
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating chemistry through nanoparticle morphology†
Lucio
Litti
 *a,
Javier
Reguera
*bcd,
F. Javier
García de Abajo
ef,
Moreno
Meneghetti
a and
Luis M.
Liz-Marzán
cdg
*a,
Javier
Reguera
*bcd,
F. Javier
García de Abajo
ef,
Moreno
Meneghetti
a and
Luis M.
Liz-Marzán
cdg
aDepartment of Chemical Sciences, University of Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: lucio.litti@unipd.it
bBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain. E-mail: javier.reguera@bcmaterials.net
cCIC biomaGUNE, Paseo de Miramón 182, 20014 Donostia-San Sebastián, Spain
dIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
eICFO-Institut de Ciencies Fotoniques, The Barcelona Institute of Science and Technology, 08860 Castelldefels, Barcelona, Spain
fICREA-Institució Catalana de Recerca I Estudis Avanc![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) ts, Passeig Lluís Companys 23, 08010 Barcelona, Spain
ts, Passeig Lluís Companys 23, 08010 Barcelona, Spain
gCIBER-BBN, Paseo de Miramón 182, 20014 Donostia-San Sebastián, Spain
First published on 7th August 2019
Abstract
We demonstrate that the protonation chemistry of molecules adsorbed at nanometer distances from the surface of anisotropic gold nanoparticles can be manipulated through the effect of surface morphology on the local proton density of an organic coating. Direct evidence of this remarkable effect was obtained by monitoring surface-enhanced Raman scattering (SERS) from mercaptobenzoic acid and 4-aminobenzenethiol molecules adsorbed on gold nanostars. By smoothing the initially sharp nanostar tips through a mild thermal treatment, changes were induced on protonation of the molecules, which can be observed through changes in the measured SERS spectra. These results shed light on the local chemical environment near anisotropic colloidal nanoparticles and open an alternative avenue to actively control chemistry through surface morphology.
New conceptsNanostructured surfaces are widely used in many fields such as sensing, catalysis and drug delivery. Surface functionalization is often adopted to enrich the material activity properties, usually through complex surface chemistry procedures. Nevertheless, molecules do not behave the same if they are free in solution or organized on a surface. The connection between the surface morphology, at the nanoscale, and the induced surface chemistry is more sophisticated than one can expect. Here we demonstrate that the protonation equilibrium of selected molecules can be tuned by operating over the nanostructured surface morphology. Surface-enhanced Raman spectroscopy, as a vibrational technique extremely sensitive to the closer surface environment, is adopted to observe the apparent-pH felt by molecules on reshaped gold nanostars. Our reported results show that it spans within a wide pH range, while the overall solution pH remains at neutrality. Thanks to the complex anisotropy of gold nanostars, SERS signals are selectively more sensitive to the tip regions, providing a unique opportunity to observe the surface chemistry where the morphological changes are more effective. In addition, the local chemical environment revealed near anisotropic colloidal nanoparticles opens an alternative avenue to control chemistry through surface morphology. |
Introduction
Surface-enhanced Raman scattering (SERS) relies on the local amplification of an incident optical field, assisted by its interaction with collective oscillations of free electrons in metallic nanostructures, known as localized surface plasmon resonances (LSPRs). These excitations emerge in the so-called hostspot regions of nanometer size where the plasmonic field is amplified.1 Hotspots can be obtained, among other strategies, through the assembly of nanoparticles leading to structures with multiple narrow gaps,2,3 or by shape control into anisotropic nanoparticles that feature sharp corners and edges.4 Molecules exposed to the intense fields present at illuminated hostpots can therefore undergo large enhancement of the Raman scattering intensity. SERS is widely employed in direct sensing,5–7 labeling assays,8–12 and development of contrast agents.13,14 In particular, vibrational SERS signals can be measured from aqueous samples, with intensities that are comparable to those of fluorescence signals, without suffering from bleaching and with much narrower spectral peaks. Gold nanostars (AuNSs) are a prominent example of colloidal SERS substrates, because their sharp tips can act