
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural product inspired optimization of a selective TRPV6 calcium channel inhibitor†
Micael Rodrigues
Cunha
 ab,
Rajesh
Bhardwaj
c,
Aline Lucie
Carrel
a,
Sonja
Lindinger
d,
Christoph
Romanin
d,
Roberto
Parise-Filho
*b,
Matthias A.
Hediger
*c and
Jean-Louis
Reymond
*a
ab,
Rajesh
Bhardwaj
c,
Aline Lucie
Carrel
a,
Sonja
Lindinger
d,
Christoph
Romanin
d,
Roberto
Parise-Filho
*b,
Matthias A.
Hediger
*c and
Jean-Louis
Reymond
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
bDepartment of Pharmacy, University of São Paulo, Prof. Lineu Prestes Avenue 580, 05508-000 São Paulo, Brazil. E-mail: roberto.parise@usp.br
cDepartment of Nephrology and Hypertension, University Hospital Bern, Inselspital, 3010 Bern, Switzerland. E-mail: matthias.hediger@ibmm.unibe.ch
dInstitute of Biophysics, Johannes Kepler University Linz, Gruberstrasse 40, 4020 Linz, Austria
First published on 16th July 2020
Abstract
Transient receptor potential vanilloid 6 (TRPV6) is a calcium channel implicated in multifactorial diseases and overexpressed in numerous cancers. We recently reported the phenyl-cyclohexyl-piperazine cis-22a as the first submicromolar TRPV6 inhibitor. This inhibitor showed a seven-fold selectivity against the closely related calcium channel TRPV5 and no activity on store-operated calcium channels (SOC), but very significant off-target effects and low microsomal stability. Here, we surveyed analogues incorporating structural features of the natural product capsaicin and identified 3OG, a new oxygenated analog with similar potency against TRPV6 (IC50 = 0.082 ± 0.004 μM) and ion channel selectivity, but with high microsomal stability and very low off-target effects. This natural product-inspired inhibitor does not exhibit any non-specific toxicity effects on various cell lines and is proposed as a new tool compound to test pharmacological inhibition of TRPV6 mediated calcium flux in disease models.
Introduction
TRPV6 is a Ca2+-selective member of the transient receptor potential vanilloid (TRPV) family, referred to as the gatekeeper of transepithelial Ca2+ transport.1–3 The channel is primarily found in the human intestine, kidney and placenta and in a number of exocrine organs such as pancreas, prostate and mammary gland.4,5 It is known that TRPV6 has an important contribution to multifactorial diseases.6,7 For instance, TRPV6-deficient mice have diminished fertility, osteopenia and reduced body weight,5,8 whilst human TRPV6 mutations, which render the channel less functional, cause transient neonatal hyperparathyroidism (TNH) and skeletal abnormalities.9,10 These pathological findings are related to tissues in which TRPV6-expression at normal levels is essential for Ca2+ homeostasis. On the other hand, TRPV6 expression was found to be abnormally upregulated in numerous cancers of breast11–13 and prostate tissues,14 compared to normal tissues.15,16We recently reported cis-22a (1, Fig. 1) as the first submicromolar small molecule inhibitor of hTRPV6 mediated Ca2+ flux.17 Compound 1 was highly selective for hTRPV6 against other Ca2+ channels, and showed a 7-fold selectivity against the closely related rTRPV5. Inhibitor 1 furthermore decreased the cell viability of a tumor cell line overexpressing TRPV6 as reported with siRNA knockdown experiments; however the effect only occurred at high concentrations (IC50 ≈ 25 μM). However, 1 also inhibited other targets such as hERG, dopamine and muscarinic receptors, and was highly unstable against microsomal degradation, implying that this inhibitor only had limited applicability as a tool compound to study hTRPV6 inhibition.
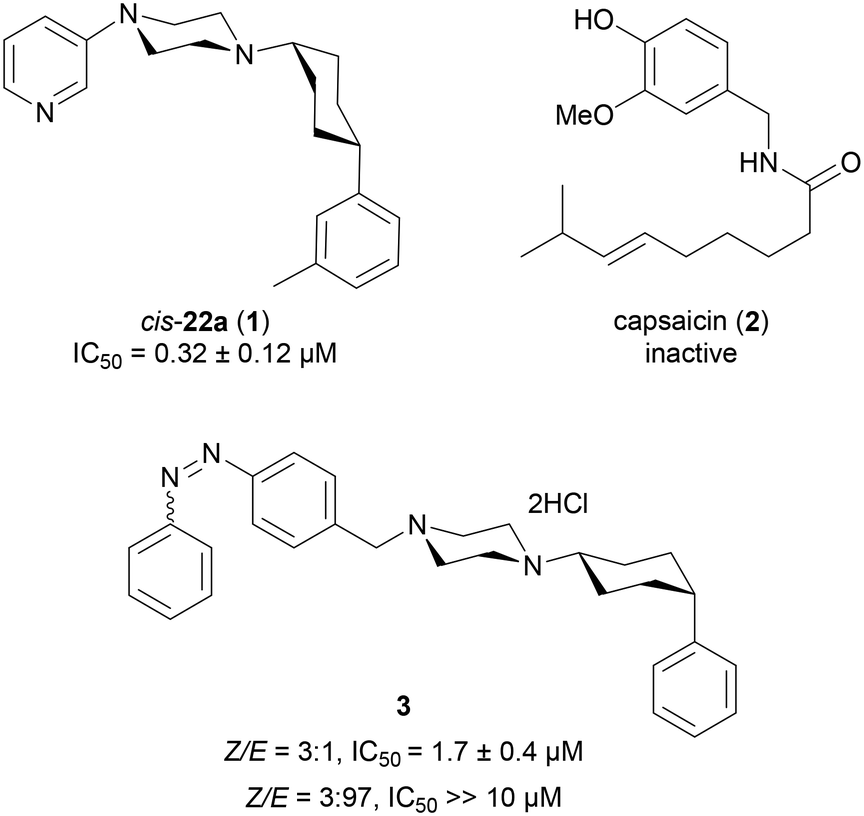 | ||
| Fig. 1 Chemical structure and hTRPV6 inhibition potency of cis-22a (1), capsaicin (2), and the photoswitchable inhibitor 3 (shown as observed in X-ray structures).17–19 | ||
Herein, we set out to search for new analogs with similar potency and selectivity but an improved pharmacological profile. To this end, we introduced the structural features of the natural product capsaicin (2),20–22 a well-known TRPV1 ligand also reported as a hTRPV6 modulator.23,24 Our aim was to modify the 3-pyridine and m-phenyl appendages of 1, which are frequent drug-type substituents and might be responsible for the undesirable off-target effects of 1. Previous structure–activity relationship studies had shown that variations in both groups were often compatible with hTRPV6 inhibition,17 such as introducing a phenyl diazo group to obtain the photoswitchable hTRPV6 inhibitor 3.18 On the other hand, modifications of the central cis-1,4-cyclohexane and piperazine groups mostly abolished the activity.17 Therefore, we maintained this central core in all derivatives of 1 investigated in the present study. We also reinvestigated 2 itself to verify the claimed activity of this natural product on hTRPV6 and extended the investigation to a series of new analogues.
