
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplate-free construction of hollow ZnFe2O4 nanotubes coated with a nano-carbon layer as a competitive anode for Li-ion batteries†
Senyang
Xu
,
Li
Qin
,
Fakhr uz
Zaman
,
Jinyang
Zhang
,
Linrui
Hou
 * and
Changzhou
Yuan
*
* and
Changzhou
Yuan
*
School of Material Science & Engineering, University of Jinan, Jinan, 250022, P. R. China. E-mail: mse_houlr@ujn.edu.cn; mse_yuancz@ujn.edu.cn; ayuancz@163.com
First published on 6th May 2020
Abstract
One-dimensional hollow ZnFe2O4 nanotubes coated with an ultrathin nano-carbon layer are delicately designed and fabricated via a facile template-free strategy and exhibit superior rate behaviors and long-term cycle stability as an anode for Li-ion batteries.
Over the past few decades, Li-ion batteries (LIBs) have attracted enormous attention owing to their high energy density and remarkable power density.1,2 Typically, graphite has been widely utilized in commercial production as a low-cost anode material for LIBs; however, it cannot fully meet the increasingly harsh requirements for higher-energy-density electrochemical devices owing to its low theoretical capacity (∼372 mA h g−1).3–5 It, therefore, has become greatly imperative to explore more promising anode materials with larger high-rate capacity along with superb cycling stability.
Recently, binary ZnFe2O4 (ZFO) has emerged as an attractive anode candidate thanks to its high specific capacity of ∼1072 mA h g−1 originating from both the conversion reaction and alloying mechanism and the cost efficiency/environmental friendliness of both elemental Zn and Fe.6–13 However, its serious volume effect during the charge/discharge process and modest electronic conductivity still need to be efficiently addressed for further commercial applications.2,3,6 Appealingly, numerous pioneering contributions reveal that the electrochemical lithium storage performance of ZnFe2O4 can be considerably improved by rational composition and/or structure design.7–13 In general, hollow structures, particularly one-dimensional (1D) hollow nanotubular architectures with rapid electronic transport, can well alleviate the volumetric expansion/shrinkage during the repeated Li+-insertion/extraction processes,5–11 which is conducive to improving the structural stability. Besides this, the elegant hybridization with carboneous materials will efficiently enhance the charge conductivity and sur/interface stability of electrode materials.6,12,13 Unfortunately, if each method mentioned above was employed alone, only a limited improvement in electrochemical Li-storage properties could be achieved. As a result, integrated avenues are always necessary to construct advanced ZFO-based hybrid anodes, especially via facile synthetic strategies that require no special equipment and stringent preparation conditions. This is undoubtedly of great significance for broad and successful industrial applications.
Herein, we report for the first time a simple, template-free fabrication of hollow ZFO nanotubes (NTs) coated with a nano-carbon coating layer (denoted as ZFO@C NTs) (more details in the experimental section, see the ESI†), as schematically illustrated in Scheme 1. Specifically, the ZnFe-PVP nanofiber precursor (Fig. S1, ESI†) was first electrospun at 25 °C and further annealed in air at 500 °C for 5 h. The resulting ZFO NTs were further coated with polydopamine (PDA). After annealing at 500 °C in N2, the ZFO@C NTs were finally achieved. As expected, when employed as an anode for LIBs, the as-fabricated hollow ZFO@C NTs exhibited even better lithium-storage behaviors in terms of reversible capacities, rate performance and cycling stability, compared to their counterpart ZFO NTs, thanks to synergistic contributions from the 1D hollow structure and ultrathin conductive carbon coating layer.
