
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrazolyl-pyrimidones inhibit the function of human solute carrier protein SLC11A2 (hDMT1) by metal chelation†
Marion
Poirier‡
a,
Jonai
Pujol-Giménez‡
 bcd,
Cristina
Manatschal
e,
Sven
Bühlmann
a,
Ahmed
Embaby
a,
Sacha
Javor
a,
Matthias A.
Hediger
*bcd and
Jean-Louis
Reymond
*a
bcd,
Cristina
Manatschal
e,
Sven
Bühlmann
a,
Ahmed
Embaby
a,
Sacha
Javor
a,
Matthias A.
Hediger
*bcd and
Jean-Louis
Reymond
*a
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
bInstitute of Biochemistry and Molecular Medicine, University of Bern, Bühlstrasse 28, 3012 Bern, Switzerland
cMembrane Transport Discovery Lab, Department of Nephrology and Hypertension, Inselspital, University of Bern Kinderklinik, Freiburgstrasse 15, 3010 Bern, Switzerland. E-mail: matthias.hediger@ibmm.unibe.ch
dDepartment of Biomedical Research, University of Bern, Murtenstrasse 35, 3008 Bern, Switzerland
eDepartment of Biochemistry, University of Zürich, Winterthurerstrasse 190, Zürich, Switzerland
First published on 2nd June 2020
Abstract
Solute carrier proteins (SLCs) control fluxes of ions and molecules across biological membranes and represent an emerging class of drug targets. SLC11A2 (hDMT1) mediates intestinal iron uptake and its inhibition might be used to treat iron overload diseases such as hereditary hemochromatosis. Here we report a micromolar (IC50 = 1.1 μM) pyrazolyl-pyrimidone inhibitor of radiolabeled iron uptake in hDMT1 overexpressing HEK293 cells acting by a non-competitive mechanism, which however does not affect the electrophysiological properties of the transporter. Isothermal titration calorimetry, competition with calcein, induced precipitation of radioactive iron and cross inhibition of the unrelated iron transporter SLC39A8 (hZIP8) indicate that inhibition is mediated by metal chelation. Mapping the chemical space of thousands of pyrazolo-pyrimidones and similar 2,2′-diazabiaryls in ChEMBL suggests that their reported activities might partly reflect metal chelation. Such metal chelating groups are not listed in pan-assay interference compounds (PAINS) but should be checked when addressing SLCs.
Introduction
Solute carrier proteins (SLCs) control fluxes of ions and other molecules across biological membranes and represent an emerging class of drug targets.1–3 Here, we investigate inhibitors of the human H+-coupled transporter of ferrous iron (Fe2+), SLC11A2 (hDMT1). This transporter is expressed in the intestinal brush border membrane, where it acts as the key mediator of dietary iron uptake. hDMT1 is also linked to pathologies such as hereditary hemochromatosis, β-thalassemia, Parkinson's disease and Alzheimer's disease, highlighting that its pharmacological inhibition may be beneficial to treat human diseases.4–9 While metal chelators have been classically used to treat metal intoxication10 and neurodegenerative diseases,11,12 a recently reported highly potent and specific inhibitor of ferroportin (SLC40), a different iron transporter acting on the same pathway as hDMT1, has been shown to have clinical efficacy against β-thalassemia.13Two families of small molecule hDMT1 inhibitors