has also been proven in PIBs in Xie’ work,47 and the key factor is also the thin and stable SEI. In our case, the KFSI-DME electrolyte efficiently increases the ICE from around 50% to 68.2%, which can be widely used for carbon-based materials. Due to the nano-size porous structure and appropriate electrolyte, the SPCS electrode not only delivers a high reversible capacity but also retains a relatively higher ICE than those of mostly reported porous carbon materials (Table S4, ESI†).
The effect of particle size and its correlation with the rate performance of the three electrodes are displayed in Fig. 3f. CS with the largest diameter (681 nm) has capacities of 208.1, 181.8, 141.3, 103.7, 81.0 and 51.2 mA h g−1 at 50, 100, 200, 500, 1000 and 2000 mA g−1, respectively. When the particle size decreases, SCS with a reduced diameter (284 nm) shows higher capacities of 257.4, 223.3, 196.5, 165.8, 134.6 and 111.6 mA h g−1 at 50, 100, 200, 500, 1000 and 2000 mA g−1, respectively. This improved rate performance can be probably attributed to the increased surface atomic energy and decreased ion diffusion pathways as the particle size decreases.48 For SPCS, the particle size is further reduced to 152 nm with a porous structure, which is highly beneficial to the contact between the electrode and electrolyte. As seen in Fig. 3f, discharge capacities of SPCS at 50, 100, 200, 500 and 1000 mA g−1 are 345.5, 271.9, 233.2, 194.9 and 171.3 mA h g−1, respectively. More strikingly, even at a high current rate of 2000 mA g−1, a high capacity of 138.4 mA h g−1 can still be maintained, which is higher than those of CS (51.2 mA h g−1) and SCS (111.6 mA h g−1), demonstrating the best rate performance of SPCS. Additionally, the effect of thermal annealing on potassium storage performance is also clarified in Fig. S7 (ESI†). SPCS-800 and SPCS-900 deliver reversible capacities of 222.9 and 215.2 mA h g−1 at 200 mA g−1 after 100 cycles, respectively, which are close to that of SPCS. Nevertheless, when the current density reaches 500 mA g−1, SPCS-800 and SPCS-900 show much lower capacities than SPCS. In Xu's work, the N and O doping contents and three N-doped models of carbon materials that changed with temperature have been reported in detail.41 As shown in Tables S2 and S3 (ESI†), due to the high N and O doping contents and higher active N-doped models (pyrrolic and pyridinic N-doped models), the SPCS electrode expects higher potassium storage capacity and greater rate performance.
The long-term cycle stability of the SPCS electrode is also investigated at high current densities of 1000 and 2000 mA g−1 (Fig. 3g). Reversible capacities of 165.2 (1000 mA g−1) and 129.7 mA h g−1 (2000 mA g−1) can still be retained over 1500 cycles with low capacity decay rates of 0.021% and 0.026% per cycle, respectively, demonstrating the excellent rate performance and stable cycle performance of the SPCS electrode. Furthermore, SEM images of the SPCS electrode after 1500 cycles at 1000 mA g−1 are shown in Fig. 4. Although some cracks occur on the electrode, the spherical morphology of SPCS remains intact, which further indicates the long cycling stability of the SPCS electrode.
