
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow double-layer carbon nanocage confined Si nanoparticles for high performance lithium-ion batteries†
Jijun
Lu
abc,
Dong
Wang
a,
Junhao
Liu
a,
Guoyu
Qian
a,
Yanan
Chen
 *c and
Zhi
Wang
*ab
*c and
Zhi
Wang
*ab
aKey Laboratory of Green Process and Engineering, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: zwang@ipe.ac.cn
bSchool of Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100039, P. R. China
cSchool of Materials Science and Engineering, Key Laboratory of Advanced Ceramics and Machining Technology of Ministry of Education, Tianjin Key Laboratory of Composite and Functional Materials, Tianjin University, Tianjin 300072, P. R. China. E-mail: yananchen@tju.edu.cn
First published on 25th May 2020
Abstract
The huge volume variation and the unstable solid electrolyte interface (SEI) of Si (Si) during the lithiation and delithiation process severely obstruct its practical application as lithium-ion battery anodes. Here, we design and fabricate a hollow structure of double-layer hybrid carbon nanocage encapsulated Si nanoparticles to address these challenges. The double-layer hybrid carbon-Si nanoarchitecture is obtained by integrating electrostatic self-assembly, seed-induced growth and heterogeneous shrinkage. The internal layer of hollow N-doped carbon of the hybrid nanoarchitecture (Si@H-NC@GC) provides limited inner space for controlling volume changes of Si nanoparticles, while the outer graphite carbon layer facilitates the formation of a stable SEI. When evaluated as anode materials for LIBs, the Si@H-NC@GC nanoarchitecture exhibits greatly enhanced electrochemical performance compared with the bare Si, Si@NC and H-NC@GC electrodes. Notably, Si@H-NC@GC delivers a reversible capacity retention of 92.5% after 550 cycles at a high current density of 1 A g−1 and a high capacity of 1081 mA h g−1 after 500 cycles at 0.5 A g−1.
1. Introduction
Lithium-ion batteries (LIBs) have been considered as one of the most important energy storage devices owing to their high energy density and lack of the memory effect.1,2 However, the commercial anode material, graphite, has got a low theoretical specific capacity (LiC6, 372 mA h g−1), and it is challenging to meet the ever-growing demands of portable electronics and electric vehicles.3,4 Therefore, to improve the energy density of current LIBs, it is urgent to develop a high specific capacity electrode material instead of commercial graphite anodes. Among the emerging anode materials, silicon (Si) is a promising candidate for next-generation LIB anodes due to its high specific capacity (Li15Si4, 3579 mA h g−1), low Li-uptake voltage (∼0.5 V vs. Li/Li+) and natural abundance (second largest resource).5–7 Unfortunately, the application of Si-based anodes has been impeded because of its inferior intrinsic conductivity and large volume effect (∼300%) during lithiation.8,9 The huge volume variation produces high mechanical stress that causes the powdering of the electrode materials andformation of an unstable solid electrolyte interphase (SEI), resulting in fast capacity fading and poor coulombic efficiency.10Researchers have developed various strategies to resolve these problems of Si-based materials. For example, the preparation of nano-sized Si can reduce the mechanical stress and suppress the pulverization of active materials caused by volume changes during the lithium alloying process.11,12 The fabrication of hollow structure materials by combining the carbon matrix can availably mitigate the volume effect of Si, improve the electrical conductivity and form a stable SEI.13–15 Besides, flexible substrates and mesoporous structures have been introduced to support the volume change of Si nanoparticles.16–19 Much effort has been devoted to controlling the volume change of Si such as constructing hollow structures and introducing buffer substrates and mesoporous structures, but there are still great challenges in solving the problem of huge volume changes and an unstable SEI.13–21 Therefore, it is meaningful to explore simple and multi-strategic coupling methods to mitigate the volume change of Si and form a stable SEI layer. Metal–organic framework compounds are constructed using metal ions/clusters and organic ligands through coordination bonding, showing great potential in energy storage.22 The application of MOFs as a derivatized carbon shell in the field of energy storage has been reported.23,24 MOFs not only heighten the mechanical integrity of materials, but also derivate the heteroatom doping and porous structure of the carbon shell, which increases the conductivity, allowing rapid transfer of lithium ions. Furthermore, MOFs can form hollow structures through heterogeneous shrinkage during high temperature heat treatment.25 Thus, it is foreseeable that the combination of Si and MOFs will be a suitable method for the preparation of multifunctional materials.
Herein, a Si-hollow double-layer hybrid carbon nanocage (Si@H-NC@GC) has been successfully prepared based on electrostatic self-assembly, seed-induced growth and heterogeneous shrinkage. The double hybrid carbon shells are obtained during heat treatment where a N-doped microporous carbon (NC) inner shell is derived from ZIF-8 and a mesoporous graphitic carbon (GC) outer shell is derived from ZIF-67.26,27 At the same time, the material is heterogeneously contracted during the high-temperature treatment to form the internal hollow, denoted as Si@H-NC@GC. In the novel structure of Si@H-NC@GC, GC has the effect of stabilizing the SEI, good electrical conductivity and rapid transfer of lithium ions, while hollow NC effectively accommodates the volume expansion of Si nanoparticles and increases lithium storage active sites.28–30 Si@H-NC@GC displays a high reversible capacity of 1052 mA h g−1 (92.5% capacity retention) after 550 cycles at a high current density of 1 A g−1 when used in LIBs. With the successful synthesis of Si@H-NC@GC, the employment of MOFs to synthesize double-layer and even multi-layer anode materials from self-templates will pave a new way for the manufacture of various anodes with high capacity and superior stability for next-generation LIBs.
2. Experimental section
2.1 Materials preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a water bath at 95 °C for 3 h with stirring. In a typical procedure, 0.2 g of the functionalized Si nanoparticles and 1.0 g of PVP were uniformly dispersed in 50 mL of methanol. Then, 4 mmol of Zn(NO3)2·6H2O was added into the above solution and stirred for 1 h to obtaine solution A. Solution B was obtained by dissolving 12 mmol 2-methylimidazole in 50 mL of methanol. Subsequently, solution B was added dropwise to solution A, and the mixed solution was allowed to stand for 24 hours at room temperature. The Si@ZIF-8 precipitate was collected by centrifugation, washed several times with methanol, and dried at 60 °C.
