
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA NiFe layered double hydroxide-decorated N-doped entangled-graphene framework: a robust water oxidation electrocatalyst†
Narugopal
Manna
ab,
Nadeema
Ayasha
ab,
Santosh K.
Singh
c and
Sreekumar
Kurungot
 *ab
*ab
aPhysical & Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune, Maharastra, India. E-mail: k.sreekumar@ncl.res.in; Fax: +91-20-25902636
bAcademy of Scientific and Innovative Research, Postal Staff College, Nehru Nagar, Ghaziabad, Uttar Pradesh-201002, India
cFaculty of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8573, Japan
First published on 3rd March 2020
Abstract
Three dimensional (3D) porous carbon materials are highly desirable for electrochemical applications owing to their high surface area and porosity. Uniformly distributed porosity in the 3D architecture of carbon support materials allows reactant molecules to access more electrochemically active centres and simultaneously facilitate removal of the product formed during electrochemical reactions. Herein, we have prepared a nitrogen-doped entangled graphene framework (NEGF), decorated with NiFe-LDH nanostructures by an in situ solvothermal method followed by freeze-drying at high vacuum pressure and low temperature. The freeze-drying method helped to prevent the restacking of the graphene sheets and the formation of a high surface area nitrogen-doped entangled graphene framework (NEGF) supported NiFe-LDHs. The incorporation of the NEGF has significantly reduced the overpotential for the electrochemical oxygen evolution reaction (OER) in 1 M KOH solution. This corresponds to an overpotential reduction from 340 mV for NiFe-LDHs to 290 mV for NiFe-LDH/NEGF to reach the benchmark current density of 10 mA cm−2. The preparation of the catalyst is conceived through a low-temperature scalable process.
Introduction
Owing to the increased energy demand and environmental concerns, there is an urgent need to focus on alternative carbon-free fuels.1 In this respect, fuel cells have emerged as a promising alternative technology to conventional heat engines.2 However, being an electrochemical energy conversion device, their sustainability, as well as eco-friendly nature, is directly dependent on the source of the fuel, e.g., H2 (having the highest gravimetric energy density, i.e., 140 MJ kg−1).3 Among the various ways of H2 generation, water splitting can be the sustainable resource of H2.4 Electrochemical water splitting involves two half-cell reactions, i.e., the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER). However, owing to four proton-coupled four electron-transfer reactions, the OER is thermodynamically unfavourable, limiting the overall energy efficiency of these vital electrochemical areas.5 Currently, precious-group metal-based, i.e., IrO2- and RuO2-based, catalysts are the benchmark in both the acidic and alkaline medium for the OER.6 However, their high cost and scarcity have triggered research toward the development of low-cost and readily available active materials for scale-up utilizations.7 In the last few years, various earth-abundant transition metal (Mn, Fe, Co, and Ni)-based oxides, hydroxides, and alloys have been explored as promising alternatives to catalyze the OER in the alkaline medium.8–10 For instance, Sumboja et al.8 designed NiMn layered double hydroxides as an efficient oxygen evolution reaction electrocatalyst. In another report, Kang et al.11 demonstrated the activity of NiFe-oxide toward the water oxidation reaction. Despite satisfactory performances, these support-free catalysts suffer from active centre agglomeration and poor electronic conductivity issues, affecting their long-term stability.11–14 A solution for the conductivity issue is to modulate the electronic structure by in situ anchoring these transition metal oxides/hydroxides over cost-effective conducting supports.15,16 Among various cost-effective conducting supports, carbon-based materials with electrochemically favourable characteristics, i.e., high electronic conductivity and surface area, have emerged as universal choices in the electrocatalysis field.15,16 For instance, in a recent report, Zhan et al.17 achieved improved activity and stability by anchoring the OER-active NiFe layered double hydroxides over nitrogen-doped graphene. Similarly, Chandrasekaran et al.18 also reported improved OER performance by anchoring NiFe layer double hydroxides over reduced graphene oxide.Though two-dimensional graphene and one-dimensional carbon nanotubes have emerged as convincing OER-catalyst supports, their in-depth micro-architecture has turned out to be the proficiency-limiting factor.16 For instance, theoretically, graphene provides a surface area as high as ∼2630 m2 g−1 as well as outstanding electrical conductivity.19,20 However, practically, the well-known restacking issue of graphene layers affects their surface area and electronic conductivity to a large extent.21 Besides, CNTs suffer from low surface area, restricting sufficient active material loading, and detachment during the reaction.20,21 Hence, tuning the micro-architectural characteristics of the catalyst support is a key contributor to increase the active centre loading as well as its accessibility to tune the overall performance.22 Among several potential candidates, three-dimensional entangled-graphene frameworks (3D-EGFs), having a high surface area as well as electronic conductivity, are gaining enormous attention as a conducting support.22,23 In spite of this, 3D-EGFs with a hierarchical porous structure also serve as a promising substrate to accommodate a huge number of exposed active materials to facilitate the seamless diffusion of the electrocatalytic reaction species.24–26 Recently, several methods have been adopted for the designing of 3D graphene including hard and soft-template-based methods,27 freeze-drying methods,30 a microporous template-based CVD method28,30,31 and a nonporous metal-based CVD method.28,29
Among all these methods, freeze-drying is found to be quite appealing as it generates plenty of homogeneous pores, and the process leads to the higher mechanical strength of the interconnected graphene network.27,32,33 Here, we focussed on alleviating the current performance-constraining issues of the low-cost best-performing-system. The primary activity degradation factors include insufficient electronic conductivity, poor active centre exposure, and hindered reaction species transport. By keeping all these performance-constraining factors in mind, herein, we introduce the anchoring of the nano-sized NiFe-layered double hydroxide (NiFe-LDH) over a N-doped entangled graphene framework (NEGF) to further improve the performance. The rationally designed NEGF acted as a useful active substrate to tune the electronic structure and the NiFe-LDH distribution, along with providing an architecture comprised of an open and exposed catalytic system.
Experimental section
Synthesis of the nitrogen doped entangled graphene framework (NEGF)
60 mg of graphene oxide (GO) was dispersed in a 20 ml (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) aqueous solution of ammonia (30% v/v), via water-bath sonication and overnight stirring. The complete solution was transferred into a Teflon-lined autoclave and kept for 12 h at 180 °C. After complete cooling to room temperature, the sample was washed with water 5–6 times to remove the excess ammonia, followed by freeze-drying to prepare the nitrogen-doped entangled graphene framework (NEGF).
:1) aqueous solution of ammonia (30% v/v), via water-bath sonication and overnight stirring. The complete solution was transferred into a Teflon-lined autoclave and kept for 12 h at 180 °C. After complete cooling to room temperature, the sample was washed with water 5–6 times to remove the excess ammonia, followed by freeze-drying to prepare the nitrogen-doped entangled graphene framework (NEGF).