have been reported in the literature as the results of high-throughput screening campaigns, namely bis-cationic isothioureas such as dibenzofurans 1 and mesitylene 2,14 as well as pyrazolyl-pyridine 3 (Fig. 1).15 In our own investigations on hDMT1, we used the inhibitors mentioned above as seeds for a ligand-based virtual screening campaign guided by 3D-shape and pharmacophore similarity16 and discovered bis-isothiourea 4 and pyrazolyl-pyrimidone 5 as two additional hDMT1 inhibitors.17 Kinetic studies, an X-ray structure of a related brominated bis-isothiourea inhibitor in complex with a bacterial analog of the transporter and mutational studies recently showed that bis-isothiourea-based compounds act as competitive inhibitors of hDMT1.18 On the other hand, pyrazolyl-pyrimidone 5, whose small size and better drug-like properties made it an attractive candidate compound, acted as a non-competitive inhibitor. Here we set out to investigate the pyrazolyl series (3 and 5) closer and understand its mechanism of action.
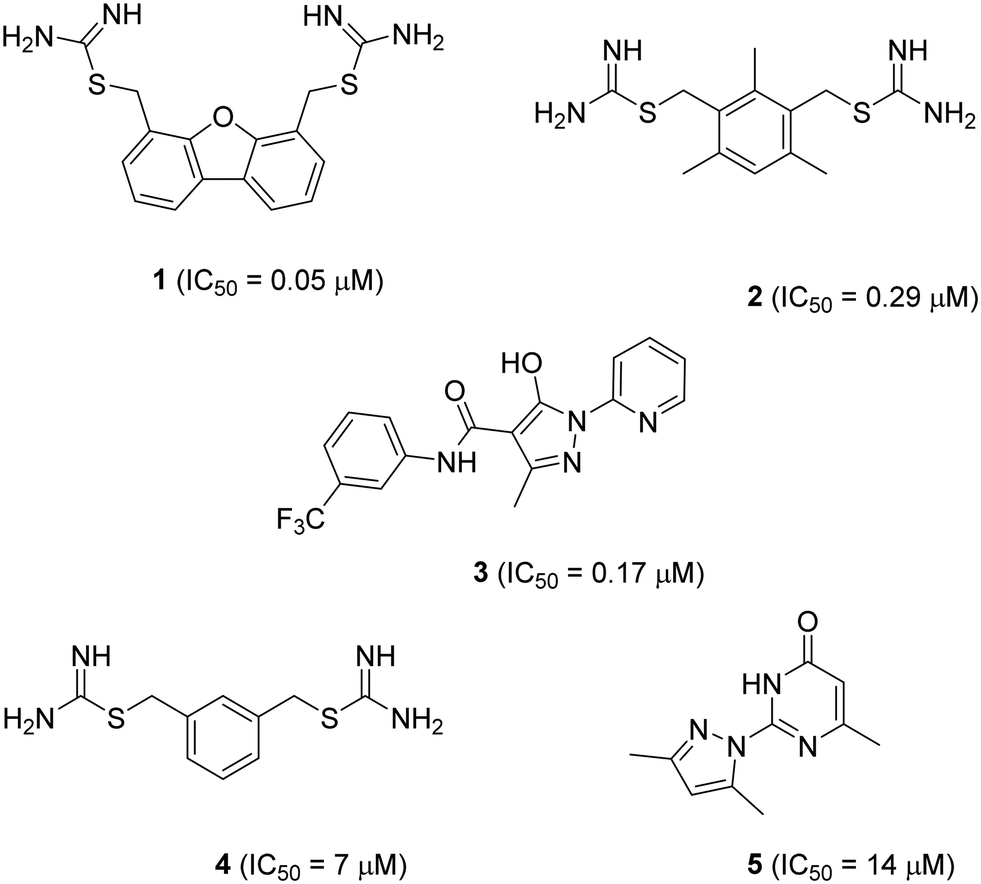 | ||
| Fig. 1 Structure and reported activity of DMT1 inhibitors. Data for inhibition of Fe2+ uptake into transfected HEK cells measured by calcein fluorescence assay (1–3) from ref. 14 and 15 or radioactive 55Fe2+ uptake assay (4 and 5) from ref. 17. | ||
Results and discussion
1. Optimization of pyrazolyl-pyrimidone 5
We prepared twenty new analogs (6–25) of pyrazolyl-pyrimidone 5 by condensing aminoguanidines with 1,3-diketones and keto-esters to form pyrazoles 6–20 and pyrazolones 21–25 using known chemistry (Scheme 1).14,15,19,20 For pyrazoles the synthesis gave only one product from symmetrical diketones (5–9, 16, 18–20). With non-symmetrical diketones synthesis also mostly yielded a single product. X-ray crystal structures of 13 and 17 showed that the product from non-symmetrical diketones was the isomer with the larger pyrazole substituent pointing away from the aminopyridone. By analogy, the same arrangement was assigned to the other pyrazoles. The synthesis of pyrazolones 21–25 only gave a single product whose structures were confirmed by an X-ray crystal structure of 24 as hydrochloride salt (Fig. 2).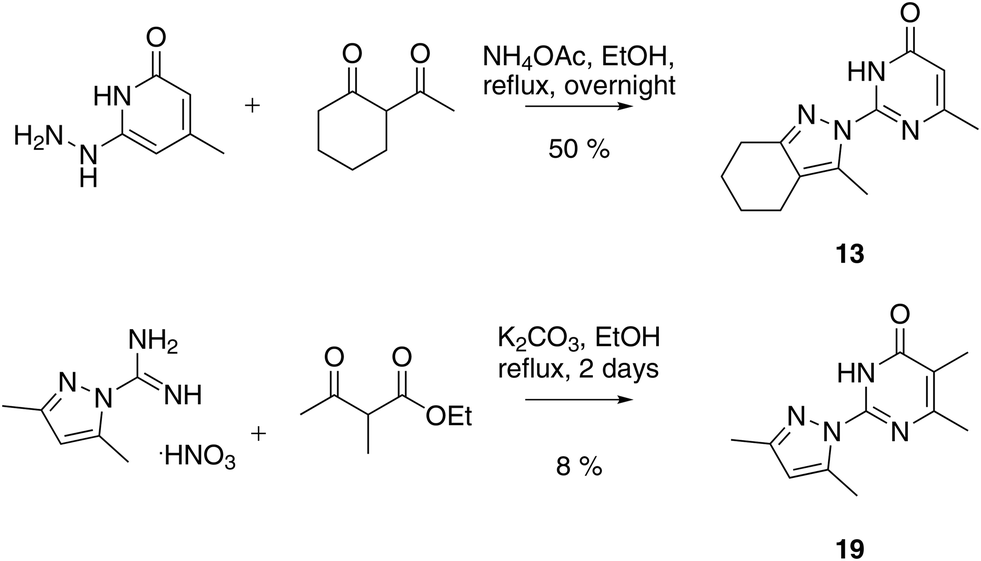 | ||
| Scheme 1 Representative synthesis of pyrazolyl-pyrimidones at the example of 13 and 19. | ||
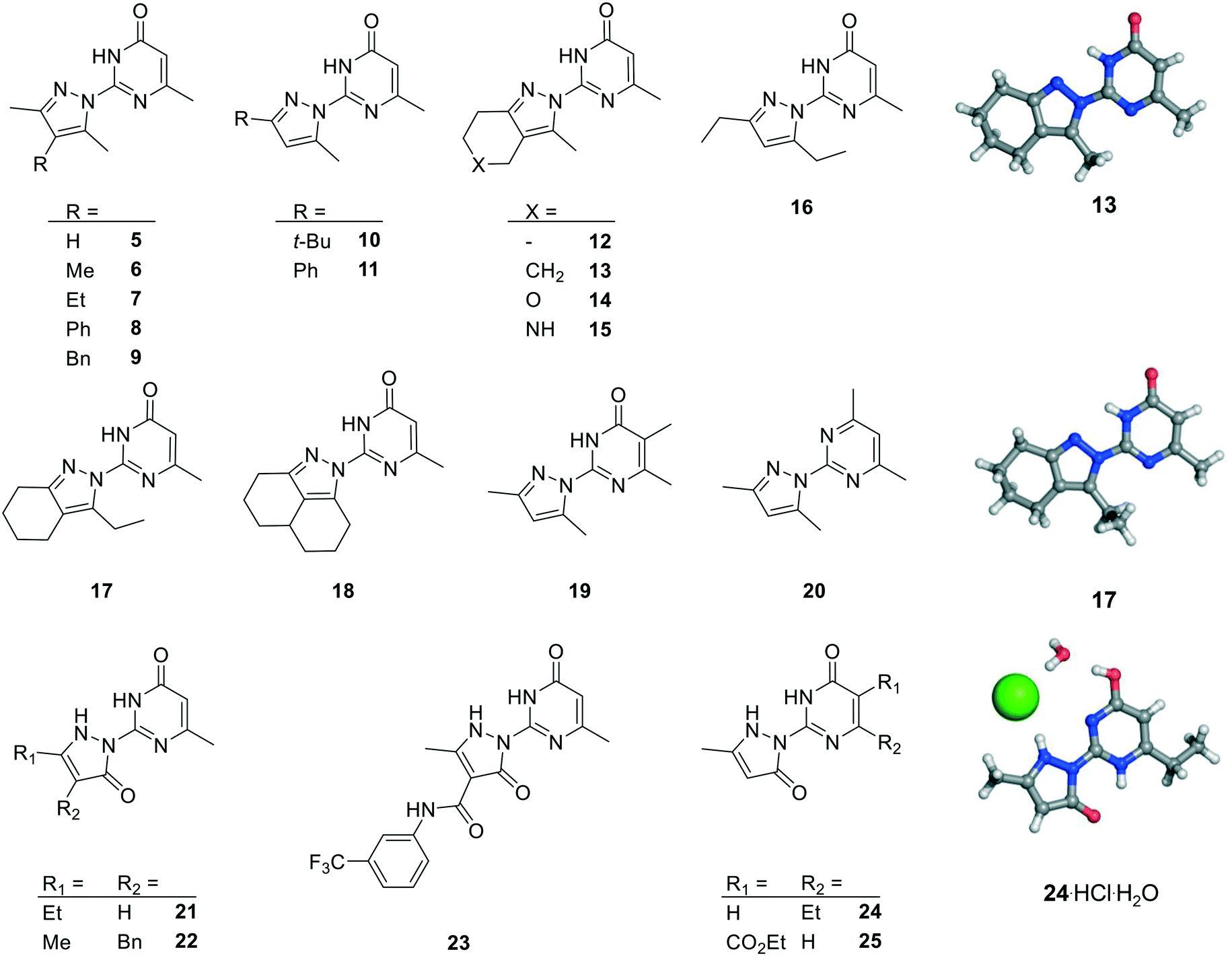 | ||
| Fig. 2 Chemical structures and X-ray crystallography of analogs of pyrazole 5. Carbon is in gray, nitrogen in blue, oxygen in red and chlorine in green. | ||
We measured hDMT1 activity using a radioactive iron uptake assay in HEK293 cells stably overexpressing hDMT1 as described previously.17 Dibenzofuran inhibitor 1 showed only micromolar potency in this assay, which is 5-fold weaker than the originally reported value based on a calcein assay, while the mesitylene inhibitor 2 showed a comparable value to the originally reported inhibition (Table 1). For the present study, we selected dibenzofuran inhibitor 1 as positive control. However, we were unable to determine an IC50 value for the known pyrazolyl-pyridine 3 due to a lack of inhibition at high compound concentration.
