
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV-induced syntheses of surfactant-free precious metal nanoparticles in alkaline methanol and ethanol†
Jonathan
Quinson‡
 *a,
Laura
Kacenauskaite‡
a,
Johanna
Schröder
b,
Søren B.
Simonsen
c,
Luise
Theil Kuhn
c,
Tom
Vosch
a and
Matthias
Arenz
*b
*a,
Laura
Kacenauskaite‡
a,
Johanna
Schröder
b,
Søren B.
Simonsen
c,
Luise
Theil Kuhn
c,
Tom
Vosch
a and
Matthias
Arenz
*b
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: jonathan.quinson@chem.ku.dk
bDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland
cDepartment of Energy Conversion and Storage, Technical University of Denmark, Fysikvej Bldg. 310, DK-2800 Kgs. Lyngby, Denmark
First published on 11th May 2020
Abstract
Surfactant-free UV-induced syntheses of Pt and Ir nanoparticles in alkaline methanol and ethanol are presented. Small size nanoparticles ca. 2 nm in diameter are obtained without surfactants in a wide range of base concentration.
Introduction
Nanoparticles (NPs) are key catalysts in many chemical processes.1 In particular, precious metal NPs are in high demand for automotive,2 energy3 or environmental applications.4 To best address specific needs and tailor specific features, simple synthesis strategies are crucial. In particular, the synthesis of NPs <10 nm in diameter made of precious metals like Pt or Ir plays a key role in catalysis.5,6 Size control at the nanoscale is important to tune catalyst properties like activity,3 selectivity7 and stability.3 In most colloidal syntheses, surfactants are used to achieve size control, stabilize the NPs and avoid agglomeration.8 The later however need to be removed for catalytic applications9 which is often performed by approaches only partially successful. In this respect surfactant-free syntheses of nanomaterials10 and colloids bear promising features.11A recently reported wet chemical surfactant-free synthesis is suitable to obtain precious metal NPs with superior catalytic activity.12–14 This was for instance demonstrated with Pt NPs for hydrogenation reaction and Ir NPs for the oxygen evolution reaction.12,15 The synthesis is performed at low temperature (<80 °C) and does not require harsh chemicals. Only a mono-alcohol (e.g. methanol or ethanol), a metal complex (e.g. H2PtCl6 or IrCl3) and a base (e.g. NaOH) are needed. So far, mainly microwave assisted synthesis has been studied and a strong focus was given to methanol as solvent. To finely tune the size of the NPs, we study here the UV-induced synthesis of Pt and Ir NPs in alkaline methanol and ethanol and compare the results with a micro-wave oven synthesis previously reported using alkaline methanol as solvent.12 The general approach is illustrated in Fig. 1a. Specific attention is given to NP size control and the stability of the colloids obtained without surfactants. The size of the NPs was evaluated by transmission electron microscopy (TEM). Insight into the reaction mechanism in different solvents is given by Fourier-transform infra-red absorption spectroscopy (FTIR) and head space gas chromatography coupled to mass spectrometry (GC-MS).