:1) in a water bath at 95 °C for 3 h with stirring. In a typical procedure, 0.2 g of the functionalized Si nanoparticles and 1.0 g of PVP were uniformly dispersed in 50 mL of methanol. Then, 4 mmol of Zn(NO3)2·6H2O was added into the above solution and stirred for 1 h to obtaine solution A. Solution B was obtained by dissolving 12 mmol 2-methylimidazole in 50 mL of methanol. Subsequently, solution B was added dropwise to solution A, and the mixed solution was allowed to stand for 24 hours at room temperature. The Si@ZIF-8 precipitate was collected by centrifugation, washed several times with methanol, and dried at 60 °C.
2.2 Materials characterization
SEM (Hitachi S4800) was carried out to study the morphologies of the composites and the electrodes. The internal structure and element distribution of the nanoarchitectures were studied by TEM (FEI Tecnai F20). The surface structural characteristics of the sample were analyzed by X-ray photoelectron spectroscopy (XPS) (Thermo Scientific K-Alpha, USA). Raman spectra were constructed through a Renishaw inVia Reflex Raman spectrometer with excitation by a 532 nm-wavelength laser. The XRD images were collected via an AXS D8 Advance diffractometer (Cu-Kα radiation, 40 mA, 40 kV) from Bruker, Inc. (Germany). Thermogravimetric analysis (TGA) was performed by using a Pyris Diamond, PerkinElmer, USA from 30 °C to 800 °C at a ramp rate of 10 °C min−1. The specific surface area and pore size distribution of the samples were measured based on the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively.2.3 Electrochemical measurements
The electrochemical properties of the samples were investigated using CR2032-type coin cells, which were composed of lithium wafers as counter electrodes, and a Celgard 2400 as a separator, and the electrolyte was 1 M LiPF6 dissolved in a mixed solvent of ethylene carbonate (EC), ethyl methyl carbonate (EMC) and dimethyl carbonate (DEC) (1:1:1 by volume). The working electrode was prepared with active materials (70%), acetylene black (20%), and PVDF (10%) dissolved in N-methyl-2-pyrrolidione. In the experiment, the diameter of the electrode was 0.6 cm and the mass loading of active materials was about 1.3 mg cm−2. The resulting slurry was coated on the copper foil and then dried in a vacuum oven at 120 °C for 12 h. The charge and discharge tests of the half battery were performed in a voltage range of 0.01–3.0 V (vs. Li/Li+). EIS was carried out between 100 kHz and 0.01 Hz with an amplitude of 5 mV.
3. Results and discussion
The synthesis strategy of Si@H-NC@GC is shown in Fig. 1 first, electronegative carboxylic functional groups are attached on the nano-Si surface by acid treatment as nucleation sites for MOF nucleation growth.31,32 The functionalized Si nanoparticles are uniformly dispersed in methanol containing PVP and Zn(NO3)2·6H2O, and Zn2+ is adsorbed on the functional group by electrostatic interaction. Subsequently, methanol containing dimethylimidazole was dropwise added to carry out the reaction, and a small ZIF-8 crystal was used as the crystal nucleus on the nano-Si surface for the growth of ZIF-8 crystals during the ageing process. The obtained Si@ZIF-8 facilitates nucleus-induced epitaxial growth of ZIF-67 to generate Si@ZIF-8@ZIF-67 materials with a uniform decahedral structure. The successful synthesis of Si@ ZIF-8@ZIF-67 is mainly attributed to the fact that ZIF-67 and ZIF-8 have a similar isoreticular structure and unit cell parameters (aZIF-8 = 16.9910 Å, aZIF-67 = 16.9589 Å).23 Finally, the target product Si@H-NC@GC was prepared after the high temperature calcination.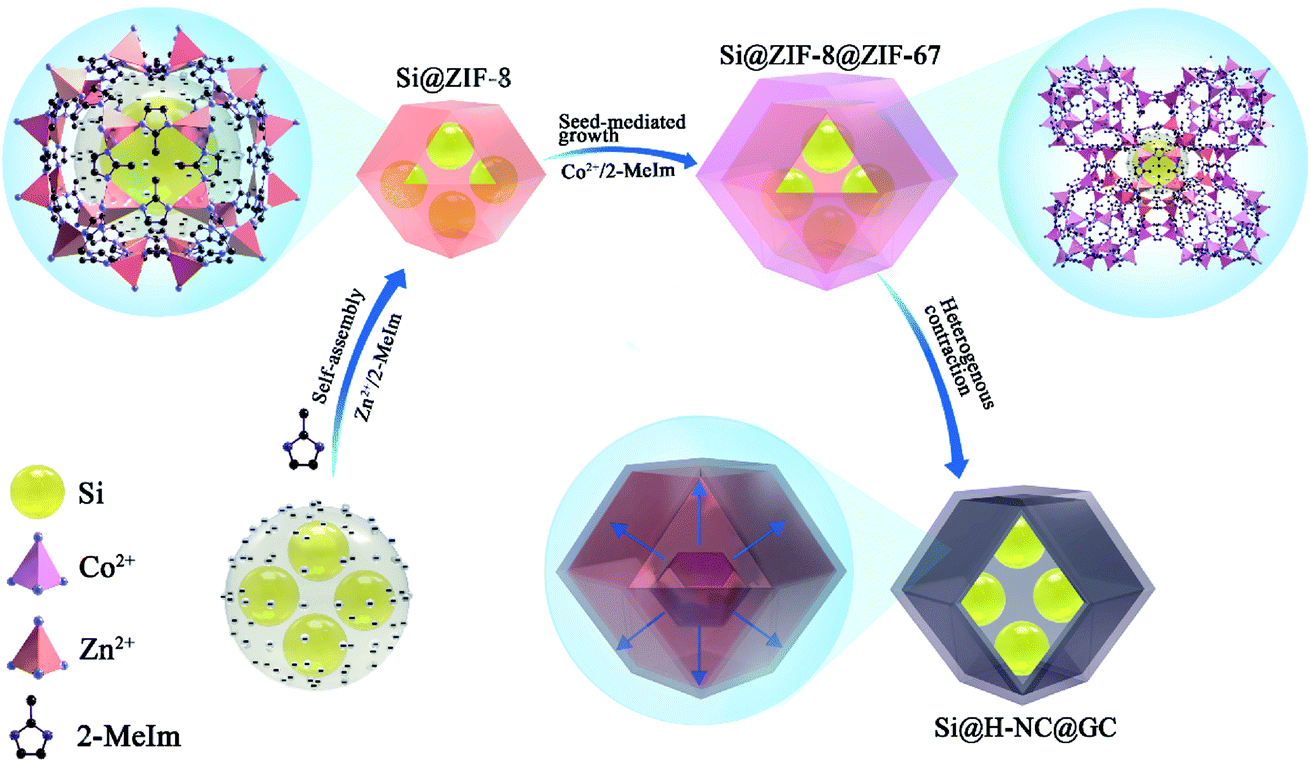 | ||
| Fig. 1 Schematic of the fabrication process for Si@H-NC@GC. | ||
The morphology and structural characteristics of the samples were demonstrated by field-emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM). Raw Si a is spherical particle with a size of 20–50 nm. The intermediate products Si@ZIF-8 and heat-treated Si@NC have a dodecahedron shape (Fig. S1a–c†). The TEM image (Fig. S1d–f†) shows that the Si nanoparticles are well coated with the carbon layer, and the interplanar spacing of the Si is 0.31 nm corresponding to the (111) crystal plane.33 The synthesized Si@ZIF-8@ZIF-67 and heat-treated Si@H-NC@GC nanoarchitectures were characterized as shown in Fig. 2a and b, which reveal that the products are uniform dodecahedral structures without collapse. The TEM image (Fig. 2c and d) shows that the Si particles are well confined within the double carbon layers. The inner shells of dense amorphous carbon are formed by ZIF-8 decomposition and subsequent reduction of metallic Zn.34 The outer shells of relatively loose carbon are formed by ZIF-67 rapid decomposition and reduction of metallic Co. The radial distribution of the elements and the double carbon layer carbon shell derived from different metals of the Si@H-NC@GC sample was confirmed by EDX linear sweep (Fig. 2e). The elemental TEM with EDX mapping of Si@H-NC@GC (Fig. 2f) shows that the material consists of C, Si, N, Co and Zn.