Synthesis of the NiFe-LDH supported nitrogen-doped entangled graphene framework (NiFe-LDH/NEGF)
60 mg of the as-prepared GO was added to a 20 ml (3:1) aqueous solution of ammonia (30% v/v), via water-bath sonication and overnight stirring. After the complete dispersion of GO, Ni(OAc)2·4H2O and Fe(OAc)2·4H2O were added to the solution and kept stirring for another 6 h. After complete mixing of the metal ions, the reaction mixture was transferred into a Teflon-lined autoclave and heated at 180 °C for 12 h; afterward the autoclave was allowed to cool down naturally, and the sample was washed with water 5–6 times to remove the excess ammonia. This was further subjected to freeze-drying for 10 h at −52 °C under high vacuum pressure. After the completion of the freeze-drying process, the sample was collected, which adopted a flaky kind of structure.
Synthesis of unsupported NiFe-LDHs
Synthesis of NiFe-LDHs was done by adding nickel acetate and iron acetate salt into a 20 ml (3:1) aqueous solution of ammonia (30% v/v), and it was kept for 6 h with constant stirring. After complete mixing, the solution was transferred into a Teflon-lined autoclave and kept for 12 h at 180 °C. After complete cooling to room temperature, the sample was washed with water 5–6 times to remove the excess ammonia. The final samples were dried in an oven at 60 °C and collected.
Synthesis of Ni(OH)2/NEGF
Synthesis of Ni(OH)2/NEGF is the same as that of NiFe-LDH/NEGF, except for addition of iron acetate; 60 mg of graphene oxide (GO) was dispersed in a 20 ml (3:1) aqueous solution of ammonia (30% v/v), via water-bath sonication and overnight stirring.
After complete dispersion of graphene oxide, nickel acetate was added to the solution and kept for 6 h with constant stirring. After complete mixing, the solution was transferred into a Teflon-lined autoclave and kept for 12 h at 180 °C. After complete cooling to room temperature, the sample was washed with water 5–6 times to remove the excess ammonia, followed by freeze-drying to prepare the Ni(OH)2/NEGF.
Synthesis of NiFe-LDH/NEGF (w/o)
To comparatively study the effect of freeze-drying and nitrogen atom doping on the carbon support, we have synthesized NiFe-LDHs over the nitrogen-doped graphene without freeze-drying, which is named NiFe-LDH/NGF (w/o). Synthesis of NiFe-LDH/NEGF (w/o) is the same as that of NiFe-LDH/NEGF, except that here instead of using freeze-drying, the sample was dried by filtration after hydrothermal treatment for 12 h at 180 °C.Synthesis of NiFe-LDH/EGF
Instead of ammonia, only water is used to study the effect of nitrogen-doping, and the sample is named NiFe-LDH/EGF. Synthesis of NiFe-LDH/EGF is the same as that of NiFe-LDH/NEGF, except that here instead of using ammonia solution only water is used as the solvent, followed by solvothermal treatment for 12 h at 180 °C and applying the freeze-drying method.Results and discussion
The preparation procedure for the homogeneously distributed NiFe-LDH over the NEGF involves two steps, as illustrated in Scheme 1. The reaction mixture was first treated solvothermally followed by lyophilization under high vacuum and low temperature (−52 °C) conditions. The adopted lyophilization process helped to prevent the graphene sheet-restacking under these experimental conditions, generating an entangled graphene framework by the crosslinking of the graphene sheets.29 The interesting role played by ammonium ions is that they help in the strengthening of the graphene hydrogel along with nitrogen doping at a temperature as low as just 180 °C under the solvothermal conditions.34 Ammonium hydroxide enhances the interaction between two graphene sheets with its alkaline hydroxyl groups and ammonium ions.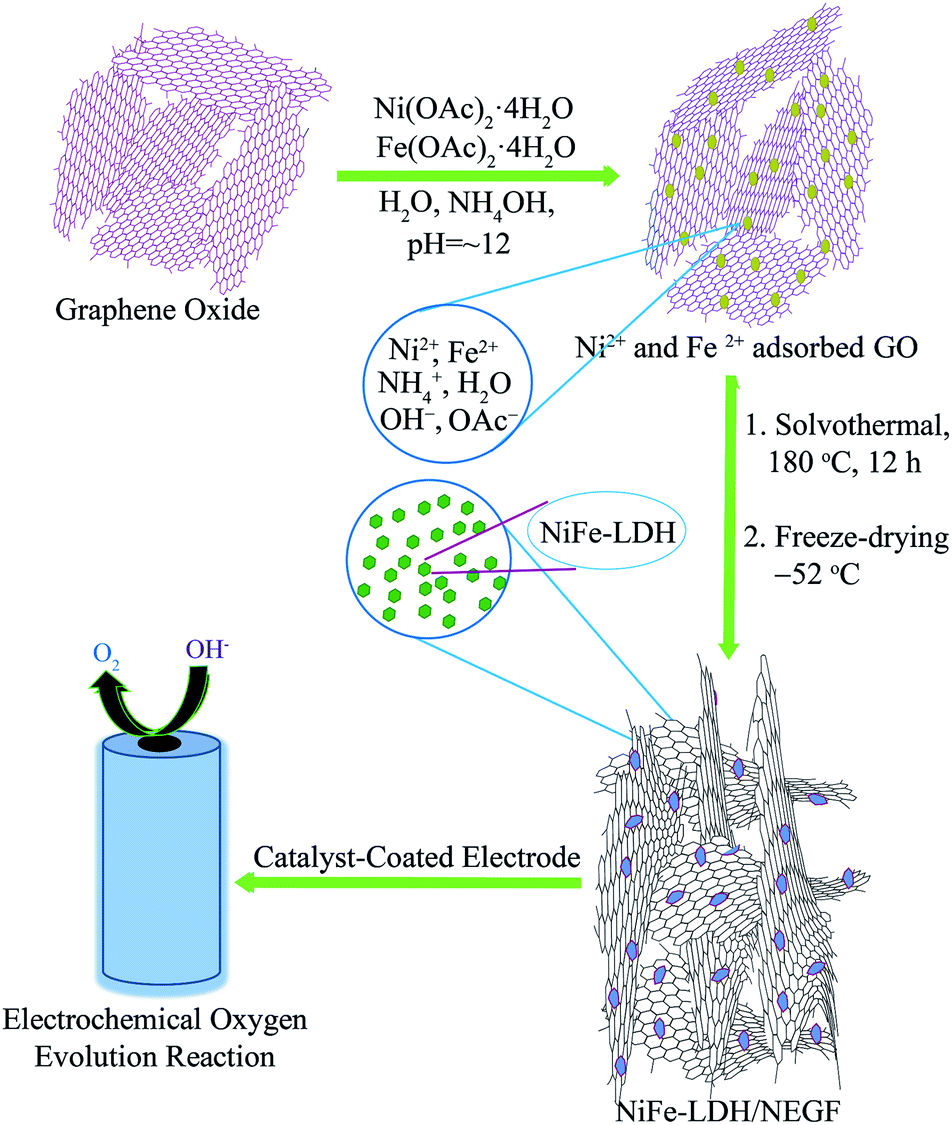 | ||
| Scheme 1 Schematic representation of the synthesis of the NiFe-LDH-anchored N-doped 3D entangled graphene framework (NiFe-LDH/NEGF) and its application as an efficient OER electrocatalyst. | ||
Formation of the hierarchical NEGF was first examined through field-emission scanning electron microscopy (FESEM). Fig. 1a presents the FESEM image of the NEGF, showing the 3D entangled network, formulated with randomly oriented graphene nanosheets. Such an entangled 3D framework not only prevented the agglomeration of the graphene nanosheets but it also provided high surface area as well as the porosity to the matrix. It, henceforth, facilitated the easy migration of reactants and gaseous products released during the electrochemical reactions.24,25 Such a type of interconnected graphene framework is also retained during the in situ loading of the active material, i.e., NiFe-LDHs, as shown in Fig. 1b. Fig. 1c shows the FESEM image of the NiFe-LDH-anchored nitrogen-doped graphene without the freeze-drying step, presenting a well-defined agglomerated morphology with thick and stacked graphene patches. Hence, FESEM analyses support the role of lyophilization to prevent the restacking of the graphene nanosheets, assisted by the water removal under the high vacuum and low-temperature conditions.