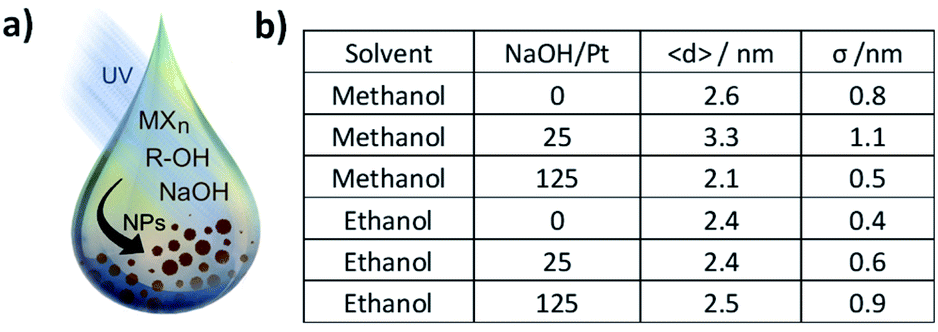 | ||
| Fig. 1 (a) Schematic of the UV-induced synthesis of Pt NPs by the mono-alcohol process. MXn represents a molecular precious metal precursor, R–OH represent a mono-alcohol solvent. (b) Average diameter 〈d〉 and standard deviation σ, obtained for different solvents and different NaOH concentration for 2 mM H2PtCl6. | ||
Results
We recently investigated the use of UV-light16,17 to obtain surfactant-free precious metal NPs like Pt, based on the polyol synthesis using ethylene glycol as solvent.18–20 UV-light is used in this context to induce the reduction of a metal complex like H2Pt(IV)Cl6 or Ir(III)Cl3 to metallic Pt(0) or Ir(0) NPs, respectively.13,21 A simple strategy to achieve size control in the surfactant-free synthesis of platinum NPs is to vary the concentration of NaOH or more precisely the NaOH/Pt molar ratio.11 To compare the mono- with the poly-alcohol process, we performed experiments in methanol and ethanol as solvents for the same NaOH concentrations and H2PtCl6 concentration as previous studies with UV-induced synthesis in alkaline ethylene glycol.16,17 This allows to investigate the effects of the different solvents for the UV-induced synthesis of Pt NPs under alkaline conditions.First, the influence of solvent and NaOH/Pt molar ratio on the size control was studied. The size obtained in methanol and ethanol is summarised in Fig. 1b. 2 mM of H2PtCl6 and concentration of NaOH of 0, 50 and 250 mM were used. If methanol is used as solvent, the average diameter size is 2.6 ± 0.8 nm, 3.3 ± 1.1 nm and 2.1 ± 0.5 nm respectively. If ethanol is used as solvent, the effect of NaOH/Pt ratio on the size is less pronounced yielding NPs with diameter of 2.4 ± 0.4 nm, 2.4 ± 0.6 nm and 2.5 ± 0.9 nm, respectively.
The weak effect of the NaOH/Pt molar ratio on size control is in agreement with previous reports for the mono-alcohol process used there but performed using a microwave synthesis.12,13 It is however in contrast with the UV-induced polyol process where the NaOH/Pt molar ratio leads to a clear size control: as the NaOH/Pt molar ratio increases, the size and size distribution decreases in alkaline ethylene glycol.11,16,17
Second, the general formation mechanism of the NPs was investigated to address these differences. The reduction of Pt(IV) in H2PtCl6 typically proceeds first via a reduction to Pt(II), followed by further reduction to Pt(0) and formation of NPs while the solvent or sacrificial agents get oxidised.22,23 Head-space GC analysis, see Fig. S1,† confirms that during the reaction, oxidation products of methanol like formic acid (in absence of NaOH), methyl formate (in presence of NaOH) and CO2 are detected. These products are consistent degradation products of formaldehyde expected to be the main product formed under the photo-degradation of H2PtCl6 in methanol, as previously reported without using a base.23 In ethanol, whether or not a base is used, the same products like acetaldehyde are detected. In parallel to the nucleation and formation of NPs, ethanol undergoes further chemical reactions evidenced by fluorescence measurements whereas methanol remains free of fluorescent species after the NPs are formed, see Fig. S2.† In respect to the formation of fluorescent species alkaline methanol seems to be a ‘cleaner’ solvent since less side reactions seem to occur.
Another important property of colloidal NPs is their stability. Without a base and for microwave synthesis, large agglomerates are formed in the mono-alcohols upon reduction of H2PtCl6. In alternative syntheses, for instance in ethanol–water mixture,24 a deposition of the Pt NPs on the cell walls is observed and no colloidal dispersions are obtained if no surfactants are used. In contrast here, using UV-induced synthesis, small size NPs with a diameter around 2.5 nm are obtained in absence of NaOH and in absence of surfactant, see Fig. 2. The size of the NPs is in agreement with NPs observed in a recent study focusing on the reduction of H2PtCl6 with methanol under UV radiation.23 More importantly, the NPs obtained here are stable as colloids. This is attributed to the milder conditions (versus microwave synthesis) for the reaction to proceed and the relatively low concentration of H2PtCl6 used.