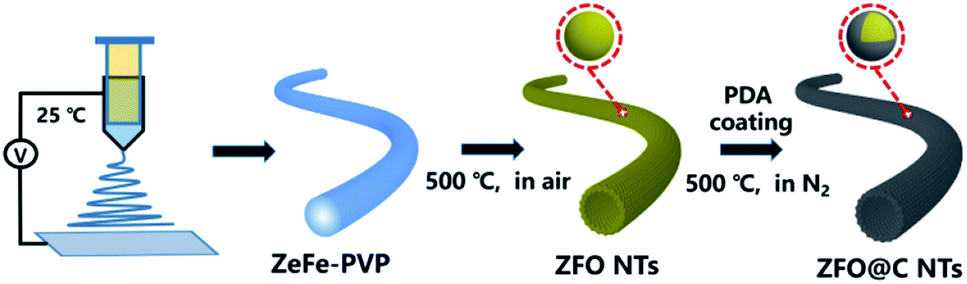 | ||
| Scheme 1 Schematic illustration of the template-free synthesis of ZFO@C NTs. | ||
X-ray diffraction (XRD) analysis was conducted to determine the crystallographic phases of the resulting ZFO and ZFO@C NTs. As shown in Fig. 1a, all the discernible diffraction peaks of the synthesized ZFO NTs (the lower panel), including not only the peak positions but also their relative intensities, can be perfectly indexed to the cubic spinel ZFO (JCPDS No. 22-1012) with the space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. As regards the ZFO@C NTs, the reflections (the upper panel) all remain the same as those of the ZFO NTs (the lower panel), indicating that the carbon coating has no obvious effect on the ZFO phase at all. One should note that the undetected representative signals for C further verify the amorphous nature of the conductive carbon in the hybrid NTs. The corresponding Raman spectrum (Fig. S2, ESI†) further evidences the existence of typical carbon with an ID/IG value (i.e., the areal ratio of the D-band to the G-band) of ∼1.19.
m. As regards the ZFO@C NTs, the reflections (the upper panel) all remain the same as those of the ZFO NTs (the lower panel), indicating that the carbon coating has no obvious effect on the ZFO phase at all. One should note that the undetected representative signals for C further verify the amorphous nature of the conductive carbon in the hybrid NTs. The corresponding Raman spectrum (Fig. S2, ESI†) further evidences the existence of typical carbon with an ID/IG value (i.e., the areal ratio of the D-band to the G-band) of ∼1.19.
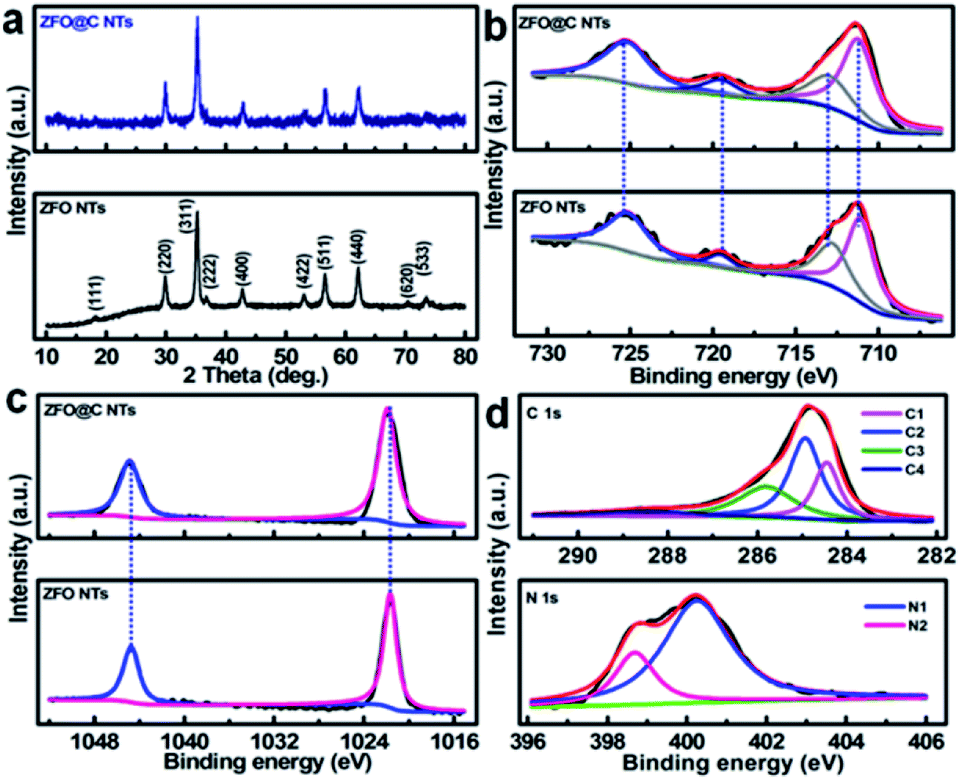 | ||
| Fig. 1 (a) XRD patterns and XPS elemental (b) Fe 2p and (c) Zn 2p spectra of ZFO and ZFO@C NTs; (d) typical C 1s (the upper panel) and N 1s (the lower panel) spectra of ZFO@C NTs. | ||
In order to gain more information about the surface elemental compositions and corresponding oxidation states of the two obtained products, X-ray photoelectron spectroscopy (XPS) was performed. Fig. 1b shows a comparison of the Fe 2p spectra and fitting curves of ZFO and ZFO@C NTs. Clearly, there is no significant difference in the bond energy (BE) between the two. The fitted peaks with BEs at ∼711.4 and ∼713.0 eV correspond to the characteristic peaks of Fe 2p3/2 in the tetrahedron (A-site) and octahedron (B-site), respectively, while those at ∼719.6 and ∼725.5 eV are related to the satellite peak and Fe 2p1/2, respectively, indicating the existence of Fe3+ in the ZFO phase.5,6,12 In the high-resolution Zn 2p spectra (Fig. 1c), the fitted peaks at BEs of ∼1021.7 and ∼1044.8 eV are ascribed to Zn 2p3/2 and 2p1/2, respectively, confirming that elemental Zn exists in the two samples in the +2 oxidation state.5,6,14Fig. 1d demonstrates the core-level XPS spectra of C 1s (the upper panel) and N 1s (the lower panel) in the ZFO@C NTs. The presence of four types of carbon can be observed. To be specific, the peaks at ∼284.5 and ∼285.0 eV are assigned to carbon in the form of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (C1) and C–C (C2), along with two other peaks at ∼286.2 and ∼288.9 eV ascribed to C–O (C3) and O–CO (C4), respectively.12 N (∼2.1 at%) is present in two forms, i.e., the quaternary N (∼400.3 eV, N1, ∼1.6 at%) and pyridinic N (∼398.7 eV, N2).6 As noted, the former favors enhanced electronic transport through the 1D ZFO@C NTs.6