 | ||
| Fig. 1 FESEM images of (a) NEGF; (b) NiFe-LDH/NEGF; (c) NiFe-LDH/NEGF (w/o); (d), (e), and (f) the TEM images of NiFe-LDH/NEGF at different magnifications. | ||
Next to FESEM, transmission electron microscopy (TEM) analysis was performed to find out the size and distribution of the decorated NiFe-LDH over the graphene sheets. Fig. 1d and e present the TEM images of NiFe-LDH/NEGF at different magnifications. The TEM analyses revealed a homogeneous distribution of the layered double hydroxide nanostructures over the NEGF. The further magnified TEM image in Fig. 1f shows that the anchored LDH nanostructures are well-resolved and amorphous in nature. The amorphous nature of the anchored NiFe-LDH is further supported by the diffused ring pattern in selected area electron diffraction (SAED) analysis (inset of Fig. 1f). The role of N-doping toward facilitating the homogeneous distribution of the metal hydroxides over the NEGF is also studied by comparatively analyzing the TEM images of NiFe-LDH-anchored N-doped and un-doped entangled graphene frameworks (EGFs). Fig. S1a and b† present the TEM image of the NiFe-LDH-supported un-doped EGF, showing the larger agglomerates of NiFe-LDHs over the support sheets. This finding shows the crucial role played by N-doping in creating plenty of metal anchoring sites along with the tuning of the size of LDH nanostructures as well as in avoiding their agglomeration.34,35 Furthermore, to study the need for supports for decorating discrete active LDH nanostructures, TEM analysis was extended to a support-free NiFe-LDH system. The comparative study showed that in the absence of any substrate, LDHs show more chances of agglomeration and acquiring a bulky morphology (Fig. S2a and b†). We have further extracted the weight % and atomic % of various species present in the sample and the corresponding data are presented in Fig. S3,† a table for respective elements shown in Table S1.†
To further understand the role of freeze-drying in improving the surface area as well as increasing the number of porous channels, specific surface area and pore size distribution were evaluated by nitrogen adsorption–desorption isotherm analysis. A BET surface area value of 547 m2 g−1 (Fig. S4†) obtained for the NEGF confirms the highly open and porous structure of the NEGF, supporting the FESEM findings. The BET surface area measurement was also carried out to find out the benefit of freeze-drying over the without freeze-drying (w/o) sample. Both NiFe-LDH/NEGF and NiFe-LDH/NG (w/o) show type-IV isotherms (Fig. S5†). However, the BET analysis of NiFe-LDH/NEGF evidenced the essential role played by freeze-drying in maintaining a highly porous and exposed surface of the catalyst. It is reflected both through the surface area obtained, i.e., 328 m2 g−1, and porosity from the pore size distribution profile, giving the distribution of pores in the range of 2 to 5 nm (Fig. 2a). These results further guarantee the individual graphene sheets into the bulk structure. Notably, the surface area of the NEGF is found to be higher than that of NiFe-LDH/NEGF (547 m2 g−1vs. 328 m2 g−1), which is obvious owing to the blockage and covering of some of the pores of the NEGF through the as-grown NiFe-LDH nanostructures. As shown in Fig. 2a, NiFe-LDH/NEGF shows the higher cumulative area in the pore size range of 2 to 5 nm, suggesting the mesoporous nature of NiFe-LDH/NEG. However, the pore-size distribution profile of NiFe-LDH/NG (w/o) showed decreased intensity in the above-mentioned pore-size region, supporting the role of freeze-drying in making open and exposed catalyst frameworks. Pore size distribution analysis is also extended on the NEGF, presenting a pore size distribution profile similar to that of NiFe-LDH/NEGF (Fig. S4b†). To make a better understanding of the contribution of different parameters towards catalytic performance, the catalyst BET surface area measurement results of all the samples are presented in Fig. S6† and the corresponding data are presented in Table S2.†
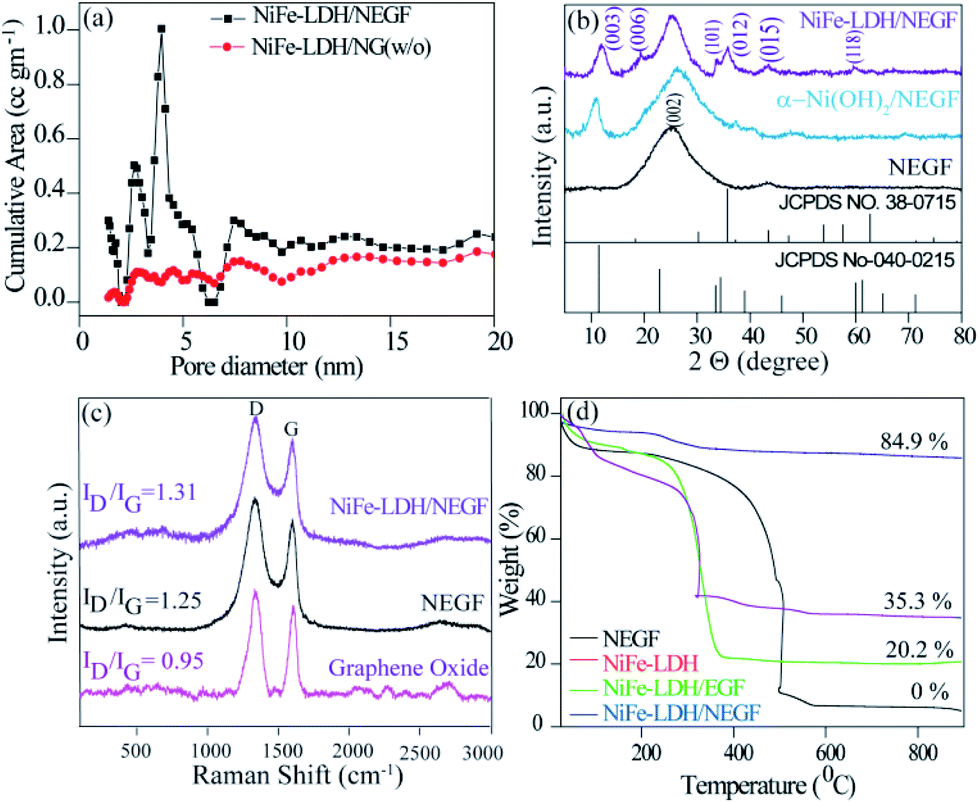 | ||
| Fig. 2 Comparative physical characterization of the as-prepared catalysts: (a) pore-size distribution profiles of the NEGF and NiFe-LDH/NEGF, (b) PXRD patterns of the NEGF, α-Ni(OH)2, and NiFe-LDH/NEGF, (c) Raman spectra of GO, the NEGF and NiFe-LDH/NEGF and (d) TGA profiles. | ||
To identify the phase and crystallinity of the as-synthesized samples, X-ray diffraction (XRD) analysis was performed. Fig. 2b consists of the comparative XRD pattern for the NEGF and NiFe-LDH/NEGF. The PXRD pattern for the NEGF shows an intense peak at a 2θ value of 26°, corresponding to the (002) diffraction peak of the reduced graphene oxide.26 The PXRD pattern of NiFe-LDH/NEGF shows a comparatively intense and prominent (00l) plane, matching well with the JCPDS Card no. 040-0215. NiFe-LDH/NEGF also shows a broad and intense (002) plane of the NEGF, attributing to the anchoring of NiFe-LDHs over the NEGF. To study the role of Fe incorporation in the Ni-hydroxide crystal structure, the growth of nickel hydroxide over the nitrogen-doped entangled graphene framework (NEGF) has been realized under the same reaction conditions and abbreviated as α-Ni(OH)2/NEGF. Comparative PXRD patterns of α-Ni(OH)2/NEGF and NiFe-LDH/NEGF suggested the increased orderness of the hydroxide layers after the Fe incorporation into the α-Ni(OH)2 crystal structure owing to the poor intensity of hydroxide peaks in the former case.36,37