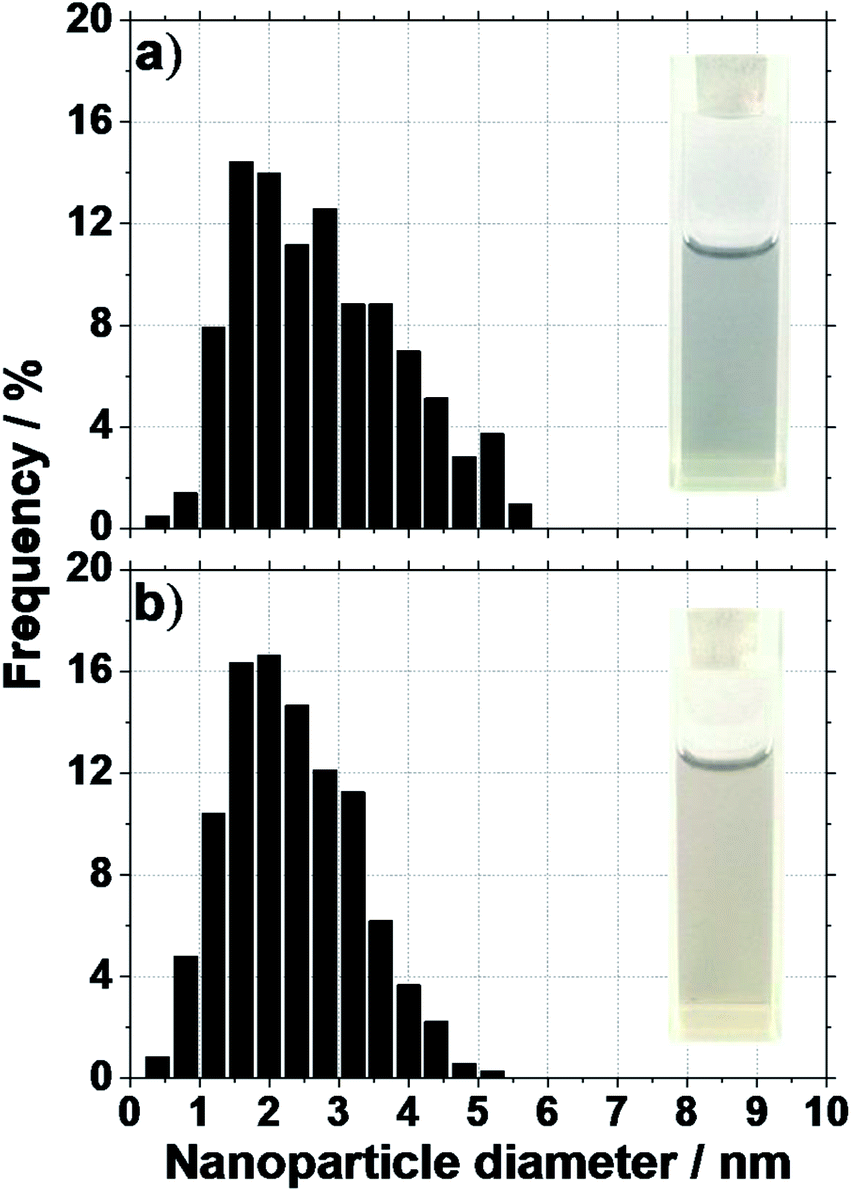 | ||
| Fig. 2 Size distribution of Pt NPs obtained after 2 hours UV irradiation of a solution of 2 mM H2PtCl6 without NaOH in (a) methanol and (b) ethanol. Images of the solution after UV-irradiation are also displayed (volume is 3 mL). | ||
In alkaline conditions, stable colloids are also obtained. In agreement with previous reports in poly-25 or mono-alcohols,13 electrostatic stabilization of the surfactant-free NPs e.g. by OH− groups account for the colloidal stability. The photo-reduction of H2PtCl6 to size controlled and stable colloidal NPs by mono-alcohols is typically performed and studied in aqueous media, in presence of photo-activators or polymers24,26–28 and typically without adding a base. The need for additives under these conditions to initialize the photo-reaction or to stabilize the colloids is largely stressed in the literature.24,26 However additives are typically detrimental for further applications e.g. in catalysis.29 Surfactants need to be removed in typically hazardous and time consuming steps.8 It is therefore a significant achievement to obtain surfactant-free colloidal NPs by photo-induced synthesis in alcohol-only solutions.