C (C1) and C–C (C2), along with two other peaks at ∼286.2 and ∼288.9 eV ascribed to C–O (C3) and O–CO (C4), respectively.12 N (∼2.1 at%) is present in two forms, i.e., the quaternary N (∼400.3 eV, N1, ∼1.6 at%) and pyridinic N (∼398.7 eV, N2).6 As noted, the former favors enhanced electronic transport through the 1D ZFO@C NTs.6
Typical field emission scanning electron microscope (FESEM) observations (Fig. 2a–c) clearly visualize a typical porous tubular structure of ZFO NTs, which are constructed with many nanoscale subunits. There is no doubt that the porous structure will facilitate the subsequent coating of each ZFO nano-grain uniformly with PDA. The hollow architecture of ZFO NTs is further confirmed by the transmission electron microscope (TEM) images (Fig. 2d, e). It can be seen from the higher-magnification TEM micrograph (the inset in Fig. 2e) that the outer diameter of ZFO NTs is approximately 110 nm, along with a thin wall of ∼12 nm in thickness. The unique ZFO NTs are typically composed of well-crystallized nano-dimensional particles with a diameter of ∼7 nm and even less (Fig. 2f). From the high-resolution TEM (HRTEM) images (Fig. 2f, g), two sets of well-defined lattice fringes with spacings of 0.49 and 0.3 nm, corresponding to the (111) and (220) planes of the spinel ZFO, respectively, can be apparently discerned. The selected area electron diffraction (SAED) pattern with multiple concentric rings confirms the polycrystalline features of ZFO NTs (Fig. 2h). The template-free formation of the unique hollow ZFO NTs mainly depends upon the applied electrospinning process. The easy volatilization of the involved ethanol solvent will result in the spontaneous outward movement of Zn2+/Fe2+ ions along the radial direction of the resultant ZnFe-PVP fibers. It is the concentration difference of metal ions that ensures the formation of NTs, along with the assistance of the subsequent calcination in air.15
 | ||
| Fig. 2 (a–c) FESEM, (d, e) TEM and (f, g) HRTEM images and (h) SAED pattern of the ZFO NTs; (i) TEM, (j, k) HRTEM, (l) STEM and corresponding EDX elemental mapping images of the ZFO@C NTs. | ||
After the subsequent carbon coating, the ZFO@C NTs well inherit the tubular structure of ZFO NTs (Fig. 2i). The enlarged TEM observation (Fig. 2j) shows that the surface of the ZFO NTs is covered evenly with an ultrathin nano-carbon layer with a thickness of about 3–4 nm. After amorphous carbon coating, the lattice fringe with a spacing of 0.3 nm, corresponding to the (220) crystal plane of ZFO, is still distinct (Fig. 2k). The scanning TEM (STEM) and corresponding elemental energy-dispersive X-ray (EDX) mapping images (Fig. 2l) explicitly corroborate the uniform distributions of Zn, Fe and O elements in ZFO@C NTs, along with the C species mainly distributed outside the ZFO NTs.