as field enhancing regions.15–17 The ability to externally modulate the local curvature (i.e., the sharpness) of such highly anisotropic nanoparticles can be conveniently used to influence and manipulate the local chemical environment, and therefore the chemical behavior, of molecular species placed on their surface. Nevertheless, careful methodologies are needed when different morphologies are to be directly compared. Other colloidal parameters (e.g., number and size of nanoparticles and capping agent concentration) should be preserved.17 Gold nanostars can be externally reshaped (e.g. rounding of the tips) by both chemical and thermal stimuli. Some examples have been reported by ageing,18 as well as by the addition of CTAB19 or NaOH,20 which have been shown to produce dramatic variations in the optical response, and therefore in the associated local environment and field enhancement experienced by molecular species adsorbed on their surfaces. Molecular interactions and behavior on flat gold surfaces have been extensively studied by computational simulations21 and also experimentally through techniques such as electrochemistry,22 surface probe microscopy, or X-ray absorption/scattering,23–26 but the situation becomes more challenging when nanoparticles or, in general, curved surfaces in the nanometer range are considered. As a good example, complete crystal characterization of functionalized gold nanoclusters has been achieved27 by using particles made of a few hundred gold atoms that were found to crystalize. In contrast, the literature is still lacking analogous examples for bigger nanoparticles, above the size of crystallizable metal nanoclusters, mainly due to insufficient size uniformity and shape anisotropy. Other techniques such as STM, 2D-NMR, and MALDI-TOF have been employed to study complex molecular arrangements on the surface of gold nanospheres.28 However, the information that can be obtained still remains partial or limited, and possible changes in the chemical state of adsorbed species are usually not taken into account. In this context, SERS offers the unique advantage of accessing the chemical states of molecules localized on metallic surfaces of varying curvature. In this context, p-mercaptobenzoic acid (MBA) has attracted high interest due to its simplicity (it is basically an aromatic molecule containing both carboxyl and thiol functional groups), as well as its relatively large Raman cross section, therefore showing a high potential in applications such as pH and ion sensing.29–33 Through careful modulation of the local curvature in gold nanostars, we demonstrate the influence on the chemical states of MBA molecules adsorbed on them. Although many studies have been carried out using MBA in various systems, the indirect effect of nanoparticle morphology on the chemical behavior (e.g., the protonation state) of MBA has not been sufficiently studied. Additionally, 4-aminobenzenethiol (ABT) and 4-acetylaminobenzenethiol (AABT) were also used to validate the effect on basic pH-sensitive (ABT) and non-pH-sensitive (AABT) molecules. By considering the SERS signals of these molecules as a function of substrate morphology, we find that a detailed analysis of the vibrational spectra is highly informative regarding the importance of the local environment on Raman transitions.Results and discussion
Reshaping was induced by heating a PVP-capped AuNS solution at 95 °C, according to the scheme presented in Fig. 1a. The plasmonic spectrum associated with gold nanostars (Fig. 1b) features two broad bands attributed to the core and tips-core hybridized LSPR modes.34,35 The first mode is close to that for a spherical particle and lies in the region of 520–560 nm. The second mode depends on the aspect ratio of the tips and is located at higher wavelengths, ranging from the red to the near-infrared region. Because the reshaping process involves smoothing and eventual removal of the tips, an LSPR blue-shift of the tip-core plasmon band occurs during the process. It is obvious in Fig. 1b that the pristine AuNSs feature an extinction maximum at about 800 nm, which gradually blueshifts, down to a final maximum around 540 nm after heating for 150 hours. Transmission electron microscopy (TEM) images of the initial AuNS show star-shaped nanostructures with tips with a variety of lengths, from a few nm up to some tens of nanometers, with tip curvature radii between 1 and 3 nm (Fig. 2a). Images of the particles resulting from heating for 150 hours (Fig. 2b) clearly show that thermal treatment resulted in gradual smoothing of the tips, which were ultimately erased, leaving rounded quasi-spherical particles, with a radius of curvature close to the nanoparticle radius. In Fig. 2c we present histograms for the TEM size distributions in the pristine and the completely reshaped AuNSs. As synthesized, AuNSs are characterized by a broad size distribution centered at ∼80 nm (Feret's diameter), which can be clearly interpreted as a consequence of almost random tip growth.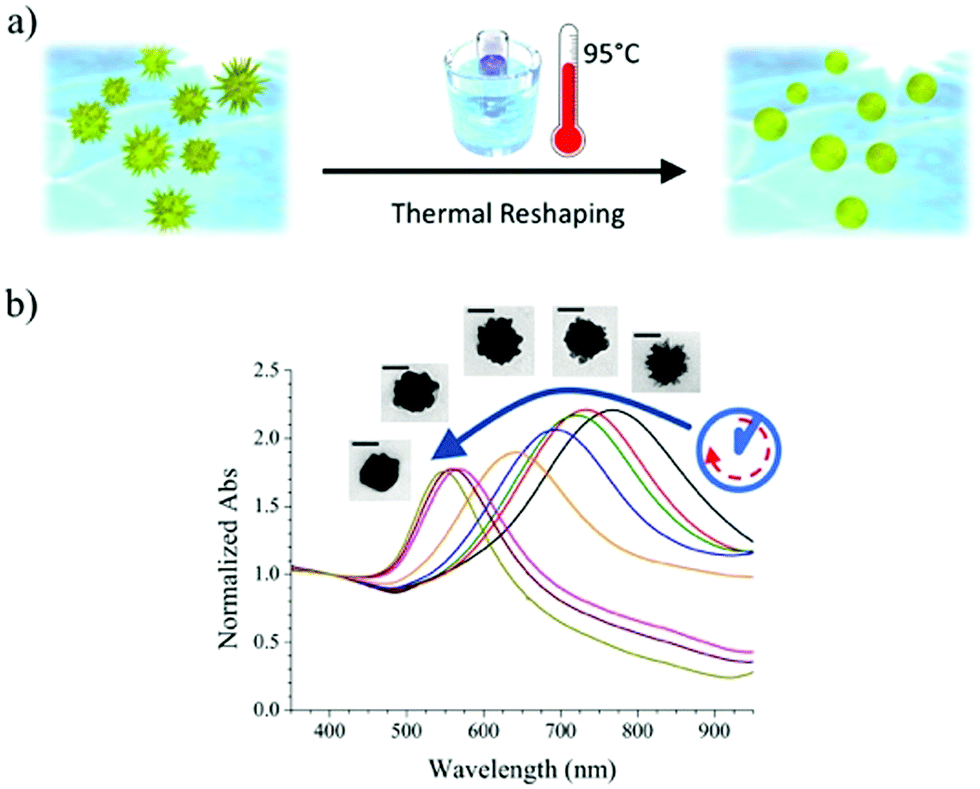 | ||