Results and discussion
1. Design and synthesis
The synthesis of all compounds investigated in this study is presented in Scheme 1. First, we replaced the pyridine ring in 1 with the O-methyl-catechol group of capsaicin while either preserving the phenyl appendage or substituting it with aliphatic groups resembling the fatty acyl group of capsaicin. Reductve alkylation of silyl protected vanillin 5 with Boc-piperazine and Boc removal, followed by a second reductive alkylation with cyclohexanone and subsequent deprotection yielded analog 6. The same reaction sequence with 4-ethyl-, 4-tert-butyl- or 4-phenyl-cyclohexanone gave the corresponding analogues 7–9. Condensation of vanillic acid (10) with N-Boc-piperazine, Boc deprotection and reductive alkylation with the same four cyclohexanones afforded analogues 11–14 including an amide linkage related to capsaicin. In a similar approach starting with 6-hydroxynicotinaldehyde (15) and 6-hydroxynicotinic acid (18), we obtained 4-phenyl- and 4-tert-butyl-cyclohexyl analogues 16–17 and 19–20 displaying a pyridone group. Pyridone can be considered as a pyridine analog containing a hydrogen-bond donor group related to the phenolic hydroxyl group of capsaicin.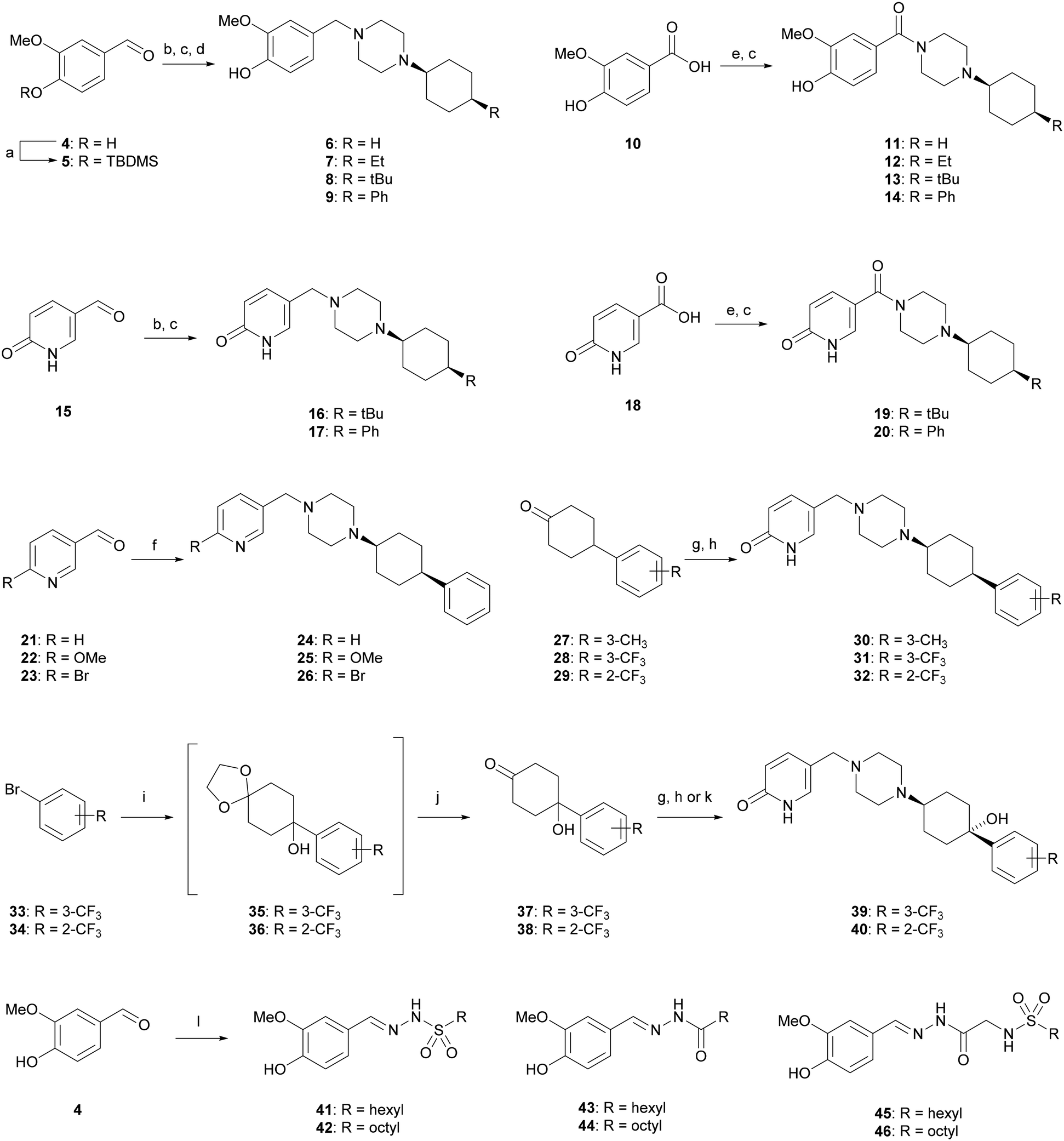 | ||
| Scheme 1 Synthesis of TRPV6 inhibitors and capsaicin analogues. Reagents and conditions. (a) TBDMSCl, DMAP, imidazole, DCM, r.t., 2 h (quant.); (b) (i) tert-butyl piperazine-1-carboxylate, AcOH, NaBH(OAc)3, DCE, r.t., 48 h; (ii) TFA, DCM, r.t., 1 h (78–45%, over 2-steps); (c) 4-R-cyclohexanones, NaBH(OAc)3, Et3N, DCE, r.t., 48 h (6–45%); (d) TBAF, THF, r.t., 3 h (30–59%, over 2-steps); (e) (i) tert-butyl piperazine-1-carboxylate, EDCl, DMAP, CH2Cl2, r.t., on; (ii) TFA, DCM, r.t., 1 h, (60%, over 2-steps); (f) 4-phenylcyclohexyl-piperazine, AcOH, NaBH(OAc)3, DCE, r.t., 48 h (6–68%); (g) (i) 1-benzylpiperazine, NaBH(OAc)3, DCE, r.t., 48 h; (ii) Pd/C, H2, AcOH, MeOH, r.t., on. (18–38%, over 2-steps); (h) 38, 15, AcOH, NaBH(OAc)3, DCE, r.t., 48 h (81%); (i) Mg, THF, Ar., r.t. to rf., 30 min; then 1,4-dioxaspiro[4.5]decan-8-one, THF, Ar., r.t. to rf., 30 min; (j) PPTS, acetone, H2O, 60 °C, 6 h (72–90%); (k) 37, 15, NaBH3CN, Et3N, MeOH, r.t., 24 h (48%); (l) hexyl- or octyl-sulfonyl-hydrazide, MeOH, r.t., 2 h (56–73%). | ||
Due to the activity of 17 (see below), we prepared further analogues of this compound. First, we modified the pyridone carbonyl group with a methoxy (25) or bromo (26) substituent by double reductive alkylation of the corresponding pyridine-carboxaldehydes 21–23, and similarly prepared a new sample of the previously reported meta-pyridine analog 24.17 We also synthesized analogues 30–32 by combining the pyridone group as a piperazine substituent with a meta-xylyl group as a cyclohexyl substituent as in 1, or with an ortho- or meta-trifluoromethyl phenyl group, by using a similar synthetic route starting from the corresponding cyclohexanones. For the particularly lipophilic trifluoromethyl derivatives, we additionally synthesized analogues 39 and 40 preserving the tertiary alcohol at the cyclohexane, which is present as an intermediate for cyclohexanone synthesis, such as to enhance water solubility.
