
Novel electro-magnetophoretic separation method for the highly sensitive detection of analytes†
Giorgia
Giovannini
 and
Francesco
De Angelis
*
and
Francesco
De Angelis
*
Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. E-mail: Francesco.deangelis@iit.it
First published on 23rd August 2019
Abstract
The detection of a low concentration of biomarkers required for an early diagnosis remains a tricky task to pursue. In this context, magnetic nanoparticles (MNPs) have been widely used to isolate/concentrate diluted samples. However, by applying the magnetic field, all MNPs are isolated, both those conjugated with the analyte and free MNPs. This excess of MNP can interfere with the analyte quantification. Herein, we propose a novel and versatile approach that ensures the selective isolation of the analyte-conjugated MNP by exploiting the electrophoretic and magnetophoretic behavior of respectively charged silica nanoparticles (SiNPs) and MNPs. In this work, the robustness and sensitivity of the developed isolation and fluorescence-based detection procedure were confirmed by comparing them with the standard method based on simple magnetic isolation of the analyte. The limit of detection (LOD) is improved by two orders of magnitude, namely, from 1 nM to 10 pM. This finding confirms the potentiality of the method and its possible application in the development of rapid and easy-to-use point-of-care devices.
New conceptsMagnetic nanoparticle (MNP)-based isolation techniques have been widely used for the isolation, concentration and quantification of targeted analyte. Despite the undoubted advantages, this approach has a fundamental drawback: the application of a magnetic field allows to isolate MNP linked to the analyte but excess of the MNP are unavoidably collected as well. This excess of free-MNP affects the precise quantification of the isolated analyte due to interferences with the method used for the detection. Different strategies has been proposed in order to overcome this limitation but all come with pros-and-cons. Herein, we present a novel and versatile approach by which the analyte-linked MNP can be selectively isolated from the excess of free-MNP simultaneously applying magnetic and electric field. By carefully choosing the properties of the nanomaterial used i.e., MNP and charge/labelled silica nanoparticles (SiNP), and ensuring their selective interaction exploiting a well-known sandwich immunoassay, we proved the efficiency and efficacy of the method proposed. Comparing this electro-magnetophoretic separation (EMPS) approach with the standard method in which the isolation of the analyte is simply achieved by applying a magnetic field, we confirmed the improved sensitivity of EMPS, for which 10 pM was found as detection limit against 1 nM determined for standard method. |
Introduction
Most diseases ranging from chronic illnesses (e.g., diabetes and cardiopathies) to cancers and bacterial infections cause alterations of the normal functionality of the body, leading to the production of biomarkers due to metabolic changes. The selective identification of such biomarkers, usually small molecules or proteins in the blood, urine, mucous and sweat are the basis of most diagnostic methods.1,2 Several devices are presently available to detect low concentration of analytes3,4 but the diagnostic market is continuously evolving.5In this context, magnetic nanoparticles (MNPs) have been widely exploited in the development of highly sensitive detection techniques.6,7 By coating the MNP surface with specific antibodies, they can be made selective for the recognition of the analyte.8,9 Although an impressive variety of approaches have been developed,10–13 most have essentially relied on the same trick: capturing the analyte of interest by using the MNP (specifically