The NPs prepared are conveniently ‘unprotected’ in the meaning of previous terminology.25 This implies that in the surfactant-free synthesis that is the mono-alcohol process, the resulting NPs are functionalized with CO and OH/OH− groups. FTIR characterization reveals that the methanol synthesized NPs express CO functional groups (clear absorption peak between 2000-2100 cm−1) whereas the ethanol based synthesis do not show a strong signal, even though a small signal appearing as ‘bump’ just below 2000 cm−1 could indicate some CO species, see Fig. 3a. This is attributed to the more facile oxidation of the one-carbon molecule that is methanol to CO, compared to the formation of CO from the two carbon molecule that is ethanol. A pronounced shift of the CO peak position in methanol-prepared NPs towards larger wavenumber is observed: shift from 2030 cm−1 with NaOH to 2086 cm−1 without NaOH. This shift is consistent with a larger NP size25,30 achieved in absence of a base and consistent with TEM characterization.
 | ||
| Fig. 3 (a) IR absorption spectra of Pt NP suspensions obtained with 2 mM H2PtCl6 without and with 250 mM NaOH in methanol and ethanol as indicated. (b–e) TEM micrographs of the Pt NPs in methanol (b) without and (c) with 250 mM NaOH and in ethanol (d) without and (e) with 250 mM NaOH. | ||
A different surface functionalization accounts for more stable colloidal solutions of Pt NPs obtained in methanol versus ethanol. Since the NPs obtained in ethanol are less ‘protected’ by CO groups they show less stability and a direct consequence is that they tend to agglomerate. An indirect effect of this different surface functionalization is observed during the preparation of TEM grids where NPs prepared in ethanol tend to form large aggregates (made of individual NPs), see Fig. 3d and e. The NPs obtained in methanol tend to be more isolated and individual on the TEM grid, see Fig. 3b and c. The NPs are also protected by OH− groups due to the alkaline conditions. As a consequence, the NPs appear more distant from each other on the TEM grids when prepared in alkaline conditions, Fig. 3b and c.
At last, size control is an important feature to take into account to design synthesis strategies. An option to achieve size control in the mono-alcohol process is to add water to the reaction mixture.12 Using a microwave reactor, adding water in the range 0–80% (volume%) lead to a size increase from ca. 2 nm to ca. 5 nm.12 This size control is more easily achieved at high (e.g. 2.5 mM) H2PtCl6 concentration but difficult to probe at low (e.g. 0.5 mM) H2PtCl6 concentration: under microwave synthesis the solvent quickly reaches reflux conditions and a fine size control is challenging to achieve.