Inspired by a report that 2 itself inhibits TRPV6,23,24 we finally prepared a series of capsaicin analogues reproducing the essential pharmacophoric features of the natural product (a methyl-catechol and a hydrophobic tail connected via an amide bond linker) by connecting vanillin with aliphatic chains via a sulfonylhydrazone (41 and 42),25N-acyl hydrazone (43 and 44)26 or sulfonylglycine hydrazone (45 and 46)27,28 as an amide replacement.29,30
2. X-ray crystallography
In all the above syntheses, the cis-1,4-cyclohexyl stereoisomer was isolated either by column chromatography or by RP-HPLC. All the compounds obtained as free bases were finally precipitated as hydrochloride salts. In the case of a freshly synthesized sample of the original inhibitor 1 and of analogues 9, 19, 31 and 40, we obtained X-ray crystal structures confirming the 1,4-cis-cyclohexane stereochemistry (Fig. 2). In these structures, the cyclohexane and piperazine rings adopt a chair conformation. Furthermore, the piperazinyl group is an axial substituent of the cyclohexane ring for 1, 9, 19, and 40, but an equatorial substituent in 31 as observed previously with the photoswitchable inhibitor 3,18 possibly reflecting crystal packing effects (Fig. S1†).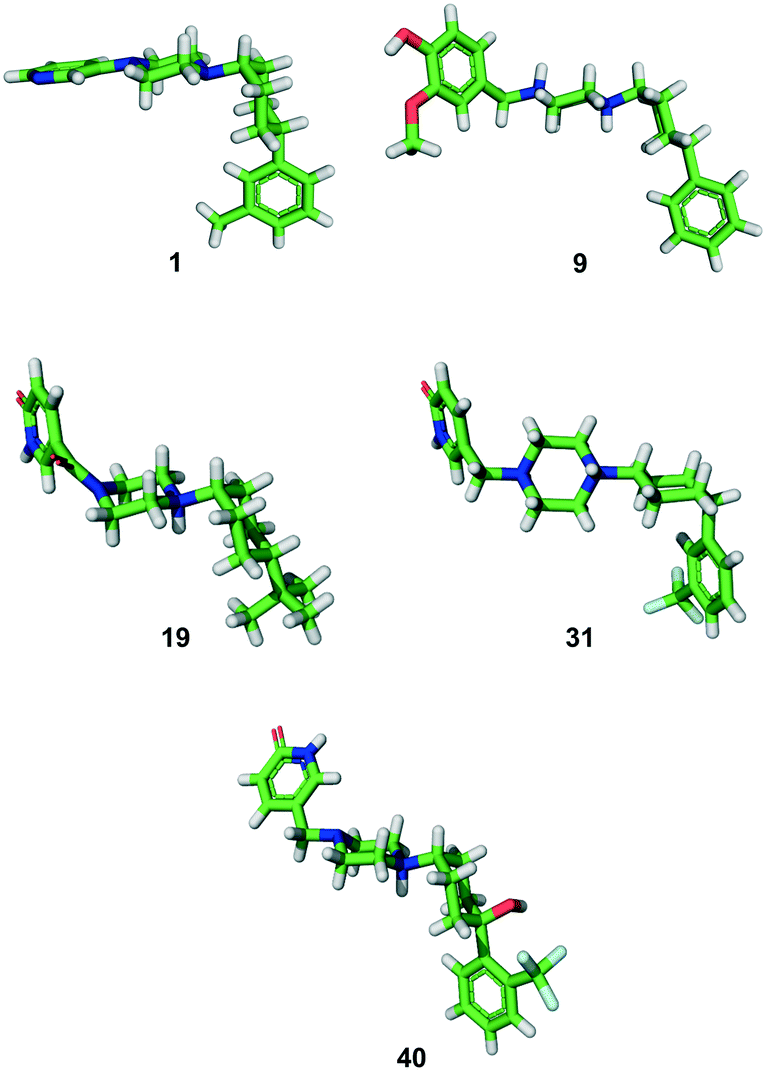 | ||
| Fig. 2 X-ray structures of compounds 1 (free base), 9 and 31 (di-HCl salts), and 19 and 40 (mono-HCl salts). | ||
3. TRPV6 activity screening
We measured the possible modulation of TRPV6 activity by detecting the uptake of Cd2+ into HEK293 cells stably overexpressing human TRPV6 (HEK-hTRPV6) using the fluorescence reporter Calcium-5 (Molecular Devices LLC) as described previously.17 The HEK-hTRPV6 cells were previously reported in our group31–33 and are routinely used for screening campaigns. We first measured the inhibition potency of reference compounds 1 and 2 in this assay (Fig. 3). The assay confirmed the submicromolar activity of our cyclohexylpiperazine inhibitor 1, showing an even slightly stronger inhibition than the originally reported value. On the other hand, we could not detect any modulation of hTRPV6 activity by 2 in this assay up to 100 μM, in contrast to the reported activity, which was determined indirectly.23,24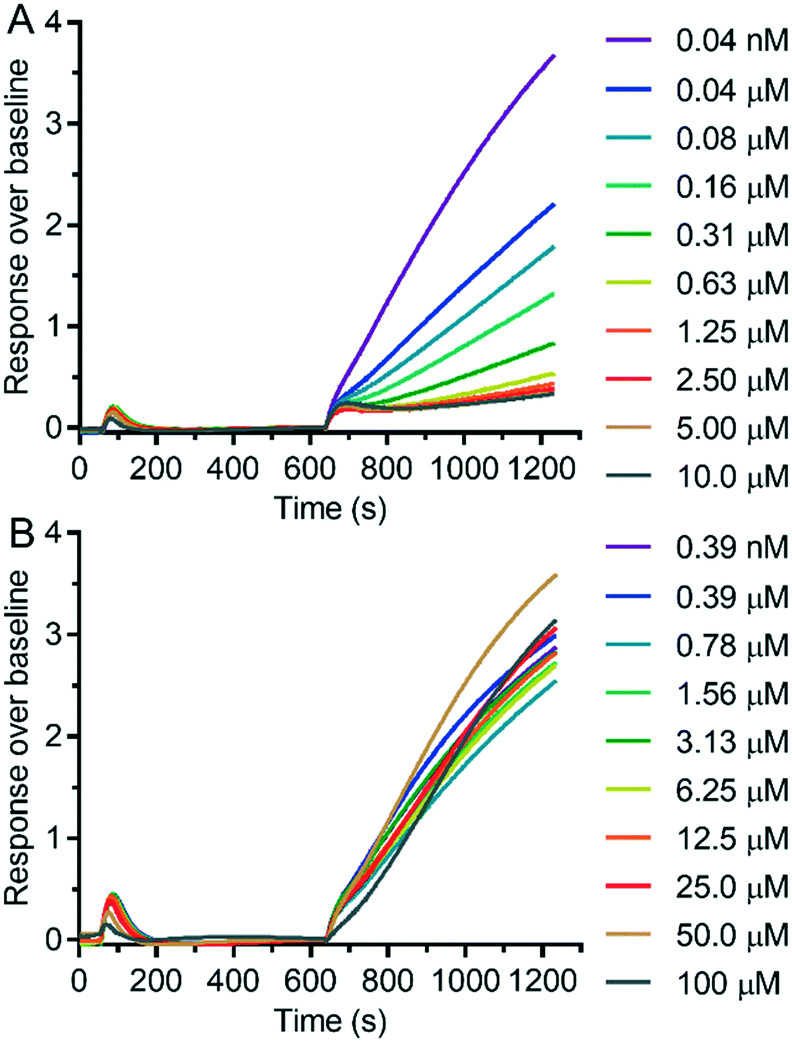 | ||
| Fig. 3 Dose–response curves of 1 (A) and 2 (B) in HEK-hTRPV6 cells, measuring Cd2+ influx. | ||
We then proceeded to test the initial series of analogues 6–9, 11–14, 19–20, 24–26, and 41–46 (Fig. 4 and Table 1). None of the capsaicin analogues 41–46 showed any activity, in line with the lack of hTRPV6 inhibition observed with 2. Nevertheless, the O-methyl-catechol group borrowed from capsaicin was suitable as a replacement for the meta-pyridine group of 1 when combined with a tert-butyl (8 and 13) or phenyl (9 and 14) group as a cyclohexyl substituent, in particular with 14 featuring a vanillic amide group, illustrating that a dibasic piperazine is not essential for activity. We also observed good hTRPV6 inhibitory activity with three of the four analogues for which the meta-pyridine group was replaced by a pyridone group (16, 17, and 20). The lower activity with 19 however showed that the tert-butyl group, although potentially interesting as a non-aromatic group, was not a very favourable replacement of the aromatic group as a cyclohexane substituent. We selected pyridone 17 (IC50 = 0.37 μM) for further optimization due to its good activity combined with a favorable ligand efficiency (LE = 0.34) and lipophilic ligand efficiency (LLE = 3.5)34 compared to all other tested compounds.
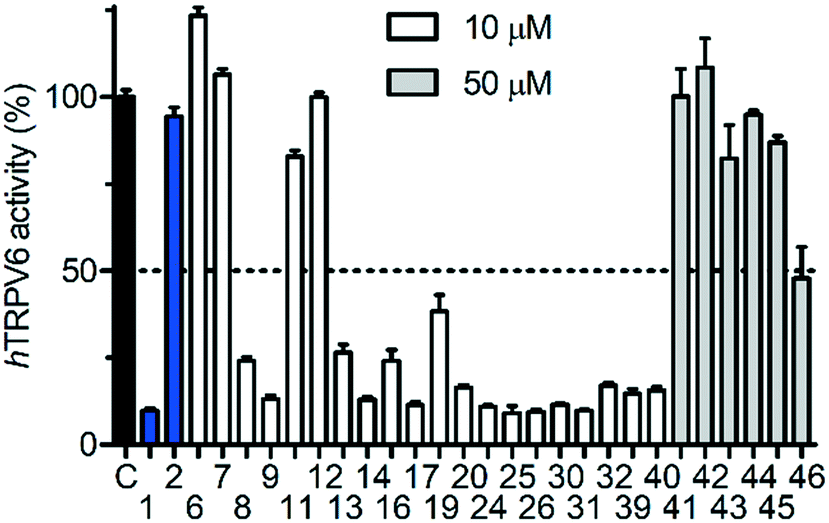 | ||
| Fig. 4 Activity screening for hTRPV6 mediated Cd2+ uptake in HEK-hTRPV6 cells. Data shown are mean + SEM (n = 3). | ||
| Cpd | IC50 (μM) (95% CI)a | HACb | LEc | LLEd |
|---|---|---|---|---|
a Data shown are mean ± SEM (n = 9/concentration) for at least 2 independent experiments or mean and 95% confidence interval (n = 6/concentration) of a single experiment.
b HAC: heavy atom count.