 | ||
| Fig. 4 SEM images of the SPCS electrode after 1500 cycles at 1000 mA g−1 at different magnifications of (a) 5000 X and (b) 10 000 X. | ||
The kinetic study of potassium storage reactions is conducted by cyclic voltammetry (CV) analysis. As shown in Fig. 5a and Fig. S10 (ESI†), CV curves with an increased scan rate from 0.1 to 2 mV s−1 are conducted. Obviously, the shape of the SPCS electrode is well maintained with a small overpotential. The potassium storage reactions can be investigated by the relationship between the peak current and scan rate:49,50
| i = avb |
 | ||
| Fig. 5 (a) CV curves of SPCS at 0.1–2 mV s−1. (b) Log(i) vs. log(v) plots of CS, SCS and SPCS. (c) Surface-controlled contribution of SPCS at 1 mV s−1. (d) Surface-controlled contributions for three electrodes at different scan rates. GITT profiles of (e) CS and (f) SPCS. Diffusion coefficients of SPCS and CS for (g) potassiation and (h) depotassiation. | ||
Here i represents the peak current and v represents the sweep rate. In general, a b value of 0.5 indicates diffusion-controlled intercalation, while a b value of 1.0 represents surface-controlled electrochemical reactions. The b value calculated for SPCS is 0.91 (Fig. 5b), which is higher than those for CS (0.72) and SCS (0.76), demonstrating that the charge–discharge process for SPCS mainly occurs on the surface. It is worth noting that b ≈ 1 is indicative of a capacitive process (either EDLC or pseudocapacitive) or a process that is not limited by ion diffusion due to the small material size/electrode thickness (nanosize effect; surface process) or specific material structure engineering.51,52 In our case, due to the high specific surface, nano-size and porous structure, a higher b value can be obtained for the SPCS electrode. Furthermore, the surface-controlled contribution ratio can be quantitatively calculated based on Dunn's method:53
| i = k1v + k2v1/2 |
To further understand the origins of the superior potassium storage of SPCS, the K+ diffusion coefficients (D) in CS and SPCS electrodes are calculated from the galvanostatic intermittent titration (GITT) measurement using Fick's second law with eqn (S1), ESI.†55 As shown in Fig. 5e and f, SPCS delivers smaller overpotentials than CS during each relaxation period, which indicates better potassiation/depotassiation kinetics for SPCS.56 According to the calculated values of D, it is clear that SPCS delivers higher diffusion coefficients than CS during both potassiation and depotassiation processes, revealing the faster reaction kinetics of SPCS.
Conclusions
In summary, unique carbon spheres with nano-size and porous structure (SPCS) have been successfully synthesized for potassium storage with enhanced performance through a facile one-step sol–gel method. Moreover, the potassium storage performance for SPCS electrodes in different conventional electrolytes is explored. Benefiting from the optimized nano-size and porous structure and the appropriate electrolyte, the SPCS electrode exhibits a high reversible capacity (347.3 m A h g−1 at 50 mA g−1), excellent rate performance (138.4 m A h g−1 at 2000 mA g−1) and superior cycling stability (165.2 mA h g−1 at 1000 mA g−1 and 129.7 mA h g−1 at 2000 mA g−1 after 1500 cycles). More importantly, a relatively high ICE of 68.2% can be achieved for the SPCS electrode at 200 mA g−1, which is much higher than those of previous hard carbon materials with a porous structure. Kinetics analysis reveals that the surface-controlled process increases along with the structure optimization, and thus the SPCS electrode delivers higher surface-controlled contribution and faster potassium diffusion coefficient than CS and SCS. This work demonstrates an efficient strategy to improve electrochemical performance while maintaining a high ICE for hard carbon electrodes, which would open a new avenue to develop carbonaceous materials for advanced PIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 21703185 and 51901013), the leading Project Foundation of Science Department of Fujian Province (Grants No. 2018H0034), Shenzhen Science and Technology Planning Project (JCYJ20170818153427106) and the “Double-First Class” Foundation of Materials and Intelligent Manufacturing Discipline of Xiamen University. Prof. H. Wang thanks the financial support from Hunan Provincial Science and Technology Plan Project of China (No. 2017TP1001 and 2018RS3009).References
- J. B. Goodenough and P. Kyu-Sung, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef CAS PubMed.
- H. K. B. Dunn and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef PubMed.
- Y. Zhou, Y. Huang, J. Pang and K. Wang, J. Power Sources, 2019, 440, 227149 CrossRef CAS.
- W. Wang, J. Yao, X. Zuo, Q. Yang, M. Wu, H. Tang, S. Jin and G. Li, Nanoscale Horiz., 2019, 4, 1211–1220 RSC.
