
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A proteome-integrated, carbon source dependent genetic regulatory network in Saccharomyces cerevisiae†
M.
Garcia-Albornoz
 a,
S. W.
Holman
b,
T.
Antonisse
c,
P.
Daran-Lapujade
d,
B.
Teusink
c,
R. J.
Beynon
b and
S. J.
Hubbard
*a
a,
S. W.
Holman
b,
T.
Antonisse
c,
P.
Daran-Lapujade
d,
B.
Teusink
c,
R. J.
Beynon
b and
S. J.
Hubbard
*a
aSchool of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester Academic Health Science Centre, Oxford Road, Manchester M13 9PT, UK. E-mail: simon.hubbard@manchester.ac.uk
bDepartment of Biochemistry, Centre for Proteome Research, Institute of Integrative Biology, University of Liverpool, Liverpool, UK
cSystems Bioinformatics, Amsterdam Institute for Molecules, Medicines and Systems, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands
dDepartment of Biotechnology, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands
First published on 23rd December 2019
Abstract
Integrated regulatory networks can be powerful tools to examine and test properties of cellular systems, such as modelling environmental effects on the molecular bioeconomy, where protein levels are altered in response to changes in growth conditions. Although extensive regulatory pathways and protein interaction data sets exist which represent such networks, few have formally considered quantitative proteomics data to validate and extend them. We generate and consider such data here using a label-free proteomics strategy to quantify alterations in protein abundance for S. cerevisiae when grown on minimal media using glucose, galactose, maltose and trehalose as sole carbon sources. Using a high quality-controlled subset of proteins observed to be differentially abundant, we constructed a proteome-informed network, comprising 1850 transcription factor interactions and 37 chaperone interactions, which defines the major changes in the cellular proteome when growing under different carbon sources. Analysis of the differentially abundant proteins involved in the regulatory network pointed to their significant roles in specific metabolic pathways and function, including glucose homeostasis, amino acid biosynthesis, and carbohydrate metabolic process. We noted strong statistical enrichment in the differentially abundant proteome of targets of known transcription factors associated with stress responses and altered carbon metabolism. This shows how such integrated analysis can lend further experimental support to annotated regulatory interactions, since the proteomic changes capture both magnitude and direction of gene expression change at the level of the affected proteins. Overall this study highlights the power of quantitative proteomics to help define regulatory systems pertinent to environmental conditions.
Introduction
For decades, the budding yeast S. cerevisiae has been a popular model organism for many aspects of systems biology research, supporting the construction of both regulatory and metabolic models.1–3 This is in part driven by its popularity as a highly industrially-relevant organism that pervades many areas of biotechnology, including its use in a wide range of industrial applications such as food, beverages, nutraceuticals, pharmaceuticals, chemicals, and fuels.4,5 It is also a model organism frequently used in biology for understanding fundamental aspects of eukaryotic biological functions.6,7 Despite this focus, a comprehensive, integrated regulatory model that brings together metabolic pathways and the underlying regulatory networks, which displays accurate and robust predictive properties, is still lacking. Progress in this area will enable a greater understanding of eukaryotic cell biology, better hypothesis generation testable in higher organisms, and more effective industrial applications.Much of our current understanding of the regulatory and metabolic networks has been built up by decades of painstaking biochemical analysis, integrated by community-driven curation and annotation efforts to construct and validate increasingly sophisticated models8–10 that capture the key features of central carbon metabolism and other biological functions.11–16 This has helped us understand, rationalise and model how S. cerevisiae is able to utilize a wide range of carbohydrates as a carbon source. Indeed, yeast has genes encoding carbohydrate-utilizing enzymes present in the genome as part of its adaptation and survival strategy, which are well characterised into sets of metabolic pathways. These in turn are coordinated by regulatory networks to regulate changes in gene expression in response to substrate availability. Here, we refer to regulatory networks principally as the relationships between transcription factors and their target genes, which likely manifest as attendant changes in their gene products, proteins, which will therefore affect biological function by changing protein concentrations. Given that chaperone proteins also can influence the final concentration of active, folded proteins, we have also considered their role too. To date, several attempts have been made to elucidate the regulatory routes involved in yeast adaptation under different growth conditions leading to proposals for general regulatory networks.11,13,17 However, all of these studies are based on transcriptomics analyses combined with, or driven by, text mining strategies.1,12,15,16,18,19 Additionally, many of these networks are built on “binary mode” data (positive or negative regulation type), excluding any quantitative information on regulatory effect type and strength.
Although compelling, these approaches do not integrate data from the true effectors of biological metabolism and function, the protein products of the genes concerned, and therefore only give a partial view of the regulatory network. Furthermore, transcription and translational levels are usually considered to be imperfectly correlated; typically, transcript abundances explain approximately one- to two-thirds of the variance in steady-state protein levels, depending on the organism,20–22 showing poorer correlations under altered environmental conditions.23 We posit, therefore, that protein levels should be an informative and complementary data source for the construction of complex and robust regulatory networks. Quantitative proteomics using mass spectrometry (MS) can fulfil this role. Label-free bottom-up shotgun MS-based proteomics can deliver protein identification and quantitative data, with genome-wide measurements now routinely possible.24–29 The elucidation of changes in the proteome under different growth conditions promises an alternative metric from which to infer integrated regulatory and metabolic interactions, and several previous studies have demonstrated its applicability for S. cerevisiae for a variety of similar biological questions.30–33
In this study, we use label-free, bottom-up MS-based proteomics, integrated with protein interaction and transcription factor target data, to derive a regulatory network for Saccharomyces cerevisiae strain CEN.PK113-7D,34 grown on minimal media with glucose, maltose, galactose or trehalose as carbon sources. This novel network therefore captures regulatory interactions supported at multiple levels, supported by direct observation of protein level changes induced by changes in carbon source with respect to the preferred reference carbon source, glucose. It captures the key biological processes controlled in central carbon metabolism, as well as some ones external to it. In particular, we noted the concerted action of a subset of chaperones and transcription factors, working in tandem to modulate protein levels and accommodate altered metabolic pathway activation. The work demonstrates the added value from integrating quantitative proteomic data into genetic regulatory networks in S. cerevisiae, since protein abundances are a direct indicator of post-transcriptional control. These new regulatory findings will have potential use in genetic engineering, bioinformatics studies, clinical research, and industrial applications.
Materials and methods
Biological samples
Saccharomyces cerevisiae strain CEN.PK113-7D (Genotype: MATa URA3 HIS3 LEU2 TRP1 MAL2-8c SUC2)34–37 was grown on minimal (Verduyn) medium38 with two monosaccharides (glucose and galactose) and two disaccharides (maltose and trehalose) as carbon sources in batch cultures with four biological replicates. Samples were grown at 30 °C, 200 rpm shaking in shake flasks, 2% (w/v) carbon source and sampling at OD600 = 1. The medium was buffered with 100 mM phthalic acid/KOH at pH 5.0. Samples were taken at mid-exponential phase. This strain was selected as the most relevant to our industrial collaborators, as it has the closest genetic background to the one they employ. Strains belonging to the CEN.PK family have three MAL loci (MAL2, MAL3 and MAL7),34,36 and use maltose efficiently, which explains why they are preferred in biotechnology.Proteomics
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C, and the supernatant fraction recovered to a low-protein binding microcentrifuge tube on ice. The pellet was resuspended with another 250 μL of 50 mM ammonium bicarbonate containing protease inhibitors. The bottom of the vial was then pierced with a hot needle, the vial placed into a clean low-protein binding microcentrifuge tube, and then centrifuged for 5 min at 4000 rpm, 4 °C. The flow-through and the originally recovered supernatant fraction were combined, the volume measured, and the protein content determined using a standard Bradford assay (Bio-Rad Laboratories Ltd, Hertfordshire, UK).
000 rpm at 4 °C, and the supernatant fraction recovered to a low-protein binding microcentrifuge tube on ice. The pellet was resuspended with another 250 μL of 50 mM ammonium bicarbonate containing protease inhibitors. The bottom of the vial was then pierced with a hot needle, the vial placed into a clean low-protein binding microcentrifuge tube, and then centrifuged for 5 min at 4000 rpm, 4 °C. The flow-through and the originally recovered supernatant fraction were combined, the volume measured, and the protein content determined using a standard Bradford assay (Bio-Rad Laboratories Ltd, Hertfordshire, UK).
One hundred μg quantities from each condition were subjected to tryptic digestion. The proteins were solubilised to a volume of 160 μL using 25 mM ammonium bicarbonate. Denaturation was achieved through the addition of 10 μL of 0.1% (w/v) RapiGest SF (Waters, Elstree, UK) followed by heating at 80 °C for 10 min. Reduction and alkylation was performed by adding 10 μL of 60 mM dithiothreitol with incubation at 60 °C for 10 min, and 10 μL of 180 mM iodoacetamide with incubation at room temperature in the dark for 30 min. Mass spectrometry grade lysyl endopeptidase (Wako Chemicals GmbH, Neuss, Germany) was solubilised to a concentration of 0.1 μg μL−1 in 10 mM acetic acid and 10 μL added to each sample. The samples were incubated at 37 °C for 4 h, after which 10 μL of mass spectrometry grade trypsin (Promega, Madison, WI, USA), also solubilised to a concentration of 0.1 μg μL−1 in 10 mM acetic acid, was added, followed by overnight incubation at 37 °C. Digestion was terminated through the addition of trifluoroacetic acid to 0.5% (v/v). The insoluble degradation products of RapiGest SF were removed by incubation at 37 °C for 2 h and centrifugation at 13000g, 4 °C for 15 min. The supernatant fraction was removed, acetonitrile was added to a final concentration of 3% (v/v) and the sample subjected to LC-MS/MS analysis.