In order to further study the pore structure and textural properties of the as-synthesized ZFO and ZFO@C NTs, nitrogen sorption tests were conducted accordingly. Typical N2 sorption isotherms show that both curves can be classified as type IV with an H3 type hysteresis loop (Fig. S3a, ESI†),2,6,11 suggesting mesoporous characteristics of ZFO and ZFO@C NTs, which can also be supported by their Barrett–Joyner–Halenda pore size distribution (PSD) plots (Fig. S3b, ESI†). In particular, the PSD plot of the ZFO NTs distinctly shifts to the smaller size range after carbon coating, verifying that the PDA-derived carbon is also located in the inherent pores of the resulting porous ZFO NTs. As a result, a pore volume (∼0.15 cm3 g−1) and average pore size (∼15.2 nm) smaller than those of ZFO NTs (Table S1, ESI†) are obtained in the ZFO@C NTs. This consequently endows the ZFO@C NTs with a specific surface area of ∼38.5 m2 g−1, somewhat higher than that of ZFO NTs (∼34.9 m2 g−1). This unique mesoporous nanotubular architecture will promote the electroactive sur/interfaces between the electrode and electrolyte and also provide enough space to accommodate huge volumetric changes over repeated lithiation/delithiation processes, particularly at high rates.6,16,17
Thanks to these attractive structural merits, as discussed above, our ZFO@C NTs can be highly anticipated to be an advanced anode for LIBs. A coin cell (CR2032) was assembled to evaluate the half-cell electrochemical behaviors of ZFO and ZFO@C NTs. The cyclic voltammetry (CV) curves of ZFO@C NTs were obtained in the voltage range of 0.01–3.0 V (vs. Li/Li+) at a sweep rate of 0.1 mV s−1 (Fig. S4, ESI†). Apparently, the shape of the first cycle is completely different from that of the subsequent cycles. In the 1st cycle, a weak cathodic peak around 0.89 V can be observed, followed by another intense peak centered at 0.61 V. These peaks correspond to the structural transformation and redox reactions that occur during potential scanning, including the formation of ZnxFeyO, and further reductive conversion into Fe0, LiZn and Li2O.5,12,15–18 For the anodic process, the broad peaks at ∼1.61 and ∼1.92 V correspond to the oxidation of Zn0/Fe0 to Zn2+/Fe3+. The well-overlapping curves of the 2nd and 3rd cycles suggest identical yet highly reversible electrochemical reactions between Fe(III)/Zn(II) and Fe(0)/Zn(0).5,15–18 Remarkably, the ZFO@C NTs exhibit large initial discharge/charge capacities of ∼1176/∼912 mA h g−1 at a current density of 300 mA g−1, corresponding to an initial coulombic efficiency (CE) of ∼77.6%, as shown in Fig. 3a. By contrast, the ZFO