c LE: ligand efficiency = (1.37 × pIC50)/HAC, where pIC50 = −log(IC50) in the molar range.34
d LLE: lipophilic ligand efficiency = pIC50 − clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, where clogP was obtained from ChemDraw v. 19.0.1.28 (PerkinElmer Informatics, Inc.).34 P, where clogP was obtained from ChemDraw v. 19.0.1.28 (PerkinElmer Informatics, Inc.).34
|
||||
| 1 | 0.050 ± 0.003 | 25 | 0.40 | 1.7 |
| 8 | 2.4 (1.4–4.2) | 26 | 0.30 | 0.3 |
| 9 | 1.6 (1.1–2.3) | 28 | 0.28 | 0.9 |
| 13 | 2.1 (1.5–2.9) | 27 | 0.29 | 1.0 |
| 14 | 0.43 (0.31–0.59) | 29 | 0.30 | 2.1 |
| 16 | 2.4 (0.30–19.0) | 24 | 0.32 | 2.1 |
| 17 | 0.37 (0.25–0.57) | 26 | 0.34 | 3.5 |
| 20 | 0.60 (0.45–0.80) | 27 | 0.32 | 3.9 |
| 24 | 0.55 (0.35–0.87) | 25 | 0.34 | 2.0 |
| 25 | 0.55 (0.40–0.77) | 27 | 0.32 | 1.3 |
| 26 | 0.17 (0.14–0.22) | 26 | 0.36 | 1.6 |
| 30 | 0.15 ± 0.04 | 27 | 0.35 | 3.2 |
| 31 | 0.064 ± 0.007 | 30 | 0.33 | 3.2 |
| 32 | 0.69 ± 0.18 | 30 | 0.28 | 2.2 |
| 39 (3OG) | 0.082 ± 0.004 | 31 | 0.31 | 4.8 |
| 40 | 0.13 ± 0.03 | 31 | 0.30 | 4.6 |
4. Optimization of pyridone 17
We investigated several changes around the phenyl group as a means to improve the activity of 17. Inhibition increased when adding a meta-CH3 group similar to 1 to form 30 (IC50 = 0.15 ± 0.04 μM). The effect was even stronger with a meta-CF3 group (31, IC50 = 0.064 ± 0.007 μM), while the inhibition decreased slightly with an ortho-CF3 group (32, IC50 = 0.69 ± 0.18 μM). Introducing a hydroxyl group at the aromatic attachment point on the cyclohexane caused a slight decrease in potency for the meta-CF3 analog 39 (IC50 = 0.082 ± 0.004 μM, Fig. 5A), but an increase in potency for the ortho-CF3 analog 40 (IC50 = 0.13 ± 0.03 μM). In both cases, the addition of the hydroxyl group also increased water solubility. Analyzing all the analogues in terms of LE and LLE led us to select 39, named 3OG, as a promising analog for further investigation.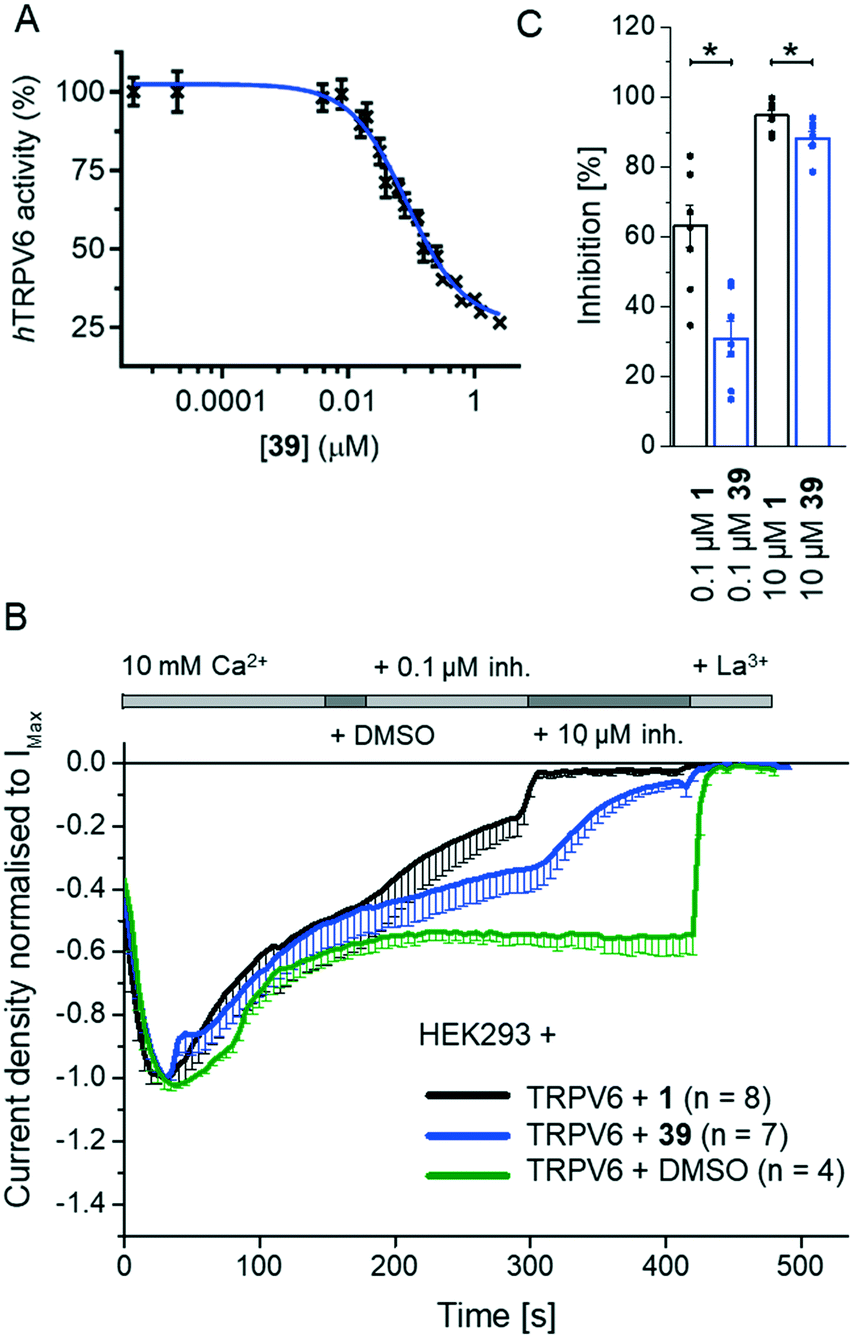 | ||
| Fig. 5 Evaluation of 39 (3OG) as a TRPV6 inhibitor. (A) Dose–response curve of 39 on Cd2+ influx into HEK-hTRPV6 cells. Data shown are mean ± SEM (n = 6/concentration) of 4 independent experiments. (B) and (C) Electrophysiological characterisation of 39. (B) Averaged time course of whole-cell current densities (mean ± SEM) from YFP-TRPV6 transfected HEK293 cells. The experiment started at 10 mM Ca2+, subsequently containing equivalent amounts of DMSO as the control (green) or 0.1 and 10 μM 39 (blue) or 1 (black), followed by La3+. (C) Bar graph (mean ± SEM and individual values) of inhibition by 0.1 and 10 μM 39 (blue) and 1 (black) on TRPV6 current densities. * indicates significant p values < 0.05. | ||
Electrophysiological experiments on YFP-TRPV6 transiently transfected HEK293 cells confirmed the activity of 39 on TRPV6. Initially, 10 mM Ca2+ solution was applied and after the currents reached a plateau, the inhibitors at 0.1 and 10 μM were added. Finally, the calcium currents were fully inhibited with La3+ (Fig. 5B). 39 induced an inhibition of 30.8 ± 5.1% and 88.2 ± 2.0%, respectively, at 0.1 μM and 10 μM (Fig. 5C). In comparison to the previously described 1, the inhibition by 39 occurred at a slower rate and was less pronounced.