Next to the crystal phase identification, Raman analysis was conducted to investigate the graphitization and defect extent in the microstructure of the synthesized graphene-based material. Fig. 2c comprises the comparative Raman spectra of graphene oxide (GO), the NEGF, and NiFe-LDH/NEGF. The spectra show that the typical peaks for all the samples are located in the range of 1300 and 1600 cm−1, which is due to the stock phonon interaction of the carbon materials created by the laser excitation.36 In all the comparative samples, the intense peak at 1320 cm−1 is due to the defective (D) nature of the carbon, including disorders in bonding, heteroatom doping and vacancies in the carbon lattice.37 As shown in Fig. 2c, another intense peak which appeared in the range of 1580 to 1600 cm−1 is attributed to the graphitic nature of the carbon,38 indicating the ordered lattice structure from the vibration of the Csp2 in the plane.39 The intensity ratio of the defective carbon peak to the graphitic carbon peak, i.e., the ID/IG ratio, helps to find out the extent of defects/disorderness and graphitization extent in carbon-based materials.34,39 The ID/IG values for the NEGF (1.31) and NiFe-LDH/NEGF (1.25) are higher than those of GO (0.95), suggesting the GO reduction to reduced graphene oxide during the hydrothermal treatment. Besides, higher intensity of the defective carbon for the NEGF and NiFe-LDH/NEGF shows a decrease in the average size of the sp2 domains due to the removal of the oxygen-containing functional groups and doping of nitrogen (N) atoms over the carbon atom.34,41 Higher defects and disorders over the carbon support help to generate plenty of nucleation sites for the nanoparticle growth, their controlled homogeneity, and size distribution.41
After confirming the highly open and entangled graphene framework characteristics of the NEGF along with the good active material dispersion over the NEGF, thermogravimetric analysis (TGA) was employed to determine total active material loading over the NEGF. TGA was carried out by annealing the sample from 25 to 900 °C with a scan rate of 5 °C per minute in an oxygen atmosphere. Fig. 2d presents the TGA curves for the NEGF, NiFe-LDH, NiFe-LDH/NEGF, and NiFe-LDH/EGF. For all the compared samples, an initial weight loss was observed at around 100–150 °C, owing to the evaporation of physisorbed water molecules. The NEGF contains only a composite of nitrogen and oxygen and major content of carbon, which starts burning at around 400 °C, and near 500 °C complete calcination is observed in an oxygen atmosphere. This complete conversion of carbon and nitrogen to mixed oxide products drawn away in the form of gas, so the rest of the sample weight becomes zero. In the case of NiFe-LDHs, no such major weight loss was observed. However, in the other two supported systems, i.e., NiFe-LDH/NEGF and NiFe-LDH/EGF, reasonable water loss is encountered owing to the presence of LDHs and porous carbon having larger accessibility for a physisorbed water molecule. In the two later samples, major weight loss in the temperature region of 350 to 500 °C might be due to the oxidation of carbon. As it is already illustrated in the Raman analysis section, nitrogen doping over the carbon surface helps to create plenty of metal-binding sites, which increases the metal support interactions.44 This can be further supported by the TGA data. As seen from Fig. 2d, NiFe-LDH/NEGF shows comparatively higher active material loading (∼35%) than NiFe-LDH/EGF (∼20%). This strongly anchored and higher active material loading may help in improving the electrocatalytic activity and durability.42
The electronic structure tuning of all the surface elements, i.e., N-doping over the carbon support, binding energy change due to the catalyst support interactions and formation of NiFe-LDHs, is thoroughly analyzed by employing X-ray photoelectron spectroscopy (XPS) measurements. Fig. 3a presents the survey spectra of the NEGF and NiFe-LDH/NEGF, confirming the presence of Ni, Fe, O, N, and C in NiFe-LDH/NEGF and N, O and C in the NEGF. In both cases, common elements are C, O, and N, having binding energy located at around ∼284.8, ∼531.5 and ∼400.5 eV, respectively.43–45 In the case of NiFe-LDH/NEGF, two new peaks are present at ∼710.8 and ∼855.6 eV corresponding to the elements Fe and Ni, respectively.46–48 From Fig. 3a, it is apparent that the surface composition of carbon and nitrogen is decreased in NiFe-LDH/NEGF, which is due to the coverage of NiFe-LDHs over the nitrogen-doped carbon. However, the O 1s peak is found to intensify after the NiFe-LDH loading, basically coming from the hydroxide moieties as well as intercalated carbonate and water species.40–42
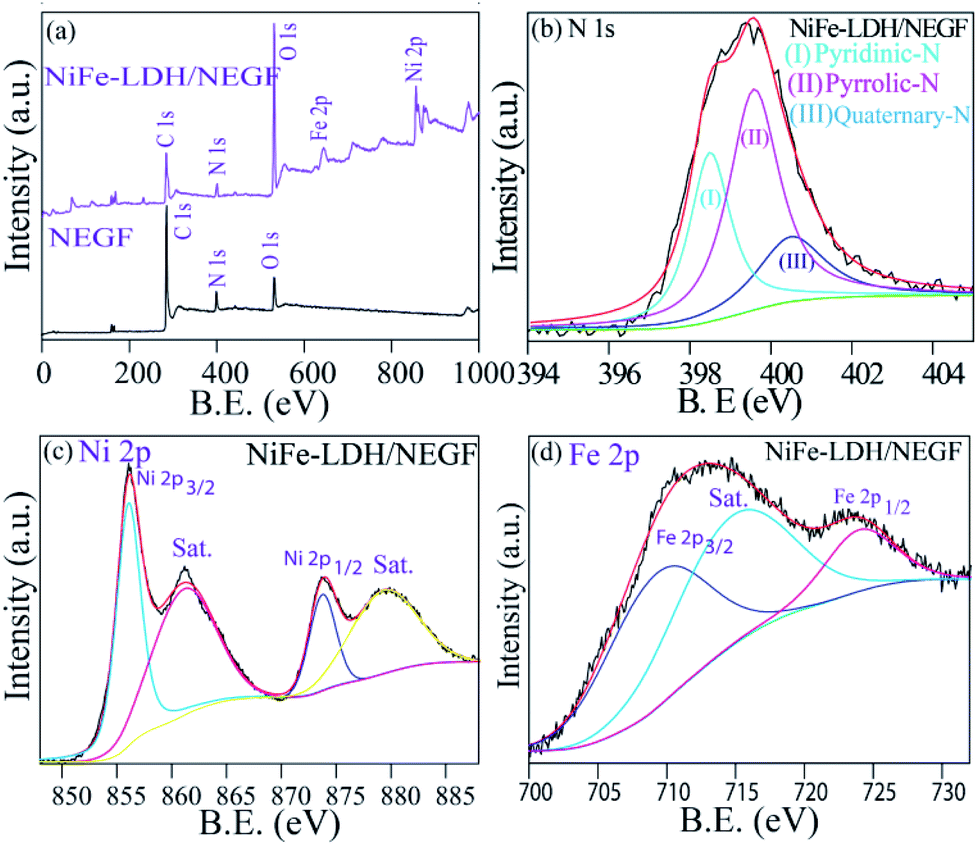 | ||
| Fig. 3 XPS analysis: (a) XPS survey scan spectra of the NEGF and NiFe-LDH/NEGF. Panels (b), (c) and (d) show the high-resolution deconvoluted spectra of N 1s, Ni 2p and Fe 2p of NiFe-LDH/NEGF, respectively. | ||
