
Colloidal PbS quantum dot stacking kinetics during deposition via printing†
Wei
Chen
 a,
Haodong
Tang
b,
Nian
Li
a,
Manuel A.
Scheel
a,
Yue
Xie
b,
Depeng
Li
b,
Volker
Körstgens
a,
Matthias
Schwartzkopf
c,
Stephan V.
Roth
cd,
Kai
Wang
b,
Xiao Wei
Sun
b and
Peter
Müller-Buschbaum
*ae
a,
Haodong
Tang
b,
Nian
Li
a,
Manuel A.
Scheel
a,
Yue
Xie
b,
Depeng
Li
b,
Volker
Körstgens
a,
Matthias
Schwartzkopf
c,
Stephan V.
Roth
cd,
Kai
Wang
b,
Xiao Wei
Sun
b and
Peter
Müller-Buschbaum
*ae
aLehrstuhl für Funktionelle Materialien, Physik-Department, Technische Universität München, James-Franck-Straße 1, 85748 Garching, Germany. E-mail: muellerb@ph.tum.de
bDepartment of Electrical and Electronic Engineering, Southern University of Science and Technology (SUSTech), Xueyuan Blvd. 1088, 518055 Shenzhen, China
cDeutsches Elektronen-Synchrotron (DESY), Notkestraße 85, 22607 Hamburg, Germany
dDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden
eHeinz Maier-Leibnitz Zentrum (MLZ), Technische Universität München, Lichtenbergstraße. 1, 85748 Garching, Germany
First published on 27th February 2020
Abstract
Colloidal PbS quantum dots (QDs) are attractive for solution-processed thin-film optoelectronic applications. In particular, directly achieving QD thin-films by printing is a very promising method for low-cost and large-scale fabrication. The kinetics of QD particles during the deposition process play an important role in the QD film quality and their respective optoelectronic performance. In this work, the particle self-organization behavior of small-sized QDs with an average diameter of 2.88 ± 0.36 nm is investigated for the first time in situ during printing by grazing-incidence small-angle X-ray scattering (GISAXS). The time-dependent changes in peak intensities suggest that the structure formation and phase transition of QD films happen within 30 seconds. The stacking of QDs is initialized by a templating effect, and a face-centered cubic (FCC) film forms in which a superlattice distortion is also found. A body-centered cubic nested FCC stacking is the final QD assembly layout. The small size of the inorganic QDs and the ligand collapse during the solvent evaporation can well explain this stacking behavior. These results provide important fundamental understanding of structure formation of small-sized QD based films prepared via large-scale deposition with printing with a slot die coater.
New conceptsColloidal PbS quantum dots (QDs) attract tremendous research interest for advanced optoelectronic applications, including photodetectors and photovoltaics. To realize the large-scale depositions of QDs via printing while maintaining their long-term storage capability, ligand capped QDs are of particular interest. In addition, small-sized QDs with suitable band-gap demonstrate better photovoltaic device performance than large-sized QDs. The present study gives new conceptual insights into the stacking behavior of ligand capped QDs during printing and can explain the energetic disorder of ligand treated QD solids. QDs experience two phase transitions. The “solvent-wet” transition is initialized by their hexagonally close-packed arrangement, which triggers the face-centered cubic (FCC) superlattice stacking afterwards. In the “wet-dry” transition, QDs reveal a superlattice distortion towards a body-centered cubic stacking with maintaining the FCC character due to the large ratio of the ligand thickness to the radius of the QDs. |
Colloidal PbS quantum dots (QDs) are attractive for being used in various optoelectronic applications due to their tunable band-gap with a large absorption wavelength range from the visible to the near-infrared region.1,2 Moreover, due to the high intrinsic permittivity, the exciton binding energy of PbS is smaller than in cadmium chalcogenide QDs and polymer materials, which is beneficial for devices driven by charge carrier generation and transport, e.g. photovoltaics and photodetectors.3–8 Colloidal QDs are commonly synthesized via hot-injection methods,9–11 in which the organic ligands in the precursor play an important role in controlling the size and shape of the QDs during the synthesis process and also for maintaining the suspension of QDs in the solvent without aggregations.12 Therefore, colloidal QDs reveal a good solubility in many solvents, which indicates their compatibility with various large-scale solution-processing thin-film deposition techniques,13,14 including spray-deposition and printing.15–17 Moreover, due to these ligands, QDs are treated as semi-soft particles with a hard-core/soft-shell configuration. The stacking behavior of QDs in a self-organization process determines the quality of as-deposited film.16 Related studies also provide the fundamental insights on the self-organization process of colloidal particles.18,19 The kinetics of PbS QDs in a slow evaporation process (over 30 min) was reported previously, in which the as-formed superlattice based on large-sized QDs (over 5 nm in diameter) and the corresponding phase transitions were well described and explained.20,21