functionalized for the selective analyte recognition) and then using an external magnetic field to isolate the analyte–MNP complex from the remaining solution. However, a significant excess of MNPs not linked to the analyte will be present at the end of the process because the MNPs must be used in large excess. To explain this concept, let us consider an assay that must provide a dynamic range of sensitivity of 4 orders of magnitude, from 10 μM to 1 nM. An initial concentration of MNPs greater than 10 μM has to be used to ensure that all of the analyte will be captured. When the real concentration of the analyte is close to the detection limit (1 nM), only a very minor fraction of MNPs is linked to the analyte (1/104) whereas the other MNPs represent the excess. Such an excess may hinder subsequent analysis and precise quantification.14 In particular, when optical methods are exploited, the excess of MNPs will cover the optical readout due to high optical absorption exhibited by MNPs in the visible range. Different strategies have been proposed to overcome these limitations,15,16 but all come with pros and cons, and a standard approach is still awaited.17,18
Herein, we present a versatile, simple and cost-effective method to improve the performance of the detection schemes based on MNPs with particular regard to sandwich assays. The concept is represented in Fig. 1. In brief, we take profit of a sandwich configuration in which the final analyte–MNP complex will have a different charge/mass ratio with respect to the free MNP (the excess). Hence, by simultaneously applying a magnetic and an electric field, analyte–MNP complexes can be selectively isolated from the excess of free MNPs. We named the proposed method electro-magnetophoretic separation (EMPS). In more detail, we developed a protocol that can be used as an additional step after running a standard isolation method (see Fig. 1, step 1). We considered a standard sandwich assay that involves the analyte, a MNP, and a third element consisting of a silica nanoparticle with a secondary recognition element and a fluorescent label (technical details provided below). As introduced above, the final pellet contains both analyte–MNP complexes and the excess of MNPs. Therefore, we repeat the separation step by applying simultaneously an electric field in addition to the magnetic field (step 2). Clearly, magnetic forces will act on all MNPs pulling them toward the magnet as in conventional magnetophoresis.19 Since all the particles are negatively charged, electric forces will simultaneously pull the particles in the opposite direction, namely toward the cathode. As a result, the particles will move in one direction or in the other depending on the balance between magnetic and electrical forces. The strength of the pulling force will depend on the charge, mass and size of the particles according to standard electrophoretic processes.20–22 When the parameters of the system are properly designed it is possible to move the free MNPs (excess) toward the anode whereas the analyte–MNP complexes are accumulated at the cathode. In this way, the analyte is extracted from the initial solution, isolated from the excess of MNPs, and labeled with fluorophore for the following optical detection. The efficacy of the proposed method was compared with a standard MNP-based isolation approach. The quantification of the analyte is achieved by measuring the fluorescent signal of the isolated fraction. Removing the excess MNPs and thus reducing the possible interference with the quantification, we improved the sensitivity by 2 orders of magnitude compared with the standard approach. Remarkably, EMPS reached a limit of detection (LOD) of 10 pM compared with that of 1 nM found for the standard method. Such an improvement confirms that the additional separation becomes crucial when a very low limit of detection (picomolar range) is combined with a broad dynamic range (>104). In the following session, we will give technical details of the methodology.