| Fig. 1 (a) Schematic representation of our reshaping experiment on AuNSs. The local curvature induces different surface chemical environments. (b) A blue shift of the nanoparticles main LSPR band from ∼800 to ∼550 nm serves to monitor reshaping, in qualitative agreement with morphological changes imaged through TEM (insets, scale bars indicate 50 nm). | ||
 | ||
| Fig. 2 (a and b) Representative TEM images of gold nanostars, before (a) and after (b) thermal reshaping. (c) Histograms obtained from TEM images showing the size distribution (Feret's diameter) for the two samples shown in (a and b), with a standard deviation over the size distribution of 26.0 nm and 16.8 nm, respectively. | ||
After reshaping, the size distribution was found to narrow down and shift to a maximum around 60 nm, due to the above-mentioned thermal reshaping mechanism, in which surface atoms migrate toward the core region, thereby removing the extra length of the nanoparticle tips, and yielding a more homogenous size distribution (the corresponding standard deviations of the distributions in Fig. 2c decrease from 26.0 to 16.8 nm). This result is similar to recent studies based on photothermal reshaping,36 where it is shown that anisotropic particles evolve towards a spherical morphology due to the higher mobility and lower melting point of atoms at corners or tips.37
Several aliquots were extracted at different times during the heating process and cooled down to quench reshaping. Each aliquot was further partitioned into three equal volumes and MBA, ABT, or 4-acetylaminobenzenthiol (AABT) were then added. This sequence ensured that all samples had the same concentration of gold, number of nanoparticles and surfactant coating density. Raman spectroscopy measurements were then performed in liquid, as a function of reshaping degree, for all three probe molecules, at both 633 and 785 nm excitation lines. As a result of the effect of tip smoothing and LSPR blueshift from the heating process, one should expect a decrease in overall SERS activity as reshaping proceeds. In Fig. 3a and b the SERS intensities for the MBA series at both 633 and 785 nm excitations are reported, expressed as the whole spectrum integral between 1000 and 1800 cm−1. According to the vis-NIR extinction spectra in Fig. 1b, the first fractions are strongly resonant at 785 nm and consequently yield higher SERS intensities than those which were heated for a longer time, which are almost fully out of resonance with the NIR excitation laser (Fig. 3b). The effect is reversed for the 633 nm excitation (Fig. 3a), where a higher signal corresponds to the most reshaped structures. This effect may appear counterintuitive regarding the sharpness of the tips, as the first fractions are still those with most pronounced tips, which should act as hotspots. In fact, simulations using the boundary element method (BEM) have shown that hotspots tend to contribute to the enhanced local fields much more than the resonant excitation.34 On the other hand, similar trends to our results have also been reported in which the plasmon resonance was the crucial parameter regarding SERS intensity.38 In the present experiment, the first fractions were obtained at relatively short times (up to 7 hours of heating), and thus show almost constant SERS intensities at 785 nm and a slight increase at 633 nm. Their extinction spectra are all partly in resonance with the NIR laser, even though the plasmon bands gradually shift toward shorter wavelengths. After this point, reshaping becomes significantly slower, and the negative contribution due to tip smoothing results in a consistent reduction of the SERS intensity at 785 nm, while the progressive blue-shift brings the samples into resonance with excitation at 633 nm.
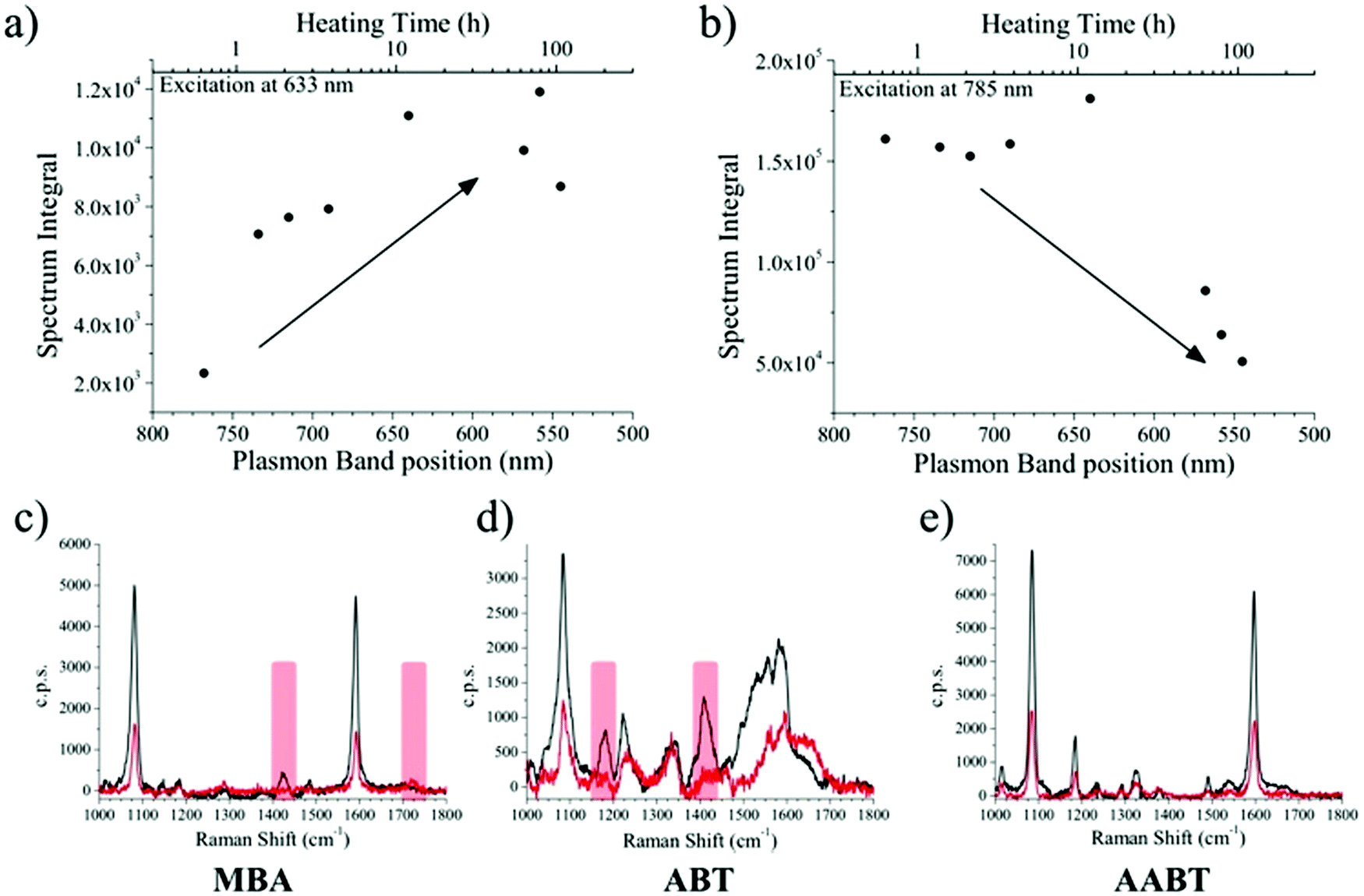 | ||