Considering that the organic shell layer thickness of the QDs is determined by the ligand thickness, which can be treated as a static value in specific solvent conditions, the semi-soft material properties are influenced by the solvent conditions as well as by the ratio of the ligand thickness (L) over the radius of the QDs (R). Thus, the stacking behavior of the QDs is determined by their overall size as seen in Scheme 1. Small-sized QDs, with a less pronounced shape contribution from their inorganic core, due to the large ratio of ligand-layer-thickness over the QD radius,22 exhibit a more sphere-like behavior during the drying process than large-sized QDs. Such differences suggest different particle kinetics during their self-assembly in a wet chemical deposition process such as printing. Moreover, from the aspect of potential applications, PbS QDs with a small average diameter demonstrate better photovoltaic performances in a planar heterojunction architecture,8,23–29 in which a large-scaled QD's deposition would also be highly desired. Although the recently developed QD-ink technologies create a simplified way for the deposition and the functionalization in a single step, these ink techniques still face many challenges. For example, ligand exchanged QDs in a solvent significantly rely on the use of costly lead halides (PbI2 or PbBr2) in combination with metal ion coordinating solvents, like butylamine, to maintain their solubility, thereby making it less compatible with current deposition methods.24,30–36 Therefore, the deposition of conventional ligand capped QDs is still promising for a low-cost large-scale device fabrication in the near future, in which the as deposited QD solids are functionalized by a post-ligand-exchange treatment as reported in previous works.23,37 Thus, the kinetics of small-sized QDs during particle stacking into a close packed configuration in a large-scale deposition process will provide a significant fundamental understanding. For example, QDs with different superlattice stacking will result in a different neighboring inter-dot distance distribution, which induces different inter-dot couplings and leads to the inner energy state disorder of QD film.16
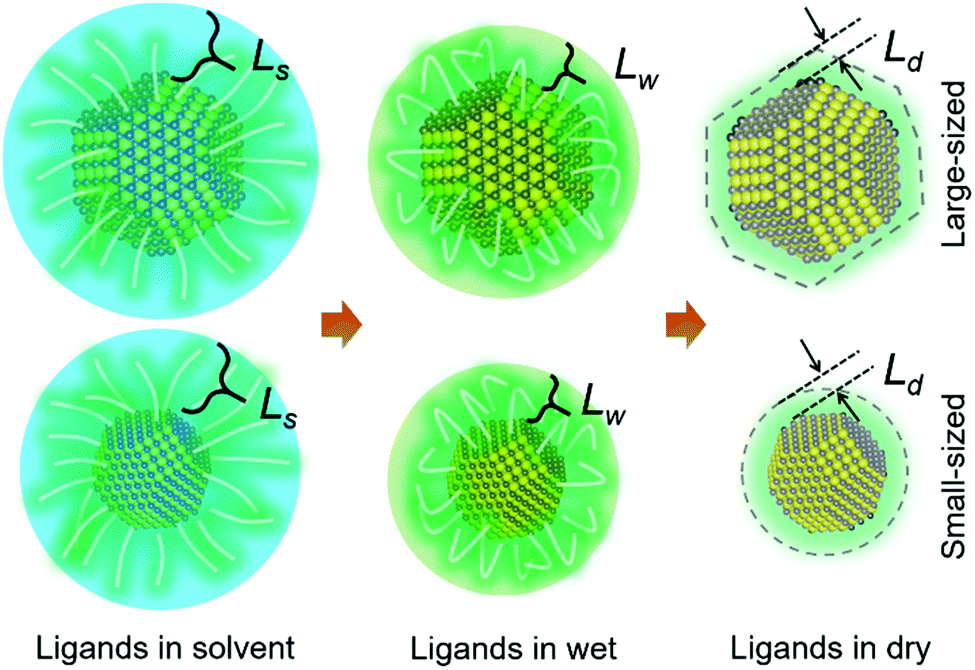 | ||
| Scheme 1 Ligand thickness L variation of QDs in different conditions during the drying process, in which Ls > Lw > Ld, corresponding to the ligand thickness for QDs in the solvent state Ls, in wet (no apparent solvent state but not fully dry) Lw and dry conditions Ld, respectively. Larger sized QDs with an apparent final shape factor are shown in the upper row and smaller sized QDs with a non-apparent shape factor in the lower row. | ||
In this work, the stacking behavior of small-sized PbS QDs is investigated during deposition via printing. We combine printing with an in situ characterization based on grazing-incidence small-angle X-ray scattering (GISAXS) at a synchrotron (P03, DESY).38 The QDs were prepared according to the literature with some modifications as seen in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 The final QDs with an average diameter of 2.88 ± 0.36 nm reveal the first exciton absorption peak at 935 nm as seen in Fig. 1. Fig. 1(a) illustrates the QDs under room-light conditions, revealing a full absorption of the visible light by the QDs. Transmission electron microscopy (TEM) images (Fig. 1(b)) indicate a good dispersion of the QDs without aggregation, due to the capping with oleic acid as surface ligands, as sketched in Fig. 1(c). The histogram data in Fig. 1(d) derived from the TEM image analysis shows the narrow size distribution of the QDs. The bandgap of the QDs (Fig. 1(e)) is well meeting with the requirements for high-performance photovoltaic applications.39,40
10 The final QDs with an average diameter of 2.88 ± 0.36 nm reveal the first exciton absorption peak at 935 nm as seen in Fig. 1. Fig. 1(a) illustrates the QDs under room-light conditions, revealing a full absorption of the visible light by the QDs. Transmission electron microscopy (TEM) images (Fig. 1(b)) indicate a good dispersion of the QDs without aggregation, due to the capping with oleic acid as surface ligands, as sketched in Fig. 1(c). The histogram data in Fig. 1(d) derived from the TEM image analysis shows the narrow size distribution of the QDs. The bandgap of the QDs (Fig. 1(e)) is well meeting with the requirements for high-performance photovoltaic applications.39,40
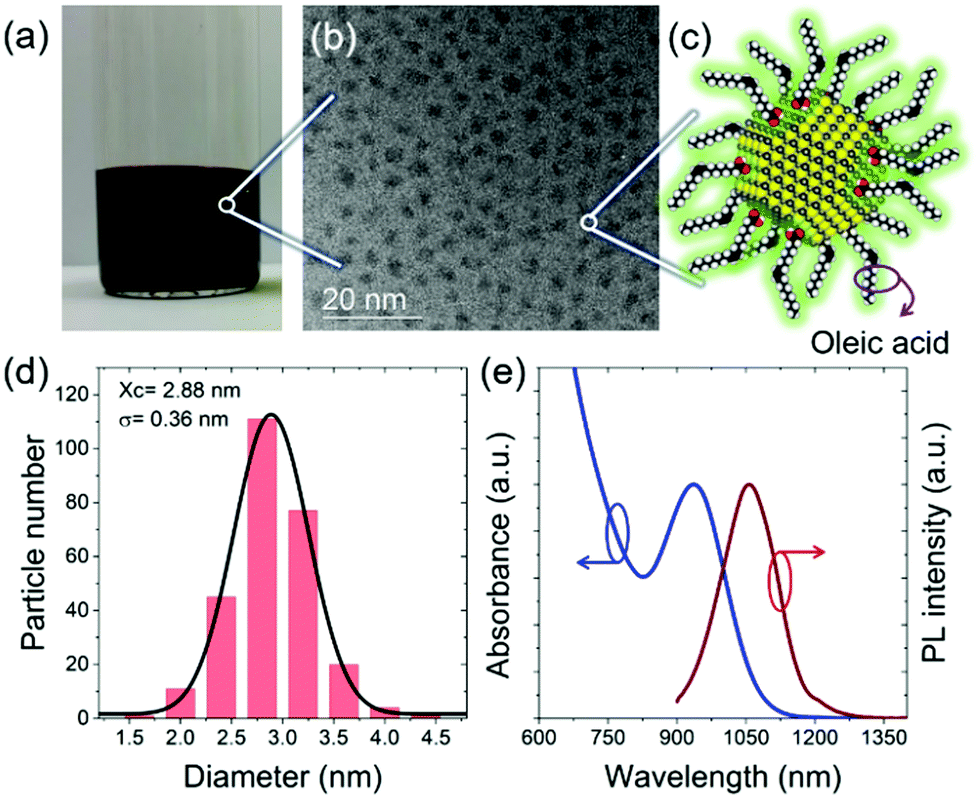 | ||