:3.8% (v/v) B (0.1% [v/v] formic acid in water:acetonitrile [80:20, v/v]) to 50% A:50% B over 90 min at a flow rate of 300 nL min−1. The column was then washed at 1% A:99% B for 8 min, and re-equilibrated to starting conditions. The nano-liquid chromatograph was operated under the control of Dionex Chromatography MS Link 2.14.
The nano-electrospray ionisation source was operated in positive polarity under the control of QExactive HF Tune (version 2.5.0.2042), with a spray voltage of 2.2 kV and a capillary temperature of 270 °C. The mass spectrometer was operated in data-dependent acquisition mode. Full MS survey scans between m/z 350–2000 were acquired at a mass resolution of 60000 (full width at half maximum at m/z 200). For MS, the automatic gain control target was set to 3 × 106, and the maximum injection time was 100 ms. The 16 most intense precursor ions with charge states of 2–5 were selected for MS/MS with an isolation window of 1.2 m/z units. Product ion spectra were recorded between m/z 200–2000 at a mass resolution of 30000 (full width at half maximum at m/z 200). For MS/MS, the automatic gain control target was set to 1 × 105, and the maximum injection time was 45 ms. Higher-energy collisional dissociation was performed to fragment the selected precursor ions using a stepped normalised collision energy of 28–30%. Dynamic exclusion was set to 20 s.
Data analysis
The resulting raw data files generated by XCalibur (version 3.1) were processed using MaxQuant software (version 1.6.0.16).40,41 The search parameters were set as follows: label free experiment with default settings; cleaving enzyme trypsin with 2 missed cleavages; Orbitrap instrument with default parameters; variable modifications: oxidation (M) and Acetyl (protein N-term); first search as default; in global parameters, the software was directed to the FASTA file; for advanced identification “Match between runs” was checked; for protein quantification we only used unique, unmodified peptides. All other MaxQuant settings were kept as default. The false discovery rate (FDR) for both accepted peptide spectrum matches and protein matches was set to 1%. The CEN.PK113-7D Yeast FASTA file was downloaded from the Saccharomyces Genome Database (SGD) (https://downloads.yeastgenome.org/sequence/strains/CEN.PK/CEN.PK113-7D/CEN.PK113-7D_Delft_2012_AEHG00000000/).The resulting MaxQuant output was then analysed using the MSstats package (version 3.5.6)42 in the R environment (version 3.3.3) to obtain differential expression fold changes with associated p values. In the first step the MaxQuant output was formatted in R into a table containing Protein name, Peptide sequence, Condition, Bioreplicate, Run, and Intensity. The MaxQtoMSstatsFormat option was used to convert the MaxQuant output tables into the compatible MSstats input table containing protein ID information from the MaxQuant proteinGroups file, intensities from the evidence file, and condition and biological replicates were included in the annotation file (see ESI†). MSstats then converts intensities into log2 values prior to data normalisation across runs using the default option for DDA data analysis, the equalizeMedians option. Summarization was then performed using the robust parameter estimation method Tukey's median polish (TMP) which works with medians across rows and columns. Subsequently, a condition comparison was performed with the groupComparison function within MSstats, which takes as input the output of the dataProcess function. Three pairwise comparisons were made, using MSstats linear mixed effect model, between glucose–galactose, glucose–maltose, and glucose–trehalose, reporting log2 fold change values with attendant p values corrected for multiple comparisons (adjusted p values) for each shared protein. A conservative significance level of an adjusted p value < 0.001, was used to report differentially abundant proteins for each comparison.
Differentially abundant proteins for each comparison (glucose–galactose, glucose–maltose and glucose–trehalose) were subjected to a GO enrichment analysis. The GO enrichment analysis was performed directly on the GO webpage (http://geneontology.org/) using PANTHER43 using the 2534 identified proteins as background obtaining the GO biological processes with default options, selecting Fisher's exact test with FDR multiple test correction for the enrichment analysis.
To build a regulatory network, proteins displaying statistically significant change (adj. p value < 0.001) in all three comparisons (glucose–galactose, glucose–maltose and glucose–trehalose) were identified and their fold change profiles clustered following K means clustering in the R environment. These 85 proteins generated three clusters, the members of which were subjected to a hypergeometric GO and TF enrichment analyses. As before, the GO enrichment analysis was performed directly on the GO webpage using PANTHER43 and using default options with the 2534 identified proteins as background. The TF enrichment analysis was conducted in R (Additional files 12, 16, and 17, ESI†) using all data obtained from YEASTRACT44 on August 29, 2018. For all 85 differentially abundant shared proteins, the list of significant transcription factors and their targets were obtained using all documented data with DNA binding and expression evidence using the 2534 identified proteins as background. The significant TF–target interactions (adj. p value < 0.05) were subsequently combined to build a general network including all interactions. Additionally, all chaperones present in the 85 statistically significant shared proteins were identified along with their targets, taken from an affinity-pulldown MS study,45 and their interactions were added to the TF network. A total of 1887 interactions including 1850 TF and 37 chaperone interactions were identified. Based on these interactions a source–target interaction network was built using Cytoscape (version 3.6.1)46 using the prefuse force directed layout.
Central carbon metabolism pathway including all metabolites and genes involved were obtained from the KEGG database47 (https://www.genome.jp/kegg-bin/show_pathway?sce00010) and literature review.48–52
Results and discussion
Label-free proteomics
To determine the yeast genes for which protein-level expression is affected by growth on alternative carbon sources, we used label-free quantification (LFQ) (Fig. 1). We measured protein abundance in S. cerevisiae when grown on minimal media with either glucose, maltose, galactose or trehalose as the sole carbon source, across four biological replicates, identifying a total of 2534 proteins from 23229 peptides across all four conditions (Table 1) covering 61% of metabolic enzymes (831 out of 1427) reported in the KEGG database (https://www.genome.jp/kegg-bin/get_htext?ko01000.keg).47 These data captured marked differences in protein abundance observed in different growth conditions, supported by excellent repeatability across biological replicates with consistent distributions of MS intensity values53 between experiments (Fig. 2). The MS intensity is a normalised, protein abundance term from MSstats analysis of peptide ion intensity values. We obtained good coverage of the proteome, with 2354 proteins detected in all four growth conditions, demonstrating the power of the label-free approach (Fig. 2C). When protein abundance (using peptide ion intensity values) was compared in a pairwise matrix of all four carbon sources, the overall proteome distributions were very similar, covering a dynamic range of between four and five orders of magnitude. Even from visual inspection, it is clear that trehalose has the largest effect on protein abundance, assessed by the large number of off-diagonal points. The glucose- and maltose-grown pairing were most similar, highlighting that trehalose, despite also being a disaccharide of two glucose molecules, is a much poorer growth substrate (Fig. 2A). Most of the off-axis points tend to the lower abundance levels. Growth on galactose elicits a greater degree of proteome discordance than that between glucose/maltose, but not as marked as that between trehalose and glucose. In terms of proteins that were uniquely identified in a single condition, the numbers were small, but the more pronounced effect of trehalose was once again in evidence, with 46 uniquely identified proteins that were not detected during growth under any of the other three sugars.