| Fig. 3 (a and b) Integrated SERS spectrum of MBA vs. heating time and LSPR band position, for excitation at 633 nm (a) and 785 nm (b). (c–e) SERS spectra recorded at 785 nm excitation, for MBA, ABT, and AABT on pristine AuNSs (black trace) and the most reshaped sample (red trace). The overall spectrum intensity decreases as the tips become smoother and the laser resonance is gradually lost. The red bars for MBA and ABT highlight the bands related to the protonation equilibrium. | ||
The corresponding overall trends were also measured for the other two model analytes, namely ABT and AABT. In Fig. 3c–e we plot the SERS spectra recorded at 785 nm, for the first and the last sample of each series, showing the overall decrease in signal intensity, as mentioned above. The evolution of the complete SERS spectra for MBA, ABT and AABT is shown in Fig. 4 as color-intensity maps of Raman shifts vs. heating time. Reported intensities were normalized and scaled to emphasize the less intense peaks. The peak at 1082 cm−1 was used to normalize the signal in all cases, because it is assigned to CS stretching, and therefore it becomes nearly insensitive to pH (Table S1, ESI†). The Raman and SERS spectra for both MBA and ABT have been reported in detail in previous studies,39–43 where it was found that their vibrational spectra can provide information regarding changes in the local environment, pH changes in particular, since the protonation equilibrium can be monitored by tracking the intensity of selected bands. The most informative bands are highlighted with red bars in Fig. 3c, d and 4a, b. We see that, along the MBA series, the signal at 1423 cm−1 decreases, while the one at 1720 cm−1 increases during reshaping. These transitions are usually attributed to the protonation of carboxyl groups.40–42 In contrast, the ABT series reveals that both bands at 1180 and 1410 cm−1 decrease during reshaping. ABT is usually considered as a suitable probe to distinguish between electromagnetic and chemical enhancement in SERS.44–46 Its dimerization into p,p′-dimercaptoazobenzene (DMAB) has been reported to occur at basic pH and/or photoinduced due to the enhanced fields produced by plasmons within hotspots.45,47,48 The main spectral changes when switching from protonated to neutral ABT and then to DMAB occur within the 1000 to 1500 cm−1 range, but recent studies have reported that neutral ABT species are particularly difficult to observe because the growing DMAB Raman bands overlap further as they get enhanced above those for neutral ABT.44,48 Nevertheless, the absence of the peaks at 1142, 1391 and 1440 cm−1, and the presence of only those at 1180 and 1410 cm−1 in Fig. 3c, should be considered as evidence of the presence of ABT rather than DMAB.44,49 Therefore, the variations in SERS spectra for ABT during the transition from pristine to reshaped gold nanostars (Fig. 3d) are consistent with those during the transition of ABT from basic to acidic environment, without the formation of DMAB. Finally, the SERS analysis of AABT was used as a control and indeed did not indicate any detectable variation, in agreement with previous literature reports.39 For both the MBA and ABT series, the variations observed are usually detected for pH values varying from 4 through 8.40,48 We observed the same behavior when pristine nanostars functionalized with MBA were titrated from pH 2 to 12 (Fig. S1, ESI†), thus showing the expected behavior for the two bands mentioned above. However, when the experiment was performed on reshaping nanostars, we did not observe any pH alteration for any morphology obtained during the heating treatment (pH of 7.1 and 6.9 for pristine and rounded AuNSs, respectively). On the basis of refs. 40, 42 and 48 and Fig. S1, we conclude that the pH variation observed in our samples (ΔpH = 0.2) should not be responsible for the SERS spectral changes observed in both MBA and ABT. Other local effects should thus act in the same manner, namely driving the protonation equilibrium of both species, induced by the particles in losing their initial star shape. To exclude any contribution from the coating agent (PVP), the MBA series was also studied using AuNSs covered with a different polymer, thiolated polyehylenglycol (PEG-SH). Whereas PVP can weakly bind to the nanoparticles through several backbone sites, PEG-SH binds at the thiolated end with a stronger bond. This polymer is also less prone to oxidation, thereby excluding chemical changes of the polymeric coating due to the reshaping process. The results, shown in Fig. S2 (ESI†), confirm a similar overall trend, with an increase of the signal at 1423 cm−1 and a decrease at 1720 cm−1 as we proceed along the reshaping process.