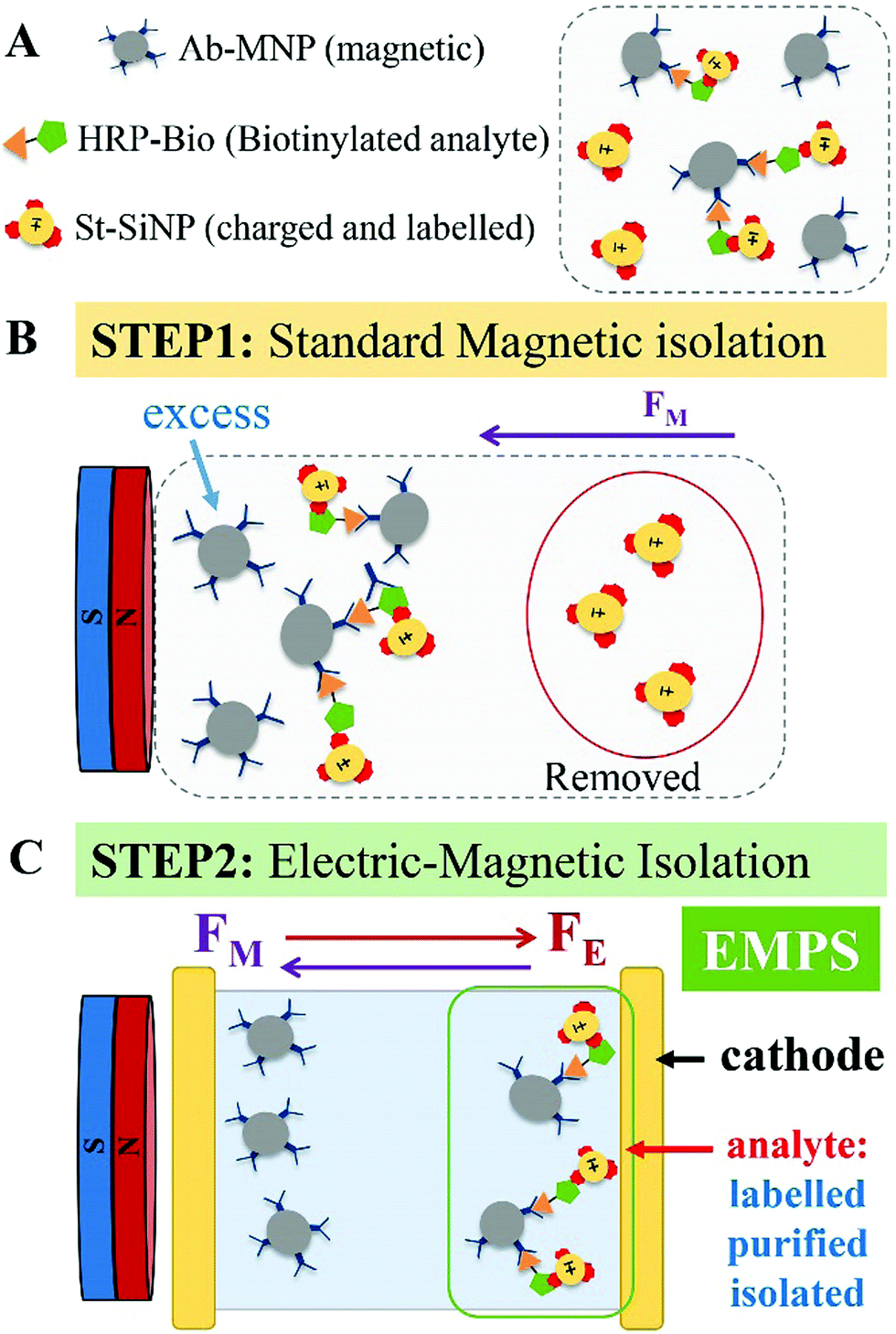 | ||
| Fig. 1 Schematic representation of the applicability of EMPS to precisely isolate the analyte of interest. (A) Once the biological assay is completed, the complexes and excess of both particles, Ab-MNP and St-SiNP, are present in the sample. (B) Upon applying a magnet, the excess labeled particles are removed. According to the standard procedure, quantification of the analyte, is accomplished by measuring the fluorescence signal of the resulting pellet. This pellet is composed of complexes (MNP–analyte–SiNP) and excess MNPs. (C) By adding the additional step proposed here, EMPS, the pellet obtained after the standard method is further purified by simultaneously applying magnetic and electric fields. In the expressly designed device, the complexes (MNP–analyte–SiNPs) can be isolated, and quantification of the analyte is performed using the fluorescence signal emitted by the isolated fraction. The additional isolation step reduces the interference related to the presence of excess-MNPs, allowing a more precise quantification of the analyte. | ||
Results and discussion
Standard MNP-based isolation approaches are widely used for the development of detection techniques. When the targeted analyte is a relatively large molecules (e.g., proteins, hormones, polypeptides) it can be selectively detected exploiting a third labeled element, which, being coated with a specific recognition element (such as a secondary antibody or other proteins), forms a sandwich assay with the analyte–MNP conjugates. The quantification of the analyte is then achieved by measuring the signal of the pellet after magnetic isolation. We notice that in the case of smaller molecules (such as bile acids, sterols, steroid-hormones, metabolites, pharmaceuticals etc.) that can only bind a single antibody, competitive assays are preferred. For the detection of small molecules, the method mentioned here would not be convenient, as would any other methods based on the sandwich approach, such as for instance the well known ELISA. However, specific surface functionalization of the particles or selective modification of the small analyte with “sandwichable” molecules could be useful in overcoming this limitation. Just to give a simple example: a small molecule such as biotin could be firstly ‘marked’ with streptavidin. Such a complex could be potentially isolated and detected by EMPS using MNPs and SiNPs functionalized with antibodies specific for streptavidin. In this work, we were interested in developing a new isolation method. The optimization of the EMPS for particular cases is doable and could become the subject of future works.EMPS: description of the method
Due to the implication of the principles of electrophoresis and magnetophoresis, the applicability and effectiveness of EMPS is strongly related to the following: (i) parameters related to the nanomaterials used (i.e., size and surface charge), (ii) environmental conditions related to the buffer (i.e., pH, ionic strength and viscosity) and (iii) electric and magnetic field applied.24,25 These parameters represent variables of the equations describing the movement of particles under specific conditions. Several theoretical studies have extensively investigated the relationship between the efficiency of electro- and magnetophoresis according to the particle or environmental parameters.26,27 We suggest the reader to refer to these works for theoretical/technical details concerning the physics of the system. Hereafter we report on the experimental work we carried out to optimise the protocol.