| Compound | IC50a (μM) | Ligand efficiency |
|---|---|---|
| a IC50 values were calculated using the radiolabeled iron uptake assay in HEK293 cells (source: American Type Culture Collection, catalog no. CRL-1573) stably overexpressing hDMT1. Absorbed radioactive iron (1 μM) was measured after 15 min of incubation in the presence of the indicated compound at different concentrations at extracellular pH 5.5. b Partial inhibition was detected at 10 μM, however apparent uptake increased at higher concentrations. Compounds 8–12, 14–15, 20–25 showed less than 20% inhibition at 10 μM in this assay. | ||
| 1 | 1.76 ± 0.06 | 0.35 |
| 2 | 0.35 ± 0.04 | 0.25 |
| 3 | ≫10 μMb | |
| 5 | 13.1 ± 0.7 | 0.46 |
| 6 | 10.0 ± 0.4 | 0.44 |
| 7 | 4.2 ± 0.4 | 0.40 |
| 13 | 1.1 ± 0.01 | 0.45 |
| 16 | 3.8 ± 0.2 | 0.45 |
| 17 | 2.26 ± 0.3 | 0.42 |
| 18 | 0.94 ± 0.2 | 0.42 |
| 19 | 12.5 ± 0.3 | 0.43 |
Activity screening of the twenty synthesized analogs of 5 for inhibition of hDMT1 revealed that seven pyrazoles (6, 7, 13, 16–19) showed similar or improved inhibition compared to 5. On the other hand, pyrazoles 8–12, 14–15 and 20 as well as pyrazolones 21–25 did not show significant inhibition of iron uptake in our assay. Across all compounds tested, pyrazole 13 stood out as one of the most potent compounds with IC50 = 1.1 ± 0.01 μM, representing a 10-fold improvement over our initial inhibitor 5 while preserving the original ligand efficiency. Note that the bis-cyclohexane analog 18 was slightly more potent, however at the cost of higher hydrophobicity.
2. Characterization of pyrazolyl-pyrimidone 13
Closer characterization of pyrazolyl-pyrimidone 13 in the radioactive 55Fe2+ uptake assay showed that this compound was a non-competitive inhibitor with similar inhibition constants Ki = 2.0 μM ([Fe2+] → 0) and Kii = 1.7 μM (saturating iron), reproducing the behavior of the previously reported less potent pyrazolyl-pyrimidone 5. By comparison, isothiourea 1 showed a competitive inhibition with inhibition constant Ki = 1.4 μM ([Fe2+] → 0), Kii = 15.6 μM (saturating iron) determined by Dixon plot analysis,21 similarly to the previously reported less potent isothiourea 4 (Fig. S1†).On the other hand, the uptake of radioactive iron in Xenopus oocytes overexpressing hDMT1 was totally inhibited by the bis-thiourea inhibitor 1, while pyrazolyl-pyrimidone 13 only inhibited 50% of the uptake (Fig. 3a). Electrophysiological recordings in this system furthermore showed that only inhibitor 1 blocked hDMT1-induced current with a potency comparable to the iron uptake experiment (IC50 = 0.14 μM), but that pyrazolyl-pyrimidone 13 had no measurable effect on the recorded currents (Fig. 3b/c).
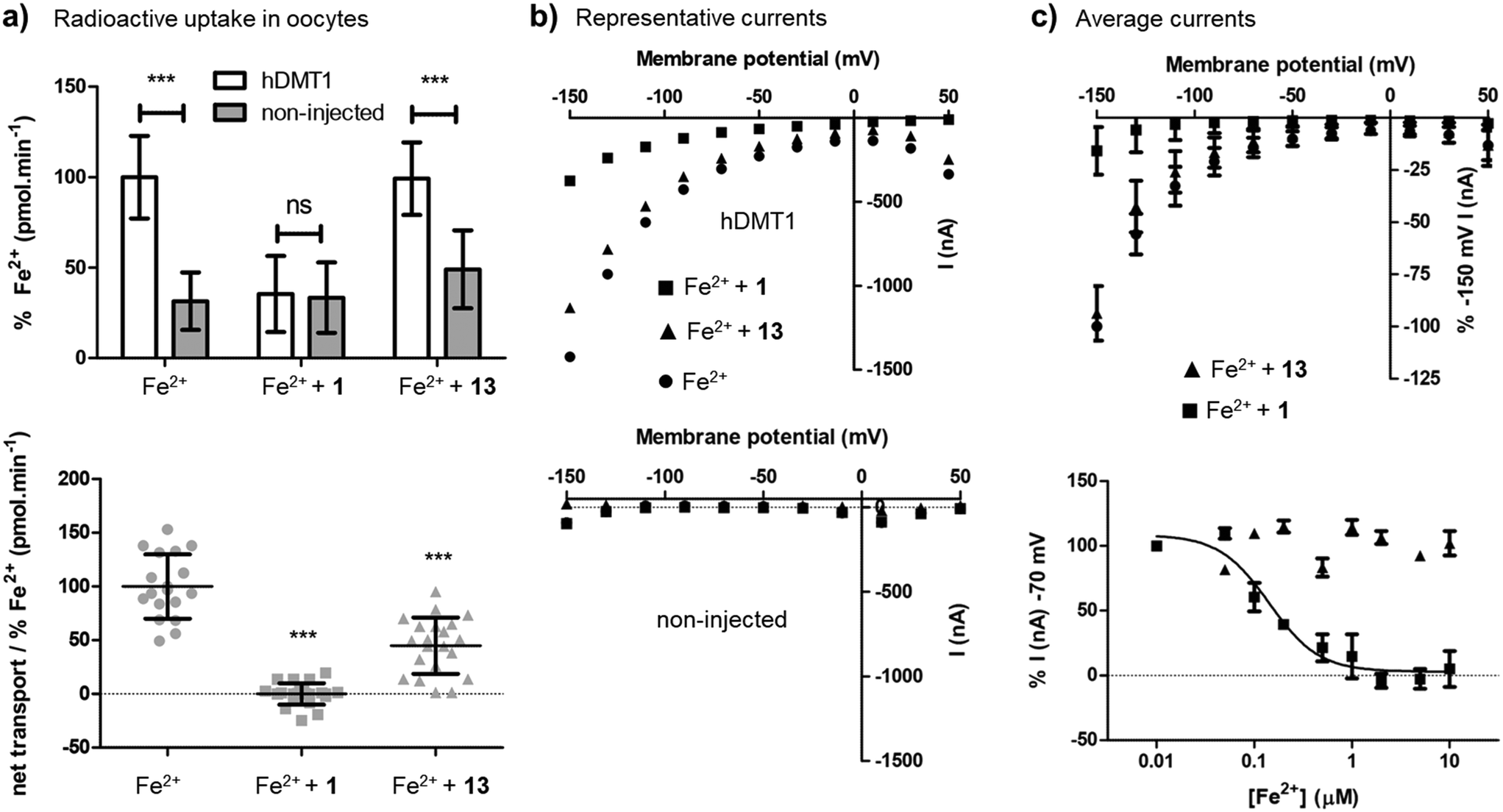 | ||
| Fig. 3 Characterization of Fe2+ transport inhibition by 1 and 13 in X. laevis oocytes (collected from frogs under license number BE60/18 conceded by the Veterinärdienst des Kantons BE, Sekretariat Tierversuche). (a) Upper panel: Average 55Fe2+-uptake (20 μM) by hDMT1-expressing and non-injected oocytes in the absence and the presence of the indicated compounds (10 μM). Data was normalized to 55Fe2+-uptake (pmol min−1) in the absence of compounds represented as mean ± SD (16–29 oocytes). Lower panel: Net uptake of 55Fe2+ (20 μM) by hDMT1 in the absence and the presence of the indicated compounds (10 μM). For each batch of oocytes, uptake values were corrected for unspecific iron uptake in non-injected oocytes. Data were normalized to the mean iron uptake by hDMT1 WT (pmol min−1) and represented as mean ± SD (14–34 oocytes). (b and c) Electrophysiology. Oocytes were held at −50 mV (Vh) and a voltage-step protocol was applied (Vm = −150 mV to +50 mV) before and after the addition of Fe2+ (20 μM) in the absence or the presence of the indicated compounds (10 μM). For each trace, recorded currents were corrected for the background currents observed without substrate. (b) Representative traces recorded with hDMT1 injected oocytes (upper panel) and non-injected oocytes (lower panel). (c) Upper panel: Average current traces of the response of hDMT1 injected oocytes to the indicated compounds. Data was normalized to Fe2+ (20 μM) evoked currents at −150 mV and represented as mean ± SD (8 to 13 oocytes). Lower panel: Average dose–response (0.01–10 μM) of the inhibition of the indicated compounds over the Fe2+ (20 μM) evoked currents at −70 mV. Data was normalized to Fe2+ (20 μM) + compound (0.01 μM) evoked currents and represented as mean ± SD (4 oocytes). Kinetic parameters were obtained by fitting experimental results to a 4-parameter sigmoidal curve (black line). Compound 13 did not fit to the equation while for compound 1 the obtained IC50 was 0.14 μM. All the experiments were performed with oocytes from at least 3 different oocyte batches. Statistical differences were assessed using T-test or Mann–Whitney U test (Fe2+-uptake by hDMT1 WT vs. non-injected oocytes or vs. compound); p > 0.05 = ns; p < 0.001 = ***. | ||