 | ||
| Fig. 2 (a, b) SEM images of Si@ZIF-8@ZIF-67 and Si@H-NC@GC; (c, d) TEM images of Si@H-NC@GC; (e) TEM images with EDX line scan corresponding to Si@H-NC@GC; and (f) TEM images with EDX mapping for Si@H-NC@GC element distribution. | ||
Moreover, the hollow structure of the products is mainly due to the diversity in the thermal stability between ZIF-8 and ZIF-67 during heat treatment, and the unstable ZIF-67 would undergo pyrolysis first to form a harsh shell of carbon around the ZIF-8 core. The rigid graphitic carbon shells of the Si@H-NC@GC nanocage are induced by the catalytic carbonization of ZIF-67, while the inner tight N-doped carbon shells are obtained from the attachment of carbonized ZIF-8.27 Therefore, the as-prepared Si@H-NC@GC nanoarchitecture is expected to have excellent electrochemical performance for lithium ions. The inner layer of hollow N-doped carbon can strongly buffer the volume change of Si, shorten the lithium-ion diffusion path and increase the lithium storage active sites in the process of lithiation and delithiation. The outer mesoporous graphite carbon facilitates the generation of a stable SEI, excellent electrical conductivity and rapid transfer of lithium ions. For comparison, the H-NC@GC material was also synthesized (Fig. S2†).
The crystalline phase and composition of the specimens were characterized through XRD patterns. It can be seen from Fig. 3a that there are distinct crystalline Si diffraction peaks in the Si@NC and Si@H-NC@GC materials.35 The absence of Zn in the Si@NC and Si@H-NC@GC materials is mainly due to the fact that metallic Zn mostly evaporated with the temperature rise.24 There are weak Co diffraction peaks for the Si@H-NC@GC material, probably due to the presence of carbon reduction of Co elements during the sintering process, and a small amount of residue after H2SO4 pickling.20,36 As shown in Fig. S3,† the TG analysis shows that the content of Si in the Si@H-NC@GC products after pickling was about 40%.
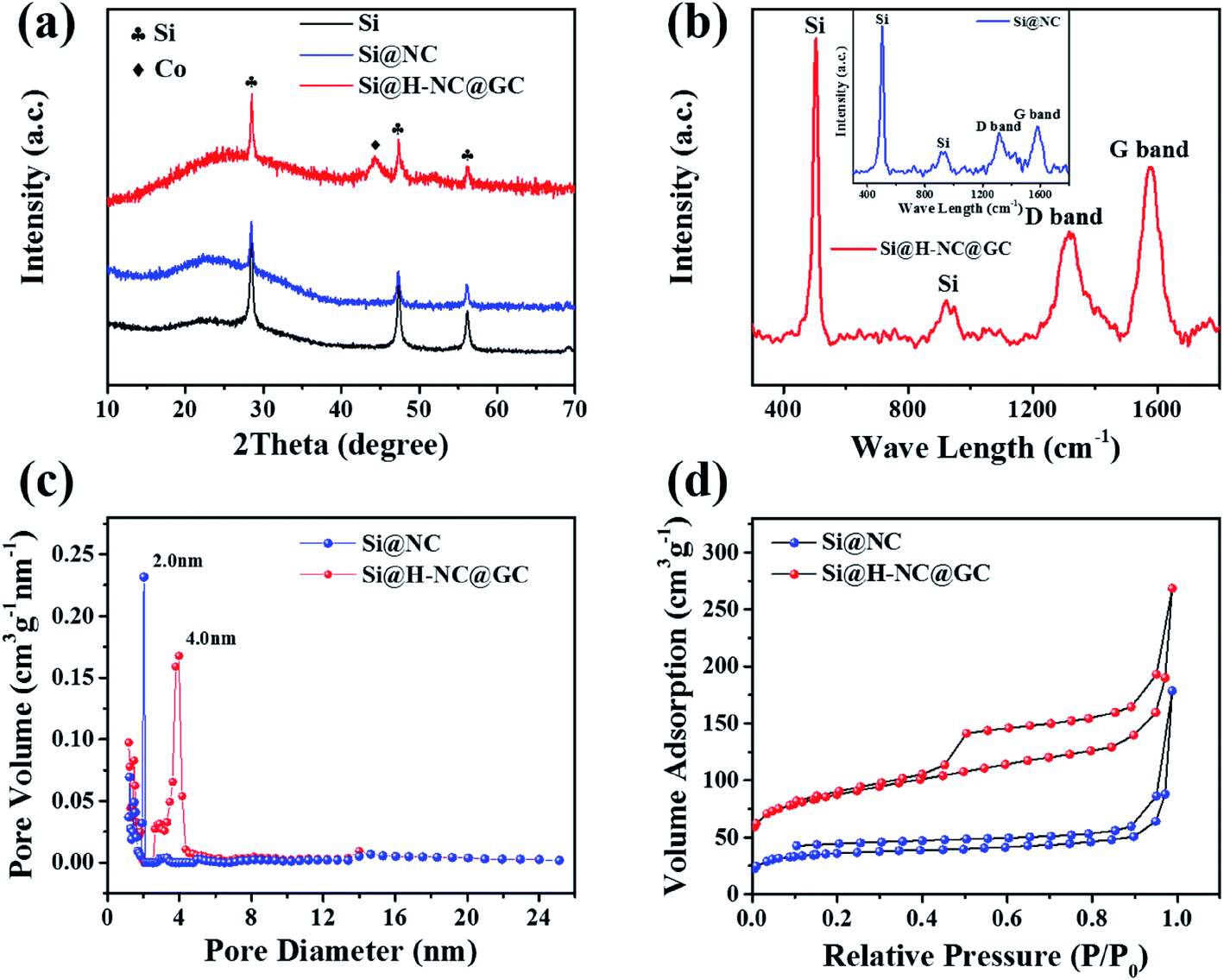 | ||
| Fig. 3 Characterization of Si@NC and Si@H-NC@GC. (a) XRD patterns of Si, Si@NC and Si@H-NC@GC; (b) Raman spectra of Si@NC and Si@H-NC@GC; (c) pore size distributions, showing the micropores in Si@NC and the mesopores in Si@H-NC@GC; (d) nitrogen adsorption–desorption isotherms of Si@NC and Si@H-NC@GC. | ||
The Raman spectrum shows the structural information of Si@NC and Si@H-NC@GC (Fig. 3b). The Raman spectrum of Si@H-NC@GC shows the wavelength of Si at 502.3 and 924.1 cm−1. The D and G bands, which are located at around 1319.0 and 1576.7 cm−1, correspond to amorphous and graphitized carbon, respectively.37 The ID/IG value is about 0.62, which indicates that the presence of short-range ordered carbon in the products. The degree of graphitization of Si@H-NC@GC is significantly higher than that of Si@NC (ID/IG ≈ 0.86), which also confirms that the ZIF-67 derived carbon layer has a high degree of graphitization, increasing the electrical conductivity of the composites. The pore size distribution of the Si@NC and Si@H-NC@GC nanoarchitecture is revealed in Fig. 3c, showing that both are mesoporous structures of 2 to 50 nm, respectively. The N2 adsorption–desorption isotherms (Fig. 3d) show that the hysteresis loop and the volume adsorption increase gradually in the range of 0.45–1.0 relative pressure, which was caused by the capillary condensation of N2 in the mesopores with broad size distribution.27 In addition, Si@H-NC@GC has a higher specific surface area of 313.8 m2 g−1 compared to Si@NC, 130.5 m2 g−1. It is beneficial to shorten the diffusion path of lithium ions, rapid transfer of lithium ions and alleviate the volume expansion of Si during the process of lithiation and delithiation.