Nanomaterial synthesis
Key aspects to be defined to successfully achieve the isolation of the wanted complexes are related to the nanomaterial itself and its behavior in the surrounding environment. Thus, both MNPs and SiNPs were characterized at each step of synthesis by transmission electron microscopy (TEM) and dynamic light scattering (DLS). The latter technique allows not only determination of the hydrodynamic diameter of the particles but also, more importantly, calculation of the surface charge of the nanomaterial itself and in relation to the surrounding environment. Carboxylate–MNP were labeled with dyes (i.e., fluorescein amine or Atto633) and were further functionalized with an antibody specific for HRP (Fig. S2.1, ESI†). SiNPs loaded with either methylene blue (MB) or fluorescein isothiocyanate (FITC) were synthesized by the microemulsion quaternary method.28,29 Both MB-SiNPs and FITC-SiNPs were further coated with glutaraldehyde. This strategy allows the exposure of aldehyde groups on the NP surface that are suitable reactive groups for the formation of covalent amide bonds with the amine groups of streptavidin (Fig. S2.3, ESI†). Modified MNPs and SiNPs were characterized by DLS, TEM and UV-vis, and fluorescence spectra were recorded (Fig. S2.4–S2.6 for MNP and Fig. S2.7 and S2.8 for SiNP, ESI†). The amount of antibody and streptavidin present on the NP surface was determined by the Bradford protein assay, and the activity of the biomolecules once on the particle's surface was determined by ABTS and the bio-FITC assay for the HRP antibody-coated and streptavidin-coated particles respectively (the protocol used can be found in Section S2.3 of the ESI†). For more details regarding the synthesis and characterization of all nanomaterials, see Section 2 of the ESI.†Evaluation of the environmental conditions for electro-magnetophoresis
Upon the application of bias across the electrodes, a double layer will be generated at their interfaces thus screening the electric field in the solution. Hence, in order to maintain the Ohmic potential drop across the interfaces, and thereby maintain the non-zero electric field in the bulk, some faradaic processes are necessary (or other mechanisms which convert ionic currents in electrical currents). Under the experimental conditions here used, we observed a constant current of about 1 mA flowing between the gold electrodes. This current is reasonably produced by some electrolytic reactions, or direct charge transfer, or adsorption of charged species into the electrodes. Since the phenomenon involved in the current production depends on the composition of the solution, it can change from case to case. Therefore, we did not investigate in details these aspects since they must be optimized depending upon the nanomaterials used and the environmental conditions chosen. However it is important to remark that an electric field must be generated and maintained across the solution in order to drive the nanoparticles. In the present work, we chose parameters only with the aim to: (i) achieve a constant flow of particle ensuring an appropriate separation, as proven by the tracking analysis (see ESI†) and (ii) to preserve the properties of nanoparticles, fluorophore and analyte used during the isolation. Further investigation and optimizations will be carried out in future works.In order to ensure a constant flow of the particles and a proper isolation of the complexes, the properties and behaviour of the nanomaterials chosen were evaluated in buffers with different ion concentrations and different pH values. This allowed determining the environmental conditions suitable for satisfactory isolation and detection of the targeted analyte. The zeta potential (ζ) and conductivity (C) were measured for MNPs and SiNPs dispersed in PBS buffers at pH 6.5 at different concentrations and in 10 mM MES buffer at different pH values to evaluate how the properties of the NP suspension change with variation in the environmental conditions. The results are presented in Section S3.1 (ESI†). Furthermore, a similar analysis was conducted using more complex buffers. The ζ and C values measured for each particle type in these different buffers were compared. The buffer that produced a better combination of results between MNPs and SiNPs was selected for tracking and quantification analysis. Fig. S3.3 (ESI†) shows the ζ and C values measured for Ab-Flu-MNPs and for St-G-MB-SiNP. To achieve good separation, buffer B-1 with 0.001% of agarose was selected as the solvent for tracking analysis. Indeed, using this solvent, Ab-Flu-MNP has a neutral ζ value, while St-G-MB-SiNP has a negative surface charge of almost −30 mV. The conductivity of both particles in this buffer is approximately 1 mS cm−1. This combination of buffer and nanomaterial was chosen for tracking analysis. Fig. S3.4 (ESI†) shows ζ and C values measured for Ab-Atto-MNPs and Ab-MNPs and for St-G-FITC-SiNPs. To achieve good separation, 10 mM MES buffer pH 4, was selected as the solvent for the quantification assay. Indeed, while MNPs (Ab-Atto-MNPs and Ab-MNPs) have a ζ value close to neutrality (5 and 0 mV, respectively) in MES pH 4, St-G-FITC-SiNPs are highly positively charged, with a value of almost 20 mV. The conductivity for both particles in this buffer is approximately 1 mS cm−1. This combination of buffer and nanomaterial was chosen for the quantification assays.
EMPS tracking analysis
Once the properties of the nanomaterials were defined and the environmental and experimental parameters were evaluated, the effectiveness of EMPS was qualitatively evaluated tracking the separation of the complexes formed from excess MNPs.Due to required washing steps (centrifugation–sonication cycles), the complexes can no longer be distinguished. However, considering that the signal of the two types of particles increases according to the same trend suggests that the particles were moving as a single entity (Fig. S4.1C, ESI†). By contrast, in control samples in which complex formation was not expected, SiNPs and MNPs eventually accumulate at the positive electrode independently, possibly indicating that they were not forming complexes (Fig. S4.1D, ESI†).