The UV-induced synthesis differs in that respect. Results presented in Fig. 4 were obtained using 0.5 mM H2PtCl6 and 10 mM NaOH. Different volume fraction of (1) 0%, (2) 20%, (3) 50%, (4) 80% and (5) 100% H2O in methanol are used. The pictures of the solutions before and after exposure time of 2 hours under UV-light, see Fig. 4a and b respectively. For 0%, 20% and 50% water the solutions turn brown upon UV-irradiation indicative of successful NP formation. The reaction mixture containing 80% is not as brown as for the other syntheses. For 100% water the reaction does not proceed. This can be attributed to the need for a suitable alcohol–Pt ratio in order to perform the reduction of the salt while the alcohol gets oxidized for the synthesis to proceed.13,14
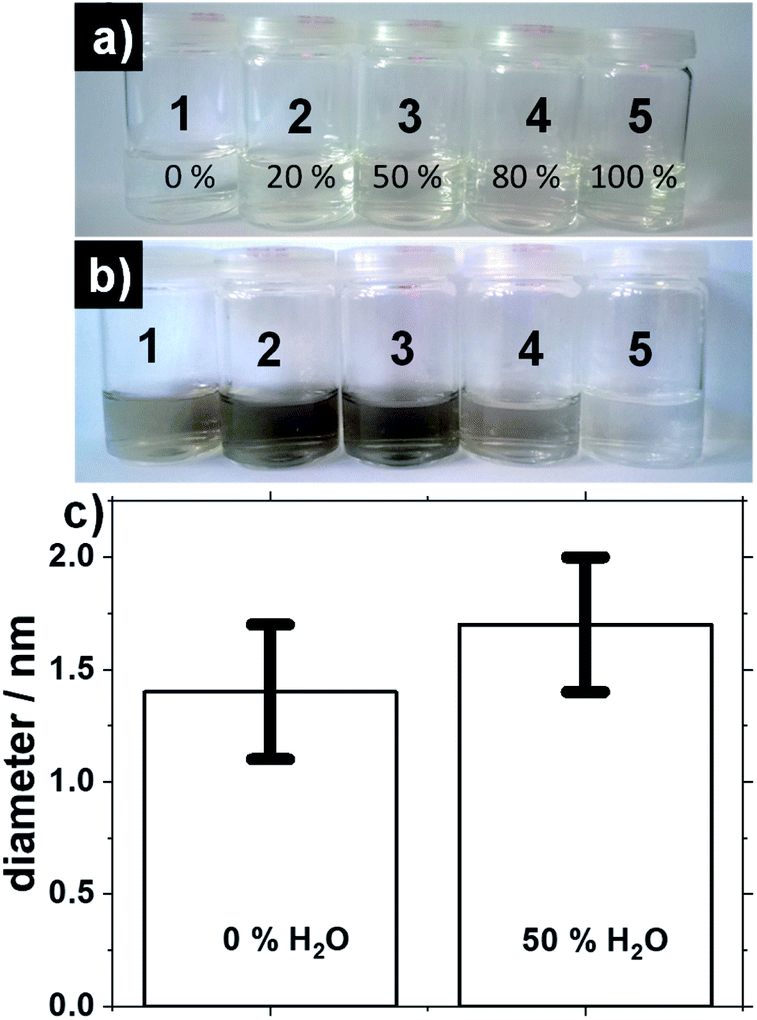 | ||
| Fig. 4 Pictures of reaction mixture containing 0.5 mM H2PtCl6 and 10 mM NaOH in methanol (a) before and (b) after 2 hours exposure to UV-light when the solvent is methanol and (1) 0%, (2) 20%, (3) 50%, (4) 80% and (5) 100% water. (c) Average diameter obtained for the experimental conditions of 0% and 50% water in alkaline methanol. | ||
For 0% water the size is 1.4 ± 0.3 nm whereas for 50% water the size is 1.7 ± 0.3 nm, see Fig. 4c. This indicates that addition of water can control to some extent the size of the NPs in the case of UV-induced synthesis, even in the case of low H2PtCl6 concentration selected here to achieve a fine size control. UV-induced synthesis offers milder reaction conditions (e.g. lower overall temperature, no need for reflux conditions) than a microwave oven which can explain why even at low Pt concentration a fine size control is achieved by adding water.