5. Blocking TRPV6 transport function with pyridone 39
We further confirmed the TRPV6 inhibitory activity of 39 by confocal microscopy of HEK-hTRPV6 cells preincubated with Leadmium™ Green, which is a Ca2+-insensitive intracellular green fluorogenic dye revealing Pb2+ and Cd2+ within the cytosol, allowing TRPV6-mediated cellular uptake independent of intracellular Ca2+ fluxes to be tracked. The cells were also stained with the wheat germ agglutinin Alexa Fluor™ 594 conjugate to mark the cytoplasmic membrane.35,36 In control cells (treated with the vehicle), the application of Cd2+ (50 μM) revealed an increase in green fluorescence after 30 min, indicating Cd2+ transport through hTRPV6 (Fig. 6A). When cells were treated with 10 μM 39 prior to the addition of Cd2+, the fluorescence was significantly less pronounced (Fig. 6B). Time-lapse imaging of Cd2+ uptake over 30 minutes furthermore showed that cells treated with inhibitor 39 had significantly reduced uptake compared to non-treated cells over the first 12 minutes, after which a plateau was reached (Fig. S2†).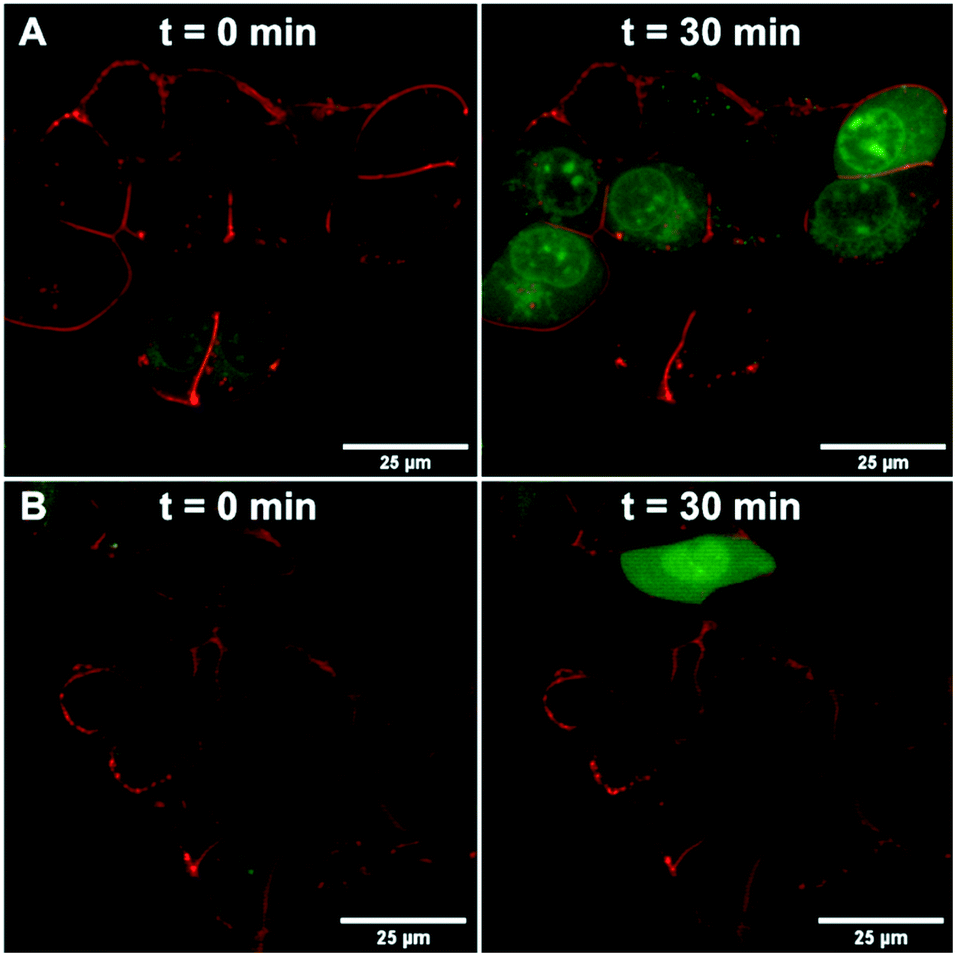 | ||
| Fig. 6 HEK-hTRPV6 cells co-stained with Leadmium™ Green and wheat germ agglutinin Alexa Fluor™ 594 conjugate. Images were collected at time 0 min and 30 min after treatment with the vehicle (A) or 10 μM 39 (B) followed by the addition of Cd2+ (50 μM) using confocal microscopy (Nikon Eclipse TE2000-E, 100×). White bars denote 25 μm scale. | ||
To assess whether pharmacological blockage of TRPV6 transport function by 39 could trigger a cellular effect, we determined the ability of 39 to reduce the Cd2+ toxicity towards HEK293 wt and HEK-hTRPV6 cell lines. Prolonged Cd2+ exposure is known to produce toxic effects on human cells, and eventually culminates in cell death.37 When we tested the viability of these cells after 24 h of treatment, we found that the Cd2+ dose–response in HEK-hTRPV6 was ∼2-fold greater than that in HEK293 wt (4.6 ± 0.1 μM and 8.3 ± 0.3 μM, respectively). These results indicate that the overexpression of hTRPV6 increased the transport of Cd2+ and consequently, the toxicity towards these cells (Fig. 7 and S3†).38 While 39 at 10 μM or 1 μM had no effect on the Cd2+ sensitivity of HEK293 wt, treating HEK-hTRPV6 cells with 39 at 10 μM significantly reduced the toxic effect of Cd2+ (Fig. 7). The reduction of Cd2+ toxicity occurring only in HEK-hTRPV6 but not in HEK293 wt indicates that 39 inhibits hTRPV6-mediated Cd2+ transport.
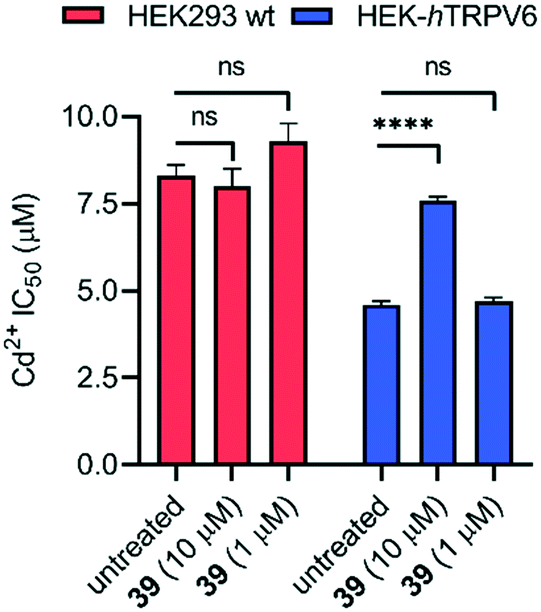 | ||
| Fig. 7 IC50 values of Cd2+ toxicity to HEK293 wt and HEK-hTRPV6 cells. Data shown are mean + SEM (n = 4/concentration) of 2 independent experiments. ****P < 0.0001; n.s., P > 0.05. | ||
6. Ion channel selectivity and off-target profiling of pyridone 39
To test if the higher polarity and LLE of 39 compared to those of 1 translated into a better selectivity profile, we investigated the activity of both compounds on the closely related calcium channels. Similar to 1, 39 showed a 7-fold selectivity for hTRPV6 (IC50 = 83 nM) against hTRPV5 (IC50 = 560 nM), low activity on L-type calcium channels at 10 μM (Fig. 8), and no detectable activity on store-operated calcium channels at 5 μM (Fig. 9). Similar to 1,1739 did not activate nor inhibit TRPV1, which is the only known TRP target of capsaicin (Fig. S2†).39,40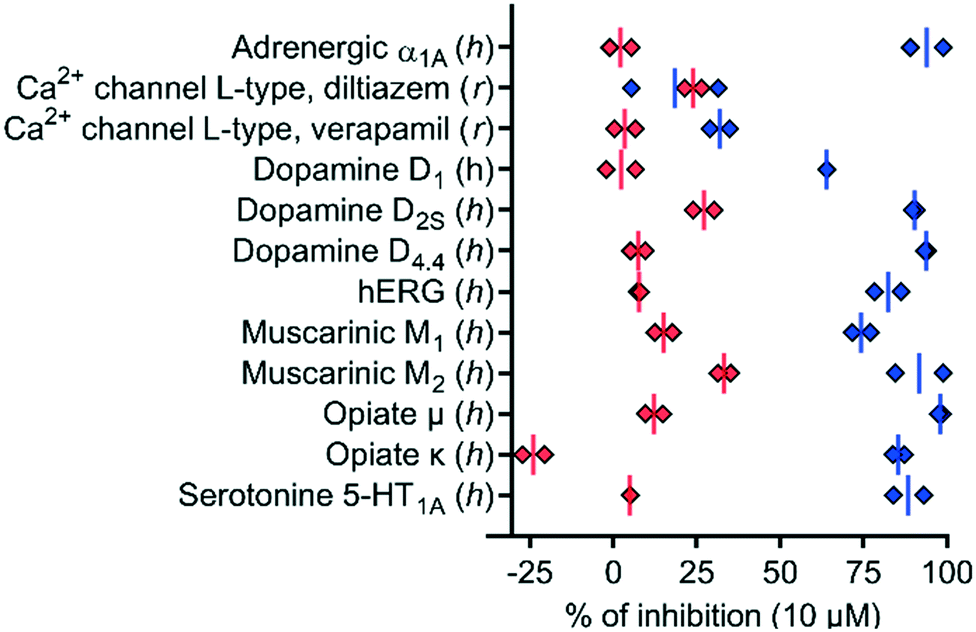 | ||
| Fig. 8 In vitro polypharmacology for selected targets of 1 (blue diamonds) and 39 (red diamonds). Data are shown for each replicate (n = 2). The data for 1 were extracted from ref. 17. The experiments were conducted by Eurofins Cerep SA, France. | ||
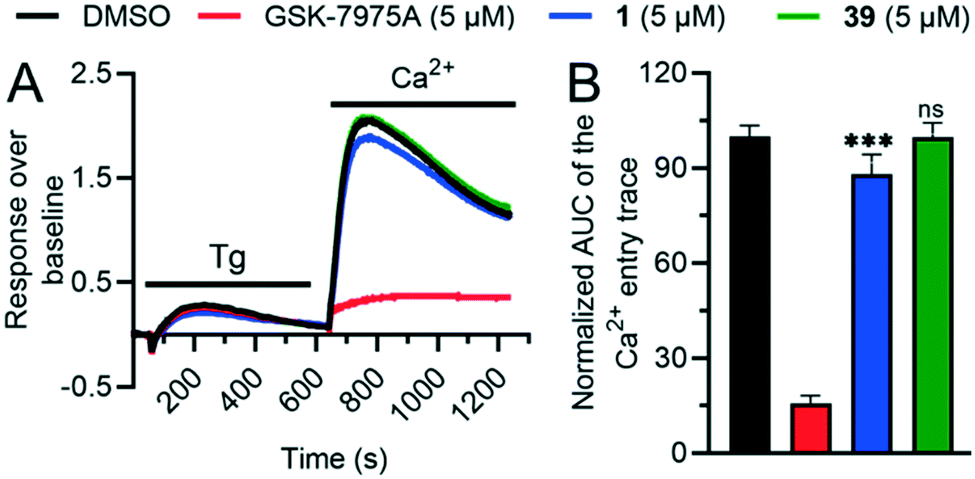 | ||