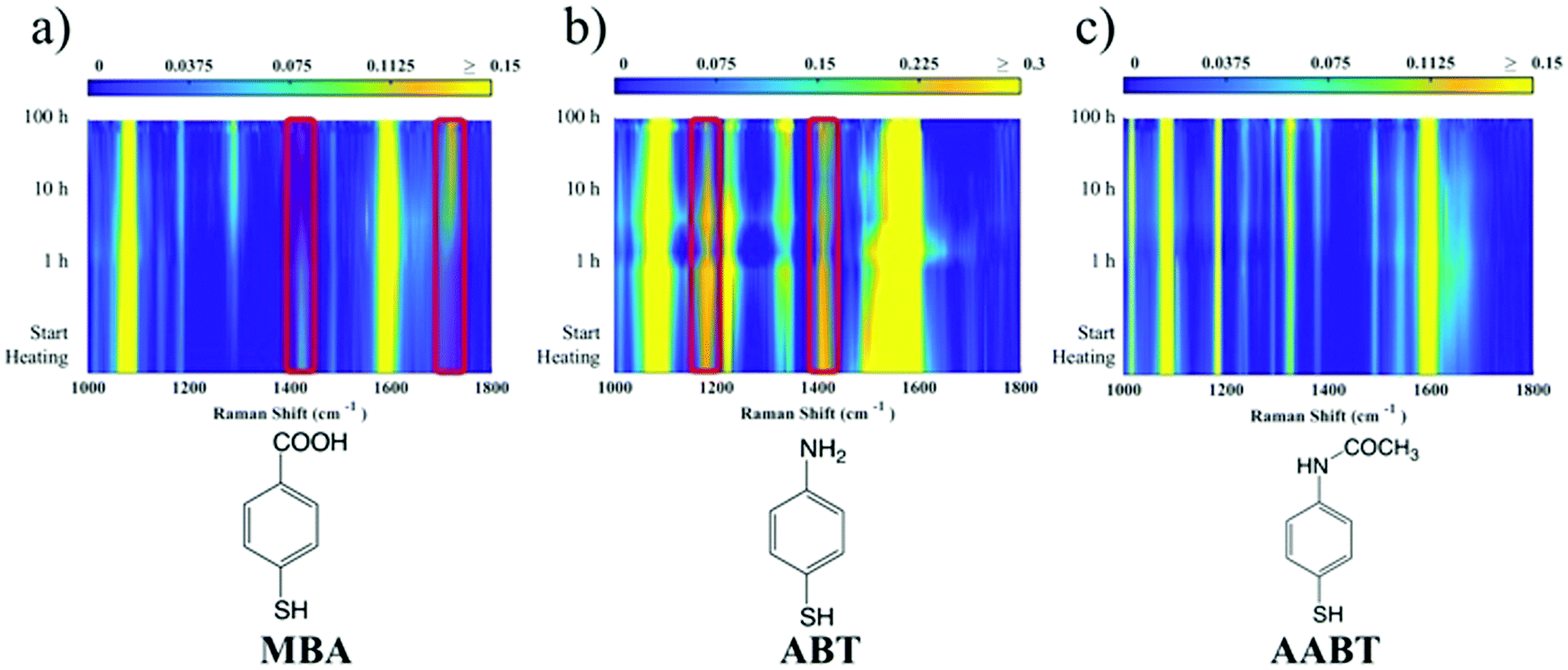 | ||
| Fig. 4 False-color maps representing measured SERS spectra at 785 nm along the reshaping series for MBA (a), ABT (b) and AABT (c). The spectra are normalized and scaled according to the color scale above each map. The red bars on the MBA and ABT series highlight the bands that change along the series; we attribute these changes to variation in protonation equilibrium. The AABT SERS map is used as a control because of the lack of sensitivity of AABT toward pH changes. | ||
Villarreal et al. recently presented a study on the complex adsorption/desorption kinetic behavior of thiolated Raman reporters (ABT included) on the surface of smooth or rough gold nanoparticles.50 In their study, the ligands were allowed to reach equilibrium for one day and then perturbations were induced by other ligands or chemicals. The authors found faster kinetics and less organized ligand adsorption on surfaces with higher curvature. A few years earlier, Wang et al. reported that a self-assembled monolayer of mercaptoundecanoic acid (MUA) shows a different pKa if functionalized on either 4 nm or 7 nm gold nanoparticles.51,52 The authors explained their results in terms of the different proximity of the carboxylic acid groups, correlated to the particle curvature. On a flatter surface, the ligands showed a tendency to be more closely packed, so equilibrium was shifted to minimize the repulsive forces. It is worth noting that the nanoparticles themselves are not neutral, but rather negatively charged (ζ-potential measured for AuNS at −15 mV and −21 mV before and after reshaping). Assuming a uniform distribution of the negative charges on the particle surface, one should consider the dependence of the potential induced by those charges on surface curvature.53 Additionally, the potential induced by any charges varies as a function of the distance from the surface in a different way depending on the local curvature of the surface; for a point charge near either a tip or a flat surface, this quantity is calculated and shown in Fig. S3 (ESI†). There is a faster potential decrease with charge-surface distance at the tip, which is consistent with the calculation reported by Pérez-Juste et al. for nanorods, argued to cause faster chemical reactions on tips than on flatter surfaces.53 The pH-like effect observed in the present study can therefore be attributed to a different environment experienced by MBA and ABT molecules on surfaces with higher or lower curvature. The peak intensity changes that are reported in Fig. 4 should be therefore considered to originate from the interplay of tip smoothing and plasmon band shift (Fig. 1b).37 As the local field enhancement at hotspots is several orders of magnitude higher than that at the rest of the nanostructure, one can assume that the acquired SERS spectra describe almost exclusively the molecules adsorbed on such hotspots, in this case the tips. Moreover, under such conditions, the molecules are less organized50 and more capable of reaching protonation equilibrium51 by orienting their polar functional groups (amine or carboxylate) toward the aqueous medium.54 The situation changes when the SERS spectra originate from molecules on flatter surfaces (i.e., after reshaping and tip smoothing), where the molecules can achieve a higher order and decrease the separation between protonable chemical groups. Then, as reshaping takes place, the interactions increase between carboxyl groups of MBA (or ABT) molecules, thereby changing their protonation states. Additionally, this change in ligand packing may also affect the interaction with the polymer coating through changes in the degree of hydrogen bond formation.55 The above considerations are schematically illustrated in Fig. 5. On high curvature surfaces, MBA and ABT are found to be in the deprotonated state, as expected in a neutral environment.40–42,48,56,57 As the surface becomes flatter, the SERS spectra begin to reveal a different configuration, in which the molecules are better organized, especially through their polar groups (i.e., carboxylic and amine).