We previously showed that Ir NPs obtained in alkaline methanol show surprisingly high catalytic activity.12,15 The general approach presented here using UV-light is also suitable to obtain Ir NPs, see Fig. S3.† Despite using different precursors (Na2IrCl6, H2IrCl6, or IrCl3), solvents (methanol, ethanol, water), amounts of water (10–25% in volume), NaOH concentrations (0–44 mM), temperatures (10–40 °C), times of synthesis (up to 24 h) and gas atmosphere (air, 5% H2 in Ar, Ar, CO) or seed-mediated growth approaches, see Fig. S4 and details in ESI,† the size of the NPs was always in the range 1.0–1.7 nm without clear size control. The synthesis in ethanol is often significantly faster than in methanol. Adding NaOH increases slightly and working in Ar containing gas atmospheres increases significantly the NP formation rate. In contrast, a CO atmosphere is slowing down the NP formation. Different duration time of synthesis and changing reaction parameters lead to the same NP size ranges regardless of the formation kinetics. It therefore seems to be a general challenge to control the size of Ir NPs. Nevertheless, UV-induced synthesis is here shown to be suitable to produce very small Ir NPs.
Conclusions
UV-induced or thermally induced syntheses of Pt and Ir NPs by the mono-alcohol surfactant-free process show similar features. In both type of synthesis, Pt NP size control is not simply achieved by varying the NaOH concentration whereas adding water seems to give a finer size control. The NPs obtained are ‘unprotected’, i.e. show mainly CO and/or OH/OH− functionalization. An advantageous feature of the UV-induced synthesis is that the reaction proceeds under milder reaction conditions (e.g. lower overall temperature). As a result, the formation of Pt NPs for instance can be achieved without NaOH. The overall synthesis is then extremely simple to obtain stable colloidal NPs of ca. 2–2.5 nm. For Ir NPs, the size control is generally more challenging to achieve while NPs with size < 2 nm are easily obtained. The mono-alcohol process combined with UV-induced synthesis is a promising synthesis route to further study NP formation and produce surfactant-free precious metal NPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Johnson, M. Santella, B. Wegge Laursen and T. Just Sørensen, all University of Copenhagen, are thanked for their help and use of equipment and N. Schären, University of Bern, for performing most of the Ir NP syntheses. M. A. acknowledges support from the Villum Foundation in form of a block stipend as well as support from the Swiss National Science Foundation (SNSF) via the project no. 200021_184742. J.S. thanks the DFG for financial support (KU 3152/6-1). J.Q. has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 703366 (SELECTRON).Notes and references
- S. Olveira, S. P. Forster and S. Seeger, J. Nanotechnol., 2014, 324089 Search PubMed.
- A. Ohma, K. Shinohara, A. Liyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2016, 181, 298 CrossRef CAS.
- T. Pradeep and Anshup, Thin Solid Films, 2009, 517, 6441 CrossRef CAS.
- B. I. Kharisov, H. V. R. Dias, O. V. Kharissova and A. Vazquez, J. Nanopart. Res., 2014, 16, 2665 CrossRef.
- M. Navlani-Garcia, D. Salinas-Torres, K. Mori, Y. Kuwahara and H. Yamashita, Catal. Surv. Asia, 2019, 23, 127 CrossRef.
- I. Schrader, S. Neumann, R. Himstedt, A. Zana, J. Warneke and S. Kunz, Chem. Commun., 2015, 51, 16221 RSC.
- Z. Q. Niu and Y. D. Li, Chem. Mater., 2014, 26, 72 CrossRef CAS.
- D. G. Li, C. Wang, D. Tripkovic, S. H. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358 CrossRef CAS.
- J. Quinson, M. Inaba, S. Neumann, A. Swane, J. Bucher, S. Simonsen, L. Theil Kuhn, J. Kirkensgaard, K. Jensen, M. Oezaslan, S. Kunz and M. Arenz, ACS Catal., 2018, 8, 6627 CrossRef CAS.
- Y. R. Ouyang, H. J. Cao, H. J. Wu, D. B. Wu, F. Q. Wang, X. J. Fan, W. Y. Yuan, M. X. He, L. Y. Zhang and C. M. Li, Appl. Catal., B, 2020, 265, 118606 CrossRef.