Quantitative evaluation of EMPS and comparison with the standard approach
In the following paragraph, the sensitivity achievable in detecting the analyte using the proposed approach, EMPS, is quantified and compared with the standard method, as schematized in Fig. 2.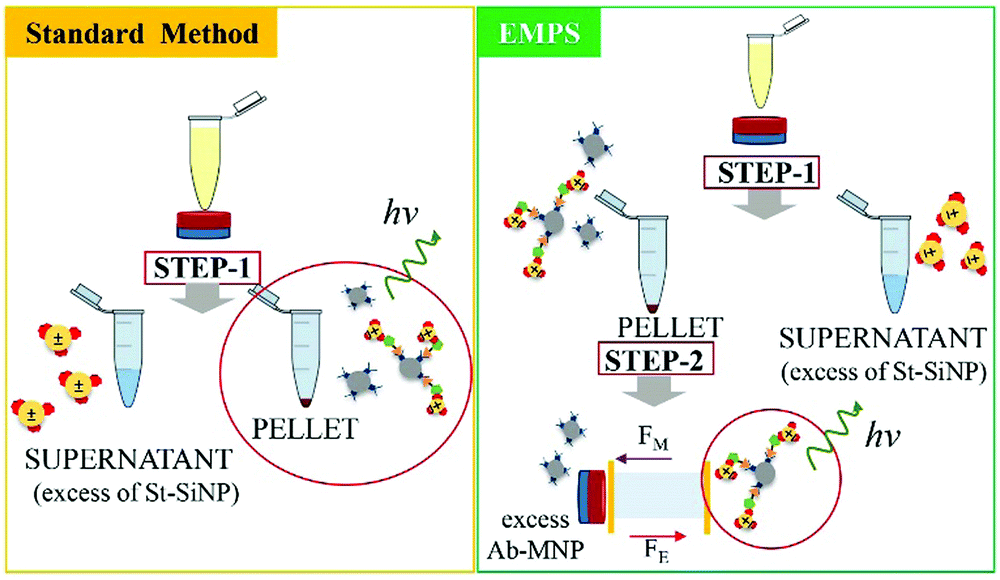 | ||
| Fig. 2 Schematic representation of the procedure followed for the comparison of the two methods. The standard method (yellow rectangle) represents the standard procedure: once the sandwich immunoassay is accomplished, the excess St-G-FITC-SiNPs are removed by applying a magnet (step 1). Quantification of the analyte is achieved by measuring the fluorescence signal given by the remaining SiNPs forming the complexes. EMPS (green rectangle): the pellet isolated by applying the magnet (step 1) is resuspended and further purified in the designed chamber where electric and magnetic fields are simultaneously applied (step 2). Quantification is accomplished by measuring the fluorescence signal of the fraction isolated at the cathode, where the complexes are expected to accumulate. | ||
For such experiments, fluorescently labeled particles were used. In particular, Atto-labeled MNPs (Ab-Atto-MNPs) and FITC-loaded SiNPs (St-G-FITC-SiNPs) have absorbance–emission peaks at 630–650 nm and 480–520 nm, respectively, allowing their discrimination during the separation. The synthesis and characterization details of the particles can be found in Section S2 (ESI†). The lower concentration of analyte used for these experiments does not allow tracking of the particles during the purification procedure (0.001–100 nM HRP-Bio). Hence, quantification was achieved by measuring the fluorescence signal of FITC-SiNPs (λem 525 nm) for the fractions isolated using the two methods. Following the standard procedure, the excess FITC-loaded streptavidin-functionalized SiNPs were removed by applying a magnet (STEP 1). The pellet was isolated and redispersed in 5 μL, and the fluorescence signal was measured. The value measured was related to the amount of SiNP-formed complexes and corresponded to the amount of analyte detected in the sample. Following EMPS, a further isolation step was added: the pellet achieved after magnetic isolation (composed of complexes and excess MNPs) was suspended and added to the designed chamber (maximum volume: 50 μL; E: Au electrodes, 0.5 V, 1 mA; B: 270 mT). Here, the complexes were purified from the excess MNPs by simultaneous exposure to magnetic and electric fields. After 20 minutes, 15 μL was withdrawn in the proximity of the cathode. This isolated fraction was then concentrated by centrifugation (8000 rpm, 5 minutes) to 5 μL, spotted on a microscope slide and analysed.