To confirm that the Si nanoparticles were well confined within the MOF shells, X-ray photoelectron spectroscopy (XPS) analysis was carried out. The Si 2p high-resolution spectrum (Fig. S4a†) shows the presence of two peaks at 102.7 and 99.3 eV, corresponding to SiOx and unoxidized Si atoms, respectively.20,33 The high-resolution C 1s peaks (Fig. S4b†) are shown at 288, 285.6 and 284.7 eV, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, and CC bond of the carbon coating, respectively.38 The high-resolution N 1s spectrum (Fig. S4c†) reveals the presence of two forms of nitrogen, corresponding to graphitic N at 400.6 eV and pyridinic N at 398.5 eV;19 Fig. S4d† shows the high-resolution Zn 2p spectrum with two distinct peaks at 1044.4 and 1021.3 eV corresponding to Zn 2p1/2 and Zn 2p3/2, respectively;35 The Co 2p XPS spectrum (Fig. S4e†) is resolved into two pairs of 2p3/2/2p1/2 doublets for metallic Co (778.1/794.7 eV) and Co2+ (779.9 eV).26 XPS of the Si@NC intermediate samples was carried out to characterize their structure and composition information, as shown in Fig. S5,† confirming that the Si nanoparticles were well encapsulated within the NC shells.
O, C–O, and CC bond of the carbon coating, respectively.38 The high-resolution N 1s spectrum (Fig. S4c†) reveals the presence of two forms of nitrogen, corresponding to graphitic N at 400.6 eV and pyridinic N at 398.5 eV;19 Fig. S4d† shows the high-resolution Zn 2p spectrum with two distinct peaks at 1044.4 and 1021.3 eV corresponding to Zn 2p1/2 and Zn 2p3/2, respectively;35 The Co 2p XPS spectrum (Fig. S4e†) is resolved into two pairs of 2p3/2/2p1/2 doublets for metallic Co (778.1/794.7 eV) and Co2+ (779.9 eV).26 XPS of the Si@NC intermediate samples was carried out to characterize their structure and composition information, as shown in Fig. S5,† confirming that the Si nanoparticles were well encapsulated within the NC shells.
Here, the electrochemical properties of Si@NC and Si@H-NC@GC have been investigated as anodes for LIBs. Fig. 4a shows the CV curves for Si@H-NC@GC at a scan rate of 0.5 mV s−1 in the potential window of 0.01–3.0 V vs. Li/Li+. It can be seen from the CV curves that a significant cathodic reduction peak near 0.6 V is attributed to the formation of the SEI during the first cycle, and the peak vanishes during continuous cycles. It shows that a stable SEI is formed on the surface of Si@H-NC@GC materials. The cathodic reduction peak near 0.2 V is associated with amorphous LixSi phases.5 The anodic peak near 0.53 V is attributed to the de-alloying process of the LixSi phase in the electrode materials and gradually becomes stronger during subsequent scanning, and it explains that the materials are further activated during the continuous cycles and more Si participates in the lithiation reaction.19 Fig. S6† shows the CV curve of Si@NC, and the results are similar to those for the Si@H-NC@GC samples. Fig. 4b exhibits the representative galvanostatic charge/discharge profiles of the Si@H-NC@GC electrode. The Si@H-NC@GC electrode delivered excellent electrochemical properties with an initial coulombic efficiency of 71.2%. The irreversible capacity loss is mainly ascribed to the formation of the SEI.14 The discharge profile of Si@H-NC@GC exhibited a distinct voltage platform around 0.2 and 0.6 V, which is consistent with the CV curve peak. The rate performance of the Si@NC and Si@H-NC@GC materials is tested (Fig. 4c) by charge and discharge cycles at different current densities. The Si@H-NC@GC nanoarchitecture exhibited higher capacities of 1640, 1500, 1307, 1090, 747, and 533 mA h g−1 at current densities of 0.1, 0.2, 0.5, 1, 2, and 3 A g−1, respectively. Specifically, when the current density returns to 0.1 A g−1, Si@H-NC@GC still has a specific capacity of 1235 mA h g−1, which confirms that Si@H-NC@GC has good rate performance.