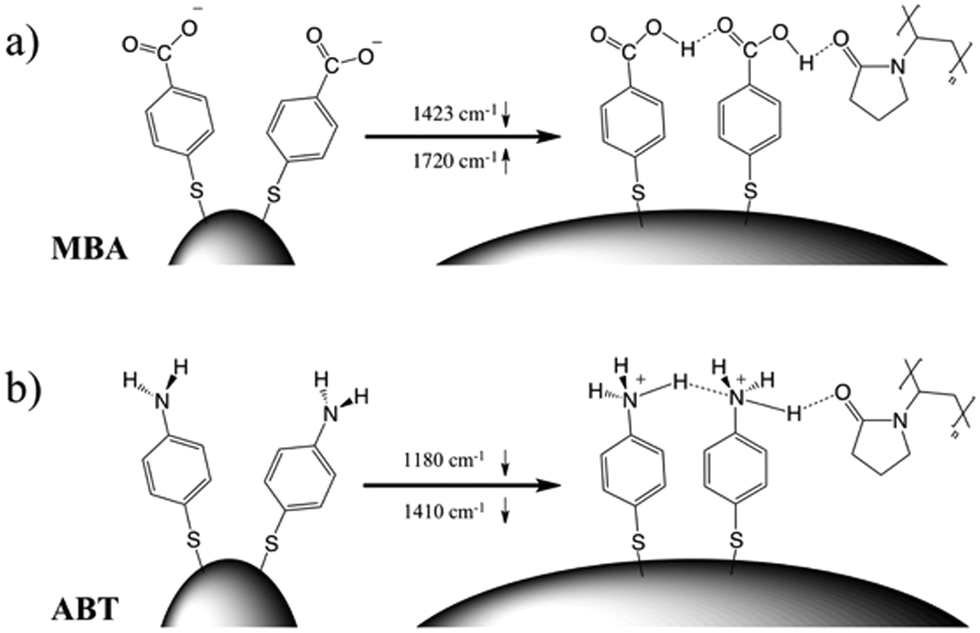 | ||
| Fig. 5 Smoothing the tip surface curvature has the effect of changing the local environment of the molecules adsorbed on the particles. The idealized situation for MBA (a) and ABT (b) is schematically shown here, including a PVP coating, illustrating different interactions and organizations that could be responsible for the SERS spectral changes observed throughout the reshaping process. | ||
The presence of weak interactions, such as hydrogen bonding between MBA or ABT molecules, or with PVP, is a reasonable assumption. This intimate interaction is likely responsible for the observed pH-like variation behavior, because even if the overall pH remains almost at neutrality, the local environment felt by the molecules can push the carboxylic and amine groups to assume a partially protonated state. The overall macroscopic effect, observed with SERS as a local probe, consists of an increase in the apparent pKa, in agreement with previous results.51 The message that emerges from the above considerations is that the same molecule near the same particle is subjected to consistent variations on its chemical equilibrium, depending on the particular site in the particle and the local surface morphology. This phenomenon is of crucial relevance in surface-chemistry-related processes, as well as in sensing applications.
Experimental
Synthesis of gold nanostars (AuNS)
The synthesis of AuNS followed a reported surfactant-free method assisted by silver ions.58 In a typical synthesis, 100 μL of citrate capped 14 nm gold nanoparticle (AuNP) seeds ([Au] = 1.5 mM) were mixed with 50 μL HAuCl4 (Sigma Aldrich) 50 mM, 10 μL HCl 1 M, 50 μL AgNO3 (Sigma Aldrich) 10 mM and 50 μL of ascorbic acid (Sigma Aldrich) 100 mM in 10 mL of pure water. The mixture was stirred for a couple of minutes. 45 μL of polyvinylpyrrolidone 10 kDa (PVP, Sigma Aldrich), or mercaptopolyethylene glycol 6 kDa (PEG-SH, Sigma Aldrich) 1 mM was then added and the solution was stirred overnight. PVP coating was selected because it prevents aggregation while allowing easy adsorption of analytes due to its relatively weak interaction with the gold surface. Furthermore, PVP has been found to be a protecting agent against chemical reshaping, such as CTAB-induced reshaping,19 and it is thermally stable (decomposition starts above 200 °C). The particles were purified by two steps of centrifugation at 6000 RCF for 10 minutes and re-dispersed ultrapure water.Thermal reshaping of AuNS
The AuNS dispersion was placed in an oil bath at 95 °C, under magnetic stirring. The first aliquot was taken before heating started, then UV-vis spectra were monitored over time (Agilent 8453 UV/vis diode-array spectrophotometer) and aliquots were sampled until no further changes in the LSPR band were observed. The various aliquots were also characterized by transmission electron microscopy (JEOL JEM-1400PLUS operating at 120 kV), the Feret's diameter (equivalent to the tip-to-tip maximum diameter of the 2D TEM projection) was used as the dimension parameter indicative of nanoparticle “spikiness”. The mechanism involving the structural rearrangement is commonly ascribed to thermally induced motion of weakly bound atoms from highly convex surfaces toward concave areas.19 Other possible reshaping mechanisms, such as Ostwald ripening, are typically discarded as no chelating molecules (CTAB, halogen analogs, etc.) are present, and no significative changes are observed in the sphere-equivalent size of the nanoparticles.Reshaped AuNS functionalization and SERS measurements
Each aliquot of partially reshaped AuNS was divided into three parts, and each of them functionalized with 30 μL of 2 mM 4-mercaptobenzoic acid (MBA, Sigma Aldrich), 30 μL of 2 mM 4-aminobenzenethiol (ABT, Sigma Aldrich) or 30 μL of 2 mM 4-acetylaminobenzenethiol (AABT, Sigma Aldrich). The amount of thiol-derivative added was estimated to be about 500 molecules per nm2, enough to ensure complete surface coverage. The reaction mixture was stirred overnight and then analyzed. SERS spectra were acquired on an inVia Renishaw μRaman, excited through a 5× Leica objective with a 785 nm diode laser at a nominal power of 60 mW, or with a 632.8 nm He–Ne laser at a nominal power of 13 mW, for 10 to 30 seconds.Conclusions
Surface-enhanced Raman scattering is a local effect, with a strong dependence on the distance from the plasmonic substrate, which renders it as a primary tool to investigate the behavior of molecules adsorbed on nanostructured surfaces. We studied the effect of thermal reshaping of gold nanostars into rounded particles on the spectroscopy of adsorbed mercaptobenzoic acid and 4-aminobenzenethiol. We found not only a strong dependence of the SERS enhancement with the position of the LSPR band, as well as the selected laser excitation frequency, but also consistent variations of the SERS spectra of both molecules during the reshaping process. We attribute such variations to changes in the protonation states of the terminal groups of the molecule, similar to those observed for the same molecules when pH is varied by more than four units. We observed that such changes take place even when the solution pH is kept constant, thus reflecting a change in the local molecular environment, which shifts their equilibrium pKa. The results can be understood as a change in the molecular interactions, both between adsorbed molecules and with the surrounding environment, induced by changes in local surface curvature. The findings presented in this study should help us to understand the complex surface chemistry of molecules, influenced by highly anisotropic nanostructured surfaces, and could be relevant for pH and ion sensing based on SERS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LL and LML-M acknowledge funding from European Commission Grant (EUSMI 731019). Funding is also acknowledged from the Spanish MINECO (MAT2017-86659-R and MDM-2017-0720 to LML-M; MAT2017-88492-R and SEV2015-0522 to JGA) and the European Research Council (Advanced Grant 787510 4DBIOSERS to LML-M; Advanced Grant 789104-eNANO to JGA).References
- R. Yu, L. M. Liz-Marzán and F. J. de Abajo, Chem. Soc. Rev., 2017, 46, 6710–6724 RSC.