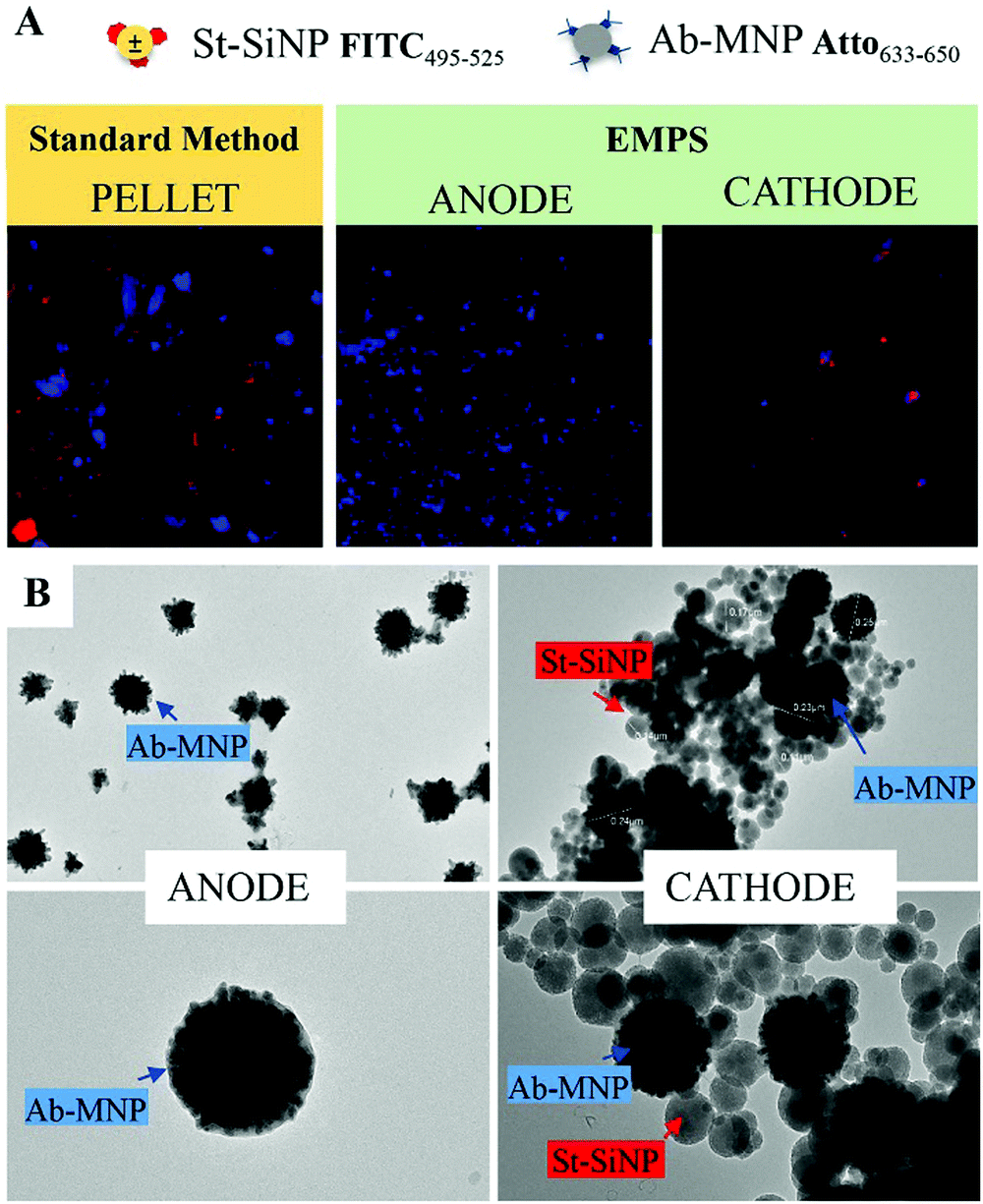 | ||
| Fig. 3 Qualitative comparison between fractions isolated by means of the standard method and EMPS. (A) The isolated fractions were imaged using a spinning disk microscope, which allows the simultaneous use of both FITC and Atto filters. Notably, the excess MNPs evident in the isolated fraction achieved by standard method can be removed by EMPS. In the latter case, indeed, free-MNPs accumulate at the anode, closer to the magnet, selectively isolating the complex at the cathode. (B) TEM images of the fraction isolated at the two electrodes confirm the effectiveness of the separation method: whereas only MNPs were visible for the fraction at the anode, both particles accumulate at the cathode. | ||
The fluorescence signal measured for the fraction isolated toward the cathode, where the complexes are expected to accumulate, was used to quantify the analyte, and this value was then compared with that obtained using the standard method. In particular, the intensity of the fraction was measured by ChemiDoc and quantified by analysing the acquired images with ImageJ software. As a reference, the same amount of the SiNP sample used for the assay was concentrated in 5 μL, and its signal was measured. By knowing the number of particles, the intensity of this signal was used to calculate the number of particles forming the complexes, considering the signal measured for the isolated fraction. The standard method and EMPS were compared by considering different parameters such as (i) selectivity, (ii) reproducibility and (iii) sensitivity.