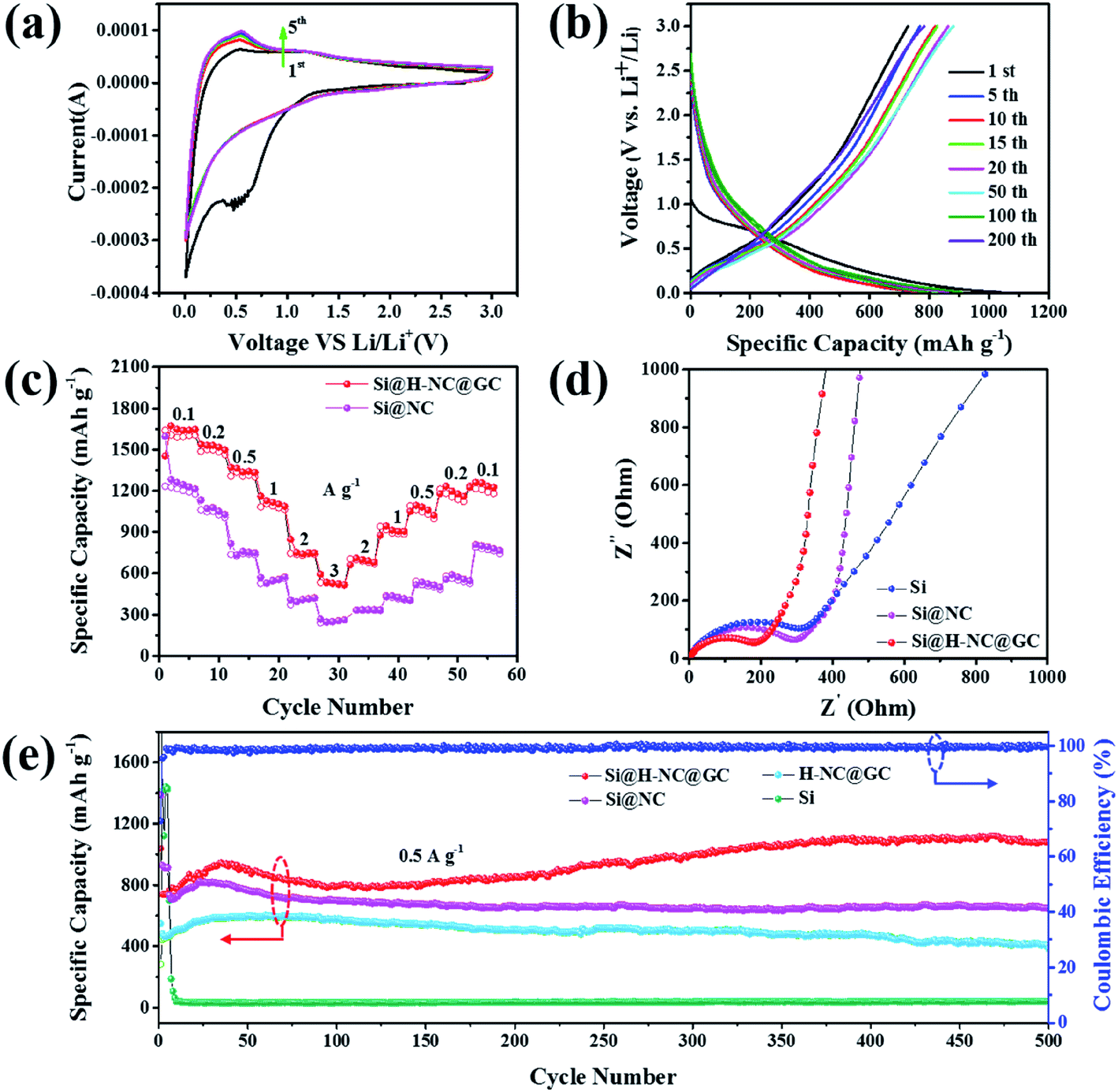 | ||
| Fig. 4 Electrochemical performance of Si@NC and Si@H-NC@GC; (a) CV curves for Si@H-NC@GC at a scan rate of 0.5 mV s−1; (b) galvanostatic charge/discharge curves of Si@H-NC@GC; (c) rate performance of Si@NC and Si@H-NC@GC; (d) Nyquist plots of Si, Si@NC and Si@H-NC@GC composites before cycling; (e) cycling performance of the four electrodes at 0.5 A g−1. | ||
In order to better understand the excellent electrochemical performance of the electrode materials, EIS measurements were performed. As shown in Fig. 4d, the Nyquist plots consist of a high frequency region (semicircle) and a low frequency region (straight line), which are ascribed to the charge transfer process and the lithium ion diffusion behavior of the electrode materials.39 In particular, the lithium ion diffusion resistance and electron transfer resistance of the Si@H-NC@GC nanoarchitectures are significantly reduced compared with those of Si@NC and bare Si, confirming their enhanced electrical conductivity. The corresponding equivalent circuit diagram (Fig. S7a†) of the Si@H-NC@GC material is obtained from the EIS test. The SEI layer resistance (Rs: 24.46 Ω) and charge transfer impedance (Rct: 48.3 Ω) were obtained. It is worth noting that the kinetics of the electrode materials are improved after 500 cycles (Fig. S7b†), demonstrating that the surface of the electrode forms a stable SEI and the electrode structure remains intact during the cycling.29 To further evaluate the lithium storage properties of the Si@H-NC@GC nanoarchitectures, electrochemical cycle performance tests were carried out at different current densities. As shown in Fig. 4e, Si@H-NC@GC exhibits an excellent reversible specific capacity of 1081 mA h g−1 after 500 cycles at a current density of 0.5 A g−1. Also, there is such a phenomenon that the capacity increases as the number of cycles increases. The initial capacity rise is manifested by the continuous activation of the electrode material; after 50 cycles, the Si particles are slightly powdered but the electrode structure is not damaged, resulting in increase in the effective contact areas of the Si/electrolyte and the reversible dissolution/generation of the polymer/gel-like film on the surface of the electrode during the discharge/charge cycles.40,41 In contrast, the Si electrode shows a rapid capacity decay, that is the capacity drops below 100 mA h g−1 after several laps of cycles. Si@NC and H-NC@GC have low reversible specific capacities of 649 and 397 mA h g−1 after 500 cycles at 0.5 A g−1. The difference in the capacity between the materials is mainly due to the poor stability of the structure, and the pure Si electrode without the carbon-coated electrode is destroyed after several cycles. Furthermore, the Si@NC material with a single layer carbon coating does not limit the volume expansion of Si and the formation of a stable SEI on its surface. Si@H-NC@GC maintains a higher reversible capacity of 1052 mA h g−1 (92.5% capacity retention) after 550 cycles at a high current density of 1 A g−1 than Si@H-NC and H-NC@GC electrodes (Fig. S8†), indicating that Si@H-NC@GC can be rapidly charged and discharged. The present work is outstanding compared to the reported literature (see Table S1†).