NTs exhibit a discharge capacity of only ∼999 mA h g−1, along with the 1st CE value of ∼72.7%. The irreversible capacity loss should be mainly ascribed to the electrolyte decomposition and formation of the solid-electrolyte-interphase (SEI) layer.5–13,27 In the 2nd charge–discharge cycle (Fig. S5, ESI†), the CE value of the ZFO@C NTs rapidly increases up to ∼97.4%, still larger than that of the ZFO NTs (∼97.1%). Fig. 3b displays the rate performance of ZFO and ZFO@C NTs at various current densities from 200 to 3200 mA h g−1. As the current density increases, the capacity of ZFO NTs decays significantly. Competitively, the ZFO@C NTs exhibit excellent rate behaviors with discharge specific capacities of ∼904, ∼836, ∼763, ∼664, and ∼539 mA h g−1, as shown in Fig. 3c, at current densities of 200, 400, 800, 1600 and 3200 mA g−1, respectively. However, the ZFO NTs only obtain reversible capacities of ∼813 and ∼240 mA h g−1 at 200 and 3200 mA g−1, respectively. Additionally, as the current density returns to 100 mA g−1, a capacity as large as ∼1013 mA h g−1 can still be recovered for the ZFO@C NTs, even higher than that of ZFO NTs (∼537 mA h g−1) in the same case. Evidently, the capacities at each current density are all larger than and/or comparable to those of other ZFO-C composites (the lower panel in Fig. 3c)10,14,19–26,28 and even ZFO/conducting polymer composites5,29,30 and ZFO nanostructures.31,32 It is worthy of note that the ZFO@C NTs possess a striking high-rate-dischargeability (HRD), which is defined as the ratio of the discharge capacity at a certain current density to that at 200 mA g−1. Obviously, the HRD of ZFO@C NTs remains at ∼60%, much higher than that of ZFO NTs (∼30%) at a larger current density of 3200 mA g−1. This highlights the remarkable rate properties of ZFO@C NTs for potential high-power applications, which should be reasonably attributed to their smaller charge transfer resistance (Fig. S6, ESI†). Furthermore, only a reversible discharge capacity of ∼479.3 mA h g−1 can be maintained by ZFO NTs after 500 consecutive cycles at a current density of 500 mA g−1. In sharp contrast, the ZFO@C NTs demonstrate superior cycling performance with a retained discharge capacity of ∼817.6 mA h g−1, revealing their exceptional electrochemical stability. As seen by TEM observation (Fig. S7, ESI†), the cycled ZFO@C NT anode maintains the 1D hollow nano-tubular structure well, demonstrating its robust structural stability for better long-duration cycle life.
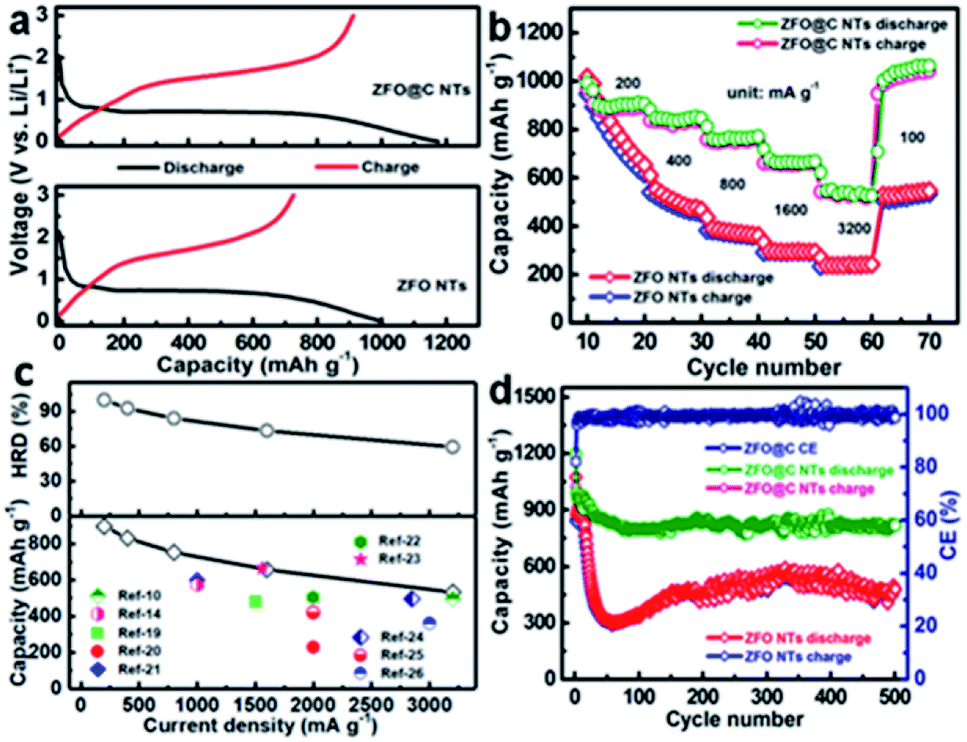 | ||