| Fig. 9 A. Recording of store-operated Ca2+ entry (SOCE) in MDA-MB-231 cells pretreated for 20 min with either DMSO or SOCE inhibitor GSK-7975A, or 1vs.39. Ca2+ store-depletion was achieved by 10 min treatment of cells with 1 μM thapsigargin (Tg) in a nominally Ca2+ free buffer. B. SOCE quantification as the area under the curve (AUC) of the 2 mM CaCl2 (Ca2+) add-back trace is shown as normalized values to the DMSO control (mean ± SD; n = 18). The P-value of the 1vs.39 pretreated cells is indicated above the respective bar as *** for p ≤ 0.001 or non-significant (n.s.) for p > 0.05. | ||
We also characterized the off-targets predicted to be potentially problematic for both compounds using the web-based target prediction tool PPB2.41 Remarkably, 39 did not show any significant off-target effects compared to 1, indicating that its higher LLE translated into reduced polypharmacology (Fig. 8). It is worth noting that the hERG activity observed with 1 was completely abolished with 39. Furthermore, we found that the half-life in human liver microsomes, which was relatively short for 1 (t1/2 = 6 min), was significantly extended for 39 (t1/2 > 60 min), which we attribute to the replacement of the aromatic meta-methyl substituent with a trifluoromethyl group.42,43 Taken together, these data showed that compound 39 had a much better selectivity and stability profile compared to the original hTRPV6 inhibitor 1.
7. Antiproliferative activity
In our initial discovery of 1, we reported that the inhibitor significantly reduced the growth rate in T47D breast cancer cells, which express high levels of TRPV6, at micromolar concentration (IC50 = 25 ± 10 μM), while SKOV3 ovarian carcinoma cells, where TRPV6 expression was not detected, were less affected (IC50 > 50 μM).17 However, these values were much higher than the submicromolar levels sufficient to block TRPV6, and occurred in the range of the off-target effects of 1 (Fig. 8).Here, we compared the effects of 1 and 39 on SKOV3 and T47D, as well as on MCF-7 and MDA-MB-231 as two additional breast cancer cell lines with low levels of TRPV6.13,44 Previous studies have shown that RNA silencing (siRNA) of TRPV6 reduces T47D cell proliferation.11,13 On the other hand, siRNA knock-down of TRPV6 does not reduce the proliferation of MCF-7 and MDA-MB-231 cells.12 We also investigated the non-cancer derived HEK293 immortalized cell line which does not express TRPV6,45,46 as well as the HEK-hTRPV6 overexpressing cell line used for activity assays.
Inhibitor 1 over 6 days caused a significant reduction in cell proliferation for all six cell lines at high concentrations and not just limited to SKOV3, T47D and HEK-hTRPV6 cells (Fig. 10A). 1 also induced changes in cell morphology for T47D cells (Fig. S4†). By contrast, we did not observe any significant decrease in proliferation with 39 (Fig. 10B) or changes in cell morphology (Fig. S4†) under similar conditions despite its comparable IC50 for TRPV6 channel function inhibition. The positive control doxorubicin (10 μM), as expected, significantly decreased the growth of all six cell lines (data not shown). Note that HEK-hTRPV6 was more susceptible to 1 toxicity than HEK293 wt cells, which might be related to the fact that HEK-hTRPV6 overexpressing cells proliferate faster than HEK293 wt cells in a Ca2+-dependent manner (Table S3†).46 These results suggest that the cytotoxic effects observed with 1 but not with 39 do not reflect TRPV6 inhibition but probably result from non-specific or off-target effects. Our findings that the pharmacological inhibition of TRPV6 channel function by 39 did not affect the viability of TRPV6 expressing cell lines is intriguing. This highlights the need for future studies to uncover the precise role of TRPV6 in cancer progression.