- C. Hamon, S. M. Novikov, L. Scarabelli, D. M. Solís, T. Altantzis, S. Bals, J. M. Taboada, F. Obelleiro and L. M. Liz-Marzán, ACS Photonics, 2015, 2, 1482–1488 CrossRef CAS PubMed.
- L. Litti and M. Meneghetti, Phys. Chem. Chem. Phys., 2019, 21, 15515–15522 RSC.
- M. A. Wall, S. Harmsen, S. Pal, L. Zhang, G. Arianna, J. R. Lombardi, C. M. Drain and M. F. Kircher, Adv. Mater., 2017, 29, 1605622 CrossRef.
- L. Litti, V. Amendola, G. Toffoli and M. Meneghetti, Anal. Bioanal. Chem., 2016, 408, 2123–2131 CrossRef CAS.
- G. Sciutto, S. Prati, I. Bonacini, L. Litti, M. Meneghetti and R. Mazzeo, Anal. Chim. Acta, 2017, 991, 104–112 CrossRef CAS.
- W. Xie and S. Schlücker, Chem. Commun., 2018, 54, 2326–2336 RSC.
- F. Biscaglia, S. Rajendran, P. Conflitti, C. Benna, R. Sommaggio, L. Litti, S. Mocellin, G. Bocchinfuso, A. Rosato and A. Palleschi, Adv. Healthcare Mater., 2017, 6, 1700596 CrossRef.
- D. Jimenez de Aberasturi, A. B. Serrano-Montes, J. Langer, M. Henriksen-Lacey, W. J. Parak and L. M. Liz-Marzán, Chem. Mater., 2016, 28, 6779–6790 CrossRef CAS.
- L. Litti, A. Ramundo, F. Biscaglia, G. Toffoli, M. Gobbo and M. Meneghetti, J. Colloid Interface Sci., 2019, 533, 621–626 CrossRef CAS PubMed.
- M. R. Willner, K. S. McMillan, D. Graham, P. J. Vikesland and M. Zagnoni, Anal. Chem., 2018, 90, 12004–12010 CrossRef CAS.
- F. Biscaglia, S. Quarta, G. Villano, C. Turato, A. Biasiolo, L. Litti, M. Ruzzene, M. Meneghetti, P. Pontisso and M. Gobbo, Mater. Sci. Eng., C, 2019, 103, 109762 CrossRef CAS.
- L. Litti, N. Rivato, G. Fracasso, P. Bontempi, E. Nicolato, P. Marzola, A. Venzo, M. Colombatti, M. Gobbo and M. Meneghetti, Nanoscale, 2018, 10, 1272 RSC.
- J. Reguera, D. J. de Aberasturi, M. Henriksen-Lacey, J. Langer, A. Espinosa, B. Szczupak, C. Wilhelm and L. M. Liz-Marzán, Nanoscale, 2017, 9, 9467–9480 RSC.
- L. Rodríguez-Lorenzo, R. A. Álvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stéphan, M. Kociak, L. M. Liz-Marzán and F. J. García de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef.
- D. M. Solís, J. M. Taboada, F. Obelleiro, L. M. Liz-Marzán and F. J. García de Abajo, ACS Photonics, 2017, 4, 329–337 CrossRef.
- J. Reguera, J. Langer, D. J. de Aberasturi and L. M. Liz-Marzán, Chem. Soc. Rev., 2017, 46, 3866–3885 RSC.
- M. M. Vega, A. Bonifacio, V. Lughi, S. Marsi, S. Carrato and V. Sergo, J. Nanopart. Res., 2014, 16, 2729 CrossRef.
- L. Rodríguez-Lorenzo, J. M. Romo-Herrera, J. Pérez-Juste, R. A. Alvarez-Puebla and L. M. Liz-Marzán, J. Mater. Chem., 2011, 21, 11544–11549 RSC.
- A. Kedia and P. S. Kumar, J. Mater. Chem. C, 2013, 1, 4540–4549 RSC.
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, Science, 2008, 321, 943–946 CrossRef CAS.
- D.-Y. Wu, J.-F. Li, B. Ren and Z.-Q. Tian, Chem. Soc. Rev., 2008, 37, 1025–1041 RSC.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS.
- F. Lamberti, D. Ferraro, M. Giomo and N. Elvassore, Electrochim. Acta, 2013, 97, 304–312 CrossRef CAS.
- R. Raval, Chem. Soc. Rev., 2009, 38, 707–721 RSC.