| Fig. 1 (a) Photograph of QDs in octane solvent (room light conditions). (b) TEM image of QDs. (c) Structure sketch of a QD with PbS core and an oleic acid acting as the organic ligand shell. (d) Size histogram from the TEM image analysis indicating the average diameter is 2.88 ± 0.36 nm. (e) Absorption spectrum of QDs in octane and corresponding PL emission spectrum. | ||
The wet-chemical deposition via printing is performed with a home-made slot-die coater, which is installed in the beamline for the time-resolved in situ GISAXS study.41 The printer head is fixed inside the printer chamber. Thus, the deposition is driven by the solvent injection and simultaneously moving of the substrate holder when the solvent flows out along the flow-guider. Notably, the morphology of printed QD films can be influenced by using different solvents and by tuning the printing parameters. We select parameters to ensure homogeneous films after printing. After the deposition of QDs, the whole printing set-up moves back by the motors to the preset position of X-ray spot and the GISAXS monitoring is started immediately. This moment is defined as t = 0 s for measuring the stacking kinetics of the deposited QDs. More details about the printing process are described in the ESI.† The related photographs of the set-up are seen in Fig. S1 (ESI†). The X-ray photon energy is 12.9 keV with a focused beam size of 25 μm × 20 μm. The sample-detector distance is set to 2591 mm to ensure that the scattering signal of the QDs is well recorded. The incident angle is 0.4°. The monitoring region on the substrate is pre-selected as a small homogeneous region (in 3 mm width), which is confirmed by the GISAXS scanning as seen in Fig. S2 (ESI†), in which a line cut mapping is shown. For in situ measurements, the region is divided into 15 points with intervals of 0.2 mm width. 0.1 s X-ray exposure is selected for each point to avoid beam damage. More detailed parameters for the in situ GISAXS experiment are provided in the ESI.† As acquired time-resolved 2D GISAXS data are seen in Fig. S3 (ESI†).
Fig. 2 shows the 2D mapping presentation (by software DPDAK42) of (a) vertical line cuts at qy = 0 nm−1 and (b) horizontal line cuts at αf = 0.2°, which is corresponding to the Yoneda peak position of the final QD film.43 The corresponding time-resolved scattering curves are seen in Fig. 2(c) and (d), respectively. In the initial stage of the QD deposition, the QD film has a disordered particle density distribution. A well-pronounced Yoneda peak of the QDs is found at αf = 0.2° (marked with blue arrow in Fig. 2(c)) at t = 20 s. Thus, the solvent has almost fully evaporated and the QDs are in a close packed configuration.44 Moreover, intensity peaks emerge at larger scattering angles due to an ordered QD layer formation (blue arrows in Fig. 2(c)).45 The scattering peak at αf = 0.75° originates mainly from the vertical layer-to-layer stacking in the as formed QD superlattice. Its position matches with a face-centered cubic (FCC) stacking with a superlattice distance d111SL (real space sketch of the superlattice as indicated in Fig. 2(e)). In the horizontal line cuts Bragg peaks emerge at larger qy values due to the formation of a well-defined inter-dot distance of neighboring QDs.46 At t = 18 s, double Bragg peaks are showing up at 1.08 nm−1 and 1.13 nm−1 (see inset in Fig. 2(d)) indicating the formation of an initial close-packed configuration in the QD assembly with two characteristic length scales. During drying, the Bragg peak at a smaller qy position is vanishing, whereas the one at a larger qy position is increasing in its scattering intensity. Such templating effect is confirmed by the corresponding simulations with the software BornAgain.47 As seen in Fig. S4 (ESI†), the hexagonal layout of QDs on the substrate plane with different inter-dot distance fits well the observed scattering peaks. Thus, the templating effect is considered to be initialized by the hexagonal arrangement of the QDs, in which the QD layout on the substrate plane corresponds to the (111)SL plane of the FCC superlattice. This hexagonal arrangement of QDs with fully stretched ligands maximizes the ensemble entropy in combination with a high degree of space filling. Moreover, the interlayer distance in the vertical direction decreases from 6.0 nm to 5.5 nm due to having a close to fully evaporated solvent. The QDs are in an ambient-condition-transition from “in solvent” to “in wet”. Upon evaporation, a further ligand collapse is continuously observed from the shift of the qy position of the Bragg peak. The final Bragg peak position of qy = 1.16 nm−1 corresponds to a final inter-QD distance dinter-dot = 5.41 nm, based on spherical models.48 We observe a homogeneous stacking of the QDs in a large region.