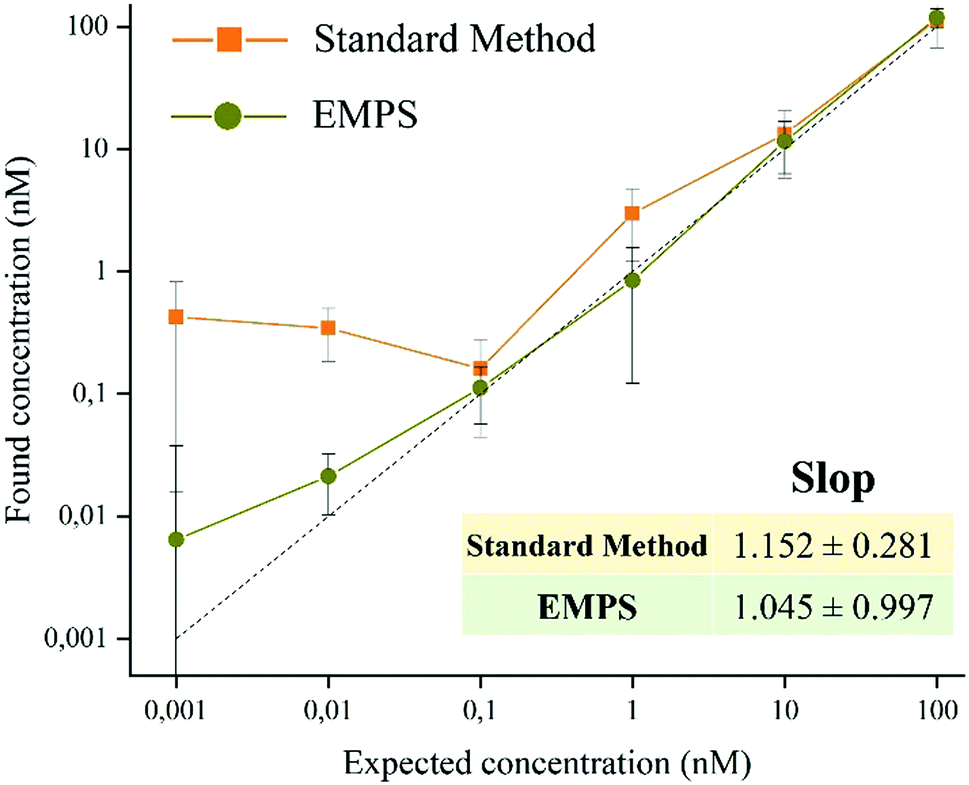 | ||
| Fig. 4 Quantitative evaluation of the sensitivity observed for the two methods. Values of the found concentrations are plotted against the expected concentrations. EMPS (green line) was shown to be clearly more sensitive at low concentrations (below 1 mM) than the standard method (orange line). The calculated slope for EMPS confirmed the sensitivity was closer to 1 than the slope of the standard method (1.045 and 1.152, respectively). The values are reported as average values (n = approximately 8) ± SD. | ||
To exclude possible interferences related to the use of labeled-MNPs, these experiments were accomplished using unlabeled-MNPs (see Section S2 of the ESI† for their synthesis and characterization and Section S5 for the experimental details). As shown in Fig. S5.4 (ESI†), similar results were found using Ab-MNP and St-G-FITC-SiNP. This confirmed the improvement achievable by using EMPS, particularly at pM concentrations, compared with the standard method in which excess MNPs led to notable errors in quantification.
Conclusions
In summary, we proposed a versatile, easy and robust method that allows better quantification of analytes in MNP-based detection schemes. In particular, when dealing with pM concentrations, further purification of the sample by means of simultaneous applications of electric and magnetic fields (EMPS method) allows selective isolation of only the analyte, achieving more precise quantification. In particular, while the LOD for the standard method is 1 nM, that for EMPS is 10 pM, leading to improvement in the detection limit by 2 orders of magnitude. The selectivity, sensitivity and reproducibility of the methods were investigated, and the results supported the claim that further purification of the sample and removing the excess MNPs decrease the interference during quantification to obtain more accurate values. As proof-of-concept, the quantification of the analyte was achieved by measuring the fluorescent signal of the labeled particles, making the method easy to use and affordable. However, choosing nanomaterials with different properties it is possible to exploit other analytical techniques. Remarkably, the presented method consists in an additional step that can be carried out at the end of standard methods, when necessary. Hence, it can be easily integrated with already existing approaches. This paves the way for further improvements, which could make this novel and promising isolation procedure a valuable analytical technique widely exploited for the development of POC devices and diagnostic tools.Conflicts of interest
There are no conflicts to declare.Notes and references
- E. G. E. de Vries, L. Kist de Ruijter, M. N. Lub-deHooge, R. A. Dierckx, S. G. Elias and S. F. Oosting, Nat. Rev. Clin. Oncol., 2019, 16, 241–255 CrossRef PubMed.
- C. H. Liu, N. D. Abrams, D. M. Carrick, P. Chander, J. Dwyer, M. R. J. Hamlet, F. Macchiarini, M. PrabhuDas, G. L. Shen, P. Tandon and M. M. Vedamony, Nat. Immunol., 2017, 18, 1175 CrossRef CAS PubMed.
- A. St. John and C. P. Price, Clin. Biochem. Rev., 2014, 35, 155–167 Search PubMed.