Furthermore, by high-resolution XPS, the NEGF and NiFe-LDH/NEGF are comparatively analyzed in the C 1s, N 1s, and O 1s core regions. Various chemical states of carbon, i.e., C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284), C–O (286), and C–N/CN (400), with different binding energy values owing to the unique chemical environment of carbon are marked in the respective figures (Fig. S7a and b†).43,44 The C 1s spectrum of NiFe-LDH/NEGF showed an additional CO32− anion peak at 289.8 eV, indirectly illustrating the growth of layer double hydroxides over the NEGF.46–48 Subsequently, a deconvoluted O 1s core level spectrum was comparatively examined for both the NEGF and NiFe-LDH/NEGF, as given in Fig. S5c and d.† In the NEGF, oxygen binds with carbon in the form of C–O and CO, and by epoxy group formation. However, in the case of NiFe-LDH/NEGF, O 1s spectra show quite good disparity owing to the surface rich layer double hydroxide (LDH), having oxygen coordinated in the form of Ni(OOH), Fe–OH, and Ni–OH and in the carbonate or lattice H2O form. Deconvoluted N 1s spectra of the NEGF and NiFe-LDH/NEGF are also examined to know the types of nitrogen present in the graphene matrix along with their total percentage. The N 1s spectrum of the NEGF in the ESI (Fig. S7e†) shows a higher percentage (42.4%) of pyrrolic-N (399.7 eV) and almost similar percentage (41.6%) of pyridinic-N (398.6 eV) nitrogen along with a lower quantity (15.9%) of quaternary-N (400.5 eV); furthermore, the N 1s spectrum of NiFe-LDH/NEGF is also deconvoluted to find out any change in the XPS spectrum after the NiFe-LDH loading over the NEGF. Fig. 3b shows the deconvoluted N 1s spectrum locating all the deconvoluted peaks which are present in the NEGF, suggesting that N-doping in the graphene matrix remains the same during the in situ loading of NiFe-LDHs. For comparison, along with the obtained data we have presented the previously reported data in Table S3 in the ESI.† Thus, deconvoluted XPS spectra of C 1s, O 1s, and N 1s clearly show the solvothermal-assisted reduced graphene oxide formation as well as layer double hydroxide growth. The survey spectrum of NiFe-LDH/NEGF also shows two extra peaks corresponding to the Ni 2p and Fe 2p core level (Fig. 3a). These core level spectra were deconvoluted to study their chemical state in the LDH system. Fig. 3c presents the deconvoluted Ni 2p spectrum of NiFe-LDH/NEGF, showing the two main spin–orbit doublets of Ni, i.e., Ni 2p3/2 (873.3 eV) and Ni 2p1/2 (855.6 eV) along with two bands. The binding energy value of these two doublets suggests the +2 oxidation state of Ni in the NiFe-LDH system.48,49
C (284), C–O (286), and C–N/CN (400), with different binding energy values owing to the unique chemical environment of carbon are marked in the respective figures (Fig. S7a and b†).43,44 The C 1s spectrum of NiFe-LDH/NEGF showed an additional CO32− anion peak at 289.8 eV, indirectly illustrating the growth of layer double hydroxides over the NEGF.46–48 Subsequently, a deconvoluted O 1s core level spectrum was comparatively examined for both the NEGF and NiFe-LDH/NEGF, as given in Fig. S5c and d.† In the NEGF, oxygen binds with carbon in the form of C–O and CO, and by epoxy group formation. However, in the case of NiFe-LDH/NEGF, O 1s spectra show quite good disparity owing to the surface rich layer double hydroxide (LDH), having oxygen coordinated in the form of Ni(OOH), Fe–OH, and Ni–OH and in the carbonate or lattice H2O form. Deconvoluted N 1s spectra of the NEGF and NiFe-LDH/NEGF are also examined to know the types of nitrogen present in the graphene matrix along with their total percentage. The N 1s spectrum of the NEGF in the ESI (Fig. S7e†) shows a higher percentage (42.4%) of pyrrolic-N (399.7 eV) and almost similar percentage (41.6%) of pyridinic-N (398.6 eV) nitrogen along with a lower quantity (15.9%) of quaternary-N (400.5 eV); furthermore, the N 1s spectrum of NiFe-LDH/NEGF is also deconvoluted to find out any change in the XPS spectrum after the NiFe-LDH loading over the NEGF. Fig. 3b shows the deconvoluted N 1s spectrum locating all the deconvoluted peaks which are present in the NEGF, suggesting that N-doping in the graphene matrix remains the same during the in situ loading of NiFe-LDHs. For comparison, along with the obtained data we have presented the previously reported data in Table S3 in the ESI.† Thus, deconvoluted XPS spectra of C 1s, O 1s, and N 1s clearly show the solvothermal-assisted reduced graphene oxide formation as well as layer double hydroxide growth. The survey spectrum of NiFe-LDH/NEGF also shows two extra peaks corresponding to the Ni 2p and Fe 2p core level (Fig. 3a). These core level spectra were deconvoluted to study their chemical state in the LDH system. Fig. 3c presents the deconvoluted Ni 2p spectrum of NiFe-LDH/NEGF, showing the two main spin–orbit doublets of Ni, i.e., Ni 2p3/2 (873.3 eV) and Ni 2p1/2 (855.6 eV) along with two bands. The binding energy value of these two doublets suggests the +2 oxidation state of Ni in the NiFe-LDH system.48,49
There is a strong interaction between the supported NiFe-LDH and the N dopant of the graphene. Due to the electronegativity difference between the N dopant and NiFe, there will be electron transfer between these two entities. The comparable XPS spectra of NiFe-LDH and NiFe-LDH/NEGF shown in Fig. S8† clearly show a shift in the binding energy of the metals evidencing the electron transfer from the nitrogen dopant to the metal, which is responsible for the reduction in B.E. Fig. 3d presents the deconvoluted XPS spectra of NiFe-LDH/NEGF in the Fe 2p core level. It exhibits two prominent spin–orbit doublets located at 710.9 and 725.1 eV corresponding to Fe 2p3/2 and Fe2p3/2, splitting for the Fe3+ oxidation state, respectively.50,51 Hence, deconvolution, as well as binding energy assignment of the different spin states of Ni and Fe core-level spectra, has helped to find out the oxidation states of the two elements in LDHs.