 | ||
| Fig. 1 Process diagram of proteome characterization for S. cerevisiae grown on four carbon sources. S. cerevisiae strain CEN.PK113-7D was grown in quadruplicate on minimal media with glucose, galactose, maltose or trehalose as carbon sources. Samples were analysed by LC-MS/MS. The resulting raw files were processed using MaxQuant software and the resulting output was then statistically analysed using MSstats to determine significant changes in abundance. | ||
| Proteins | Peptides | |
|---|---|---|
| All | 2534 | 23229 |
| Galactose | 2477 | 21663 |
| Glucose | 2426 | 20604 |
| Maltose | 2442 | 21029 |
| Trehalose | 2486 | 21836 |
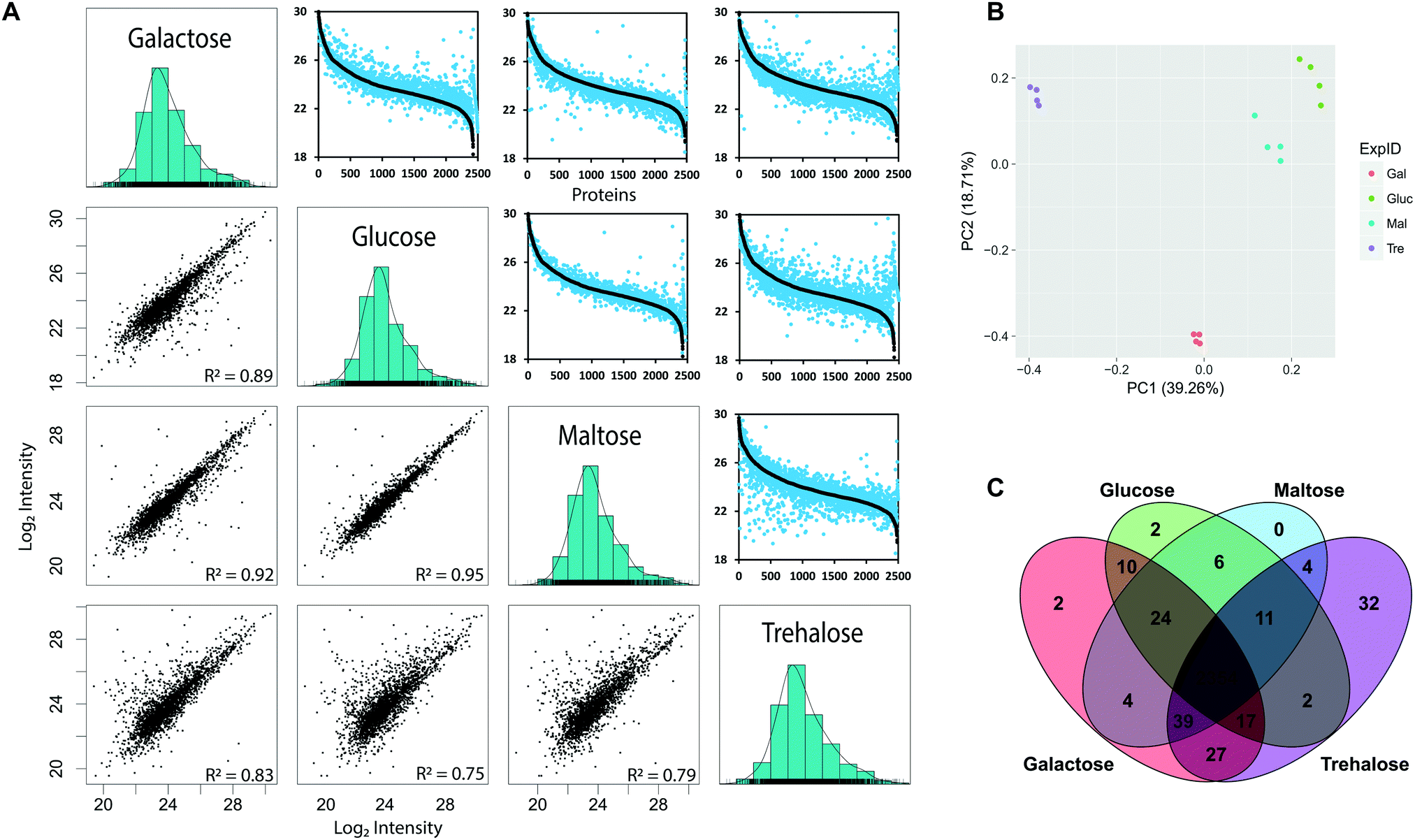 | ||
| Fig. 2 Overview of protein mass spectrometry results. (A) Panels in the lower left corner present scatterplots showing the correlation between median protein MS intensities between the four different carbon sources. Protein intensity is presented as the log10 transformed median MS intensities. Panels in the upper right present a comparison in the dynamic range of detected proteins between the four carbon sources. Proteins were ranked according to their log10 transformed median MS intensities. In black are those proteins from the carbon source indicated on the left and in blue from those on the bottom. (B) PCA analysis for MS intensities in corresponding carbon sources. (C) Overlap of identified proteins between carbon sources. | ||
MS ion intensity data from MaxQuant were analysed using the MSstats package42 to identify significant changes in proteome abundance between strains grown on different carbon sources, compared to glucose. Since cells can sense glucose levels in the media resulting in a cascade of signalling pathways,54–56 it is expected that these changes would reflect changes to carbon metabolism and related metabolic pathways. For this analysis, protein abundance fold changes (FC) with respect to glucose growth conditions and adjusted p values were determined using MSstats describing increased (red) or decreased (blue) levels for proteins grown on the non-glucose carbon sources when compared to glucose (Fig. 3A). In total, 892 proteins show statistically significant differential abundance for the pairwise comparisons (glucose–galactose, glucose–maltose and glucose–trehalose) when considering a highly conservative adjusted p value threshold <0.001 (Fig. 3A and B). Notably, only 69 (up) and 16 (down) proteins change in a coherent fashion under all three comparisons according to this criterion.
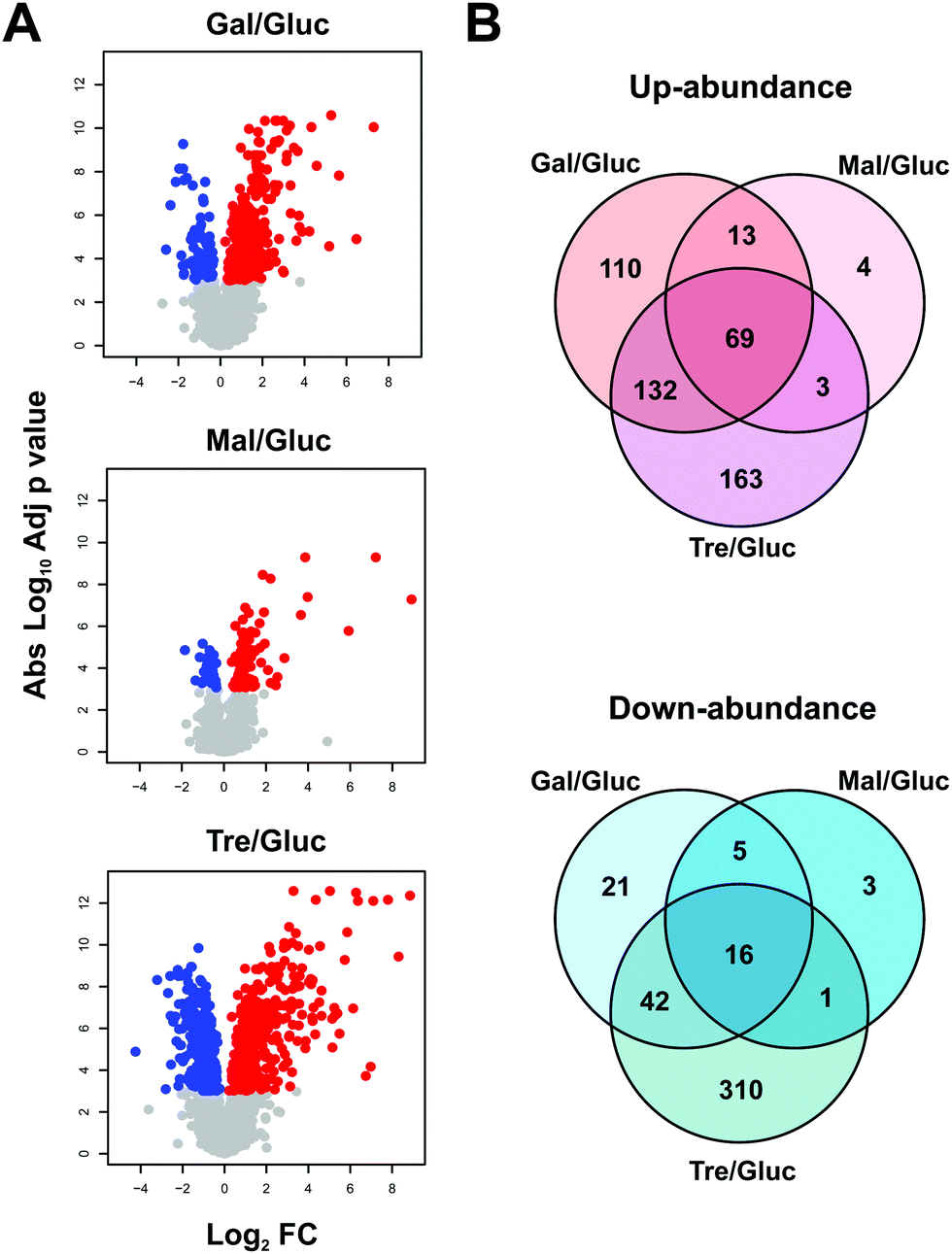 | ||
| Fig. 3 Comparative protein abundance between glucose and galactose, maltose and trehalose. (A) Volcano plots of pairwise carbon source comparisons with glucose, showing the adjusted p value (log10) versus fold change (log2). Volcano plots show those proteins with statistically significant differential abundance between the two carbon sources which were determined from a baseline of an adjusted p value threshold of 0.001. The plots show a noticeable difference in the number of differentially abundant proteins between carbon source comparisons. Red dots indicate positive fold changes and blue dots indicate negative fold changes. (B) Overlap of statistically significant differentially abundant proteins between carbon sources. | ||
Carbon source comparisons
From the lists of differentially abundant proteins for each pairwise comparison with glucose, a GO biological process enrichment analysis was performed (Fig. 4). As would be expected, proteins linked to TCA cycle, respiratory electron transport chain, and carbohydrate metabolic processes increase in abundance, whilst amino acid biosynthetic proteins levels are correspondingly down. This is consistent with the lower observed growth rates of all three carbohydrates (galactose 0.19 h−1, maltose 0.30 h−1 and trehalose 0.08 h−1) when compared to that of glucose (0.36 h−1); growth curves are shown in the Additional files (ESI†). Proteins which increase in abundance in galactose growth compared to glucose show enrichment in carbohydrate catabolic process, TCA cycle, mitochondrial electron transport, and cellular protein modification process. Proteins increasing under maltose growth compared to glucose show enrichment in respiratory electron transport chain, ATP synthesis, tricarboxylic acid cycle, cation transport, and carbohydrate metabolic process, with the maltose metabolism related proteins Mal11p, and Mal32p notable among them. The proteins increasing in abundance in trehalose growth compared to glucose are largely involved in respiratory electron transport chain, ATP synthesis, tricarboxylic acid cycle, translation, and fatty acid metabolic process, with several others involved in the cell stress response. It is therefore highly likely that we were able to identify a group of growth-condition sensitive proteins modulating important signalling processes related to trehalose metabolism and cell stress. Along with this, our data shows an abundance increase in the proteins involved in oxidative phosphorylation while low level changes in enzymes related to glycolysis are observed. These findings are consistent with previous studies reporting the regulation of the mitochondrial respiratory chain pathway by the trehalose pathway through Tps1p and Hxk2p in Saccharomyces cerevisiae.33,57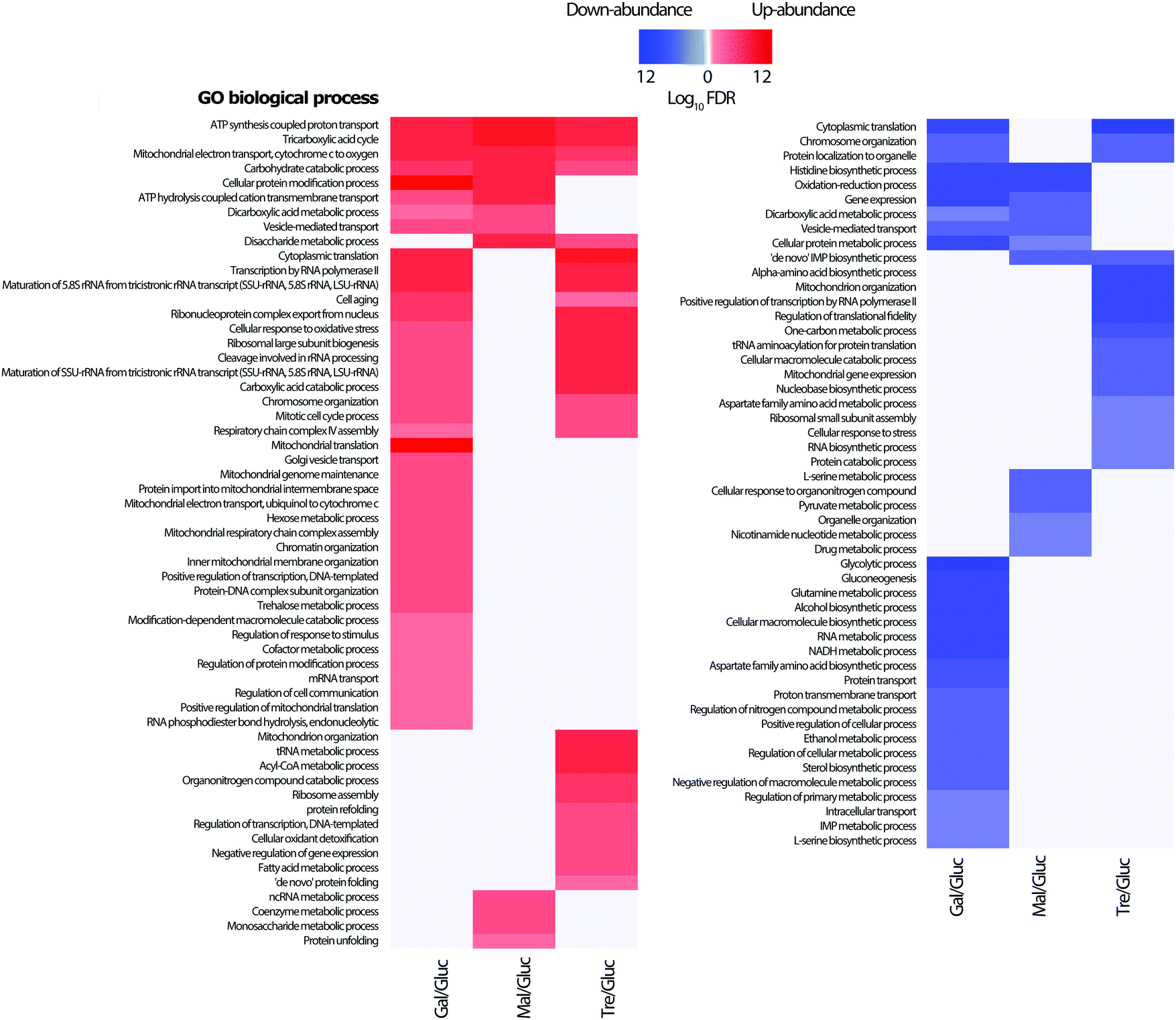 | ||