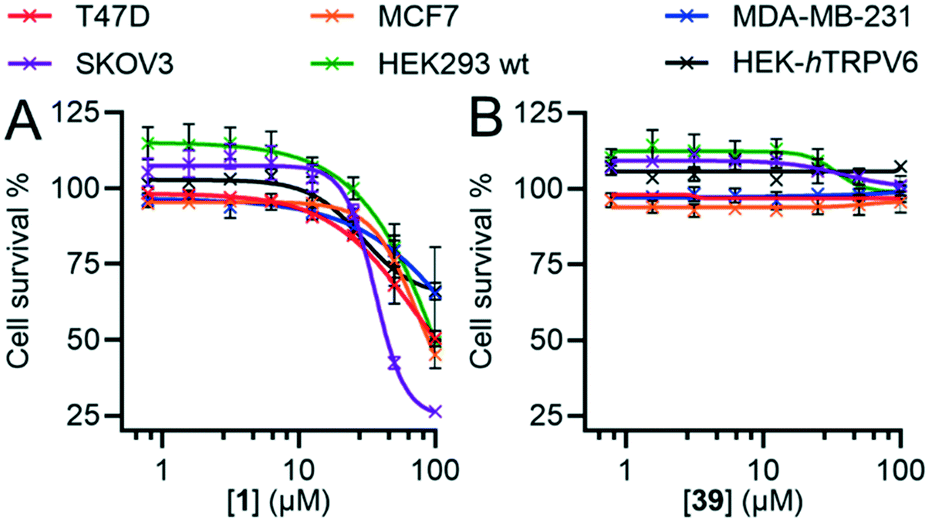 | ||
| Fig. 10 Dose–response curves of 1 (A) and 39 (B) in different breast and ovarian cancer cells and HEK293. Relative cell survival compared to the DMSO control is shown as a function of inhibitor concentration. Data shown are mean ± SEM (n = 8/concentration) of 3 independent experiments. | ||
8. Overview of TRPV6 inhibitor development
The overall development of our inhibitors is illustrated here with an interactive tree-map (TMAP)47 representing each molecule as a point color-coded by TRPV6 inhibitory potency (Fig. 11). In this map, molecules are connected by a minimum spanning tree to their most similar analogs as measured by the extended connectivity fingerprint MHFP6.48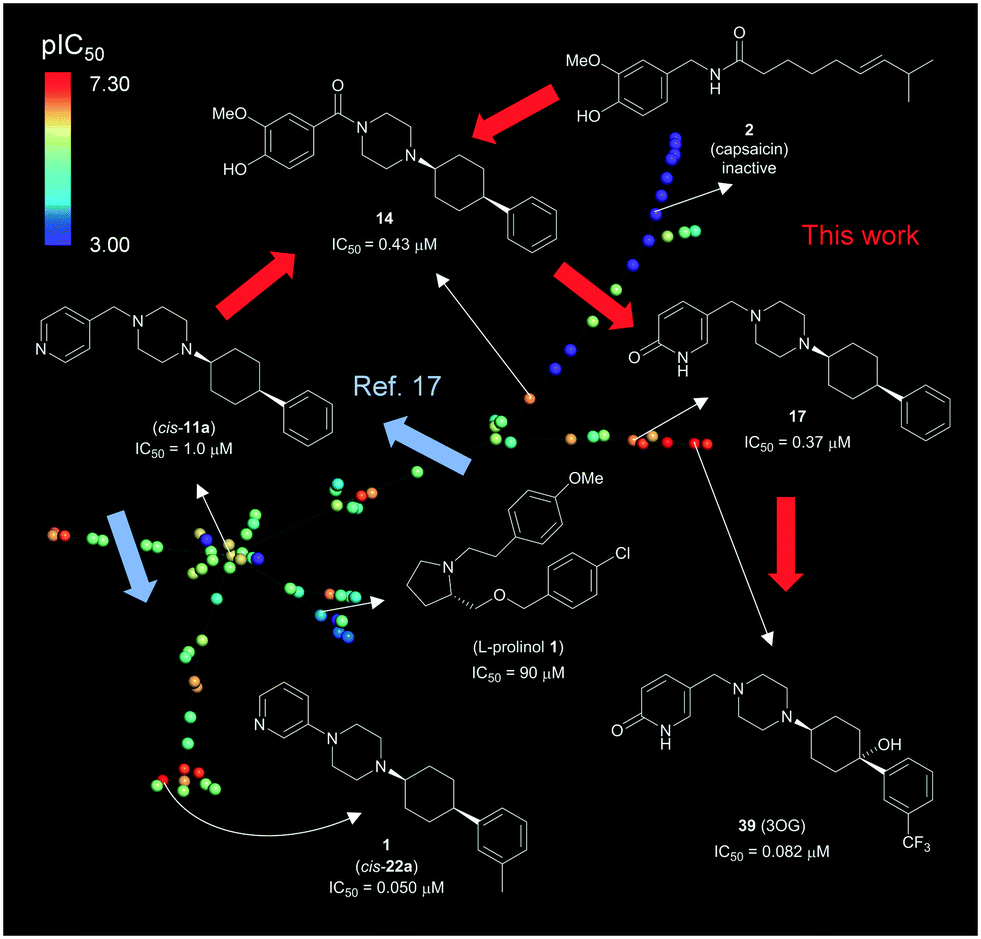 | ||
| Fig. 11 Overview of TRPV6 inhibitor development. The interactive version of the map is accessible at http://tm.gdb.tools/trpv6-inhibitors/. For color-coding, IC50 values were used as reported, or estimated from reported percentage inhibition at 5 μM or 10 μM. Inactive compounds were assigned IC50 = 1 mM. Compound numbers in parentheses are from ref. 17. The map was prepared using the public website and instructions at https://try-tmap.gdb.tools/. | ||
The lower left portion of the TMAP illustrates our initial study (blue arrows),17 in which we searched for scaffold-hopping analogues of known, weakly active TRPV6 inhibitors including an L-prolinol derivative. We discovered a first hit compound (cis-11a), which we optimized to 1 (cis-22a). The upper right branch contains capsaicin and its analogs 41–48, which were inactive. This branch also contains 14 which combines elements from capsaicin and cis-22a. The further optimization of 14 by introducing a pyridone to form 17 and its further optimization to inhibitor 39 (3OG) appears as an additional side branch.
Throughout these explorations, we found that a (4-arylcyclohexyl)-piperazine with cis-stereochemistry on the cyclohexane was critical to give strong TRPV6 inhibition. On the other hand, TRPV6 inhibition was compatible with variations in the second piperazine substituent and to a certain extent with substitutions on the aromatic cyclohexane substituent. This suggests that further improvements in inhibitory potency and in compound properties might be achievable with further variations at these positions.
Conclusions
To improve the properties of the previously reported TRPV6 inhibitor 1, we surveyed analogues incorporating structural features inspired by the natural product capsaicin such as aliphatic and oxygen-containing functional groups. Although we found that, contrary to previous reports, capsaicin does not have any inhibitory effect on TRPV6, our strategy led us to identify the new inhibitor 39 (3OG), which incorporates a pyridone group and a tertiary alcohol as typical natural product-like features. Inhibitor 39 shows similar potency against TRPV6 and ion channel selectivity to 1 but much better microsomal stability and much lower off-target effects, in particular suppressed hERG inhibition. Inhibitor 39 blocks TRPV6 transport function in cells as assessed by the reduction of Cd2+ toxicity in HEK-hTRPV6. However, even at high concentration, 39 does not display any measurable cellular toxicity on various cell lines, expressing TRPV6 or not. Structural and mutagenesis studies based on the recently published structure of hTRPV6 (ref. 3 and 49) and showing how 1 and 39 inhibit TRPV6 will be reported in the near future. This new tool compound should be useful to decipher the role of TRPV6 mediated calcium flux in various disease models.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. L. R. acknowledges the financial support from the Swiss National Science Foundation (SNF), NCCR TransCure. R. B. and M. A. H. acknowledge the SNF Sinergia grants (grant numbers CRSII5_180326 and CRSII3_160782) for funding support of this work. R. P. F acknowledges the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant numbers: 2013/19311-6 and 2017/00689-0) for financial support. MRC thanks the Swiss Excellence Scholarship for Foreign Students and Scholars (ESKAS – 2017.0670) for the academic exchange program in Switzerland. The authors thank Dr. Daniel Probst for help in assembling the TMAP in Fig. 11. This work was also supported in part by the Austrian Science Funds FWF P33283 (to C. R.).References
- J.-B. Peng, X.-Z. Chen, U. V. Berger, S. Weremowicz, C. C. Morton, P. M. Vassilev, E. M. Brown and M. A. Hediger, Biochem. Biophys. Res. Commun., 2000, 278, 326–332 CrossRef CAS PubMed.
- R. Vennekens, J. G. J. Hoenderop, J. Prenen, M. Stuiver, P. H. G. M. Willems, G. Droogmans, B. Nilius and R. J. M. Bindels, J. Biol. Chem., 2000, 275, 3963–3969 CrossRef CAS PubMed.
- L. L. McGoldrick, A. K. Singh, K. Saotome, M. V. Yelshanskaya, E. C. Twomey, R. A. Grassucci and A. I. Sobolevsky, Nature, 2018, 553, 233–237 CrossRef CAS PubMed.
- J. G. J. Hoenderop, EMBO J., 2003, 22, 776–785 CrossRef CAS PubMed.
- C. Fecher-Trost, F. Lux, K.-M. Busch, A. Raza, M. Winter, F. Hielscher, T. Belkacemi, B. van der Eerden, U. Boehm, M. Freichel and P. Weissgerber, J. Bone Miner. Res., 2019, 34, e3646 CrossRef PubMed.
- T. Nijenhuis, J. G. J. Hoenderop, B. Nilius and R. J. M. Bindels, Pflügers Arch., 2003, 446, 401–409 CrossRef CAS PubMed.
- C. Fecher-Trost, U. Wissenbach and P. Weissgerber, Cell Calcium, 2017, 67, 116–122 CrossRef CAS PubMed.
- F. Chen, B. Ni, Y. O. Yang, T. Ye and A. Chen, Cell. Physiol. Biochem., 2014, 33, 796–809 CrossRef CAS PubMed.
- Y. Suzuki, D. Chitayat, H. Sawada, M. A. Deardorff, H. M. McLaughlin, A. Begtrup, K. Millar, J. Harrington, K. Chong, M. Roifman, K. Grand, M. Tominaga, F. Takada, S. Shuster, M. Obara, H. Mutoh, R. Kushima and G. Nishimura, Am. J. Hum. Genet., 2018, 102, 1104–1114 CrossRef CAS PubMed.
- S. Yamashita, H. Mizumoto, H. Sawada, Y. Suzuki and D. Hata, J. Endocr. Soc., 2019, 3, 602–606 CrossRef CAS PubMed.
- K. A. Bolanz, M. A. Hediger and C. P. Landowski, Mol. Cancer Ther., 2008, 7, 271–279 CrossRef CAS PubMed.