Electrochemical oxygen evolution reaction
All the employed physical characterization techniques conclude the formation and favorable characteristics of NiFe-LDH/NEGF in catalyzing the water oxidation; half-cell electrochemical analyses were carried out by employing various techniques, e.g., electrochemical surface area (ECSA) measurement, linear sweep voltammetry (LSV) and chronoamperometry. All the analyses were carried out using Hg/HgO as a reference electrode in 1 M KOH, which was further calibrated to RHE using a standard calibration method.49 All the polarization curves were recorded after an ohmic potential drop (iR-drop) compensation by 65%. Comparative ECSA values were first examined to find out the accessible active center density of the various employed catalysts. For platinum-free systems, double-layer capacitance (Cdl) in the non-faradaic region is the reasonable parameter to measure the ECSA.50Fig. 4a and S9† show the higher double-layer capacitance (Cdl) values possessed by NiFe-LDH/NEGF (7.9 mF cm−2), as compared to those of the other two samples, i.e., for NiFe-LDH/NG (w/o) (3.7 mF cm−2) and NiFe-LDH (1.0 mF cm−2). This suggests higher charge accumulation as well as electrolyte infiltration on the highly exposed and open texture of the catalyst system (NiFe-LDH/NEGF). Fig. 4b presents the polarization curves recorded for the oxygen evolution reaction (OER). The overpotential observed at a current density of 10 mA cm−2 for NiFe-LDH/NEGF (290 mV) is lower than that of NiFe-LDHs (350 mV), NiFe-LDH/NG (w/o) (340 mV), and 20% RuO2/C (310 mV). The higher electrocatalytic activity of NiFe-LDH/NEGF toward the OER, as compared to that of NiFe-LDHs clearly reveals the effect of the NEGF support on distributing the active NiFe-LDH homogeneously and also alleviates the active center agglomeration. Moreover, the effect of freeze-drying is also reflected from the polarization plots, representing poor activity of the without freeze-drying sample (i.e., NiFe-LDH/NG (w/o)). All the catalysts composed of Ni has a characteristic oxidation peak just before starting the OER process but due to higher OER current in LSV these peaks are not prominently visible. However, after zooming the oxidative region these peaks can be visualized prominently as shown in Fig. S11.† The oxidation peak at 1.4 V is derived from the oxy hydroxyl formation of the NiFe active sites. This peak appears mostly in the Ni based systems. When Ni is mixed with Fe, the intensity of the peak is enhanced by the redox active metal centers. As the conductivity and activity of the NiFe system become low, during oxy hydroxyl ion formation, current generated will not be much significant. In the case of the carbon supported systems viz. NiFe-LDH/NG (w/o) and NiFe-LDH/NEGF, intense peaks should appear. In the case of the without freeze drying system, NiFe-LDH/NG (w/o), activity is too low compared to NiFe-LDH/NEGF. Hence, the peak at 1.4 V for the NiFe-LDH/NG (w/o) system is not a prominent one. On the other hand, in the case of NiFe-LDH/NEGF, the activity is high and so is the intensity of the peak at 1.4 V when all the synergy works well for this system. Hence, in the case of NiFe-LDH/NG (w/o), the self-assembly texture gets collapsed, restricting the active center accessibility. Further, the role of N-doping in designing a robust catalyst can also be studied by comparatively analyzing the OER activity for NiFe-LDH/NEGF and NiFe-LDH/EGF (w/o) (without N-doping in the carbon support matrix). Fig. S10† presents the lower performance of NiFe-LDH/EGF (370 mV) as compared to that of NiFe-LDH/NEGF (290 mV). The reason for such a performance variation substantiates the role of nitrogen doping, which provides better interaction of the active material with the support as well as its better active material dispersion properties. Hence, N-doping helps to improve the electrocatalytic activity by controlling the particle size as well as facilitating a higher catalytically active center loading. Table S3 in the ESI† summarizes the performance of the various employed catalysts.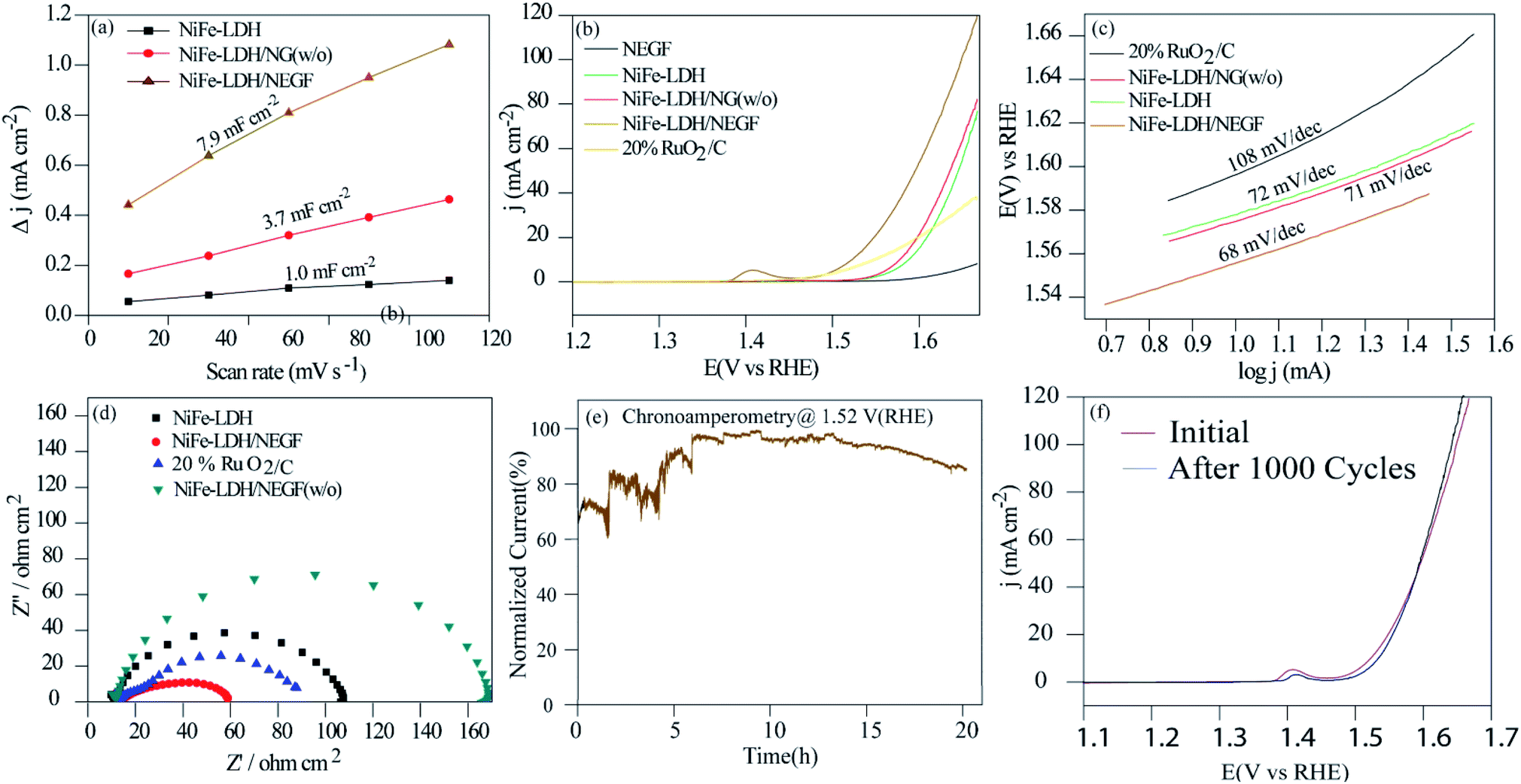 | ||
| Fig. 4 Electrochemical analysis of the as-synthesized catalysts in 1 M KOH: (a) comparative plots of the scan rate dependent double layer capacitive current density (Cdl) at 0.91 V vs. RHE; (b) the OER polarization curves recorded at the 10 mV s−1 scan rate and 1600 rpm of the working electrode; (c) Tafel plots; (d) Nyquist plots recorded for NiFe-LDH, NiFe-LDH/NEGF, 20% RuO2/C and NiFe-LDH/NEGF (w/o) at an applied potential of 1.57 V in the AC frequency range between 100 kHz and 0.1 Hz; (e) chronoamperometric stability test for NiFe-LDH/NEGF; (f) LSV plots recorded over NiFe-LDH/NEGF before and after the 1000 CV cycles. | ||