- S. A. Claridge, W.-S. Liao, J. C. Thomas, Y. Zhao, H. H. Cao, S. Cheunkar, A. C. Serino, A. M. Andrews and P. S. Weiss, Chem. Soc. Rev., 2013, 42, 2725–2745 RSC.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- Q. Ong, Z. Luo and F. Stellacci, Acc. Chem. Res., 2017, 50, 1911–1919 CrossRef CAS.
- Y. Wang, W. Ji, H. Sui, Y. Kitahama, W. Ruan, Y. Ozaki and B. Zhao, J. Phys. Chem. C, 2014, 118, 10191–10197 CrossRef CAS.
- A. M. Schwartzberg, T. Y. Oshiro, J. Z. Zhang, T. Huser and C. E. Talley, Anal. Chem., 2006, 78, 4732–4736 CrossRef CAS PubMed.
- S. J. Lee and M. Moskovits, Nano Lett., 2010, 11, 145–150 CrossRef PubMed.
- J. P. Scaffidi, M. K. Gregas, V. Seewaldt and T. Vo-Dinh, Anal. Bioanal. Chem., 2009, 393, 1135–1141 CrossRef CAS PubMed.
- X. Han, H. Wang, X. Ou and X. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 5811–5814 CrossRef CAS.
- L. Rodríguez-Lorenzo, R. A. Alvarez-Puebla, F. J. de Abajo and L. M. Liz-Marzán, J. Phys. Chem. C, 2009, 114, 7336–7340 CrossRef.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- D. Liu, C. Li, F. Zhou, T. Zhang, H. Zhang, X. Li, G. Duan, W. Cai and Y. Li, Sci. Rep., 2015, 5, 7686 CrossRef CAS.
- H. Vanrompay, E. Bladt, W. Albrecht, A. Béché, M. Zakhozheva, A. Sánchez-Iglesias, L. M. Liz-Marzán and S. Bals, Nanoscale, 2018, 10, 22792–22801 RSC.
- N. Pazos-Perez, L. Guerrini and R. A. Alvarez-Puebla, ACS Omega, 2018, 3, 17173–17179 CrossRef CAS.
- K. Kim, D. Shin, J.-Y. Choi, K. L. Kim and K. S. Shin, J. Phys. Chem. C, 2011, 115, 24960–24966 CrossRef CAS.
- A. Jaworska, L. E. Jamieson, K. Malek, C. J. Campbell, J. Choo, S. Chlopicki and M. Baranska, Analyst, 2015, 140, 2321–2329 RSC.
- A. Pallaoro, G. B. Braun, N. Reich and M. Moskovits, Small, 2010, 6, 618–622 CrossRef CAS PubMed.
- Y. Liu, H. Yuan, A. M. Fales and T. Vo-Dinh, J. Raman Spectrosc., 2013, 44, 980–986 CrossRef CAS.
- W. Ji, N. Spegazzini, Y. Kitahama, Y. Chen, B. Zhao and Y. Ozaki, J. Phys. Chem. Lett., 2012, 3, 3204–3209 CrossRef CAS PubMed.
- D.-Y. Wu, X.-M. Liu, Y.-F. Huang, B. Ren, X. Xu and Z.-Q. Tian, J. Phys. Chem. C, 2009, 113, 18212–18222 CrossRef CAS.
- L. Cao, P. Diao, L. Tong, T. Zhu and Z. Liu, ChemPhysChem, 2005, 6, 913–918 CrossRef CAS.
- M. Osawa, N. Matsuda, K. Yoshii and I. Uchida, J. Phys. Chem., 1994, 98, 12702–12707 CrossRef CAS.
- P. Pienpinijtham, S. Vantasin, Y. Kitahama, S. Ekgasit and Y. Ozaki, J. Phys. Chem. C, 2016, 120, 14663–14668 CrossRef CAS.
- S. Zong, Z. Wang, J. Yang and Y. Cui, Anal. Chem., 2011, 83, 4178–4183 CrossRef CAS.
- T. Zhang, Y. Sun, L. Hang, H. Li, G. Liu, X. Zhang, X. Lyu, W. Cai and Y. Li, ACS Appl. Mater. Interfaces, 2018, 10, 9792–9801 CrossRef CAS.
- E. Villarreal, G. G. Li, Q. Zhang, X. Fu and H. Wang, Nano Lett., 2017, 17, 4443–4452 CrossRef CAS.
- D. Wang, R. J. Nap, I. Lagzi, B. Kowalczyk, S. Han, B. A. Grzybowski and I. Szleifer, J. Am. Chem. Soc., 2011, 133, 2192–2197 CrossRef CAS PubMed.
- D. A. Walker, E. K. Leitsch, R. J. Nap, I. Szleifer and B. A. Grzybowski, Nat. Nanotechnol., 2013, 8, 676 CrossRef CAS PubMed.
- J. Pérez-Juste, L. M. Liz-Marzán, S. Carnie, D. Y. Chan and P. Mulvaney, Adv. Funct. Mater., 2004, 14, 571–579 CrossRef.
- G. Lu, B. Shrestha and A. J. Haes, J. Phys. Chem. C, 2016, 120, 20759–20767 CrossRef CAS.
- V. I. Teberekidis and M. P. Sigalas, THEOCHEM, 2007, 803, 29–38 CrossRef CAS.
- S. W. Bishnoi, C. J. Rozell, C. S. Levin, M. K. Gheith, B. R. Johnson, D. H. Johnson and N. J. Halas, Nano Lett., 2006, 6, 1687–1692 CrossRef CAS.
- K. Kim, K. L. Kim, D. Shin, J.-Y. Choi and K. S. Shin, J. Phys. Chem. C, 2012, 116, 4774–4779 CrossRef CAS.
- R. García-Alvarez, M. Hadjidemetriou, A. Sánchez-Iglesias, L. M. Liz-Marzan and K. Kostarelos, Nanoscale, 2017, 10, 1256–1264 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00456d |
| This journal is © The Royal Society of Chemistry 2020 |