- G.-T. Xia, C. Li, K. Wang and L.-W. Li, Sci. Adv. Mater., 2019, 11, 1079–1086 CrossRef CAS.
- J. C. Pramudita, D. Sehrawat, D. Goonetilleke and N. Sharma, Adv. Energy Mater., 2017, 7, 1602911 CrossRef.
- A. Eftekhari, Z. Jian and X. Ji, ACS Appl. Mater. Interfaces, 2016, 9, 4404–4419 CrossRef PubMed.
- L. Xue, Y. Li, H. Gao, W. Zhou, X. Lü, W. Kaveevivitchai, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 2164–2167 CrossRef CAS PubMed.
- A. Eftekhari, ACS Sustainable Chem. Eng., 2019, 7, 3684–3687 CrossRef CAS.
- H. He, D. Huang, Q. Gan, J. Hao, S. Liu, Z. Wu, W. K. Pang, B. Johannessen, Y. Tang and J.-L. Luo, ACS Nano, 2019, 13, 11843–11852 CrossRef CAS PubMed.
- X. Wu, D. P. Leonard and X. Ji, Chem. Mater., 2017, 29, 5031–5042 CrossRef CAS.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- D. Stevens and J. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- Z. Jian, S. Hwang, Z. Li, A. S. Hernandez, X. Wang, Z. Xing, S. Dong and X. Ji, Adv. Funct. Mater., 2017, 27, 1700324 CrossRef.
- C. Chen, Z. Wang, Z. Bao, M. Ling, C. Jie, L. Peng, Y. Huang, J. Jiang, Y. Huang and L. Zhang, Energy Storage Mater., 2017, 8, 161–168 CrossRef.
- H. He, D. Huang, Y. Tang, Q. Wang, X. Ji, H. Wang and Z. Guo, Nano Energy, 2019, 57, 728–736 CrossRef CAS.
- M. Chen, W. Wang, X. Liang, S. Gong, J. Liu, Q. Wang, S. Guo and H. Yang, Adv. Energy Mater., 2018, 8, 1800171 CrossRef.
- Y. Li, Y. S. Hu, M. M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef.
- J. Ding, H. Wang, Z. Li, K. Cui, D. Karpuzov, X. Tan, A. Kohandehghan and D. Mitlin, Energy Environ. Sci., 2015, 8, 941–955 RSC.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- J. B. Cook, H. S. Kim, Y. Yan, J. S. Ko, S. Robbennolt, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2016, 6, 1501937 CrossRef.
- D. Qiu, J. Guan, M. Li, C. Kang, J. Wei, Y. Li, Z. Xie, F. Wang and R. Yang, Adv. Funct. Mater., 2019, 1903496 CrossRef.
- L. Liu, Y. Chen, Y. Xie, P. Tao, Q. Li and C. Yan, Adv. Funct. Mater., 2018, 28, 1801989 CrossRef.
- S. Zhuo, M. Tang, Y. Wu, Y. Chen, S. Zhu, Q. Wang, C. Xia and C. Wang, Nanoscale Horiz., 2019, 4, 1092–1098 RSC.
- Y. Wang, Z. Zhang, G. Wang, X. Yang, Y. Sui, F. Du and B. Zou, Nanoscale Horiz., 2019, 4, 1394–1401 RSC.
- D. Sun, B. Luo, H. Wang, Y. Tang, X. Ji and L. Wang, Nano Energy, 2019, 64, 103937 CrossRef CAS.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- J. Liu, S. Qiao, H. Liu, J. Chen, A. Orpe, D. Zhao and G. Lu, Angew. Chem., Int. Ed., 2011, 50, 5947–5951 CrossRef CAS PubMed.
- A. B. Fuertes, P. Valle-Vigón and M. Sevilla, Chem. Commun., 2012, 48, 6124–6126 RSC.