Taken together, these experiments showed that 13 was indeed a more potent version of the non-competitive inhibitor 5. However, in contrast to the well-behaved bis-isothiourea 1, the inhibitory effect of 13 was only detected in iron uptake experiments and was not observed in electrophysiological recordings. A similar effect had been reported for the literature inhibitor 3.15
3. Chemical evidence for metal complex formation
At this stage we suspected that pyrazolyl-pyrimidone 13 might inhibit hDMT1 indirectly by chelating iron, although this possibility was judged unlikely for the related inhibitor 3 by lack of conclusive evidence.15 These inhibitors contain an N:N(:C)–C:N substructure which is potentially capable of forming a 5-membered ring chelate with metal ions via the two terminal sp2 nitrogen atoms, although this substructure is present in all the synthesized analogs including those that did not show any inhibition. The formation of such 5-membered ring chelate has been recently established by X-ray crystallography of a ruthenium complex containing both a pyrazolyl-pyridine and a pyrazolyl-pyridone ligand and investigated as C–H oxidation catalyst.22 Unfortunately, UV-vis titration of 13 could not be used to provide any evidence for metal complexation since the UV-vis spectrum did not significantly change upon titration with Fe2+ and Cd2+. Attempts to crystallize the iron complex of 13 as well as investigation by 1H-NMR and mass spectrometry similarly did not yield any indication of complex formation.A first indication that pyrazolyl-pyrimidone 13 might interact with divalent metal ions was obtained by isothermal titration calorimetry (ITC) with Cd2+, which we used as a redox stable analog of Fe2+ less prone to hydroxide formation at pH = 7.4 and which is also transported by DMT1. At a pH of 7.4, the ITC data indicated an apparent dissociation constant of KD = 6.4 μM with n = 0.54, consistent with the formation of a 2:1 ligand–metal complex (Fig. 4a). At pH 5.5 under which the hDMT1 transport experiment is carried out, the ITC data could not be interpreted with certainty. Upon titration of Cd2+ into a solution of 13, the exothermic signal seems to saturate after the first injections, but later results in a noisy signal, indicating precipitation (Fig. 4b). The less potent pyrazolyl-pyrimidone DMT1 inhibitor 5 also interacted with Cd2+ at pH 7.4, although binding was much weaker (KD ∼ 250 μM, n = 0.74).
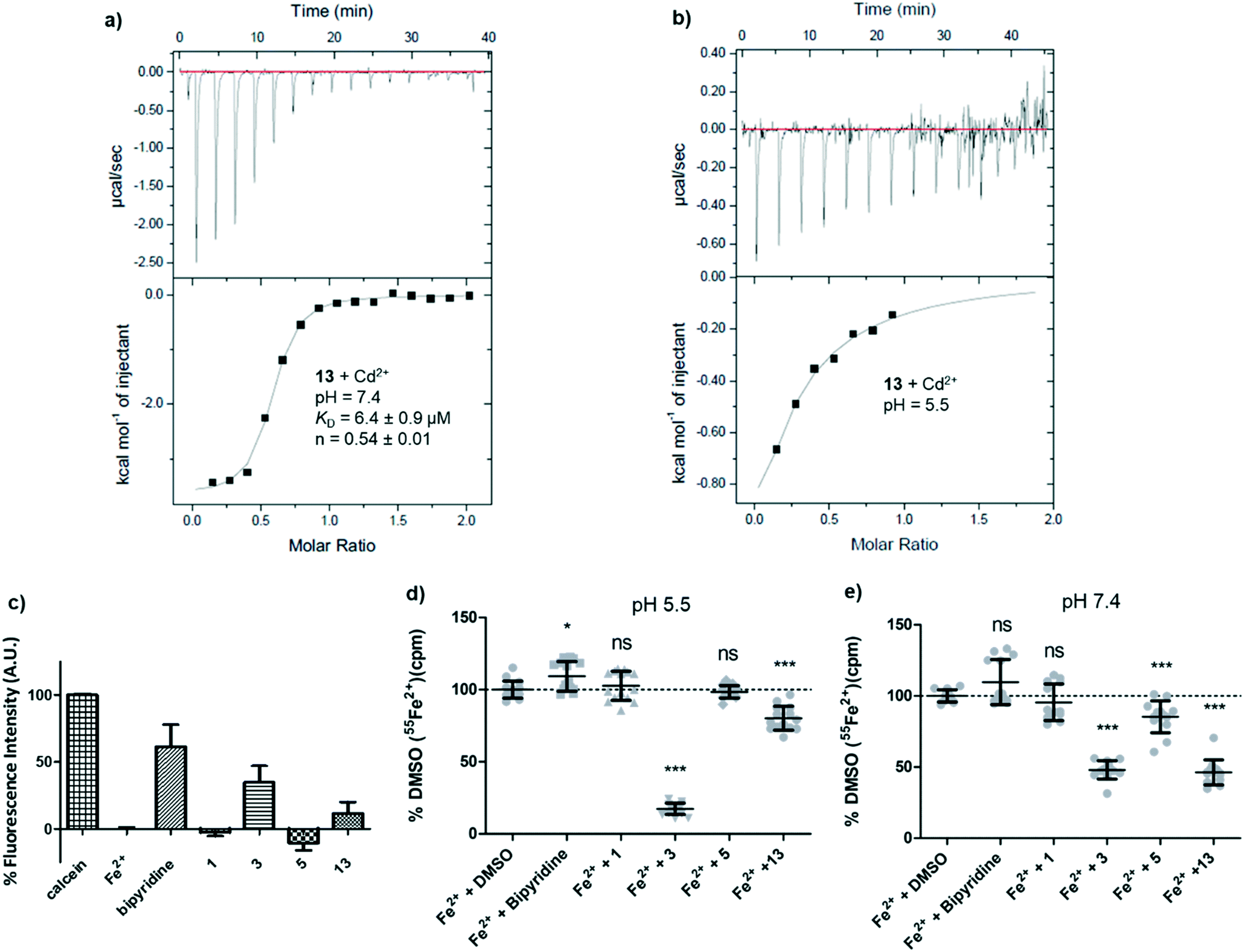 | ||
| Fig. 4 Chemical evidence of divalent metal complexation by hDMT1 inhibitors. (a and b) Isothermal titration calorimetry of pyrazole 13 (0.4 mM) with Cd2+ (5 mM) in aqueous buffer pH 7.4 and pH 5.5, 25 °C. (c) Calcein quenching assay in aqueous buffer pH 5.5 (2 μM Fe2+, 10 μM ligand, 1 μM calcein). Fe2+ (2 μM), ascorbic acid (200 μM) and compounds (20 μM) were incubated for 5 min before the addition of calcein (1 μM) in Krebs buffer at pH 5.5. Fluorescence was measured after 5 min incubation. (d and e) Removal of radioactive 55Fe2+ from buffers upon incubation with ligands and centrifugation. 55Fe2+ (1 μM) precipitation upon interaction with the indicated compounds (10 μM). 55Fe2+ and the indicated compounds were incubated for 15 min, then, an aliquot of the solution supernatant was taken for determination of the remaining 55Fe2+. Data was normalized to the values obtained for the Fe2+ + DMSO (0.1%) solution and represented as mean ± SD (N = 15; 2 independent experiments). Statistical differences were assessed using T-test or Mann–Whitney U test (Fe2++ DMSO vs. each compound); p > 0.05 = ns; p < 0.05 = *; p < 0.001 = ***. | ||
A calcein competition assay to detect Fe2+ chelation at pH 5.5 also indicated a significant level of iron chelation by 13, although the effect was weaker than with the positive control bipyridine or the literature inhibitor 3, and pyrazolyl-pyrimidone 5 did not show any iron chelation in this assay (Fig. 4c). Tracking the radioactivity of 55Fe2+ in solutions after incubation with the various inhibitors indicated that pyrazolyl-pyrimidone 13 as well as the literature inhibitor 3 induced significant precipitation of Fe2+ at pH 7.4, while bis-isothiourea 1, bipyridine or pyrazolyl-pyrimidone 5 had no effect. On the other hand, at pH 5.5, we only observed precipitation with the literature inhibitor 3 corresponding to the pH of the assay with hDMT1 (Fig. 4d and e). Such precipitation might indicate chelation of iron to form an insoluble complex. Taken together, these data pointed to potential iron chelating abilities of our inhibitors, however the evidence was inconclusive and did not match well with the observed inhibitory potencies.