| Fig. 4 GO enrichment analysis. All differentially abundant proteins for all three comparisons (glucose–galactose, glucose–maltose and glucose–trehalose) were selected and GO biological process enrichment was calculated from the Gene Ontology database, performed separately for up- (red) and down-regulated proteins (blue), with −logFDR values indicated in the scale. | ||
Genetic regulatory network
In a subsequent analysis, we used the conservative set of 85 statistically significant proteins (adj. p value < 0.001) from the three comparisons to explore common patterns of protein expression. Since it has been recently argued that protein abundance profiles offer a buffered and more potent predictor of gene function than transcript levels alone,20,58 a K means clustering was performed which partitioned the proteins into three cluster groups displaying coherent abundance changes (Fig. 5). Notably, groups 1 and 3 included proteins with increasing protein abundance relative to glucose, while group 2 showed the opposite, putatively down-regulated, changes in protein abundances. | ||
| Fig. 5 Heatmap showing the log2 fold changes in relative abundance between glucose and galactose, glucose and maltose and glucose and trehalose for 85 proteins with significant changes (adjusted p value < 0.001) in protein abundance in comparison with glucose media. Rows are organized by groups following a K means clustering which yielded three clusters, with results of a GO enrichment analysis on biological process terms indicated alongside each cluster group, showing attendant FDR values. | ||
The groups contain proteins with common and significant functional enrichment according to Gene Ontology biological process terms.59 Those proteins in group 1 (showing increases in abundance) were enriched in carbohydrate metabolic process, while group 3 (showing more modest increases) in respiratory electron transport chain, ATP synthesis, tricarboxylic acid cycle, cation transport, mitochondrial transport, acyl-CoA metabolic process, protein unfolding, and gene expression. On the other hand, group 2 (showing abundance decreases) were enriched in cellular amino acid biosynthetic process. These trends are entirely consistent with expectation, with increases in processes related to carbohydrate metabolism and respiration accompanied by decreases in processes related to amino acid biosynthesis. Amino acid biosynthesis would be expected to decrease when yeast is grown on alternate carbon sources compared to glucose, as fermentative capacity and growth rate decreases; the latter was confirmed from growth rate curves (see Additional files, ESI†).
Whilst we recognise we have not directly measured regulatory relationships between transcription factors and their targets, we examined the three protein groups for common, apparently coordinated, regulation on the different carbon sources. We looked for transcription factors (TF) associated with the clusters, dependent on protein fold changes, searching for cluster-wise enrichment of known TF targets defined according to the YEASTRACT database.44 The enrichment analysis yielded 50 unique TFs whose targets were significantly (adj. p value < 0.05) over-represented in the differentially abundant protein set, which were considered to be regulating all 85 differentially expressed proteins. This approach to define TF–target networks is particularly valuable since TF proteins are generally low abundance molecules and difficult to measure directly with shotgun MS methods.39 An additional merit of this approach, despite its indirect nature, is that it does not rely on single TF–target interactions. Instead, it detects concerted action on a group of genes which would typify regulons that are often controlled by a common TF. By similar arguments, the concerted action of multiple TFs on a single gene can also be detected. When experimental data is available, this approach will add a new dimension to text mining analysis for specific TF–gene interactions, since the enrichment analysis encompasses concerted regulatory action on common gene clusters. In addition to the transcription factor analysis, we also noted that 7 out of the 85 differentially expressed proteins were chaperones (out of 60 chaperones reported in the study of Gong et al.)45 and that 16 out of the 85 proteins were mapped chaperone targets (out of 1710 reported targets in the same study). Given that transcription factors and chaperones play key roles in the response to environmental change we constructed a carbon source dependent genetic regulatory network inferred from the protein clusters and integrated data. In total, the regulatory network includes 1887 interactions involving 1850 TF and 37 chaperone interactions (Table A13, ESI†). Strikingly, the regulatory network (Fig. 6) has a highly coherent clustering for cluster 1 that mirrors the protein profile clusters in Fig. 5. The protein profile clustering also provides directionality of the changes in protein expression and therefore the effect type (up or down-regulation) as well as the magnitude of the effect (weak or strong) (Fig. 6). For example, cluster 2 shows proteins whose abundance is reduced under alternate carbon sources, and is significantly enriched for targets of the following transcription factors (e.g., RAP1, ROX1, FHL1, YAP6, all with significant enrichment of targets in cluster 2, padjust < 0.05). Collectively, this information provides an integrated picture of a complex regulatory network characterizing the adaptation in yeast at the protein level to changes in carbon source; and is better informed than a simple binary interaction network.