- M. S. Strano, V. C. Moore, M. K. Miller, M. J. Allen, E. H. Haroz, C. Kittrell, R. H. Hauge and R. E. Smalley, J. Nanosci. Nanotechnol., 2003, 3, 81–86 CrossRef CAS PubMed.
- C. Tondre and C. Caillet, Adv. Colloid Interface Sci., 2001, 93, 115–134 CrossRef CAS PubMed.
- J. Leng, Z. Wang, J. Wang, H.-H. Wu, G. Yan, X. Li, H. Guo, Y. Liu, Q. Zhang and Z. Guo, Chem. Soc. Rev., 2019, 48, 3015 RSC.
- L. Zhao, H.-H. Wu, C. Yang, Q. Zhang, G. Zhong, Z. Zheng, H. Chen, J. Wang, K. He, B. Wang, T. Zhu, X. Zeng, M. Liu and M.-S. Wang, ACS Nano, 2018, 12, 12597–12611 CrossRef CAS PubMed.
- Z. Jian, Z. Xing, C. Bommier, Z. Li and X. Ji, Adv. Energy Mater., 2016, 6, 1501874 CrossRef.
- M. Sevilla and A. B. Fuertes, Carbon, 2013, 56, 155–166 CrossRef CAS.
- C. Portet, G. Yushin and Y. Gogotsi, Carbon, 2007, 45, 2511–2518 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- N. Xiao, W. D. Mcculloch and Y. Wu, J. Am. Chem. Soc., 2017, 139, 9475–9478 CrossRef CAS PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu and V. Barone, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef PubMed.
- M. Ihsan-Ul-Haq, H. Huang, J. Wu, J. Cui, S. Yao, W. G. Chong, B. Huang and J.-K. Kim, Nano Energy, 2020, 71, 104613 CrossRef CAS.
- J. Jang, Y. Kim, O. B. Chae, T. Yoon, S. M. Kim, H. S. Kim, H. Park, J. H. Ryu and S. M. Oh, Angew. Chem., Int. Ed., 2014, 53, 10654–10657 CrossRef CAS PubMed.
- Q. Shen, L. Xiao, M. L. Sushko, K. S. Han and J. Liu, Adv. Energy Mater., 2017, 7, 1700403 CrossRef.
- J. Zhang, D.-W. Wang, W. Lv, S. Zhang, Q. Liang, D. Zheng, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 370–376 RSC.
- J. Xie, X. Li, H. Lai, Z. Zhao, J. Li, W. Zhang, W. Xie, Y. Liu and W. Mai, Angew. Chem., Int. Ed., 2019, 131, 14882–14889 CrossRef.
- S. R. Sivakkumar, J. Nerkar and A. Pandolfo, Electrochim. Acta, 2010, 55, 3330–3335 CrossRef CAS.
- A. Veronica, C. Jérémy, M. A. Lowe, K. Jong Woung, T. Pierre-Louis, S. H. Tolbert, H. D. AbrunA, S. Patrice and D. Bruce, Nat. Mater., 2013, 12, 518–522 CrossRef PubMed.
- S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan and S. Yu, Nat. Commun., 2016, 7, 13318 CrossRef CAS PubMed.
- Y. Jiang and J. Liu, Energy Environ. Sci., 2019, 2, 30–37 Search PubMed.
- J. Xie, P. Yang, Y. Wang, T. Qi, Y. Lei and C. M. Li, J. Power Sources, 2018, 401, 213–223 CrossRef CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146 CrossRef CAS PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- B. Cao, Q. Zhang, H. Liu, B. Xu, S. Zhang, T. Zhou, J. Mao, W. K. Pang, Z. Guo and A. Li, Adv. Energy Mater., 2018, 8, 1801149 CrossRef.
- Y. Xu, Y. Zhu, Y. Liu and C. Wang, Adv. Energy Mater., 2013, 3, 128–133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00018c |
| This journal is © The Royal Society of Chemistry 2020 |