Kinetics for the adsorption of the reactant and intermediates as well as desorption of the product, i.e., O2 over the catalytic site, can be examined by Tafel analysis. A lower Tafel slope defines faster reaction kinetics, low overpotential, and higher kinetic current density.51,52 The Tafel slope was obtained by plotting the log of current density (logj) vs. potential (V) in the OER potential range. The comparative Tafel plots for NiFe-LDH/NEGF (68 mV dec−1), NiFe-LDH (72 mV dec−1), NiFe-LDH/NEGF (w/o) (72 mV dec−1) and 20% RuO2/C (68 mV dec−1) have been shown in Fig. 4c. The lower Tafel value for NiFe-LDH/NEGF indicates better OER kinetics as compared to that of the other catalysts. To further determine the kinetics of the electron transport in various employed catalysts, the faradaic impedance analysis at a particular potential (1.57 V) was performed. N-doping in the graphitic carbon matrix is expected to give better electrochemical activity compared to the system without nitrogen doping due to electronic structure modulation of the catalysts. In the revised manuscript, we have included the electrochemical impedance measurement data in Fig. S12 in the revised ESI,† which clearly shows a smaller charge transfer resistance (RCT) for NrGO. This result stands out as direct evidence of faster charge transfer during the catalytic OER process. The comparative Nyquist plots given in Fig. 4d show the lowest charge-transfer resistance (Rct = 46 ohm cm2) in the case of NiFe-LDH/NEGF as compared to other catalysts, RuO2 (Rct = 86 ohm cm2), NiFe-LDH (Rct = 107 ohm cm2), and NiFe-LDH/NEGF (Rct = 159 ohm cm2). The lower Rct value indicated better electronic transport throughout the catalyst system owing to a better interaction of NiFe-LDHs with the N-doped entangled framework of graphene.
The durability of the catalyst is another vital parameter to define its robustness, basically its lifetime. The durability of the catalyst was measured by performing chronoamperometry and cycling durability. The chronoamperometric test was performed at a constant potential (a potential needed to achieve a current density of 10 mA cm−2) with a continuous rotation of the working electrode as 1600 rpm. Fig. 4e presents quite good stability of the designed catalyst and shows nearly 80% performance retention even after 20 h of continuous performance. Fig. 4f shows the LSV plots before and after the 1000 CV stability cycles, further supporting the higher durability of the designed catalyst. A slight increment in the current density after the cycle durability is basically due to the exposure of more and more catalytic active centers during the electrochemical cycling. Such good durability of the catalyst is attributed to the better interaction of the N-doped entangled graphene with NiFe-LDHs.
Conclusions
In this work, a solvothermal treatment followed by the freeze-drying method is proposed for the synthesis of NiFe-LDHs on the surface of the nitrogen-doped entangled graphene framework (NEGF). In the presence of ammonium hydroxide, the formation of layered double hydroxides and nitrogen doping on the graphitic carbon skeleton are realized in a single step. Furthermore, freeze-drying helped to maintain the stable, interconnected structure of the graphene sheets, leading to the formation of the entangled structure of graphene. Establishment of high surface area N-doped entangled graphene supported NiFe-LDH has been confirmed by FESEM, TEM, XRD and BET surface area measurements. High surface area and well-maintained porous graphene has shown higher accessibility of gaseous reactants and electrolytes. Homogeneously distributed NiFe-LDH over nitrogen-doped graphene exhibited excellent performance for the oxygen evolution reaction. This promising OER catalytic activity can be ascribed to the following reasons: (1) the uniform distribution of the NiFe-LDH nanostructure in the presence of the anchoring sites on the NEGF, (2) the synergistic effect of the bimetallic double hydroxide layer and N-doped graphene and (3) homogeneous distribution of the pores over the N-doped graphene. The catalyst also exhibited outstanding catalytic stability even after 20 h of continuous operation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NM acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for Research Fellowship. KS acknowledges the CSIR, New Delhi, for project funding (TLP003526).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed.