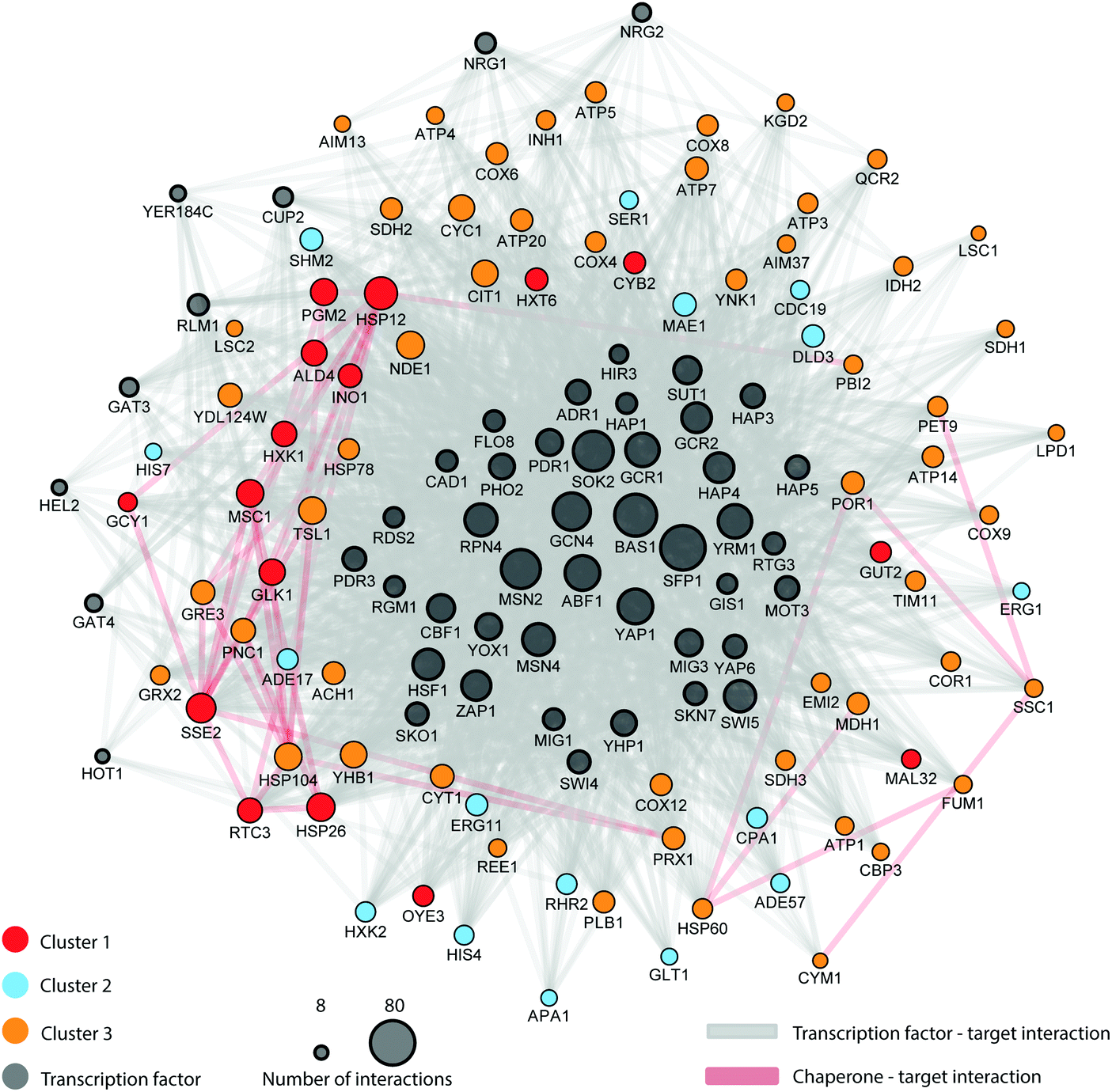 | ||
| Fig. 6 Interaction-based network connecting transcription factors, chaperones and target interactions. All statistically significant shared proteins for all three comparisons (glucose–galactose, glucose–maltose and glucose–trehalose) were paired to their TF and/or chaperone interactions as described in the methods. Gene pairs were subsequently connected in a network using Cytoscape. | ||
The regulatory network is complex, involving several control points (e.g., transcription factors and chaperones) widely spread across several metabolic pathways. This controlling network is not just limited to central carbon metabolism; its influence extends to control points in several connected metabolic pathways including amino acid metabolism, alcohol fermentation, ergosterol biosynthesis, glycerol biosynthesis, nucleotide metabolism, stress response, mitochondrial transport, and oxidative phosphorylation. Indeed, Fig. 6 shows the direct role in which the proteostatic mechanism epitomised by chaperones plays in the regulation of this broad range of general biosynthetic and metabolic processes; notably Hsp26 and Hsp12 (both cluster 1 members) are known to be major actors under dietary restriction and heat shock,60 whilst mitochondrially-active chaperones (Hsp60, Hsp78, Ssc1) lie in cluster 3.
This analysis can lead to the understanding of the spatial organization on the interaction of proteins within the regulatory network. The network reveals the specificity of given TFs acting on amino acid biosynthesis (cluster 1), as well as those proteins related to glycolysis, trehalose metabolism and respiration. Additionally, the network highlights the specificity of the chaperones and target interactions and their localization in the architecture of the regulatory network. The attendant changes in the proteome under different carbon sources were then cast onto the yeast CC metabolism pathway to provide an alternative view of these changes; Fig. 7 summarises the routes whereby changes in nutrient manifest as responses at the proteome level. For example, the switch to galactose precipitates down-regulation of glucose transporter HXT1 with attendant up-regulation of GAL genes as would be expected, and maltose growth causes a similar switch to maltose import (MAL11) and catabolism (MAL32). Genes involved in the regulatory network (highlighted in blue on the figure) are involved in expected regulatory positions inside the CC metabolism network, such as at the initiation of glycolysis, in the shift to alcoholic fermentation, along the TCA cycle, and in starch metabolism (related to stress condition responses).
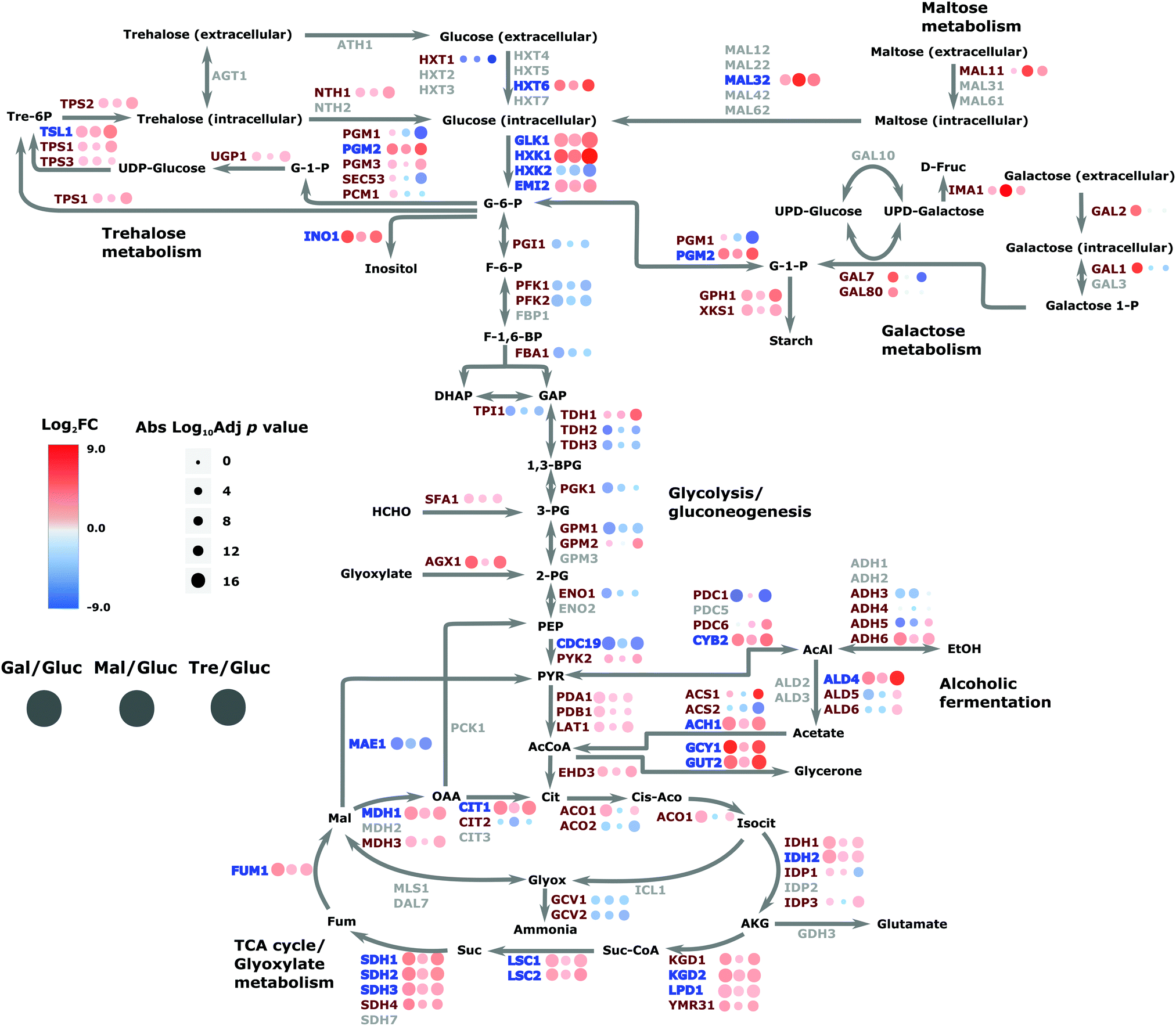 | ||
| Fig. 7 Comparative protein abundance in central carbon metabolism in S. cerevisiae. Diagram of the central carbon metabolism pathway showing the interactions between metabolites and enzymes in which all quantified protein SGD names are indicated in red and those proteins included in the regulatory network in blue; unidentified protein names are in grey. Each dot represents the comparative protein abundance across all three comparisons (glucose–galactose, glucose–maltose and glucose–trehalose). Dot colour indicates the log2 fold changes in abundance and size indicates the adjusted p value, with larger dots indicating higher significance. | ||
Results compared to previously published results
The regulatory network presents a novel integration of quantitative proteomics and existing interactomic data, but bears some comparison with previously published proteomics and transcriptomics studies involved in regulatory networks and nutrient metabolism in yeast. A previous literature-based reconstruction of the nutrient-controlled transcriptional regulatory network regulating metabolism in S. cerevisiae from Herrgard et al.,13 shares 14 TFs with the current study with 36 unique TFs described in our study and 46 in Herrard's. For TF targets, 59 targets are shared with 26 unique targets in our study and 691 in Herrard's (different in silico methods were used to obtain this network). The Gygi group have published two quantitative proteomic studies of yeast grown on different carbon sources.33,61 In the first, 128 TFs were quantified on samples growing on glucose, galactose and raffinose33 of which 30 were identified in our enrichment set. In the more recent study61 considering 10 different carbon sources, when comparing against those proteins with a p value < 0.001 for the cultures growing on glucose–fructose–sucrose against those growing on the other carbon sources, we share 76 out of the 85 differentially abundant proteins in our study. Both these studies used stable isotope labelling via isobaric tagging, whilst our current study used a simpler label free approach to define protein changes. Our study is conceptually very similar, though we have introduced a stringent quality control step and built an integrated network to extend this further.The only direct comparison supported with the Gygi study33 is with our glucose–galactose carbon source proteome changes, which both labs measured. To enable a direct comparison, we estimated log2 fold changes for proteins in their dataset to compare with ours. The overall correlation between estimated fold changes was positive but modest, with an R2 of 0.26 for proteins considered to be differentially abundant (p < 0.01). This suggests the differences in strain, experimental procedures and analytical protocols lead to divergence in results. However, we observed a higher degree of concordance when we compared the TFs whose targets are enriched in the two differentially abundant proteomes, where 31 TFs possess significant enrichments in both data sets (see Additional file A18, ESI†), though again, we stress that methodological differences make this a challenging comparison. Regardless, several notable key TFs involved in central carbon metabolism and glycolysis were present in both datasets. This also included TFs related to general stress responses S. cerevisiae, as would be expected when the organism adapts to growth on a sub-optimal carbon source. This includes Yap1 (and its homolog YAP6) which is a controller of a large specialized oxidative stress response regulon involving several proteins,62 Sok2 which regulates yeast transition to filamentous pseudohyphal growth due to glucose depletion,63,64 and Rpn4 whose induced gene expression is critical for cell viability under stress conditions through a negative feedback loop in the expression of proteasome subunits and other genes.65
Some further well-known TF complexes were also represented in both inferred proteome networks, including Gcr1 and its activator Gcr2, whose complex regulates the expression of several glycolytic genes and have also been found to have effects on genes encoding the TCA cycle and respiration.66 These previously known results are in accordance with our own findings with both TFs being found actively linked to all three groups. Another TF complex found in our study is the HAP complex which coordinates mitochondrial and nuclear gene expression related to the respiratory chain and other mitochondrial processes that define the metabolic state of the cell.67