4. Cross-inhibition of human SLC11A2 (hDMT1) and SLC39A8 (hZIP8)
Complex formation with Fe2+ should result in non-selective inhibition of different metal transporters. To test this hypothesis, we investigated whether human SLC39A8 (hZIP8), a transporter of Zn2+ and Fe2+ unrelated to SLC11 family, might also be inhibited by our hDMT1 inhibitors. Both SLCs can be tested by inhibition of radioactive iron uptake into HEK293 cells expressing the transporter, at pH 5.5 for hDMT1 and at pH 7.4 for hZIP8.Indeed, activity screening showed that bipyridine, a well-known iron chelator, non-specifically inhibited both transporters. Similarly, the literature pyrazolyl-pyridine 3, our optimized pyrazolyl-pyrimidone 13, and to a lesser extent pyrazolyl-pyrimidone 5, all inhibited both transporters, although the inhibition by 3 was not consistently observed at higher concentrations, probably due to adsorption of the complex to the cell material and assay plate, resulting in apparent uptake. On the other hand, bis-isothiourea 1, which inhibits hDMT1 by competing in the iron binding site, only inhibited hDMT1 and had no measurable activity on hZIP8 (Fig. 5a). A precise determination of the inhibitory effects with pyrazolyl-pyrimidone 13 and with the known iron chelator bipyridine showed that these compounds had comparable IC50 values on both transporters (13: IC50 ∼ 1.1 μM, bipyridine: IC50 ∼ 6.5 μM, Fig. 5b/c).
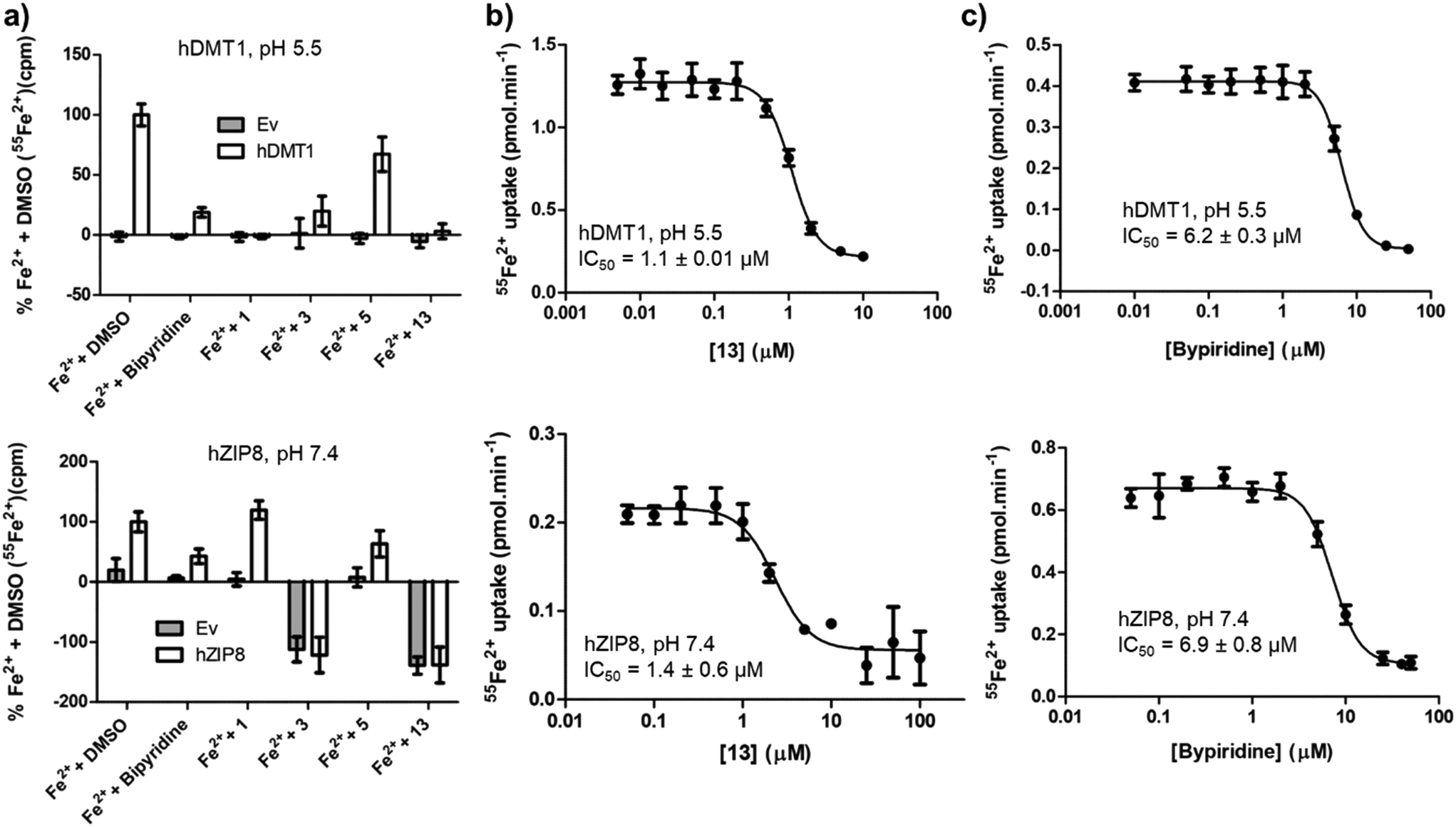 | ||
| Fig. 5 Cross reactivity of hDMT1 inhibitors over the functional activity of the divalent metal transporter hZIP8. (a) 55Fe2+ uptake (1 μM) by HEK293T cells (source: American Type Culture Collection, catalog no. CRL-3216) transiently transfected with the empty vector (Ev), hDMT1 or hZIP8. Uptake assay was performed at the optimal pH for the functional activity of each transporter (pH 5.5 and pH 7.4 respectively). The indicated compounds (10 μM) were pre-incubated for 5 min, and then, for 15 min with both compound and 55Fe2+. Measured 55Fe2+ was corrected by subtracting the background iron uptake measured in non-transfected cells. The data was normalized to the Fe2+ uptake measured in the presence of DMSO (0.1%) and represented as mean ± SD (N = 5–16; obtained from 2 independent experiments). IC50 determination for pyrazolyl-pyrimidone 13 (b) and bipyridine (c) in HEK293T cells transiently transfected with hDMT1 (left panels) or hZIP8 (right panels). 55Fe2+ uptake (1 μM) was measured in the presence of the indicated compound concentration ranges, and the measurements were corrected by subtracting the background iron uptake measured in non-transfected cells. IC50 values were calculated by fitting the data to a sigmoidal 4-parameter equation. Representative experiments are depicted, while IC50 values were calculated as mean ± SD of 3 different experiments. | ||
5. Mechanistic model