- I. Dhennin-Duthille, M. Gautier, M. Faouzi, A. Guilbert, M. Brevet, D. Vaudry, A. Ahidouch, H. Sevestre and H. Ouadid-Ahidouch, Cell. Physiol. Biochem., 2011, 28, 813–822 CrossRef CAS PubMed.
- A. A. Peters, P. T. Simpson, J. J. Bassett, J. M. Lee, L. Da Silva, L. E. Reid, S. Song, M.-O. Parat, S. R. Lakhani, P. A. Kenny, S. J. Roberts-Thomson and G. R. Monteith, Mol. Cancer Ther., 2012, 11, 2158–2168 CrossRef CAS PubMed.
- L. Zhuang, J.-B. Peng, L. Tou, H. Takanaga, R. M. Adam, M. A. Hediger and M. R. Freeman, Lab. Invest., 2002, 82, 1755–1764 CrossRef CAS PubMed.
- J.-B. Peng, L. Zhuang, U. V. Berger, R. M. Adam, B. J. Williams, E. M. Brown, M. A. Hediger and M. R. Freeman, Biochem. Biophys. Res. Commun., 2001, 282, 729–734 CrossRef CAS PubMed.
- V. Lehen'kyi, M. Raphaël and N. Prevarskaya, J. Physiol., 2012, 590, 1369–1376 CrossRef PubMed.
- C. Simonin, M. Awale, M. Brand, R. Van Deursen, J. Schwartz, M. Fine, G. Kovacs, P. Häfliger, G. Gyimesi, A. Sithampari, R. P. Charles, M. A. Hediger and J. L. Reymond, Angew. Chem., Int. Ed., 2015, 54, 14748–14752 CrossRef CAS PubMed.
- M. R. Cunha, R. Bhardwaj, S. Lindinger, C. Butorac, C. Romanin, M. A. Hediger and J.-L. Reymond, ACS Med. Chem. Lett., 2019, 10, 1341–1345 CrossRef CAS PubMed.
- W. I. F. David, K. Shankland, K. Shankland and N. Shankland, Chem. Commun., 1998, 931–932 RSC.
- G. J. V. Pereira, M. T. Tavares, R. A. Azevedo, B. B. Martins, M. R. Cunha, R. Bhardwaj, Y. Cury, V. O. Zambelli, E. G. Barbosa, M. A. Hediger and R. Parise-Filho, Bioorg. Med. Chem., 2019, 27, 2893–2904 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- M. Grigalunas, A. Burhop, A. Christoforow and H. Waldmann, Curr. Opin. Chem. Biol., 2020, 56, 111–118 CrossRef CAS PubMed.
- J. Chow, M. Norng, J. Zhang and J. Chai, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773, 565–576 CrossRef CAS PubMed.
- J. K. Lau, K. C. Brown, A. M. Dom, T. R. Witte, B. A. Thornhill, C. M. Crabtree, H. E. Perry, J. M. Brown, J. G. Ball, R. G. Creel, C. L. Damron, W. D. Rollyson, C. D. Stevenson, W. E. Hardman, M. A. Valentovic, A. B. Carpenter and P. Dasgupta, Apoptosis, 2014, 19, 1190–1201 CrossRef CAS PubMed.
- M. R. Cunha, M. T. Tavares, C. F. Carvalho, N. A. T. Silva, A. D. F. Souza, G. J. V. Pereira, F. F. Ferreira and R. Parise-Filho, ACS Sustainable Chem. Eng., 2016, 4, 1899–1905 CrossRef CAS.
- T. F. Silva, W. Bispo Júnior, M. S. Alexandre-Moreira, F. N. Costa, C. Monteiro, F. Furlan Ferreira, R. C. R. Barroso, F. Noël, R. T. Sudo, G. Zapata-Sudo, L. M. Lima and E. Barreiro, Molecules, 2015, 20, 3067–3088 CrossRef PubMed.
- B. Tian, M. He, S. Tang, I. Hewlett, Z. Tan, J. Li, Y. Jin and M. Yang, Bioorg. Med. Chem. Lett., 2009, 19, 2162–2167 CrossRef CAS PubMed.
- B. Tian, M. He, Z. Tan, S. Tang, I. Hewlett, S. Chen, Y. Jin and M. Yang, Chem. Biol. Drug Des., 2011, 77, 189–198 CrossRef CAS PubMed.
- S. Thota, D. A. Rodrigues, P. de S. M. Pinheiro, L. M. Lima, C. A. M. Fraga and E. J. Barreiro, Bioorg. Med. Chem. Lett., 2018, 28, 2797–2806 CrossRef CAS PubMed.
- J. de O. Carneiro Brum, T. C. C. França and J. D. F. Villar, Mini-Rev. Med. Chem., 2020, 20, 342–368 CrossRef PubMed.
- G. Kovacs, T. Danko, M. J. Bergeron, B. Balazs, Y. Suzuki, A. Zsembery and M. A. Hediger, Cell Calcium, 2011, 49, 43–55 CrossRef CAS PubMed.
- A. Hofer, G. Kovacs, A. Zappatini, M. Leuenberger, M. A. Hediger and M. Lochner, Bioorg. Med. Chem., 2013, 21, 3202–3213 CrossRef CAS PubMed.
- G. Kovacs, N. Montalbetti, A. Simonin, T. Danko, B. Balazs, A. Zsembery and M. A. Hediger, Cell Calcium, 2012, 52, 468–480 CrossRef CAS PubMed.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- L. M. Malaiyandi, H. Sharthiya and K. E. Dineley, BioMetals, 2016, 29, 625–635 CrossRef CAS PubMed.
- P. Lundberg, M. Magzoub, M. Lindberg, M. Hällbrink, J. Jarvet, L. E. G. Eriksson, Ü. Langel and A. Gräslund, Biochem. Biophys. Res. Commun., 2002, 299, 85–90 CrossRef CAS PubMed.
- G. Bertin and D. Averbeck, Biochimie, 2006, 88, 1549–1559 CrossRef CAS PubMed.
- G. Kovacs, N. Montalbetti, M.-C. Franz, S. Graeter, A. Simonin and M. A. Hediger, Cell Calcium, 2013, 54, 276–286 CrossRef CAS PubMed.
- L. Zubcevic, M. A. Herzik, B. C. Chung, Z. Liu, G. C. Lander and S.-Y. Lee, Nat. Struct. Mol. Biol., 2016, 23, 180–186 CrossRef CAS PubMed.
- G. D. Smith, M. J. Gunthorpe, R. E. Kelsell, P. D. Hayes, P. Reilly, P. Facer, J. E. Wright, J. C. Jerman, J.-P. Walhin, L. Ooi, J. Egerton, K. J. Charles, D. Smart, A. D. Randall, P. Anand and J. B. Davis, Nature, 2002, 418, 186–190 CrossRef CAS PubMed.
- M. Awale and J.-L. Reymond, J. Chem. Inf. Model., 2019, 59, 10–17 CrossRef CAS PubMed.
- N. Murayama, N. Imai, T. Nakane, M. Shimizu and H. Yamazaki, Biochem. Pharmacol., 2007, 73, 2020–2026 CrossRef CAS PubMed.
- A. Parikh, E. M. J. Gillam and F. P. Guengerich, Nat. Biotechnol., 1997, 15, 784–788 CrossRef CAS PubMed.
- K. A. Bolanz, G. G. Kovacs, C. P. Landowski and M. A. Hediger, Mol. Cancer Res., 2009, 7, 2000–2010 CrossRef CAS PubMed.
- D. Hirnet, J. Olausson, C. Fecher-Trost, M. Bödding, W. Nastainczyk, U. Wissenbach, V. Flockerzi and M. Freichel, Cell Calcium, 2003, 33, 509–518 CrossRef CAS PubMed.
- E. C. Schwarz, U. Wissenbach, B. A. Niemeyer, B. Strauß, S. E. Philipp, V. Flockerzi and M. Hoth, Cell Calcium, 2006, 39, 163–173 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 12 Search PubMed.
- D. Probst and J.-L. Reymond, J. Cheminf., 2018, 10, 66 CAS.
- A. K. Singh, K. Saotome, L. L. McGoldrick and A. I. Sobolevsky, Nat. Commun., 2018, 9, 2465 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3, Fig. S1–S4, protocols for FLIPR assays, confocal imaging and antiproliferative assays, preparation, crystal structure reports, HRMS results, 1H, 13C and 19F NMR spectra of all new compounds, and purity of final compounds. CCDC 1997201–1997205. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0md00145g |
| This journal is © The Royal Society of Chemistry 2020 |