According to a previous report, the Faraday contribution (pseudocapacitance) of the lithium ion adsorption/desorption process can boost the storage capacity of LIBs at a large current density during the discharge/charge cycles.42 To understand the contribution ratios of the pseudocapacitance and diffusion-controlled capacities to the total storage capacity of the electrode materials, Dunn et al.43 proposed an important method to investigate the distribution of capacitance by testing CV at different scan rates. Fig. 5a shows the CV curves of the Si@H-NC@GC electrode material tested at a sweep rate of 1, 2, 3, 5, and 10 mV s−1. The CV data were analyzed at various sweep rates using the power law formula (eqn (1)):
| i = avb | (1) |
| i(V) = k1v + k2v1/2 | (2) |
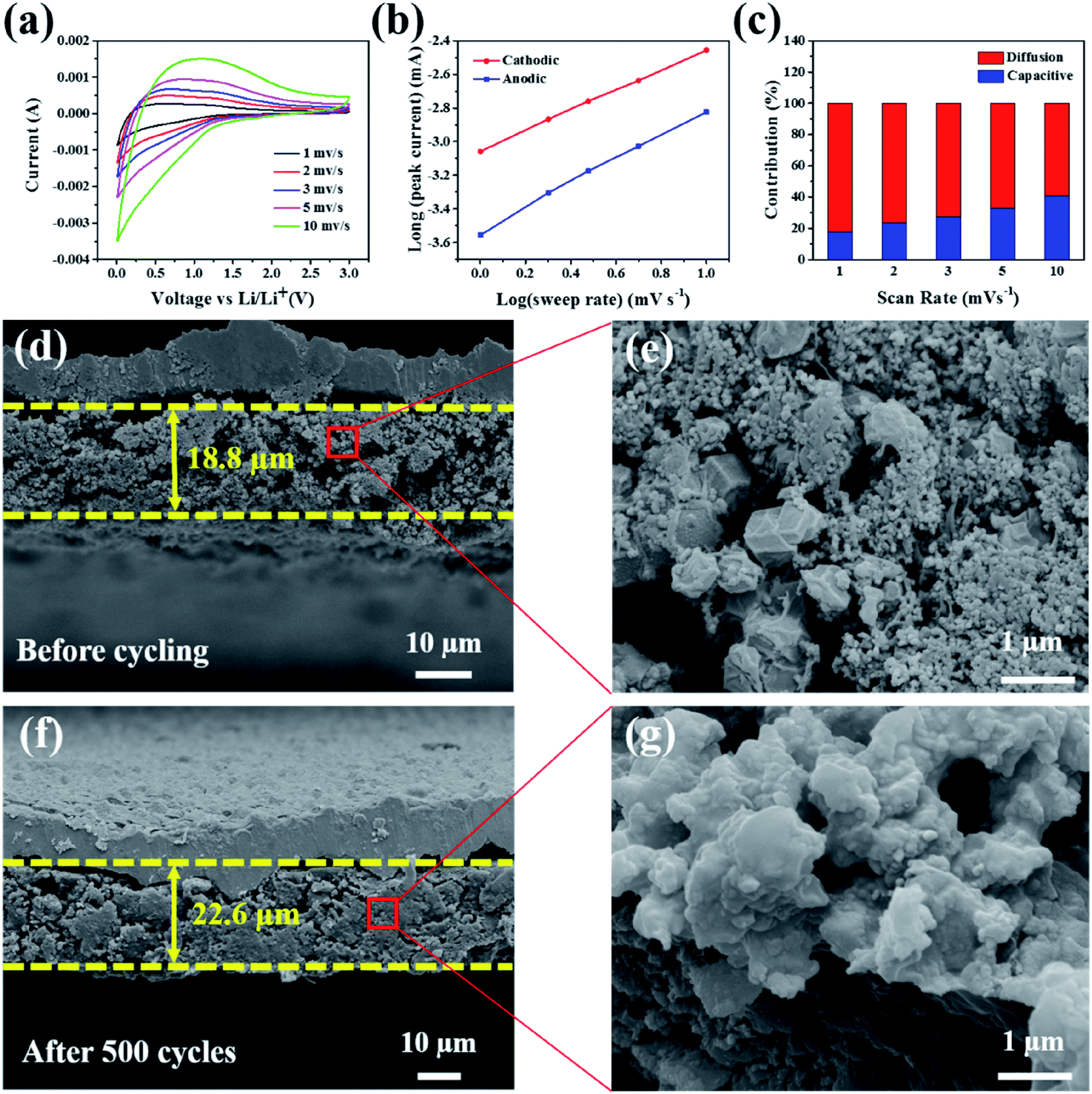 | ||
| Fig. 5 (a) CV curves of the Si@H-NC@GC electrode at different scan rates; (b) log(i) versus log(v) plots for different cathodic/anodic peaks; (c) the contribution ratios of the capacitive (blue) and diffusion-controlled (red) capacities in the Si@H-NC@GC electrode; (d) cross-sectional SEM images of Si@H-NC@GC; (e) partial enlargement of active materials in the Si@H-NC@GC electrode before cycling; (f) cross-sectional SEM images of the Si@H-NC@GC electrode after 500 cycles at a current density of 0.5 A g−1; (g) partial enlargement of the active materials in Si@H-NC@GC after 500 cycles. | ||
As exhibited in Fig. 5c, it is clear that the Si@H-NC@GC electrode demonstrates 18% of the capacitive contribution at a scan rate of 1 mV s−1. As the scan rate increases to 2, 3, 5, and 10 mV s−1, respectively, the contribution of the capacitance gradually increases to 24%, 27%, 33%, and 41%. It is confirmed that the pseudocapacitive behavior plays a non-negligible role in the charge storage process of Si@H-NC@GC electrode materials, especially at large current densities. This is mainly because of the fact that Si@H-NC@GC has a rich specific surface area which can be conducive to the diffusion of lithium-ions and the N-doped carbon has a rich defect that is beneficial to accommodate more lithium.38
To further understand the good integrity of the electrode, the SEM cross-sectional image of the Si@H-NC@GC electrode was obtained before and after 500 cycles at a 0.5 A g−1 current density (Fig. 5d–g). Before and after 500 cycles, the thickness of the electrode active materials is 18.8 μm and 22.6 μm, respectively, and the volume expansion is about 20.2%. However, the SEM cross sectional image of the Si@NC electrode before and after 500 cycles is shown in Fig. S9,† and its volume expansion is about 92.3%. The Si@H-NC@GC electrode has a small volume expansion mainly due to the hollow inner and outer mesoporous carbon coating in the materials, which can effectively accommodate the volume expansion of Si during the lithiation and delithiation process. Besides, the electrode material’s section is partially enlarged, as shown in Fig. 5e and g. The electrode materials have a dodecahedral shape of Si@H-NC@GC before the cycling, and the active materials are tightly connected after the cycling, confirming that the material has good retention integrity. To further verify that a stable SEI was formed on the electrode surface, the electrode surface after 500 cycles at a 0.5 A g−1 current density was observed by SEM (Fig. S10†), revealing that the surface of the Si@H-NC@GC electrode was smooth without obvious cracks.37 However, there are many gullies on the surface of the Si@NC electrode, which is due to the surface rupture to regenerate the SEI. Furthermore, the composition changes on the surface of the Si@H-NC@GC electrode after the cycle were analyzed by XPS (Fig. S11†). The F 1s spectrum shows that there is a strong peak at 685.6 eV corresponding to LiF.47 The two compounds in the P 2p spectrum correspond to fluoride and LiPF6 at 134.6 and 137.2 eV, respectively.48 Fluoride and LiF are the main components of the SEI formed by the decomposition of LiPF6. It was further confirmed that a stable SEI was built on the surface of the Si@H-NC@GC electrode.31 The excellent electrode structure properties are mainly due to the fact that the internal cavity of the Si@H-NC@GC nanoarchitecture can effectively buffer the volume expansion of Si to stabilize the electrode structure, and the outer graphite carbon is favorable for the formation of the SEI.