- G. W. Pirjo Hedberg, Clin. Chem. Lab. Med., 2009, 47, 376 Search PubMed.
- R. Ranjan, E. N. Esimbekova and V. A. Kratasyuk, Biosens. Bioelectron., 2017, 87, 918–930 CrossRef CAS PubMed.
- R. Di Corato, N. C. Bigall, A. Ragusa, D. Dorfs, A. Genovese, R. Marotta, L. Manna and T. Pellegrino, ACS Nano, 2011, 5, 1109–1121 CrossRef CAS PubMed.
- M. Essandoh and R. A. Garcia, Chemosphere, 2018, 206, 502–512 CrossRef CAS.
- Z. Rong, C. Wang, J. Wang, D. Wang, R. Xiao and S. Wang, Biosens. Bioelectron., 2016, 84, 15–21 CrossRef CAS PubMed.
- C. Murray, E. Pao, P. Tseng, S. Aftab, R. Kulkarni, M. Rettig and D. Di Carlo, Small, 2016, 12, 1891–1899 CrossRef CAS PubMed.
- G. Rizzi, J.-R. Lee, P. Guldberg, M. Dufva, S. X. Wang and M. F. Hansen, Biosens. Bioelectron., 2017, 93, 155–160 CrossRef CAS PubMed.
- C.-C. Huang, X. Zhou and D. A. Hall, Sci. Rep., 2017, 7, 45493 CrossRef CAS PubMed.
- H. Lee, T.-J. Yoon and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 5657–5660 CrossRef CAS PubMed.
- M. Pannetier-Lecoeur, L. Parkkonen, N. Sergeeva-Chollet, H. Polovy, C. Fermon and C. Fowley, Appl. Phys. Lett., 2011, 98, 2011–2014 CrossRef.
- T. Wang, Y. Zhou, C. Lei, J. Luo, S. Xie and H. Pu, Biosens. Bioelectron., 2017, 90, 418–435 CrossRef CAS PubMed.
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS PubMed.
- W. U. Dittmer, T. H. Evers, W. M. Hardeman, W. Huijnen, R. Kamps, P. de Kievit, J. H. M. Neijzen, J. H. Nieuwenhuis, M. J. J. Sijbers, D. W. C. Dekkers, M. H. Hefti and M. F. W. C. Martens, Clin. Chim. Acta, 2010, 411, 868–873 CrossRef CAS.
- G. A. R. Y. Suaifan, C. Esseghaier, A. Ng and M. Zourob, Analyst, 2013, 138, 3735–3739 RSC.
- F. Salis, A. B. Descalzo, E. Benito-Peña, M. C. Moreno-Bondi and G. Orellana, Small, 2018, 14, 1–10 CrossRef.
- J. Lim, C. Lanni, E. R. Evarts, F. Lanni, R. D. Tilton and S. A. Majetich, ACS Nano, 2011, 5, 217–226 CrossRef CAS PubMed.
- A. I. López-Lorente, B. M. Simonet and M. Valcárcel, TrAC, Trends Anal. Chem., 2011, 30, 58–71 CrossRef.
- G. Ruan, G. Vieira, T. Henighan, A. Chen, D. Thakur, R. Sooryakumar and J. O. Winter, Nano Lett., 2010, 10, 2220–2224 CrossRef CAS.
- T. Vissers, A. Imhof, F. Carrique, Á. V. Delgado and A. van Blaaderen, J. Colloid Interface Sci., 2011, 361, 443–455 CrossRef CAS.
- S. Puertas, P. Batalla, M. Moros, E. Polo, P. del Pino, J. M. Guisán, V. Grazú and J. M. de la Fuente, ACS Nano, 2011, 5, 4521–4528 CrossRef CAS.
- N. Lewpiriyawong, K. Kandaswamy, C. Yang, V. Ivanov and R. Stocker, Anal. Chem., 2011, 83, 9579–9585 CrossRef CAS.
- U. Pyell, Electrophoresis, 2010, 31, 814–831 CrossRef CAS.
- S. A. Allison, Y. Xin and H. Pei, J. Colloid Interface Sci., 2007, 313, 328–337 CrossRef CAS.
- R. Gerber and R. R. Birss, High gradient magnetic separation, Research Studies Press, 1983 Search PubMed.
- G. Giovannini, F. Kunc, C. C. Piras, O. Stranik, A. A. Edwards, A. J. Hall and V. Gubala, RSC Adv., 2017, 7, 19924–19933 RSC.
- G. Giovaninni, C. J. Moore, A. J. Hall, H. J. Byrne and V. Gubala, Colloids Surf., B, 2018, 169, 242–248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Material and methods and additional details. See DOI: 10.1039/c9nh00279k |
| This journal is © The Royal Society of Chemistry 2020 |