We interpret the non-selective inhibition of bipyridine and 13 on both hDMT1 and hZIP8 as an indication that inhibition is caused by chelation of iron without specific interactions with the transporters. Metal chelation most likely involves a five-membered ring chelate with the two cyclic nitrogen atoms within the N:N(:C)–C:N substructures, similar to the well-known chelation by bipyridine forming a tight 3:1 complex with Fe2+. Acid–base titration of 3, 5 and 13 shows in each case two ionizable groups (3: pKa1 = 3.3, pKa2 = 6.7; 5: pKa1 = 3.6, pKa2 = 6.5; 13: pKa1 = 3.3, pKa2 = 7.0, Fig. 6a). The transition at neutral pH (pKa2) can be attributed to the pyrazole hydroxyl group for 3 and to the pyrimidone NH group for 5 and 13, resulting in a transition from the neutral form below pH 7 to a mono-anionic form above pH 7 (Fig. 6a). Both forms possess an N:N(:C)–C:N substructure and should have metal chelating abilities, the neutral form existing at pH 5.5 where hDMT1 transport is measured, and the anionic form existing at pH 7.4 where hZIP8 transport is measured (Fig. 6b).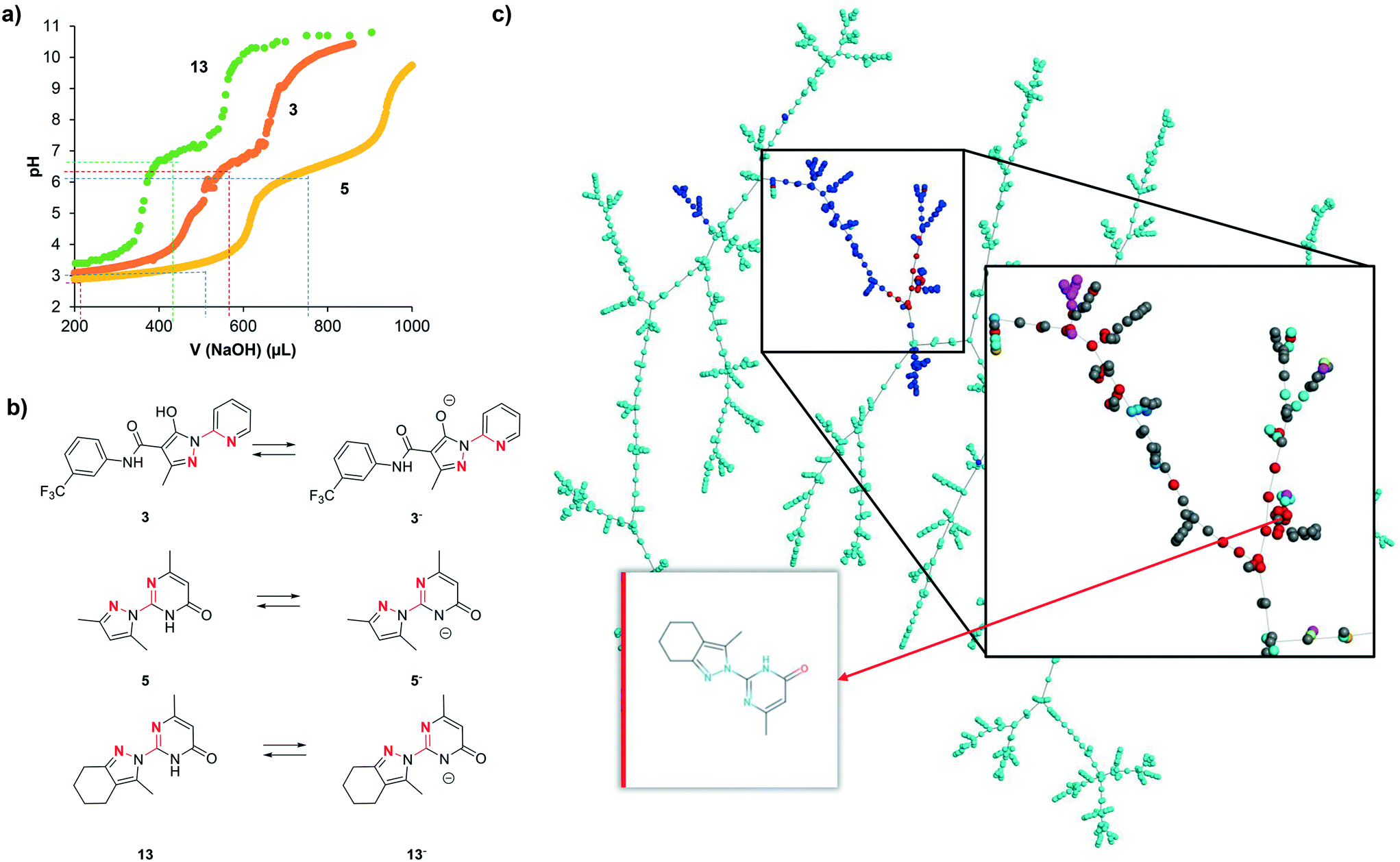 | ||
| Fig. 6 (a) Acid–base titration of 3, 5, 13 and neutral and basic forms of 3, 5, 13. (20 μmol of compound in 10 mL (10% DMSO, ∼2 mM), 3 and 5 were acidified with HCl (50 μmol)). (b) Acid-base equilibria for inhibitors 3, 5 and 13 highlighting potentially metal chelating nitrogen atoms in red. (c) TMAP of our inhibitors combined with 169 pyrazolyl-pyrimidones and 1717 pyrazolyl-pyridines found in ChEMBL, color coded by source: cyan = pyrazolyl-pyridines from ChEMBL, blue = pyrazolyl-pyrimidones from ChEMBL, red = this work. Insert: Close-up view around pyrazolyl-pyrimidones color-coded by target class: red = transporter, cyan = enzyme, magenta = unclassified protein, grey = unclassified target. The interactive TMAP including further color-codes (target classes, QED, etc.) is accessible at: http://tm.gdb.tools/dmt1_inhibitors/pyrimidones_pyridines/ | ||
However, while all pyrazolyl-pyrimidones and pyrazolyl-pyridines tested here should have comparable metal chelating abilities, only a few of them actually showed activity in the iron uptake assays with hDMT1 and hZIP8. We therefore postulate that inhibition only occurs either if metal complexation is tight, as for the reference chelator bipyridine, or if metal complexation results in the formation of an insoluble complex, as observed using radioactive 55Fe2+ with 3 and 13. Note that, while iron uptake into hDMT1 expressing cells was partially reduced with the literature inhibitor 3, we did not observe inhibition at higher concentrations (Table 1), probably because complexed iron was in this case absorbed on the cell material and assay plate, and resulted in apparent iron uptake. This is in line with the Fe2+ precipitation experiments at pH 5.5, where we only observe high precipitation of iron in the presence of compound 3. The lack of inhibition by most of the compounds tested might therefore either reflect partial iron chelation or formation of an iron complex that partially absorbs on the cells, resulting in apparent iron uptake and no net inhibition.