4. Conclusion
In summary, Si nanoparticles encapsulated in a double-shell hybrid carbon nanocage were successfully synthesized by electrostatic self-assembly, seed-induced growth and heterogeneous shrinkage, to address the volume expansion of Si and create a stable SEI. The obtained Si@H-NC@GC nanoarchitecture has an inner hollow and outer double layer carbon coating (NC inner shells and GC outer shells). This structure has the following advantages: first, hollow N-doped carbon provides effective buffer space for volume expansion of Si during the charging and discharging; second, the graphite carbon outer layer is beneficial in improving the electrical conductivity of the materials and forming a stable SEI on the electrode surface. In addition, the rich specific surface area of the carbon nanocage can greatly improve the diffusion kinetics of lithium ions. Consequently, the Si@H-NC@GC nanoarchitecture delivers high reversible capacities of 1081 mA h g−1 after 500 cycles at 0.5 A g−1 and 92.5% capacity retention after 550 cycles at a high current density of 1 A g−1.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially funded by National Key R&D Program of China (2018YFC1901801) and the National Natural Science Foundation of China (No. U1702251).References
- Q. He, J. Liu, Z.-H. Li, Q. Li, L. Xu, B.-X. Zhang, J.-S. Meng, Y.-Z. Wu and L.-Q. Mai, Small, 2017, 13, 1701504 CrossRef PubMed
.
- X.-T. Hu, W.-J. Qiang and B.-X. Huang, Energy Storage Materials, 2017, 8, 141–146 CrossRef
- F. Luo, D.-T. Ma, Y.-L. Li, H.-W. Mi, P.-X. Zhang and S. Luo, Electrochim. Acta, 2019, 299, 173e181 CrossRef
- H. Yuan, J.-Q. Huang, H.-J. Peng, M.-M. Titirici, R. Xiang, R.-J. Chen, Q.-B. Liu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1802107 CrossRef
- Y.-L. An, H.-F. Fei, G.-F. Zeng, L.-J. Ci, S.-L. Xiong, J.-K. Feng and Y.-T. Qian, ACS Nano, 2018, 12, 4993–5002 CrossRef CAS PubMed
- C.-F. Sun, H. Zhu, M. Okada, K. Gaskell, Y. Inoue, L.-B. Hu and Y.-H. Wang, Nano Lett., 2015, 15, 703–708 CrossRef CAS PubMed
- J.-Y. Li, Q. Xu, G. Li, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Mater. Chem. Front., 2017, 1, 1691–1708 RSC
- X. Chen, P. Hu, J.-W. Xiang, R.-Y. Zhang and Y.-H. Huang, ACS Appl. Energy Mater., 2019, 2, 5214–5218 CrossRef CAS
- N. Liu, Z.-D. Lu, J. Zhao, M. McDowell, H. Lee, W.-T. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed
- Q. Xu, J.-K. Sun, J.-Y. Li, Y.-X. Yin and Y.-G. Guo, Energy Storage Materials, 2018, 12, 54–60 CrossRef
- X.-H. Liu, L. Zhong, S. Huang, S.-X. Mao, T. Zhu and J.-Y. Huang, ACS Nano, 2012, 6(2), 1522–1531 CrossRef CAS PubMed
- X.-L. Li, P.-F. Yan, B.-W. Arey, W. Luo, X.-L. Ji, C.-M. Wang, J. Liu and J.-G. Zhang, Nano Energy, 2016, 20, 68–75 CrossRef CAS
- N. Liu, H. Wu, M.-T. McDowell, Y. Yao, C.-M. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed
- C. Yang, Y.-L. Zhang, J.-H. Zhou, C.-F. Lin, F. Lv, K. Wang, J.-R. Feng, Z.-K. Xu, J.-B. Li and S.-J. Guo, J. Mater. Chem. A, 2018, 6, 8039–8046 RSC
- M. Huang, K. Mi, J.-H. Zhang, H.-L. Liu, T.-T. Yu, A.-H. Yuan, Q.-H. Kong and S.-L. Xiong, J. Mater. Chem. A, 2017, 5, 266–274 RSC
- B. Li, S.-B. Yang, S.-M. Li, B. Wang and J.-H. Liu, Adv. Energy Mater., 2015, 5, 1500289 CrossRef
- H.-W. Mi, X.-D. Yang, Y.-L. Li, P.-X. Zhang and L.-N. Sun, Chem. Eng. J., 2018, 351, 103–109 CrossRef CAS
- H.-W. Mi, Y.-L. Li, P.-Y. Zhu, X.-Y. Chai, L.-N. Sun, H.-T. Zhuo, Q.-L. Zhang, C.-X. He and J.-Y. Liu, J. Mater. Chem. A, 2014, 2, 11254–11260 RSC
- H.-W. Mi, X.-D. Yang, F. Li, X.-Q. Zhuang, C.-X. Chen, Y.-L. Li and P.-X. Zhang, J. Power Sources, 2019, 412, 749–758 CrossRef CAS