- J. O. M. Bockris, Science, 1972, 176, 1323 CrossRef CAS PubMed.
- T. E. Mallouk, Nat. Chem., 2013, 5, 362–363 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. F. d. Araújo, T. Reier, S. Dresp, B. Paul, R. krahnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- A. Sumboja, J. Chen, Y. Zong, P. S. Lee and Z. Liu, Nanoscale, 2017, 9, 774–780 RSC.
- S. K. Singh, D. Kumar, V. M. Dhavale, S. Pal and S. Kurungot, Adv. Mater. Interfaces, 2016, 3, 1600532 CrossRef.
- Q. Wang, L. Shang, R. Shi, X. Zhang, Y. Zhao, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2017, 1700467 CrossRef.
- B. K. Kang, M. H. Woo, J. Lee, Y. H. Song, Z. Wang, Y. Guo and D. H. Yoon, J. Mater. Chem. A, 2017, 5, 4320–4324 RSC.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 11973–12000 RSC.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- S. Chen, S. S. Thind and A. Chen, Electrochem. Commun., 2016, 63, 10–17 CrossRef CAS.
- N. Chandrasekaran and S. Muthusamy, Langmuir, 2017, 33, 2–10 CrossRef CAS PubMed.
- Y. Li, H. He, W. Fu, C. Mu, X. Z. Tang, Z. Liu, D. Chi and X. Hu, Chem. Commun., 2016, 52, 1439–1442 RSC.
- T. Zhan, X. Liu, S. Lu and W. Hou, Appl. Catal., B, 2017, 205, 551–558 CrossRef CAS.
- N. Chandrasekaran and S. Muthusamy, Langmuir, 2017, 3, 2–10 CrossRef PubMed.
- Q. Quan, X. Lin, N. Zhang and Y. J. Xu, Nanoscale, 2017, 9, 2398–2416 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- W. Ng, Y. Yang, K. van der Veen, G. Rothenberg and N. Yan, Carbon, 2018, 129, 293–300 CrossRef CAS.
- X. Tang, Y. Zeng, L. Cao, L. Yang, Z. Wang, D. Fang, Y. Gao, Z. Shao and B. Yi, J. Mater. Chem. A, 2018, 6, 15074–15082 RSC.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2013, 52, 13567–13570 CrossRef CAS PubMed.
- Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- Y. Ma, L. Sun, W. Huang, L. Zhang, J. Zhao, Q. Fan and W. Huang, J. Phys. Chem. C, 2011, 115, 24592–24597 CrossRef CAS.
- Y. Ito, Y. Tanabe, K. Sugawara, M. Koshino, T. Takahashi, K. Tanigaki, H. Aoki and M. Chen, Phys. Chem. Chem. Phys., 2018, 20, 6024–6033 RSC.
- Q. Shi, Y. Cha, Y. Song, J. Lee, I. Zhu, C. Li, X. Song, M. K. Du, D. Lin and Y. Lin, Nanoscale, 2016, 8, 15414–15447 RSC.
- L. Zhang, T. Wu, H. Na, C. Pan, X. Xu, G. Huang, Y. Liu, J. Gao and L. Zhang, Ind. Eng. Chem. Res., 2016, 55, 6553–6562 CrossRef CAS.
- A. Kong, A. Kong, X. Zhu, Z. Han, Y. Yu, Y. Zhang, B. Dong and Y. Shan, ACS Catal., 2014, 4, 1793–1800 CrossRef CAS.
- L. Jiang and Z. Fan, Nanoscale, 2014, 6, 1922–1945 RSC.
- X. Zhang, Z. Sui, B. Xu, S. Yue, Y. Luo, W. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494 RSC.
- S. H. Lee, H. W. Kim, J. O. Hwang, W. J. Lee, J. Kwon, C. W. Bielawski, R. S. Ruoff, S. O. Kim and S. H. Lee, Angew. Chem., Int. Ed., 2010, 49, 10084–10088 CrossRef CAS PubMed.
- S. Kabir, K. Artyushkova, A. Serov, P. Atanassov and S. Kabir, ACS Appl. Mater. Interfaces, 2018, 10, 11623–11632 CrossRef CAS PubMed.
- R. Karunagaran, C. Coghlan, C. Shearer, D. Tran, K. Gulat, T. T. Tung, C. Doonan and D. Losic, Materials, 2018, 11, 205 CrossRef PubMed.
- N. Ayasha, V. M. Dhavale and S. Kurungot, Nanoscale, 2017, 9, 12590 RSC.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 442–451 CrossRef CAS PubMed.
- I. S. Amiinu, J. Zhang, Z. Kou, X. Liu, O. K. Asare, H. Zhou, K. Cheng, H. Zhang, L. Mai, M. Pan and S. Mu, ACS Appl. Mater. Interfaces, 2016, 8, 29408–29418 CrossRef CAS PubMed.
- A. Nadeema, S. Walko Priyanka, D. R. Nandini and K. Sreekumar, ACS Appl. Energy Mater., 2018, 1, 5500–5510 CrossRef CAS.
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 21138–21149 CrossRef CAS PubMed.
- S. K. Singh, V. Kashyap, N. Manna, S. N. Bhange, R. Soni, R. Boukherroub, S. Szunerits and S. Kurungot, ACS Catal., 2017, 7, 6700–6710 CrossRef CAS.
- Q. Xiang, Y. Liu, X. Zou, B. Hu, Y. Qiang, D. Yu, W. Yin and C. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 10842–10850 CrossRef CAS PubMed.
- S. N. Bhange, S. M. Unni and S. Kurungot, J. Mater. Chem. A, 2016, 4, 6014–6020 RSC.
- C. Xie, Y. Wang, K. Hu, L. Tao, X. Huang, J. Huo and S. Wang, J. Mater. Chem. A, 2017, 5, 87–91 RSC.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 442–451 CrossRef CAS PubMed.
- D. Mullangi, V. Dhavale, S. Shalini, S. Nandi, S. Collins, T. Woo, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1600110 CrossRef.
- P. K. Gangadharan, S. M. Unni, N. Kumar, P. Ghosh and S. Kurungot, ChemElectroChem, 2017, 4, 2643–2652 CrossRef CAS.
- V. A. Alves, L. A. da Silva and J. F. C. Boodts, Electrochim. Acta, 1998, 44, 1525–1534 CrossRef CAS.
- V. Kashyap and S. Kurungot, ACS Catal., 2018, 8, 3715–3726 CrossRef CAS.
- V. M. Dhavale, S. S. Gaikwad, L. George, R. N. Devi and S. Kurungot, Nanoscale, 2014, 6, 13179–13187 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00808j |
| This journal is © The Royal Society of Chemistry 2020 |