6. Pyrazolyl-pyrimidones and pyrazolyl-pyridines in chemical space
To better understand our compound series, we surveyed the ChEMBL database, which lists 1.82 million bioactive molecules and their annotated targets,23 and found that 0.86% of these (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 585 molecules) contain a potentially metal chelating 2,2′-diazabiaryl group, including 1717 pyrazolyl-pyridines related to 3 and 169 pyrazolyl-pyrimidones related to 5 and 13 and annotated with bioactivities (Table 2). Among the 9.2 million screening compounds available in stock from commercial providers as listed in the ZINC database,24 we similarly found 0.87% (80153 molecules) 2,2′-diazabiaryls, however with relatively fewer pyrazolyl-pyridines (4673 molecules) but more abundant pyrazolyl-pyrimidones (1960 molecules), which suggests that such 2,2′-diazabiraryl compounds are relatively easy to synthesize and drug-like. The abundance of 2,2′-diazabiaryls was significantly lower in theoretically enumerated chemical space databases such as FDB17,25 GDBMedChem26 and GDBChEMBL,27 reflecting the fact that these databases contain a much higher fraction of non-aromatic molecules.
585 molecules) contain a potentially metal chelating 2,2′-diazabiaryl group, including 1717 pyrazolyl-pyridines related to 3 and 169 pyrazolyl-pyrimidones related to 5 and 13 and annotated with bioactivities (Table 2). Among the 9.2 million screening compounds available in stock from commercial providers as listed in the ZINC database,24 we similarly found 0.87% (80153 molecules) 2,2′-diazabiaryls, however with relatively fewer pyrazolyl-pyridines (4673 molecules) but more abundant pyrazolyl-pyrimidones (1960 molecules), which suggests that such 2,2′-diazabiraryl compounds are relatively easy to synthesize and drug-like. The abundance of 2,2′-diazabiaryls was significantly lower in theoretically enumerated chemical space databases such as FDB17,25 GDBMedChem26 and GDBChEMBL,27 reflecting the fact that these databases contain a much higher fraction of non-aromatic molecules.
| Database | ChEMBL24 | ZINCinStock | FDB17 | GDBMedChem | GDBChEMBL |
|---|---|---|---|---|---|
|
a Compounds were extracted using the corresponding SMARTS patterns. A TMAP of the 15585 2,2′-diazabiaryls from ChEMBL24 is available at http://tm.gdb.tools/dmt1_inhibitors/diazabiaryls/. See also Fig. 6c–f and Table S1† for SMARTS patterns.
|
|||||
| Number of molecules | 1820035 |
9238092 |
10101204 |
9994112 |
9978095 |
| 2,2′-Diazabiaryls | 15585 |
80153 |
13880 |
6241 | 2629 |
| % of database | 0.86% | 0.87% | 0.14% | 0.06% | 0.03% |
| Pyrazolyl-pyridines | 1717 | 4673 | 110 | 72 | 65 |
| % of 2,2′-diazabiaryls | 11% | 5.8% | 0.8% | 1.2% | 2.5% |
| Pyrazolyl-pyrimidones | 169 | 1960 | 0 | 26 | 0 |
| % of 2,2′-diazabiaryls | 1.1% | 2.4% | 0.0% | 0.4% | 0.0% |
We recently developed interactive chemical space visualization tools that provide useful insights into polypharmacology and structure–activity relationships.28,29 To obtain a closer insight into the selected 2,2′-diazabiaryls, we created a tree-map (TMAP)30 representing pyrazolyl-pyridines and pyrazolyl-pyrimidones from ChEMBL (Fig. 6c). The TMAP can be interactively analyzed in-browser by loading its layout using the program Faerun.31 In this map each compound is represented by a point that can be color-coded according to selected properties, and the molecular structure is shown upon mouse-over by the program Smilesdrawer.32 The compounds are connected in a tree topology according to their structural similarities as measured by the MHFP6 molecular fingerprint.33 The TMAP illustrates the diversity of pyrazolyl-pyrimidones and pyrazolyl-pyridines in ChEMBL. The pyrazolyl-pyrimidones investigated here belong to the more drug-like molecules in the series, as can be appreciated by color-coding for drug-likeness using the quantitative estimate of drug-likeness (QED).34
The activities reported for these molecules in ChEMBL cover a diversity of target types, spanning from transporters and ion channels to enzymes and uncharacterized targets. We found 45 2,2′-diazabiaryl including 18 pyrazolyl-pyrimidones closely related to our compounds and reported with transporter activity in ChEMBL (Table S2†). The ChEMBL record indicates, among various targets, inhibition of glucose and hexose transporters identified in antiparasitic screening campaigns against Leishmania mexicana, tuberculosis, and Plasmodium falciparum.35 However, the screens performed were not transport assays but simple cytotoxicity tests. Considering that metal chelation is known to be a mechanism of toxicity against these parasites,36,37 one can speculate that the reported transporter inhibitory activity of these pyrazolyl-pyrimidones might in fact reflect toxicity as a result of metal chelation. Further activities reported for pyrazolyl-pyrimidones and pyrazolyl-pyridines might similarly be the consequence of metal chelation rather than the attributed activity, including a recently reported activity of 3 and 5 against cancer cells.38 Note that potentially metal chelating 2,2′-diazabiaryls such as pyrazolyl-pyrimidones are not listed in PAINS (pan-assay interference compounds).39–41 Indeed a systematic checking of our compounds with the PAINS filter from RDkit does not flag any molecule in the series.
Conclusion
To date the pharmacology of divalent metal transporters is extremely limited. In view of the increasing importance of SLCs as drug targets, we investigated the inhibition of hDMT1 mediated iron transport by two previously reported inhibitors, namely pyrazolyl-pyridine 3 and pyrazolyl-pyrimidone 5. Optimization of 5 uncovered analog 13 showing potent inhibition of hDMT1 (IC50 = 1.1 μM). However, this inhibitor did not modulate hDMT1-induced currents in Xenopus oocytes and exerted cross-inhibition of the unrelated iron transporter hZIP8.Our combined experiments suggest that this non-selective inhibition reflects chelation of Fe2+ in the assay medium by these molecules to form an insoluble precipitate. Although metal chelation is often exploited as a mechanism of drug action,10–12 metal chelation is in principle an assay interference. This effect might occur across thousands of bioactive molecules reported in ChEMBL that share the same potentially chelating substructure and explain some of their reported bioactivities. Pyrazolyl-pyrimidones and related potentially metal chelating 2,2′-diazabiaryls are not considered in the list of pan-assay interference compounds (PAINS), however these should be checked whenever screening SLC targets.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Swiss National Science Foundation, grants no. 51NF40-185544 (JLR) and 310030_182272 (MH). We thank the group of Chemical Crystallography of the University of Bern (PD Dr. P. Macchi) for the X-ray structure solution and the Swiss National Science Foundation (R'equip project 206021_128724) for co-funding the single crystal X-ray diffractometer at the department of Chemistry and Biochemistry of the University of Bern.References
- A. César-Razquin, B. Snijder, T. Frappier-Brinton, R. Isserlin, G. Gyimesi, X. Bai, R. A. Reithmeier, D. Hepworth, M. A. Hediger, A. M. Edwards and G. Superti-Furga, Cell, 2015, 162, 478–487 CrossRef PubMed.