Major stress response transcriptional activators were also enriched, such as Msn2 and Msn4 which play major roles in the general stress response by mediating the transcription of hundreds of genes; however, there remains a lack of information regarding the entirety of the network of proteins that regulate its activity.68 Therefore, we believe the proposed regulatory network expands the knowledge on these (and other) TFs via the direct observation of changes at the proteome level. For example, when performing an enrichment analysis with the observed differentially abundant proteins regulated by Msn2/4 they showed enrichment in several processes such as trehalose metabolism in response to stress, cellular glucose homeostasis, carbohydrate phosphorylation, tricarboxylic acid cycle, and glycolytic process. These results can guide future research related to those proteins and processes involved in cell stress response and their control by the Msn2/4 TFs.
A final noteworthy TF is Gcn4, the transcriptional activator linked to amino acid synthesis, and whose substrates were strongly enriched in the 85 protein set (p < 3.4 × 10−6). This TF is derepressed in amino acid-deprived cells, leading to transcriptional induction of nearly all genes encoding amino acid biosynthetic enzymes.69 This regulatory response enables cells to limit their amino acid consumption and divert resources instead into amino acid biosynthesis when facing nutrient-poor environments, and the enrichment of its substrates in the differentially abundant protein pool is consistent with expectation.
Although many TFs were enriched in both datasets, some were exclusive to our proteomics data. Of note, members of the Hap complex (Hap3 and Hap5) were found, which are known regulators of respiratory genes that are activated in glucose-depleted media. Similarly, Nrg2 targets were enriched in our data, a transcription factor activated in stochastic pulses of nuclear localization in response to low glucose.70 We also detected enrichment for the histone regulators Hel2 and Hir3,71,72 and the yeast homeobox Yhp1,73,74 as well as Yap6 another known stress factor.75
Conclusions
In response to the change of carbon source, a widespread reorganisation of the protein complement is observed in yeast. This is underpinned by a common regulatory network that controls the response of the cell based on carbon source availability. In this study, we used a label-free MS-based strategy to define key changes in the proteome, integrating this with existing regulatory relationships, and constructing a carbon source-dependent genetic regulatory network for S. cerevisiae. The proteomic analysis is in good accordance with previously published works33,39,61 and the attendant regulatory network brings together diverse functions including stress defence, amino acid synthesis and central carbon metabolism. Rather than transcriptomic data, our network is built on, and derived from, observed protein abundance changes. This constitutes an alternative approach to add value to existing regulatory network data since protein changes can buffer from co-expression artefacts at the transcript level, such as those arising from proximal expression patterns.20 Indeed, the yeast response to alternate carbon sources has been very well characterised at the global transcriptome level;11–13,15–17 however, to the best of our knowledge, no large-scale proteome studies have yet generated or refined a network and this is the first such attempt, using label free proteomics. We suggest that our protein-centric approach may therefore offer additional advantages, by focussing on the ‘end product’ (the proteins) of a regulatory pathway, integrating the complex interactions between TFs, chaperones and key proteins in carbon source metabolism.The regulatory network is a complex network involving 1887 interactions including 1850 TF–target and 37 chaperone–target interactions with control points on different metabolic pathways such as, amino acid metabolism, alcohol fermentation, ergosterol biosynthesis, glycerol biosynthesis, central carbon metabolism, nucleotide metabolism, stress response, mitochondrial protein biosynthesis, and oxidative phosphorylation. While many genetic regulatory networks informing on carbon source availability have been previously described, these networks have been based on indirect text mining computational strategies and transcriptomics analyses.1,12,15,16,18,19 Additionally, in the present work we provide evidence from which regulatory effect type and strength can be predicted, from the attendant protein abundance changes in terms of direction (up or down) and size. This provides supporting evidence as to whether the putative regulation is weak or strong, and whether it appears to be activation or suppression of the affected protein. We recognise that we do not directly observe the regulatory mechanism itself, since we do not measure transcriptional or translational control directly, nor protein turnover; however, the observed proteome changes can be judged to be in accordance with previously characterised and known TF–target and chaperone–target interactions linked to dietary stress, lending confidence to the network. Whilst we recognise that an increase/decrease in intracellular enzyme concentration might not always lead to a directly equivalent change in glycolytic flux (for example see ref. 76 and 77), we do see attendant changes in the metabolic map (see Fig. 7). Consequently, we believe this adds to understanding of signalling pathways in yeast central carbon metabolism, and the integrated MS-informed network approach offers a convenient tool to construct complex cell signalling networks and has wide applicability for genome-scale metabolic modelling which considers the proteome.
Availability of supporting data
The raw mass spectrometry data has been deposited to the ProteomeXchange Consortium with the identifier PXD009420. For R script, Maxquant and MSstats output files, see Additional files (ESI†).Funding
We thank Biotechnology and Biological Sciences Research Council (Manchester: BB/M025748/1, Liverpool: BB/M025756/1) for funding.Authors' contributions
TA, PDL, BT, RJB and SJH conceived and designed the experiments. SWH prepared samples and generated the MS data. MGA performed bioinformatics analyses. MGA, SWH, RJB and SJH wrote and edited the paper.Conflicts of interest
All authors declare no potential conflict of interest.Acknowledgements
We thank the members of the EcoYeast/EraSysApp, and Dr Philip Brownridge for instrument support. We gratefully acknowledge our industrial partners Tania Gerard from Roquette and Liang Wu from DSM for their assistance and helpful comments on the manuscript.References
- B. A. Blount, T. Weenink and T. Ellis, Construction of synthetic regulatory networks in yeast, FEBS Lett., 2012, 586, 2112–2121 CrossRef CAS PubMed.
- T. Laomettachit, K. C. Chen, W. T. Baumann and J. J. Tyson, A Model of Yeast Cell-Cycle Regulation Based on a Standard Component Modeling Strategy for Protein Regulatory Networks, PLoS One, 2016, 11, e0153738 CrossRef PubMed.
- T. Osterlund, I. Nookaew, S. Bordel and J. Nielsen, Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling, BMC Syst. Biol., 2013, 7, 36 CrossRef PubMed.
- M. Garcia-Albornoz and J. Nielsen, Application of genome-scale metabolic models in metabolic engineering, Ind. Biotechnol., 2013, 9, 203–214 CrossRef CAS.
- T. Osterlund, I. Nookaew and J. Nielsen, Fifteen years of large scale metabolic modeling of yeast: developments and impacts, Biotechnol. Adv., 2012, 30, 979–988 CrossRef PubMed.
- D. Botstein and G. R. Fink, Yeast: an experimental organism for 21st Century biology, Genetics, 2011, 189, 695–704 CrossRef CAS PubMed.
- S. Feyder, J. O. De Craene, S. Bar, D. L. Bertazzi and S. Friant, Membrane trafficking in the yeast Saccharomyces cerevisiae model, Int. J. Mol. Sci., 2015, 16, 1509–1525 CrossRef CAS PubMed.
- F. Emmert-Streib, M. Dehmer and B. Haibe-Kains, Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks, Front. Cell Dev. Biol., 2014, 2, 38 Search PubMed.
- M. S. Halfon, Perspectives on Gene Regulatory Network Evolution, Trends Genet., 2017, 33, 436–447 CrossRef CAS PubMed.
- L. T. Macneil and A. J. Walhout, Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression, Genome Res., 2011, 21, 645–657 CrossRef CAS PubMed.
- S. M. Fendt, A. P. Oliveira, S. Christen, P. Picotti, R. C. Dechant and U. Sauer, Unraveling condition-dependent networks of transcription factors that control metabolic pathway activity in yeast, Mol. Syst. Biol., 2010, 6, 432 CrossRef CAS PubMed.
- L. Geistlinger, G. Csaba, S. Dirmeier, R. Kuffner and R. Zimmer, A comprehensive gene regulatory network for the diauxic shift in Saccharomyces cerevisiae, Nucleic Acids Res., 2013, 41, 8452–8463 CrossRef CAS PubMed.
- M. J. Herrgard, B. S. Lee, V. Portnoy and B. O. Palsson, Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae, Genome Res., 2006, 16, 627–635 CrossRef CAS PubMed.
- H. Lavoie, H. Hogues and M. Whiteway, Rearrangements of the transcriptional regulatory networks of metabolic pathways in fungi, Curr. Opin. Microbiol., 2009, 12, 655–663 CrossRef CAS PubMed.
- B. Turcotte, X. B. Liang, F. Robert and N. Soontorngun, Transcriptional regulation of nonfermentable carbon utilization in budding yeast, FEMS Yeast Res., 2010, 10, 2–13 CrossRef CAS PubMed.
- K. Weinhandl, M. Winkler, A. Glieder and A. Camattari, Carbon source dependent promoters in yeasts, Microb. Cell Fact., 2014, 13, 5 CrossRef PubMed.
- Z. Hu, P. J. Killion and V. R. Iyer, Genetic reconstruction of a functional transcriptional regulatory network, Nat. Genet., 2007, 39, 683–687 CrossRef CAS PubMed.
- T. R. Hughes and C. G. de Boer, Mapping yeast transcriptional networks, Genetics, 2013, 195, 9–36 CrossRef CAS PubMed.
- D. Kim, M. S. Kim and K. H. Cho, The core regulation module of stress-responsive regulatory networks in yeast, Nucleic Acids Res., 2012, 40, 8793–8802 CrossRef CAS PubMed.
- G. Kustatscher, P. Grabowski and J. Rappsilber, Pervasive coexpression of spatially proximal genes is buffered at the protein level, Mol. Syst. Biol., 2017, 13, 937 CrossRef PubMed.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Global quantification of mammalian gene expression control, Nature, 2011, 473, 337–342 CrossRef PubMed.
- C. Vogel and E. M. Marcotte, Insights into the regulation of protein abundance from proteomic and transcriptomic analyses, Nat. Rev. Genet., 2012, 13, 227–232 CrossRef CAS PubMed.
- J. B. Smirnova, J. N. Selley, F. Sanchez-Cabo, K. Carroll, A. A. Eddy, J. E. McCarthy, S. J. Hubbard, G. D. Pavitt, C. M. Grant and M. P. Ashe, Global gene expression profiling reveals widespread yet distinctive translational responses to different eukaryotic translation initiation factor 2B-targeting stress pathways, Mol. Cell. Biol., 2005, 25, 9340–9349 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Mass-spectrometric exploration of proteome structure and function, Nature, 2016, 537, 347–355 CrossRef CAS PubMed.
- D. Kappei, M. Scheibe, M. Paszkowski-Rogacz, A. Bluhm, T. I. Gossmann, S. Dietz, M. Dejung, H. Herlyn, F. Buchholz, M. Mann and F. Butter, Phylointeractomics reconstructs functional evolution of protein binding, Nat. Commun., 2017, 8, 14334 CrossRef CAS PubMed.
- M. C. Kuang, P. D. Hutchins, J. D. Russell, J. J. Coon and C. T. Hittinger, Ongoing resolution of duplicate gene functions shapes the diversification of a metabolic network, eLife, 2016, 5, e19027 CrossRef PubMed.
- T. K. Sato, M. Tremaine, L. S. Parreiras, A. S. Hebert, K. S. Myers, A. J. Higbee, M. Sardi, S. J. McIlwain, I. M. Ong, R. J. Breuer, R. Avanasi Narasimhan, M. A. McGee, Q. Dickinson, A. La Reau, D. Xie, M. Tian, J. L. Reed, Y. Zhang, J. J. Coon, C. T. Hittinger, A. P. Gasch and R. Landick, Directed Evolution Reveals Unexpected Epistatic Interactions That Alter Metabolic Regulation and Enable Anaerobic Xylose Use by Saccharomyces cerevisiae, PLoS Genet., 2016, 12, e1006372 CrossRef PubMed.
- E. Shishkova, A. S. Hebert and J. J. Coon, Now, More Than Ever, Proteomics Needs Better Chromatography, Cell Syst., 2016, 3, 321–324 CrossRef CAS PubMed.
- J. A. Stefely, N. W. Kwiecien, E. C. Freiberger, A. L. Richards, A. Jochem, M. J. P. Rush, A. Ulbrich, K. P. Robinson, P. D. Hutchins, M. T. Veling, X. Guo, Z. A. Kemmerer, K. J. Connors, E. A. Trujillo, J. Sokol, H. Marx, M. S. Westphall, A. S. Hebert, D. J. Pagliarini and J. J. Coon, Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling, Nat. Biotechnol., 2016, 34, 1191–1197 CrossRef CAS PubMed.
- S. Liu, L. Li, C. Tong, Q. Zhao, P. A. Lukyanov, O. V. Chernikov and W. Li, Quantitative proteomic analysis of the effects of a GalNAc/Man-specific lectin CSL on yeast cells by label-free LC-MS, Int. J. Biol. Macromol., 2016, 85, 530–538 CrossRef CAS PubMed.
- A. L. Mosley, L. Florens, Z. Wen and M. P. Washburn, A label free quantitative proteomic analysis of the Saccharomyces cerevisiae nucleus, J. Proteomics, 2009, 72, 110–120 CrossRef CAS PubMed.
- A. L. Mosley, M. E. Sardiu, S. G. Pattenden, J. L. Workman, L. Florens and M. P. Washburn, Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes, Mol. Cell. Proteomics, 2011, 10, M110 000687 CrossRef PubMed.
- J. A. Paulo, J. D. O'Connell, A. Gaun and S. P. Gygi, Proteome-wide quantitative multiplexed profiling of protein expression: carbon-source dependency in Saccharomyces cerevisiae, Mol. Biol. Cell, 2015, 26, 4063–4074 CrossRef CAS PubMed.
- J. F. Nijkamp, M. van den Broek, E. Datema, S. de Kok, L. Bosman, M. A. Luttik, P. Daran-Lapujade, W. Vongsangnak, J. Nielsen, W. H. Heijne, P. Klaassen, C. J. Paddon, D. Platt, P. Kotter, R. C. van Ham, M. J. Reinders, J. T. Pronk, D. de Ridder and J. M. Daran, De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology, Microb. Cell Fact., 2012, 11, 36 CrossRef CAS PubMed.
- K. D. Entian and P. Kötter, 25 Yeast Genetic Strain and Plasmid Collections, Methods Microbiol., 2007, 36, 629–666 CAS.
- A. N. Salazar, A. R. Gorter de Vries, M. van den Broek, M. Wijsman, P. de la Torre Cortes, A. Brickwedde, N. Brouwers, J. G. Daran and T. Abeel, Nanopore sequencing enables near-complete de novo assembly of Saccharomyces cerevisiae reference strain CEN.PK113-7D, FEMS Yeast Res., 2017, 17(7), fox074 CrossRef PubMed.
- J. P. van Dijken, J. Bauer, L. Brambilla, P. Duboc, J. M. Francois, C. Gancedo, M. L. Giuseppin, J. J. Heijnen, M. Hoare, H. C. Lange, E. A. Madden, P. Niederberger, J. Nielsen, J. L. Parrou, T. Petit, D. Porro, M. Reuss, N. van Riel, M. Rizzi, H. Y. Steensma, C. T. Verrips, J. Vindelov and J. T. Pronk, An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains, Enzyme Microb. Technol., 2000, 26, 706–714 CrossRef CAS PubMed.
- C. Verduyn, E. Postma, W. A. Scheffers and J. P. Van Dijken, Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation, Yeast, 1992, 8, 501–517 CrossRef CAS PubMed.
- C. Lawless, S. W. Holman, P. Brownridge, K. Lanthaler, V. M. Harman, R. Watkins, D. E. Hammond, R. L. Miller, P. F. Sims, C. M. Grant, C. E. Eyers, R. J. Beynon and S. J. Hubbard, Direct and Absolute Quantification of over 1800 Yeast Proteins via Selected Reaction Monitoring, Mol. Cell. Proteomics, 2016, 15, 1309–1322 CrossRef CAS PubMed.
- J. Cox and M. Mann, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS PubMed.
- S. Tyanova, T. Temu and J. Cox, The MaxQuant computational platform for mass spectrometry-based shotgun proteomics, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS PubMed.
- M. Choi, C. Y. Chang, T. Clough, D. Broudy, T. Killeen, B. MacLean and O. Vitek, MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments, Bioinformatics, 2014, 30, 2524–2526 CrossRef CAS PubMed.
- H. Mi, X. Huang, A. Muruganujan, H. Tang, C. Mills, D. Kang and P. D. Thomas, PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements, Nucleic Acids Res., 2017, 45, D183–D189 CrossRef CAS PubMed.
- M. C. Teixeira, P. T. Monteiro, M. Palma, C. Costa, C. P. Godinho, P. Pais, M. Cavalheiro, M. Antunes, A. Lemos, T. Pedreira and I. Sa-Correia, YEASTRACT: an upgraded database for the analysis of transcription regulatory networks in Saccharomyces cerevisiae, Nucleic Acids Res., 2018, 46, D348–D353 CrossRef CAS PubMed.
- Y. Gong, Y. Kakihara, N. Krogan, J. Greenblatt, A. Emili, Z. Zhang and W. A. Houry, An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell, Mol. Syst. Biol., 2009, 5, 275 CrossRef PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- M. Kanehisa, Y. Sato, M. Kawashima, M. Furumichi and M. Tanabe, KEGG as a reference resource for gene and protein annotation, Nucleic Acids Res., 2016, 44, D457–D462 CrossRef CAS PubMed.
- R. Costenoble, P. Picotti, L. Reiter, R. Stallmach, M. Heinemann, U. Sauer and R. Aebersold, Comprehensive quantitative analysis of central carbon and amino-acid metabolism in Saccharomyces cerevisiae under multiple conditions by targeted proteomics, Mol. Syst. Biol., 2011, 7, 464 CrossRef PubMed.
- J. Francois and J. L. Parrou, Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae, FEMS Microbiol. Rev., 2001, 25, 125–145 CrossRef CAS PubMed.
- M. Jules, V. Guillou, J. Francois and J. L. Parrou, Two distinct pathways for trehalose assimilation in the yeast Saccharomyces cerevisiae, Appl. Environ. Microbiol., 2004, 70, 2771–2778 CrossRef CAS PubMed.
- H. Phenix, K. Morin, C. Batenchuk, J. Parker, V. Abedi, L. Yang, L. Tepliakova, T. J. Perkins and M. Kaern, Quantitative epistasis analysis and pathway inference from genetic interaction data, PLoS Comput. Biol., 2011, 7, e1002048 CrossRef CAS PubMed.
- F. Tripodi, R. Nicastro, V. Reghellin and P. Coccetti, Post-translational modifications on yeast carbon metabolism: regulatory mechanisms beyond transcriptional control, Biochim. Biophys. Acta, 2015, 1850, 620–627 CrossRef CAS PubMed.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ, Mol. Cell. Proteomics, 2014, 13, 2513–2526 CrossRef CAS PubMed.
- S. Busti, P. Coccetti, L. Alberghina and M. Vanoni, Glucose signaling-mediated coordination of cell growth and cell cycle in Saccharomyces cerevisiae, Sensors, 2010, 10, 6195–6240 CrossRef CAS PubMed.
- F. Rolland, J. Winderickx and J. M. Thevelein, Glucose-sensing and -signalling mechanisms in yeast, FEMS Yeast Res., 2002, 2, 183–201 CrossRef CAS PubMed.
- G. M. Santangelo, Glucose signaling in Saccharomyces cerevisiae, Microbiol. Mol. Biol. Rev., 2006, 70, 253–282 CrossRef CAS PubMed.
- A. Noubhani, O. Bunoust, B. M. Bonini, J. M. Thevelein, A. Devin and M. Rigoulet, The trehalose pathway regulates mitochondrial respiratory chain content through hexokinase 2 and cAMP in Saccharomyces cerevisiae, J. Biol. Chem., 2009, 284, 27229–27234 CrossRef CAS PubMed.
- J. Wang, Z. Ma, S. A. Carr, P. Mertins, H. Zhang, Z. Zhang, D. W. Chan, M. J. Ellis, R. R. Townsend, R. D. Smith, J. E. McDermott, X. Chen, A. G. Paulovich, E. S. Boja, M. Mesri, C. R. Kinsinger, H. Rodriguez, K. D. Rodland, D. C. Liebler and B. Zhang, Proteome Profiling Outperforms Transcriptome Profiling for Coexpression Based Gene Function Prediction, Mol. Cell. Proteomics, 2017, 16, 121–134 CrossRef CAS PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nat. Genet., 2000, 25, 25–29 CrossRef CAS PubMed.
- A. F. Jarnuczak, M. G. Albornoz, C. E. Eyers, C. M. Grant and S. J. Hubbard, A quantitative and temporal map of proteostasis during heat shock in Saccharomyces cerevisiae, Mol. Omics, 2018, 14, 37–52 RSC.
- J. A. Paulo, J. D. O'Connell, R. A. Everley, J. O'Brien, M. A. Gygi and S. P. Gygi, Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources, J. Proteomics, 2016, 148, 85–93 CrossRef CAS PubMed.
- J. Lee, C. Godon, G. Lagniel, D. Spector, J. Garin, J. Labarre and M. B. Toledano, Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast, J. Biol. Chem., 1999, 274, 16040–16046 CrossRef CAS PubMed.
- P. J. Cullen and G. F. Sprague, Jr., Glucose depletion causes haploid invasive growth in yeast, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13619–13624 CrossRef CAS PubMed.
- X. Pan and J. Heitman, Sok2 regulates yeast pseudohyphal differentiation via a transcription factor cascade that regulates cell–cell adhesion, Mol. Cell. Biol., 2000, 20, 8364–8372 CrossRef CAS PubMed.
- X. Wang, H. Xu, S. W. Ha, D. Ju and Y. Xie, Proteasomal degradation of Rpn4 in Saccharomyces cerevisiae is critical for cell viability under stressed conditions, Genetics, 2010, 184, 335–342 CrossRef CAS PubMed.
- H. Sasaki and H. Uemura, Influence of low glycolytic activities in gcr1 and gcr2 mutants on the expression of other metabolic pathway genes in Saccharomyces cerevisiae, Yeast, 2005, 22, 111–127 CrossRef CAS PubMed.
- S. Buschlen, J. M. Amillet, B. Guiard, A. Fournier, C. Marcireau and M. Bolotin-Fukuhara, The S. Cerevisiae HAP complex, a key regulator of mitochondrial function, coordinates nuclear and mitochondrial gene expression, Comp. Funct. Genomics, 2003, 4, 37–46 CrossRef CAS PubMed.
- A. Sadeh, N. Movshovich, M. Volokh, L. Gheber and A. Aharoni, Fine-tuning of the Msn2/4-mediated yeast stress responses as revealed by systematic deletion of Msn2/4 partners, Mol. Biol. Cell, 2011, 22, 3127–3138 CrossRef CAS PubMed.
- A. G. Hinnebusch, Translational regulation of GCN4 and the general amino acid control of yeast, Annu. Rev. Microbiol., 2005, 59, 407–450 CrossRef CAS PubMed.
- A. Kummel, J. C. Ewald, S. M. Fendt, S. J. Jol, P. Picotti, R. Aebersold, U. Sauer, N. Zamboni and M. Heinemann, Differential glucose repression in common yeast strains in response to HXK2 deletion, FEMS Yeast Res., 2010, 10, 322–332 CrossRef CAS PubMed.
- M. A. Osley and D. Lycan, Trans-acting regulatory mutations that alter transcription of Saccharomyces cerevisiae histone genes, Mol. Cell. Biol., 1987, 7, 4204–4210 CrossRef CAS PubMed.
- R. K. Singh, M. Gonzalez, M. H. Kabbaj and A. Gunjan, Novel E3 ubiquitin ligases that regulate histone protein levels in the budding yeast Saccharomyces cerevisiae, PLoS One, 2012, 7, e36295 CrossRef CAS PubMed.
- E. Kaufmann, In vitro binding to the leucine tRNA gene identifies a novel yeast homeobox gene, Chromosoma, 1993, 102, 174–179 CrossRef CAS PubMed.
- T. Kunoh, Y. Kaneko and S. Harashima, YHP1 encodes a new homeoprotein that binds to the IME1 promoter in Saccharomyces cerevisiae, Yeast, 2000, 16, 439–449 CrossRef CAS PubMed.
- G. Liu, D. Bergenholm and J. Nielsen, Genome-Wide Mapping of Binding Sites Reveals Multiple Biological Functions of the Transcription Factor Cst6p in Saccharomyces cerevisiae, mBio, 2016, 7(3), e00559-16 CrossRef PubMed.
- J. Heinisch, Isolation and characterization of the two structural genes coding for phosphofructokinase in yeast, Mol. Gen. Genet., 1986, 202, 75–82 CrossRef CAS PubMed.
- I. Schaaff, J. Heinisch and F. K. Zimmermann, Overproduction of glycolytic enzymes in yeast, Yeast, 1989, 5, 285–290 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A1 MaxQuant settings, A2 MaxQuant annotation table, A3 MaxQuant evidence output table, A4 MaxQuant proteinGroup output table, A5 R script to run MSstats analysis, A6 MSstats quantData output table, A7 MSstats RunlevelData output table, A8 MSstats comparisons output table, A9 log2FCs and adjusted p values for all three comparisons, A10 differentially abundant proteins by comparison with their GO enriched biological terms, A11 differentially abundant shared proteins divided by Kmean clusters with their GO enriched biological terms, A12 TF enrichment analysis, A13 Fig. 6 edges to run Cytoscape figure including all TF–target and chaperone–target interactions, A14 Fig. 6 nodes to run Cytoscape figure, A15 Fig. 6 Cytoscape file, A16 enrichment analysis output table, A17 R script to run the TF enrichment analysis, CENPK113-7D fasta file, MaxQuant mqpar file. A18 TF enrichment analysis Gygi vs. 4C. See DOI: 10.1039/c9mo00136k |
| This journal is © The Royal Society of Chemistry 2020 |