- J. Quinson, S. Neumann, T. Wannmacher, L. Kacenauskaite, M. Inaba, J. Bucher, F. Bizzotto, S. B. Simonsen, L. T. Kuhn, D. Bujak, A. Zana, M. Arenz and S. Kunz, Angew. Chem., Int. Ed., 2018, 57, 12338 CrossRef CAS PubMed.
- J. Quinson, L. Kacenauskaite, J. Bucher, S. B. Simonsen, L. T. Kuhn, M. Oezaslan, S. Kunz and M. Arenz, ChemSusChem, 2019, 12, 1229 CrossRef CAS PubMed.
- J. Quinson, J. Bucher, S. B. Simonsen, L. T. Kuhn, S. Kunz and M. Arenz, ACS Sustainable Chem. Eng., 2019, 7, 13680 CrossRef CAS.
- F. Bizzotto, J. Quinson, A. Zana, J. Kirkensgaard, A. Dworzak, M. Oezaslan and M. Arenz, Catal. Sci. Technol., 2019, 9, 6345 RSC.
- L. Kacenauskaite, J. Quinson, H. Schultz, J. J. K. Kirkensgaard, S. Kunz, T. Vosch and M. Arenz, ChemNanoMat, 2017, 2, 89 CrossRef.
- J. Quinson, L. Kacenauskaite, T. L. Christiansen, T. Vosch, M. Arenz and K. M. Ø. Jensen, ACS Omega, 2018, 3, 10351 CrossRef CAS PubMed.
- H. Dong, Y. C. Chen and C. Feldmann, Green Chem., 2015, 17, 4107 RSC.
- F. Fievet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J.-Y. Piquemal, L. Sicarda and G. Viaub, Chem. Soc. Rev., 2018, 47, 5187 RSC.
- Y. Wang, J. W. Ren, K. Deng, L. L. Gui and Y. Q. Tang, Chem. Mater., 2000, 12, 1622 CrossRef CAS.
- T. S. Rodrigues, M. Zhao, T. H. Yang, K. D. Gilroy, A. G. M. da Silva, P. H. C. Camargo and Y. N. Xia, Chem.–Eur. J., 2018, 24, 16944 CrossRef CAS PubMed.
- S. M. Chen, Q. Y. Yang, H. H. Wang, S. Zhang, J. Li, Y. Wang, W. S. Chu, Q. Ye and L. Song, Nano Lett., 2015, 15, 5961 CrossRef CAS PubMed.
- M. Wojnicki and P. Kwolek, J. Photochem. Photobiol., A, 2016, 314, 133 CrossRef CAS.
- M. Harada and H. Einaga, Langmuir, 2006, 22, 2371 CrossRef CAS PubMed.
- I. Schrader, J. Warneke, S. Neumann, S. Grotheer, A. A. Swane, J. J. K. Kirkensgaard, M. Arenz and S. Kunz, J. Phys. Chem. C, 2015, 119, 17655 CrossRef CAS.
- N. Toshima, K. Nakata and H. Kitoh, Inorg. Chim. Acta, 1997, 265, 149 CrossRef CAS.
- M. Harada, K. Okamoto and M. Terazima, Langmuir, 2006, 22, 9142 CrossRef CAS PubMed.
- M. Harada and Y. Kamigaito, Langmuir, 2012, 28, 2415–2428 CrossRef CAS PubMed.
- M. Cargnello, C. Chen, B. T. Diroll, V. V. T. Doan-Nguyen, R. J. Gorte and C. B. Murray, J. Am. Chem. Soc., 2015, 137, 6906 CrossRef CAS PubMed.
- E. A. Baranova, C. Bock, D. Ilin, D. Wang and B. MacDougall, Surf. Sci., 2006, 600, 3502 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, GC-MS, fluorescence measurements and detailed Ir investigation. See DOI: 10.1039/d0na00218f |
| ‡ These authors equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2020 |