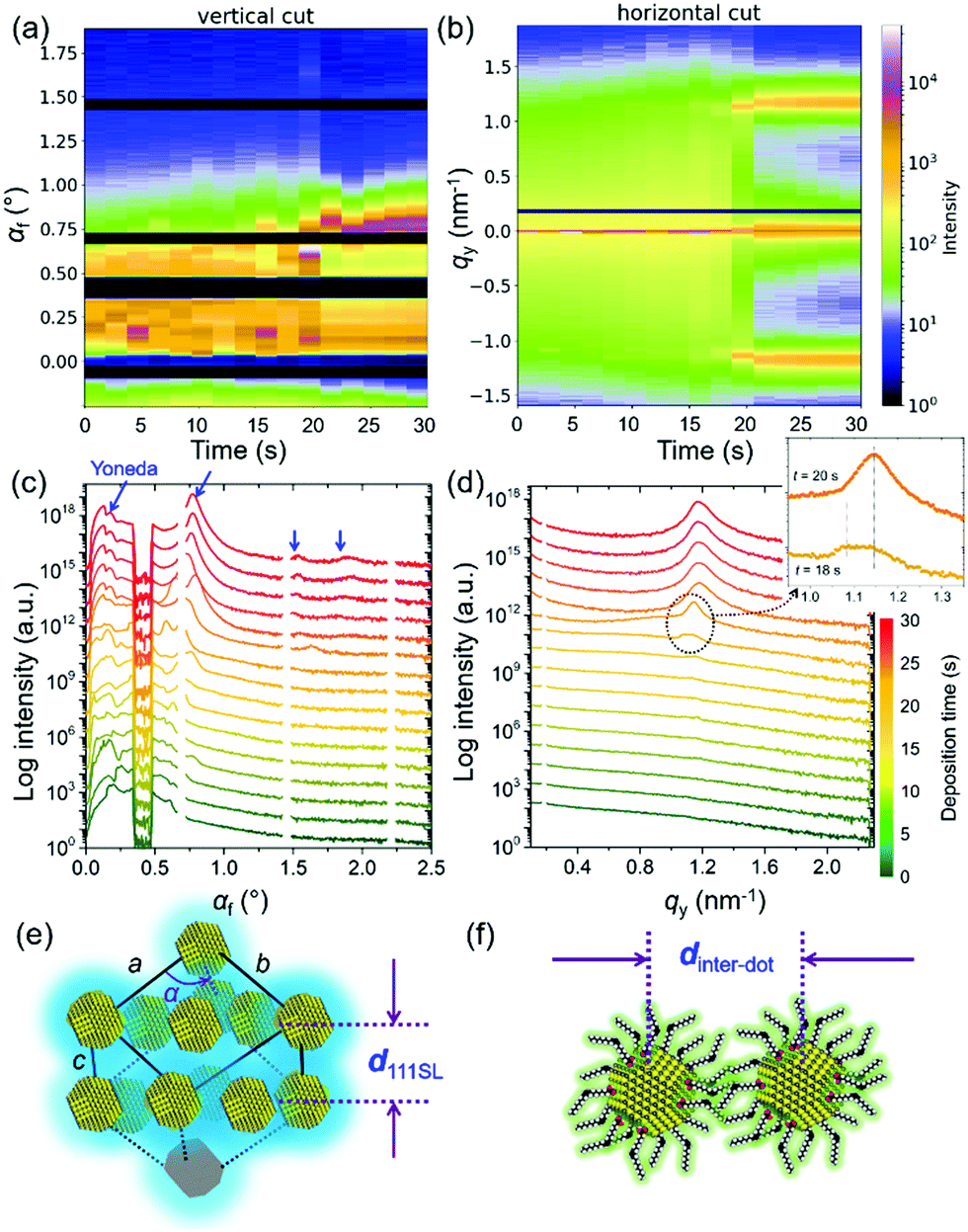 | ||
| Fig. 2 2D mapping presentation of (a) vertical line cuts and (b) horizontal line cuts of the 2D GISAXS data. In the corresponding curves in (c) vertical direction the specular beam is shielded and in (d) horizontal direction the resolution limit is not shown. Arrows mark scattering features in vertical (blue) and horizontal direction (black) as described in the text. Scheme for the (e) inter-lattice distance in vertical direction, in which for example d111SL from an FCC configuration causes a scattering peak at αf = 0.75° and for the (f) inter-dot distance in horizontal direction corresponding to the in-plane Bragg peak at around qy = 1.2 nm−1. | ||
To better understand the stacking process of QDs, the superlattice based specific lattice orientation is further analyzed from the sector integration of the 2D GISAXS data and the corresponding tube cuts as indicated in Fig. 3(a). During the drying process of the as-deposited film, the QD superlattice is gradually forming, as seen by the development of the Bragg peaks in the integrated data (Fig. 3(b)). The QD film transforms from a disordered state to an ordered state (superlattice state) at t = 18 s by the driving forces of an increased QD concentration in the deposited film. The uniform structure factor originates from the size mono-dispersity of the QDs and from the surface ligand shell with a defined thickness. The peaks are indexed with the QD superlattice (SL), in which the 200SL peak from the FCC stacking demonstrates a full scattering peak signal, without hidden information on the detector, with an intensity, which is used for a further orientation distribution analysis. Thus, the tube cuts are performed along the χ direction in a range of 1.45 nm−1 < q < 1.55 nm−1 to cover the time-resolved 200SL peak evolution (Fig. 3(c)). A Gaussian function is used to fit the 200SL peak in the tube-cuts and the results are shown in Fig. 3(d). The 200SL peak starts to show up at t = 20 s resembling an orientation peak at χ = 49.5°. Afterwards, the peak moves to higher scattering angles. It reaches χ = 52° at t = 24 s. Notably, this value is approaching the theoretical value of 54.7°, which is the intersection angle between (200)SL and (111)SL planes in a standard cubic model, in which the (111)SL plane is parallel to the substrate. That the theoretical value cannot be fully reached in the grazing-incidence configuration, even though the superlattice is perfectly aligned, is attributed to refraction effects.49 During the further drying process, the 200SL peak moves to a decreased χ value in the time range of 26 s < t < 30 s, which indicates a superlattice distortion. The ligand collapse is supposed to be the reason for this distortion.50 Instead of the original spherical shape introduced by the ligand capping, a truncated octahedron shape from the inorganic core of the QDs is dominating in dry conditions. Thus, the stacking behavior of QDs undergoes a transition from FCC (for spheres) to a BCC (for truncated octahedrons) packing according to their specific close packed configurations during drying, respectively.40 Consequently, the particle self-organized system intrinsically reveals a decreased total surface area and thus an increased entropy.22
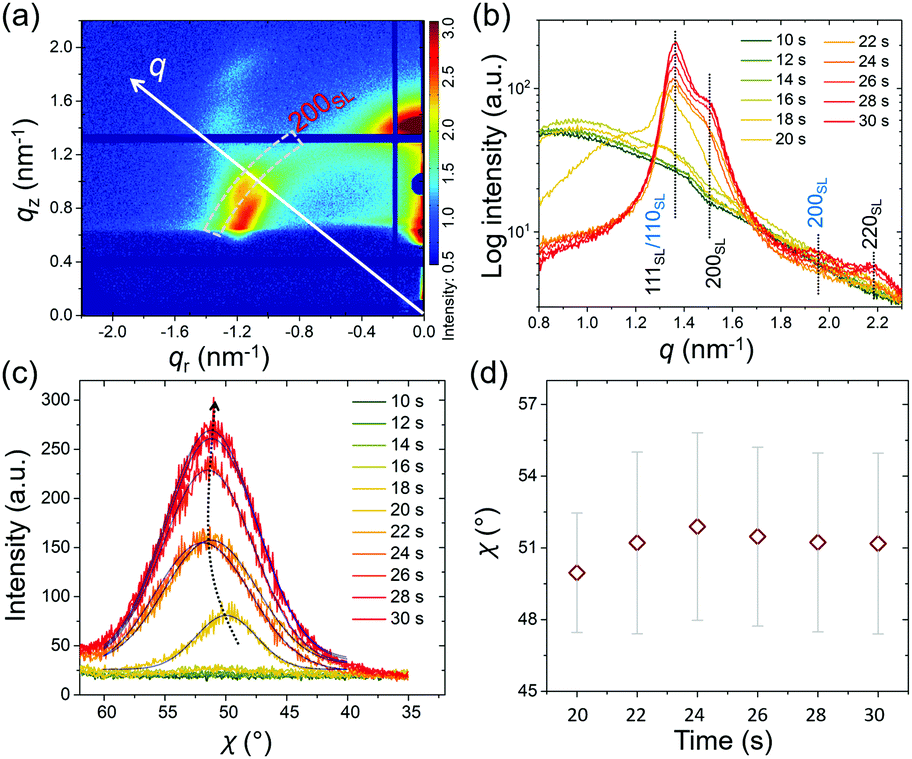 | ||
| Fig. 3 Diagram for the superlattice orientation analysis in the (a) 2D GISAXS data indicated by the q direction and χ direction along the 200SL superlattice peak (dashed box). (b) Sector integration in a range of 70° > χ > 20° with dashed lines marking expected Bragg peaks from the superlattice. The FCC superlattice is indexed in black and the BCC superlattice in blue. (c) Orientation distribution of the 200SL peak from the FCC superlattice fitted by Gaussian function. (d) Corresponding peak positions from the fits with the dispersity represented by Sigma from the corresponding Gaussian function shown by the bar. The experimental error is shown by the symbol size. | ||
To describe the particle kinetics in more detail and accuracy, model lattice calculations based on standard superlattice models are performed with the software GIXSGUI,51 to compare with the measured GISAXS patterns. Selected GISAXS data with calculated positions of Bragg peaks are seen in Fig. 4(a)–(c). Apparently, before t = 18 s the QDs have a disordered packing as seen by the absence of structure factor contributions in the GISAXS data. At t = 18 s, initial Bragg peaks show up at qy = 1.1 nm−1 with a weak intensity as seen in Fig. 4(a). At this stage, the solvent evaporation increased the QD concentration reaching a transition between the “solution phase” and the “wet phase”. QDs with stretched ligands are in an isotropically close-packed configuration, giving rise to a uniform structure factor. Moreover, due to the ligand stretching in the residual solvent, the QDs still behave like spheres as indicated in Scheme 1. Thus, the QDs are supposed to stack in an FCC packing due to the increased entropy. Sphere models prefer to better occupy space in order to decrease the total surface energy. Therefore, an FCC superlattice model with lattice parameters a = b = c = 9.40 nm and α = β = γ = 90° is used in the calculation of the scattering peaks (Fig. 4(a), red circles for transmission, white circles for reflection). We observe a good agreement with the detected scattering peaks, in which the (111)SL plane is oriented parallel to the substrate. The phase transition happens rapidly in the following 2 s. The FCC lattice decreases slightly from 9.40 ± 0.10 nm to 9.00 ± 0.10 nm (see calculations in Fig. 4(b)). Moreover, the scattering from the FCC superlattice is dominating the 2D GISAXS intensity distribution, meaning that all QDs are aligned in superlattice structures. The calculation indicates that the stacking of the QDs is getting denser. The obtained superlattice distance of (220)SL is 6.36 nm, which can infer that the ligand thickness of the QDs is around 1.74 nm considering the average diameter of the QDs from the TEM image analysis. The superlattice undergoes a further collapse during the next 2 s with a lattice spacing of 8.60 nm, meaning that the thickness of the ligand shell decreases to 1.60 nm (Fig. S5, ESI†). Further variations of the superlattice parameters are also found to give rise to additional Bragg peaks showing up as seen in Fig. 4(c). A body-centered cubic (BCC) superlattice model with lattice parameters of a = b = c = 6.60 nm and α = β = γ = 90° is found to match these additional scattering peaks very well, especially for the 200SL peak in the BCC superlattice, in which the (110)SL plane is parallel to the substrate. The agreement indicates that the final QDs have nested superlattice structures with both FCC and BCC packing as indicated in Fig. 4(e). Moreover, the effective ligand thickness of the QDs in the BCC structure is further decreased to 1.42 nm, which is calculated based on the superlattice distance of (111)SL in the BCC superlattice. Notably, these results differ slightly from the in-plane line cut analysis of the GISAXS data due to the used superlattice approximation. QDs are treated as spherical particles in the superlattice stacking as a simplification in the ligand thickness calculation. The crystal lattice orientation analysis (Fig. S7, ESI†) indicates that QDs reveal a specific orientation of the facets, which is in good agreement with previous research.20,40 This BCC packing originates from the shape of the QDs in dry conditions. Since the ligands are collapsed and the QDs reveal more a behavior like truncated octahedrons, a BCC stacking tendency is found. However, the QDs superlattice in the final dry film still mainly remains an FCC stacking character. This means that the QDs still mainly have a sphere-like stacking behavior due to the large ratio of ligand thickness over the average radius of the QDs.
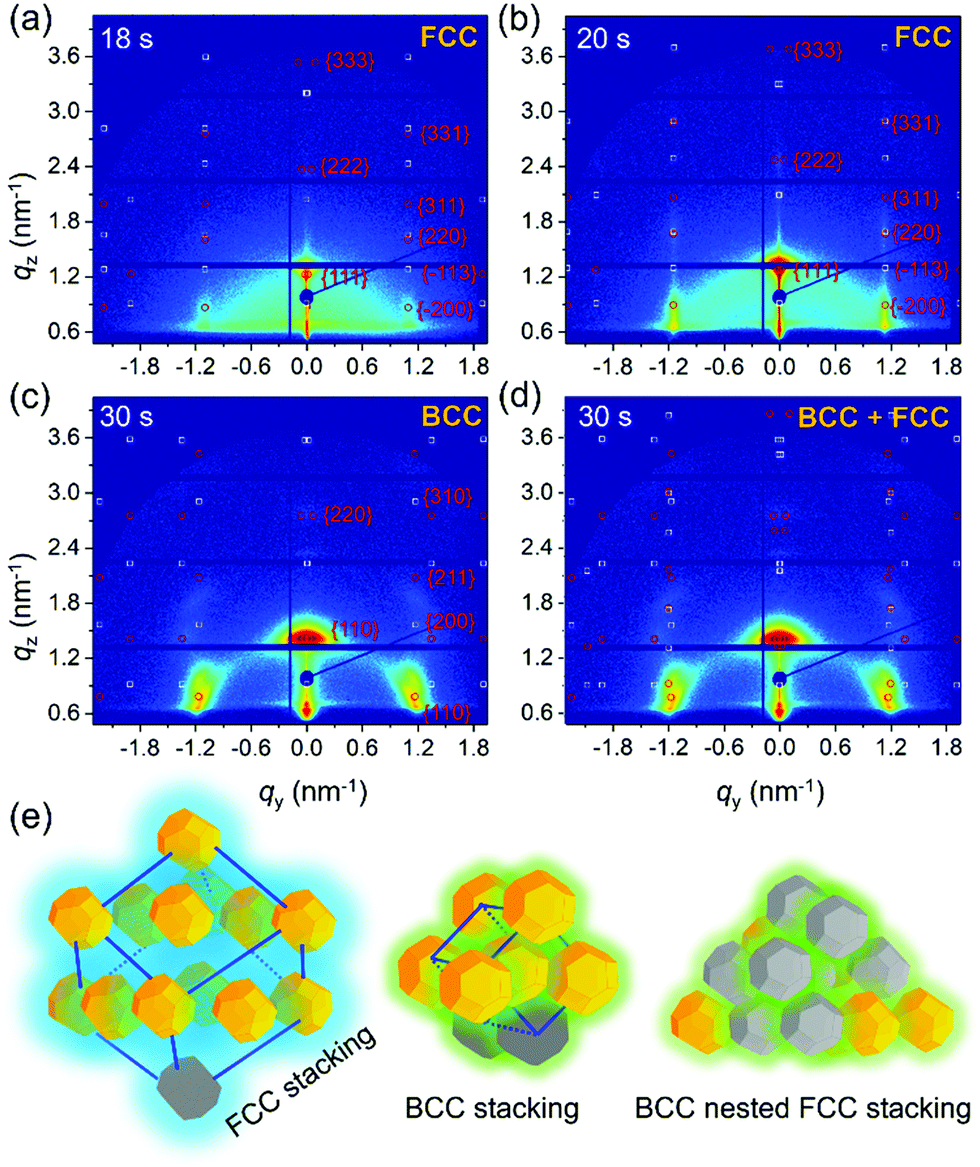 | ||
| Fig. 4 2D GISAXS data at (a) t = 18 s and (b) t = 20 s with calculated scattering peaks based on an FCC superlattice. 2D GISAXS data of t = 30 s with a (c) BCC and (d) BCC nested FCC superlattice Bragg peaks (red dots: transmitted scattering peaks; white dots: reflected scattering peaks). Scheme for the FCC superlattice with (111)SL plane parallel to the substrate, BCC superlattice with (110)SL plane parallel to the substrate and BCC nested superlattice with (111)SL plane of FCC lattice parallel to the substrate and (110)SL plane of BCC lattice parallel to the substrate. | ||
A quite similar stacking behavior of the QDs is found when the printing is done on a different substrate using the same printing parameters as seen in Fig. S9 (ESI†).
Conclusions
In conclusion, in situ monitoring of the kinetics of the QD assembly during printing with in situ GISAXS allows to derive the nanoscale structure formation including a superlattice analysis. The QDs undergo a fast superlattice formation on the second time-scale and a phase transition process during the deposition and solvent evaporation. The stacking behavior of the QDs is initialized by a hexagonal templating effect and their subsequent FCC stacking behavior originates from the large ratio of the ligand thickness over the radius of the QDs due to their small size. With ongoing solvent evaporation, the ligand shell exhibits a further collapse, which impacts on the shape of the QDs. The effective shape of the QDs is considered to play an important role in the finally formed superlattice. With further decreased inter-QD distance and distorted orientation distribution, a BCC nested FCC superlattice finally forms. These results provide important fundamental understandings of the structure formation of thin QD films via the large-scale deposition method printing, based on small-sized QDs. Moreover, the ligand-dependent kinetics of the QDs also infers that a control of the stacking behavior of QDs via a ligand engineering will also be possible.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2089/1 – 390776260 (e-conversion), TUM.solar in the context of the Bavarian Collaborative Research Project Solar Technologies Go Hybrid (SolTech), the Center for NanoScience (CeNS) and the International Research Training Group 2022 Alberta/Technical University of Munich International Graduate School for Environmentally Responsible Functional Hybrid Materials (ATUMS). K. W. acknowledges funding supports from National Natural Science Foundation of China (No. 61875082, No. 61674074), National Key Research and Development Program (No. 2017YFE0120400), and Natural Science Foundation of Guangdong (No. 2017B030306010). W. C. and N. L. are grateful for the support from the China Scholarship Council (CSC). We thank Dr He Huang (LMU, Munich) for the TEM measurements of QD superlattice. Parts of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association (HGF).References
- H. Lu, G. M. Carroll, N. R. Neale and M. C. Beard, ACS Nano, 2019, 13, 939–953 CAS.
- J. Z. Fan, N. T. Andersen, M. Biondi, P. Todorovic, B. Sun, O. Ouellette, J. Abed, L. K. Sagar, M. J. Choi, S. Hoogland, F. P. G. de Arquer and E. H. Sargent, Adv. Mater., 2019, 31, e1904304 CrossRef.
- Y. Xia, S. Liu, K. Wang, X. Yang, L. Lian, Z. Zhang, J. He, G. Liang, S. Wang, M. Tan, H. Song, D. Zhang, J. Gao, J. Tang, M. C. Beard and J. Zhang, Adv. Funct. Mater., 2019, 30, 1907379 CrossRef.
- R. J. Ellingson, M. C. Beard, J. C. Johnson, P. Yu, O. I. Micic, A. J. Nozik, A. Shabaev and A. L. Efros, Nano Lett., 2005, 5, 865–871 CrossRef CAS PubMed.
- O. Voznyy, B. R. Sutherland, A. H. Ip, D. Zhitomirsky and E. H. Sargent, Nat. Rev. Mater., 2017, 2, 17026 CrossRef CAS.
- W. Chen, J. Castro, S. Ahn, X. Li and O. Vazquez-Mena, Adv. Mater., 2019, 31, e1807894 CrossRef PubMed.
- S. Pradhan, F. Di Stasio, Y. Bi, S. Gupta, S. Christodoulou, A. Stavrinadis and G. Konstantatos, Nat. Nanotechnol., 2019, 14, 72–79 CrossRef CAS PubMed.
- H. Lim, D. Kim, M. J. Choi, E. H. Sargent, Y. S. Jung and J. Y. Kim, Adv. Energy Mater., 2019, 9, 1901938 CrossRef CAS.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- J. Zhang, R. W. Crisp, J. Gao, D. M. Kroupa, M. C. Beard and J. M. Luther, J. Phys. Chem. Lett., 2015, 6, 1830–1833 CrossRef CAS PubMed.
- W. K. Bae, J. Joo, L. A. Padilha, J. Won, D. C. Lee, Q. Lin, W. K. Koh, H. Luo, V. I. Klimov and J. M. Pietryga, J. Am. Chem. Soc., 2012, 134, 20160–20168 CrossRef CAS PubMed.
- S. Ghosh and L. Manna, Chem. Rev., 2018, 118, 7804–7864 CrossRef CAS PubMed.
- H. Zhang, B. R. Hyun, F. W. Wise and R. D. Robinson, Nano Lett., 2012, 12, 5856–5860 CrossRef CAS PubMed.
- Y. Liu, M. Liu, D. Yin, D. Zhu and M. T. Swihart, Nanoscale, 2018, 11, 136–144 RSC.
- M. J. Choi, Y. Kim, H. Lim, E. Alarousu, A. Adhikari, B. S. Shaheen, Y. H. Kim, O. F. Mohammed, E. H. Sargent, J. Y. Kim and Y. S. Jung, Adv. Mater., 2019, 31, e1805886 CrossRef PubMed.
- I. J. Kramer, J. C. Minor, G. Moreno-Bautista, L. Rollny, P. Kanjanaboos, D. Kopilovic, S. M. Thon, G. H. Carey, K. W. Chou, D. Zhitomirsky, A. Amassian and E. H. Sargent, Adv. Mater., 2015, 27, 116–121 CrossRef CAS.
- A. Yousefi Amin, N. A. Killilea, M. Sytnyk, P. Maisch, K. C. Tam, H. J. Egelhaaf, S. Langner, T. Stubhan, C. J. Brabec, T. Rejek, M. Halik, K. Poulsen, J. Niehaus, A. Kock and W. Heiss, ACS Nano, 2019, 13, 2389–2397 CAS.
- K. Whitham, D.-M. Smilgies and T. Hanrath, Chem. Mater., 2017, 30, 54–63 CrossRef.
- J. J. Geuchies, C. van Overbeek, W. H. Evers, B. Goris, A. de Backer, A. P. Gantapara, F. T. Rabouw, J. Hilhorst, J. L. Peters, O. Konovalov, A. V. Petukhov, M. Dijkstra, L. D. A. Siebbeles, S. van Aert, S. Bals and D. Vanmaekelbergh, Nat. Mater., 2016, 15, 1248–1254 CrossRef CAS.
- M. C. Weidman, D. M. Smilgies and W. A. Tisdale, Nat. Mater., 2016, 15, 775–781 CrossRef CAS.
- R. Li, K. Bian, T. Hanrath, W. A. Bassett and Z. Wang, J. Am. Chem. Soc., 2014, 136, 12047–12055 CrossRef CAS.
- P. Ziherl and R. D. Kamien, J. Phys. Chem. B, 2001, 105, 10147–10158 CrossRef CAS.
- C. H. Chuang, P. R. Brown, V. Bulovic and M. G. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS.
- M. Liu, O. Voznyy, R. Sabatini, F. P. Garcia de Arquer, R. Munir, A. H. Balawi, X. Lan, F. Fan, G. Walters, A. R. Kirmani, S. Hoogland, F. Laquai, A. Amassian and E. H. Sargent, Nat. Mater., 2017, 16, 258–263 CrossRef CAS.
- R. Wang, X. Wu, K. Xu, W. Zhou, Y. Shang, H. Tang, H. Chen and Z. Ning, Adv. Mater., 2018, 30, 1704882 CrossRef PubMed.
- M. Liu, Y. Chen, C. S. Tan, R. Quintero-Bermudez, A. H. Proppe, R. Munir, H. Tan, O. Voznyy, B. Scheffel, G. Walters, A. P. T. Kam, B. Sun, M. J. Choi, S. Hoogland, A. Amassian, S. O. Kelley, F. P. Garcia de Arquer and E. H. Sargent, Nature, 2019, 570, 96–101 CrossRef CAS PubMed.
- X. Lan, O. Voznyy, F. P. Garcia de Arquer, M. Liu, J. Xu, A. H. Proppe, G. Walters, F. Fan, H. Tan, M. Liu, Z. Yang, S. Hoogland and E. H. Sargent, Nano Lett., 2016, 16, 4630–4634 CrossRef CAS.