| Fig. 3 Electrochemical evaluation of ZFO and ZFO@C NTs: (a) the first discharge–charge plots (300 mA g−1); (b) rate behaviors in the current range from 200 to 3200 mA g−1; (c) discharge capacities and HRD as a function of current density; and (d) cycling behaviors at 500 mA g−1. | ||
In summary, in this work, we devised a bottom-up template-free method towards scalable fabrication of 1D ZFO NTs for the first time, and then coated them with a PDA-derived ultrathin nano-carbon layer to obtain the hollow ZFO@C NTs. The underlying formation process of the hollow ZFO NTs was tentatively proposed here. Thanks to its unique 1D hollow mesoporous architecture, robust structural stability, and small charge-transfer resistance, the as-synthesized ZFO@C NT electrode displayed remarkable electrochemical properties in terms of reversible capacity, rate behaviors and long-term cycle lifespan, particularly at large current densities, when utilized as a competitive anode for LIBs. Our contribution here will provide better guidance for future rational design of hollow binary and even ternary metal oxide NTs with a conducting carbon coating for advanced LIBs and beyond.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (No. 51772127 and 51772131), Taishan Scholars (No. ts201712050), Major Program of Shandong Province Natural Science Foundation (ZR2018ZB0317) and Collaborative Innovation Center of Technology and Equipment for Biological Diagnosis and Therapy in Universities of Shandong.Notes and references
- Y. Liu, Z. H. Sun, X. Sun, Y. Lin, K. Tan, J. F. Sun, L. W. Liang, L. R. Hou and C. Z. Yuan, Angew. Chem., Int. Ed., 2020, 59, 2473 CrossRef CAS PubMed.
- M. B. Zheng, H. Tang, L. L. Li, Q. Hu, L. Zhang, H. G. Xue and H. Pang, Adv. Sci., 2018, 5, 1700592 CrossRef PubMed.
- W. S. Weng, J. Lin, Y. C. Du, X. F. Ge, X. S. Zhou and J. C. Bao, J. Mater. Chem. A, 2018, 6, 10168 RSC.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. D. Chen, Chem. Soc. Rev., 2015, 44, 5926 RSC.
- L. R. Hou, R. Q. Bao, D. K. Denis, X. Sun, J. Y. Zhang, F. U. Zaman and C. Z. Yuan, Electrochim. Acta, 2019, 306, 198 CrossRef CAS.
- C. Z. Yuan, H. Cao, S. Q. Zu, H. Hua and L. R. Hou, J. Mater. Chem. A, 2015, 3, 20389 RSC.
- Z. B. Fang, L. N. Zhang, H. Qi, H. J. Yue, T. Zhang, X. S. Zhao, G. Chen, Y. J. Wei, C. Z. Wang and D. Zhang, J. Power Sources, 2018, 762, 480 CAS.
- C. Yan, G. Chen, X. Zhou, J. Sun and C. Lv, Adv. Funct. Mater., 2016, 26, 1428 CrossRef CAS.
- W. Liu, L. Xu, K. Sheng, C. Chen, X. Y. Zhou, B. Dong, X. Bai, S. Zhang, G. Y. Lu and H. W. Song, J. Mater. Chem. A, 2018, 6, 10976 RSC.
- X. J. Wang, Y. Li, T. Jin, J. Meng, L. F. Jiao, M. Zhu and J. Chen, Nano Lett., 2017, 17, 7989 CrossRef CAS PubMed.