- N.-T. Liu, J. Liu, D.-Z. Jia, Y.-D. Huang, J. Luo, X. Mamat, Y. Yu, Y.-M. Dong and G.-Z. Hu, Energy Storage Materials, 2019, 18, 165–173 CrossRef
- L. Zhang, H.-P. Guo, R. Rajagopalan, X.-L. Hu, Y.-H. Huang, S.-X. Dou and H.-K. Liu, J. Mater. Chem. A, 2016, 4, 4056–4061 RSC
- R. Zhao, Z.-B. Liang, R.-Q. Zou and Q. Xu, Joule, 2018, 19, 1–25 Search PubMed
- Y.-Z. Han, P.-F. Qi, S.-W. Li, X. Feng, J.-W. Zhou, H.-W. Li, S.-Y. Su, X.-G. Li and B. Wang, Chem. Commun., 2014, 50, 8057–8060 RSC
- Y.-Z. Han, P.-F. Qi, X. Feng, S.-W. Li, X.-T. Fu, H.-W. Li, Y.-F. Chen, J.-W. Zhou, X.-G. Li and B. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 2178–2182 CrossRef CAS PubMed
- Z.-X. Cai, Z.-L. Wang, J. Kim and Y. Yamauchi, Adv. Mater., 2019, 31, 1804903 CrossRef PubMed
- S.-H. Liu, Z.-Y. Wang, S. Zhou, F.-J. Yu, M.-Z. Yu, C. Y. Chiang, W.-Z. Zhou, J.-J. Zhao and J.-S. Qiu, Adv. Mater., 2017, 29, 1700874 CrossRef PubMed
- J. Tang, R. Salunkhe, J. Liu, N. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed
- S.-Q. Chen, L.-F. Shen, P. Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1605650 CrossRef PubMed
- J.-L. Wu, J.-H. Liu, Z. Wang, X.-Z. Gong, M.-P. Qi and Y. Wang, J. Mater. Chem. A, 2019, 7, 11347–11354 RSC
- Q. Xu, J.-Y. Li, Y.-X. Yin, Y.-M. Kong, Y.-G. Guo and L.-J. Wan, Chem.–Asian J., 2016, 11, 1205–1209 CrossRef CAS PubMed
- G. Huang, F.-F. Zhang, X.-C. Du, Y.-L. Qin, D.-M. Yin and L.-M. Wang, ACS Nano, 2015, 9(2), 1592–1599 CrossRef CAS PubMed
- Y.-L. Liu, C. Hsu, M.-L. Wang and H.-S. Chen, Nanotechnology, 2003, 14(7), 813–819 CrossRef CAS
- B.-R. Wang, W.-W. Li, T. Wu, J. Guo and Z.-Y. Wen, Energy Storage Materials, 2018, 15, 139–147 CrossRef
- H.-B. Wu, S.-Y. Wei, L. Zhang, R. Xu, H.-H. Hng and X.-W. Lou, Chem. – Eur. J., 2013, 19, 10804–10808 CrossRef CAS PubMed
- T. Yoon, T. Bok, C. Kim, Y. Na, S. Park and K.-S. Kim, ACS Nano, 2017, 11, 4808–4815 CrossRef CAS PubMed
- R.-B. Wu, D.-P. Wang, X.-H. Rui, B. Liu, K. Zhou, A. Law, Q.-Y. Yan, J. Wei and Z. Chen, Adv. Mater., 2015, 27, 3038–3044 CrossRef CAS PubMed
- Z. Yi, N. Lin, Y.-Y. Zhao, W.-W. Wang, Y. Qian, Y.-C. Zhu and Y.-T. Qian, Energy Storage Materials, 2019, 17, 93–100 CrossRef
- Y.-Q. Fu, Q.-L. Wei, G.-X. Zhang, X.-M. Wang, J.-H. Zhang, Y.-F. Hu, D.-N. Wang, L. Zuin, T. Zhou, Y.-C. Wu and S.-H. Sun, Adv. Energy Mater., 2018, 1801445 CrossRef
- S.-C. Guo, X. Hu, Y. Hou and Z.-H. Wen, ACS Appl. Mater. Interfaces, 2017, 9, 42084–42092 CrossRef CAS PubMed
- J.-L. Wu, J.-H. Liu, Z. Wang, X.-Z. Gong and Y. Wang, Chem. Eng. J., 2019, 370, 565–572 CrossRef CAS
- J.-J. Deng, X.-L. Yu, X.-Y. Qin, B.-L. Liu, Y.-B. He, B.-H. Li and F.-Y. Kang, Energy Storage Materials, 2018, 11, 184–190 CrossRef
- Y.-T. Chu, L.-Y. Guo, B.-J. Xi, Z.-Y. Feng, F.-F. Wu, Y. Lin, J.-C. Liu, D. Sun, J.-K. Feng, Y.-T. Qian and S.-L. Xiong, Adv. Mater., 2018, 30, 1704244 CrossRef PubMed
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed
- W. Tian, H. Hu, Y.-X. Wang, P. Li, J.-Y. Liu, J.-L. Liu, X.-B. Wang, X.-D. Xu, Z.-T. Li, Q.-S. Zhao, H. Ning, W.-T. Wu and M.-B. Wu, ACS Nano, 2018, 12, 1990–2000 CrossRef CAS PubMed
- J.-L. Liu, J. Wang, C.-H. Xu, H. Jiang, C.-Z. Li, L.-L. Zhang, J.-Y. Lin and Z.-X. Shen, Adv. Sci., 2018, 5, 1700322 CrossRef PubMed
- R.-T. Xu, G. Wang, T.-F. Zhou, Q. Zhang, H.-P. Cong, S. Xin, J. Rao, C.-F. Zhang, Y.-K. Liu, Z.-P. Guo and S.-H. Yu, Nano Energy, 2017, 39, 253–261 CrossRef CAS
- D. Agyeman, K. Song, G.-H. Lee, M. Park and Y.-M. Kang, Adv. Energy Mater., 2016, 6, 1600904 CrossRef
- O. Park, J. Lee, M. Chun, J. Yeon, S. Yoo, S. Choi, N. Choi and S. Park, RSC Adv., 2013, 3, 2538–2542 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00297f |
| This journal is © The Royal Society of Chemistry 2020 |