- X. Zhang, C. Hägglund and E. M. J. Johansson, Energy Environ. Sci., 2017, 10, 216–224 RSC.
- A. Stavrinadis, S. Pradhan, P. Papagiorgis, G. Itskos and G. Konstantatos, ACS Energy Lett., 2017, 2, 739–744 CrossRef CAS.
- Z. Ning, H. Dong, Q. Zhang, O. Voznyy and E. H. Sargent, ACS Nano, 2014, 8, 10321–10327 CrossRef CAS.
- J. Z. Fan, M. Liu, O. Voznyy, B. Sun, L. Levina, R. Quintero-Bermudez, M. Liu, O. Ouellette, F. P. Garcia de Arquer, S. Hoogland and E. H. Sargent, ACS Appl. Mater. Interfaces, 2017, 9, 37536–37541 CrossRef CAS.
- J. Kim, O. Ouellette, O. Voznyy, M. Wei, J. Choi, M. J. Choi, J. W. Jo, S. W. Baek, J. Fan, M. I. Saidaminov, B. Sun, P. Li, D. H. Nam, S. Hoogland, Z. H. Lu, F. P. Garcia de Arquer and E. H. Sargent, Adv. Mater., 2018, 30, e1803830 CrossRef PubMed.
- M. Gu, Y. Wang, F. Yang, K. Lu, Y. Xue, T. Wu, H. Fang, S. Zhou, Y. Zhang, X. Ling, Y. Xu, F. Li, J. Yuan, M. A. Loi, Z. Liu and W. Ma, J. Mater. Chem. A, 2019, 7, 15951–15959 RSC.
- M. Liu, F. Che, B. Sun, O. Voznyy, A. Proppe, R. Munir, M. Wei, R. Quintero-Bermudez, L. Hu, S. Hoogland, A. Mandelis, A. Amassian, S. O. Kelley, F. P. García de Arquer and E. H. Sargent, ACS Energy Lett., 2019, 4, 1225–1230 CrossRef CAS.
- R. Sliz, M. Lejay, J. Z. Fan, M. J. Choi, S. Kinge, S. Hoogland, T. Fabritius, F. P. Garcia de Arquer and E. H. Sargent, ACS Nano, 2019, 13, 11988–11995 CrossRef CAS PubMed.
- A. Kiani, B. R. Sutherland, Y. Kim, O. Ouellette, L. Levina, G. Walters, C.-T. Dinh, M. Liu, O. Voznyy, X. Lan, A. J. Labelle, A. H. Ip, A. Proppe, G. H. Ahmed, O. F. Mohammed, S. Hoogland and E. H. Sargent, Appl. Phys. Lett., 2016, 109, 183105 CrossRef.
- B. D. Chernomordik, A. R. Marshall, G. F. Pach, J. M. Luther and M. C. Beard, Chem. Mater., 2016, 29, 189–198 CrossRef.
- A. Buffet, A. Rothkirch, R. Döhrmann, V. Körstgens, M. M. Abul Kashem, J. Perlich, G. Herzog, M. Schwartzkopf, R. Gehrke, P. Müller-Buschbaum and S. V. Roth, J. Synchrotron Radiat., 2012, 19, 647–653 CrossRef CAS PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- W. Chen, J. Zhong, J. Li, N. Saxena, L. P. Kreuzer, H. Liu, L. Song, B. Su, D. Yang, K. Wang, J. Schlipf, V. Körstgens, T. He, K. Wang and P. Müller-Buschbaum, J. Phys. Chem. Lett., 2019, 10, 2058–2065 CrossRef CAS PubMed.
- K. S. Wienhold, V. Körstgens, S. Grott, X. Jiang, M. Schwartzkopf, S. V. Roth and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2019, 11, 42313–42321 CrossRef CAS PubMed.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Müller-Buschbaum, M. Trebbin, S. Förster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- S. V. Roth, G. Herzog, V. Körstgens, A. Buffet, M. Schwartzkopf, J. Perlich, M. M. Abul Kashem, R. Döhrmann, R. Gehrke, A. Rothkirch, K. Stassig, W. Wurth, G. Benecke, C. Li, P. Fratzl, M. Rawolle and P. Müller-Buschbaum, J. Phys.: Condens. Matter, 2011, 23, 254208 CrossRef CAS PubMed.
- A. Hexemer and P. Müller-Buschbaum, IUCrJ, 2015, 2, 106–125 CrossRef CAS.
- G. Santoro, A. Buffet, R. Döhrmann, S. Yu, V. Körstgens, P. Müller-Buschbaum, U. Gedde, M. Hedenqvist and S. V. Roth, Rev. Sci. Instrum., 2014, 85, 043901 CrossRef CAS PubMed.
- N. Paul, E. Metwalli, Y. Yao, M. Schwartzkopf, S. Yu, S. V. Roth, P. Müller-Buschbaum and A. Paul, Nanoscale, 2015, 7, 9703–9714 RSC.
- G. Pospelov, W. Van Herck, J. Burle, J. M. Carmona Loaiza, C. Durniak, J. M. Fisher, M. Ganeva, D. Yurov and J. Wuttke, J. Appl. Crystallogr., 2020, 53, 262–276 CrossRef CAS.
- H. Tang, J. Zhong, W. Chen, K. Shi, G. Mei, Y. Zhang, Z. Wen, P. Müller-Buschbaum, D. Wu, K. Wang and X. W. Sun, ACS Appl. Nano Mater., 2019, 2, 6135–6143 CrossRef CAS.
- P. Busch, M. Rauscher, D. M. Smilgies, D. Posselt and C. M. Papadakis, J. Appl. Crystallogr., 2006, 39, 433–442 CrossRef CAS.
- K. Bian, J. J. Choi, A. Kaushik, P. Clancy, D. M. Smilgies and T. Hanrath, ACS Nano, 2011, 5, 2815–2823 CrossRef CAS PubMed.
- Z. Jiang, J. Appl. Crystallogr., 2015, 48, 917–926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00008f |
| This journal is © The Royal Society of Chemistry 2020 |