- C. J. Niu, J. S. Meng, X. P. Wang, C. H. Han, M. Y. Yan, K. G. Zhao, X. M. Xu, W. H. Ren, Y. L. Zhao, L. Xu, Q. J. Zhang, D. Y. Zhao and L. Q. Mai, Nat. Commun., 2015, 6, 7402 CrossRef PubMed.
- D. Cai, B. Qu, Q. Li, H. Zhan and T. Wang, J. Alloys Compd., 2017, 716, 30 CrossRef CAS.
- Z. J. Jiang, S. Cheng, H. B. Rong, Z. Q. Jiang and J. L. Huang, J. Mater. Chem. A, 2017, 5, 23641 RSC.
- L. H. Zhang, T. Wei, J. Q. Yue, L. Z. Sheng, Z. M. Jiang, D. R. Yang, L. B. Yuan and Z. J. Fan, J. Mater. Chem. A, 2017, 5, 11188 RSC.
- W. Luo, X. L. Hu, Y. M. Sun and Y. H. Huang, J. Mater. Chem., 2012, 22, 8916 RSC.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622 CrossRef CAS PubMed.
- L. R. Hou, L. Lian, L. H. Zhang, G. Pang, C. Z. Yuan and X. G. Zhang, Adv. Funct. Mater., 2015, 25, 238 CrossRef CAS.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999 RSC.
- L. R. Hou, R. Q. Bao, Y. R. Zhang, X. Sun, J. Y. Zhang, H. Dou, X. G. Zhang and C. Z. Yuan, J. Mater. Chem. A, 2018, 6, 17947 RSC.
- J. J. Cai, C. Wu, Y. Zhu, P. K. Shen and K. L. Zhang, Electrochim. Acta, 2016, 187, 584 CrossRef CAS.
- L. M. Yao, X. H. Hou, S. J. Hu, J. Wang, M. Li, C. Su, M. O. Tade, Z. P. Shao and X. Liu, J. Power Sources, 2014, 258, 305 CrossRef CAS.
- X. Y. Yao, J. H. Kong, C. Y. Zhao, D. Zhou, R. Zhou and X. H. Lu, Electrochim. Acta, 2014, 146, 464 CrossRef CAS.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513 CrossRef CAS.
- R. M. Thankachan, M. M. Rahman, I. Sultana, A. M. Glushenkov, S. Thomas, N. Kalarikkal and Y. Chen, J. Power Sources, 2015, 282, 462 CrossRef CAS.
- C. Y. Yang, M. Q. Sun, L. Zhang, P. Y. Liu, P. Wang and H. B. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 14713 CrossRef CAS PubMed.
- D. Das, A. Mitra, S. Jena, S. B. Majumder and R. N. Basu, ACS Sustainable Chem. Eng., 2018, 6, 17000 CrossRef CAS.
- S. S. Zhang, InfoMat, 2019 DOI:10.1002inf2.12058.
- B. B. Jiang, C. P. Han, B. Li, Y. J. He and Z. Q. Lin, ACS Nano, 2016, 10, 2728 CrossRef CAS PubMed.
- H. Qiao, R. Li, Y. Yu, Z. Xia, L. Wang, Q. Wei, K. Chen and Q. Qiao, Electrochim. Acta, 2018, 273, 282 CrossRef CAS.
- X. R. Sun, H. W. Zhang, L. Zhou, X. D. Huang and C. Z. Yu, Small, 2016, 12, 3732 CrossRef CAS PubMed.
- L. L. Peng, Z. W. Fang, J. Li, L. Wang, A. M. Bruk, Y. Zhu, Y. M. Zhang, K. J. Takeuchi, A. C. Marschilok, E. A. Stach, E. S. Takeuchi and G. H. Yu, ACS Nano, 2018, 12, 820 CrossRef CAS PubMed.
- Y. J. Gao, L. Yin, S. J. Kim, H. Yang, I. Jeon, J. P. Kim, S. Y. Jeong, H. W. Lee and C. R. Cho, Electrochim. Acta, 2019, 296, 565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00294a |
| This journal is © The Royal Society of Chemistry 2020 |