- L. Lin, S. W. Yee, R. B. Kim and K. M. Giacomini, Nat. Rev. Drug Discovery, 2015, 14, 543–560 CrossRef CAS PubMed.
- T. Schumann, J. König, C. Henke, D. M. Willmes, S. R. Bornstein, J. Jordan, M. F. Fromm and A. L. Birkenfeld, Pharmacol. Rev., 2020, 72, 343–379 CrossRef PubMed.
- N. F. Olivieri, N. Engl. J. Med., 1999, 341, 99–109 CrossRef CAS PubMed.
- R. Mariani, P. Trombini, M. Pozzi and A. Piperno, Mediterr. J. Hematol. Infect. Dis., 2009, 1, e2009006 Search PubMed.
- A. Shawki, P. B. Knight, B. D. Maliken, E. J. Niespodzany and B. Mackenzie, Curr. Top. Membr., 2012, 70, 169–214 CAS.
- N. Montalbetti, A. Simonin, G. Kovacs and M. A. Hediger, Mol. Aspects Med., 2013, 34, 270–287 CrossRef CAS PubMed.
- L.-N. Lu, Z.-M. Qian, K.-C. Wu, W.-H. Yung and Y. Ke, Mol. Neurobiol., 2017, 54, 5213–5224 CrossRef CAS PubMed.
- X. Wang, M. Zhang, S. R. L. Flores, R. R. Woloshun, C. Yang, L. Yin, P. Xiang, X. Xu, M. D. Garrick, S. Vidyasagar, D. Merlin and J. F. Collins, Mol. Ther., 2019, 27, 493–506 CrossRef CAS PubMed.
- S. J. S. Flora and V. Pachauri, Int. J. Environ. Res. Public Health, 2010, 7, 2745–2788 CrossRef CAS PubMed.
- T. A. Sales, I. G. Prandi, A. A. de Castro, D. H. S. Leal, E. F. F. da Cunha, K. Kuca and T. C. Ramalho, Int. J. Mol. Sci., 2019, 20, 1829 CrossRef CAS PubMed.
- Y. P. Singh, A. Pandey, S. Vishwakarma and G. Modi, Mol. Diversity, 2019, 23, 509–526 CrossRef CAS PubMed.
- V. Manolova, N. Nyffenegger, A. Flace, P. Altermatt, A. Varol, C. Doucerain, H. Sundstrom and F. Dürrenberger, J. Clin. Invest., 2020, 130, 491–506 CrossRef CAS PubMed.
- Z. Zhang, V. Kodumuru, S. Sviridov, S. Liu, M. Chafeev, S. Chowdhury, N. Chakka, J. Sun, S. J. Gauthier, M. Mattice, L. G. Ratkay, R. Kwan, J. Thompson, A. B. Cutts, J. Fu, R. Kamboj, Y. P. Goldberg and J. A. Cadieux, Bioorg. Med. Chem. Lett., 2012, 22, 5108–5113 CrossRef CAS PubMed.
- J. A. Cadieux, Z. Zhang, M. Mattice, A. Brownlie-Cutts, J. Fu, L. G. Ratkay, R. Kwan, J. Thompson, J. Sanghara, J. Zhong and Y. P. Goldberg, Bioorg. Med. Chem. Lett., 2012, 22, 90–95 CrossRef CAS PubMed.
- C. Simonin, M. Awale, M. Brand, R. van Deursen, J. Schwartz, M. Fine, G. Kovacs, P. Hafliger, G. Gyimesi, A. Sithampari, R. P. Charles, M. A. Hediger and J. L. Reymond, Angew. Chem., Int. Ed., 2015, 54, 14748–14752 CrossRef CAS PubMed.
- N. Montalbetti, A. Simonin, C. Simonin, M. Awale, J.-L. Reymond and M. A. Hediger, Biochem. Pharmacol., 2015, 96, 216–224 CrossRef CAS PubMed.
- C. Manatschal, J. Pujol-Giménez, M. Poirier, J.-L. Reymond, M. A. Hediger and R. Dutzler, eLife, 2019, 8, e51913 CrossRef PubMed.
- A. V. Erkin and V. I. Krutikov, Russ. J. Gen. Chem., 2009, 79, 1204–1209 CrossRef CAS.
- J. Orrego-Hernández, J. Cobo and J. Portilla, Eur. J. Org. Chem., 2015, 2015, 5064–5069 CrossRef.
- W. J. Dixon, Annu. Rev. Pharmacol. Toxicol., 1980, 20, 441–462 CrossRef CAS PubMed.
- C.-B. Bo, Q. Bu, X. Li, G. Ma, D. Wei, C. Guo, B. Dai and N. Liu, J. Org. Chem., 2020, 85, 4324–4334 CrossRef CAS PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- R. Visini, M. Awale and J. L. Reymond, J. Chem. Inf. Model., 2017, 57, 700–709 CrossRef CAS PubMed.
- M. Awale, F. Sirockin, N. Stiefl and J.-L. Reymond, Mol. Inf., 2019, 38, 1900031 CrossRef CAS PubMed.
- S. Bühlmann and J.-L. Reymond, Front. Chem., 2020, 8, 46 CrossRef PubMed.
- M. Poirier, M. Awale, M. A. Roelli, G. T. Giuffredi, L. Ruddigkeit, L. Evensen, A. Stooss, S. Calarco, J. B. Lorens, R.-P. Charles and J.-L. Reymond, ChemMedChem, 2019, 14, 224–236 CrossRef CAS PubMed.
- C. Delalande, M. Awale, M. Rubin, D. Probst, L. C. Ozhathil, J. Gertsch, H. Abriel and J.-L. Reymond, Eur. J. Med. Chem., 2019, 166, 167–177 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, Aust. J. Chem., 2020, 12, 12 Search PubMed.
- D. Probst and J.-L. Reymond, Bioinformatics, 2018, 34, 1433–1435 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, J. Chem. Inf. Model., 2018, 58, 1–7 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, Aust. J. Chem., 2018, 10, 66 CAS.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- F.-J. Gamo, L. M. Sanz, J. Vidal, C. de Cozar, E. Alvarez, J.-L. Lavandera, D. E. Vanderwall, D. V. S. Green, V. Kumar, S. Hasan, J. R. Brown, C. E. Peishoff, L. R. Cardon and J. F. Garcia-Bustos, Nature, 2010, 465, 305–310 CrossRef CAS PubMed.
- C. Raventos-Suarez, S. Pollack and R. L. Nagel, Am. J. Trop. Med. Hyg., 1982, 31, 919–922 CrossRef CAS PubMed.
- R. J. Wall, S. Moniz, M. G. Thomas, S. Norval, E.-J. Ko, M. Marco, T. J. Miles, I. H. Gilbert, D. Horn, A. H. Fairlamb and S. Wyllie, Antimicrob. Agents Chemother., 2018, 62, e00235-18 CrossRef PubMed.
- A. L. Turcu, A. Versini, N. Khene, C. Gaillet, T. Cañeque, S. Müller and R. Rodriguez, Chem. – Eur. J., 2020 DOI:10.1002/chem.202000159.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- S. J. Capuzzi, E. N. Muratov and A. Tropsha, J. Chem. Inf. Model., 2017, 57, 417–427 CrossRef CAS PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods for bioassays, details of kinetic studies, SMARTS pattern analysis, chemical synthesis procedures and compound characterization, X-ray deposition information, HPLC purity table, NMR spectra of final compounds, and SMILES list of final compounds with activities and of compounds extracted from ChEMBL. CCDC 1976759, 1976832 and 1976862 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0md00085j |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |