
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of novel 2,4-disubstituted arylthiazoles against T. brucei†
Markos-Orestis
Georgiadis
 a,
Violeta
Kourbeli
a,
Ioannis P.
Papanastasiou
*a,
Andrew
Tsotinis
a,
Martin C.
Taylor
b and
John M.
Kelly
b
a,
Violeta
Kourbeli
a,
Ioannis P.
Papanastasiou
*a,
Andrew
Tsotinis
a,
Martin C.
Taylor
b and
John M.
Kelly
b
aDivision of Pharmaceutical Chemistry, Department of Pharmacy, School of Health Sciences, National and Kapodistrian University of Athens, Panepistimioupoli-Zografou, 157 84 Athens, Greece. E-mail: papanastasiou@pharm.uoa.gr
bDepartment of Pathogen Molecular Biology, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1 E7HT, UK
First published on 19th December 2019
Abstract
The design, synthesis and pharmacological evaluation of the 4-substituted-2-[3-(adamant-1-yl)-4-fluorophenyl]thiazoles 1a–j, the 4-substituted-2-[4-(adamant-1-yl)phenyl]thiazoles 2a–h, the 2-substituted-4-[4-(adamant-1-yl)phenyl]thiazoles 3a–e, the N-substituted 2-phenylthiazol-4-ethylamides 4a, b and the N-substituted 4-phenylthiazol-2-ethylamides 4c, d is described. Compounds 1a and 2a exhibit trypanocidal activity in the range of IC50 = 0.42 μM and IC50 = 0.80 μM, respectively. Both of these derivatives bear a lipophilic end, which consists of a 4-(1-adamantyl) phenyl or a 3-(1-adamantyl)phenyl moiety, a 1,3-thiazole ring and a functional end, which comprises of an alkylamine and can be considered as promising candidates for the treatment of Trypanosoma brucei infections.
Introduction
The African sleeping sickness and the Chagas disease are two of the major neglected tropical diseases (NTDs). The trypanosomiases are vector-borne parasitic infections caused by flagellated protozoa of the class Kinetoplastida.1 There are two species of human-infectious trypanosomes, Trypanosoma brucei, that causes human African trypanosomiasis (HAT), and Trypanosoma cruzi, which is responsible for the Chagas disease. HAT is prevalent in sub-Saharan Africa, transmitted by the bite of a tsetse fly infected with one of the two subspecies, Trypanosoma brucei gambiense or Trypanosoma brucei rhodesiense. The Chagas disease is spread predominantly in Latin and Central America by Triatominae bugs infected with T. cruzi.2,3 Trypanosomiases, as with other NTDs, are becoming public health problems in non-endemic countries, as a result of travel and migration. New drugs are urgently required, as those that are currently available are characterized by side-effects and treatment failures.4,5Various initiatives6–8 have led to the discovery of promiscuous trypanocidal derivatives from phenotypic high-throughput screening of a number of compound libraries. These have been further refined and optimized to enhance drug-like properties. The Walter and Eliza Hall Institute (WEHI), in partnership with the Drugs for Neglected Diseases initiative (DNDi), and the Genomics Institute of the Novartis Research Foundation (GNF) have described the amide and urea derivatives of thiazolethylamines I, II and sulfonamides III, shown in Fig. 1, as potent trypanocidals.6,7
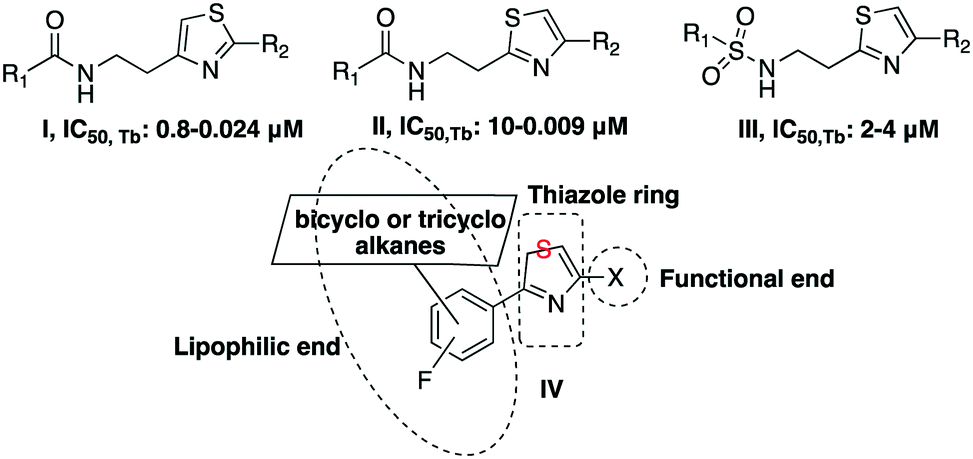 | ||
| Fig. 1 General type scaffolds with trypanocidal activity. | ||
Based on these findings and our involvement in the adamantane chemistry,9–20 we report herein on the chemistry and biology of thiazole derivatives of the general type scaffold IV. The thiazole moiety is an important pharmacophore in many compounds used against several tropical infectious diseases.21
Scaffold IV includes a 1,3-thiazole moiety, which is 2,4-disubstituted. One substituent is the lipophilic end of the scaffold, which consists of a phenyl ring bearing fluoro- and 1-adamantyl-functionalities. The 4-(1-adamantyl)phenyl substituent has been proven to be well tolerated and is endowed with trypanocidal properties.22 The thiazole ring bears a variety of functional groups (Fig. 2).
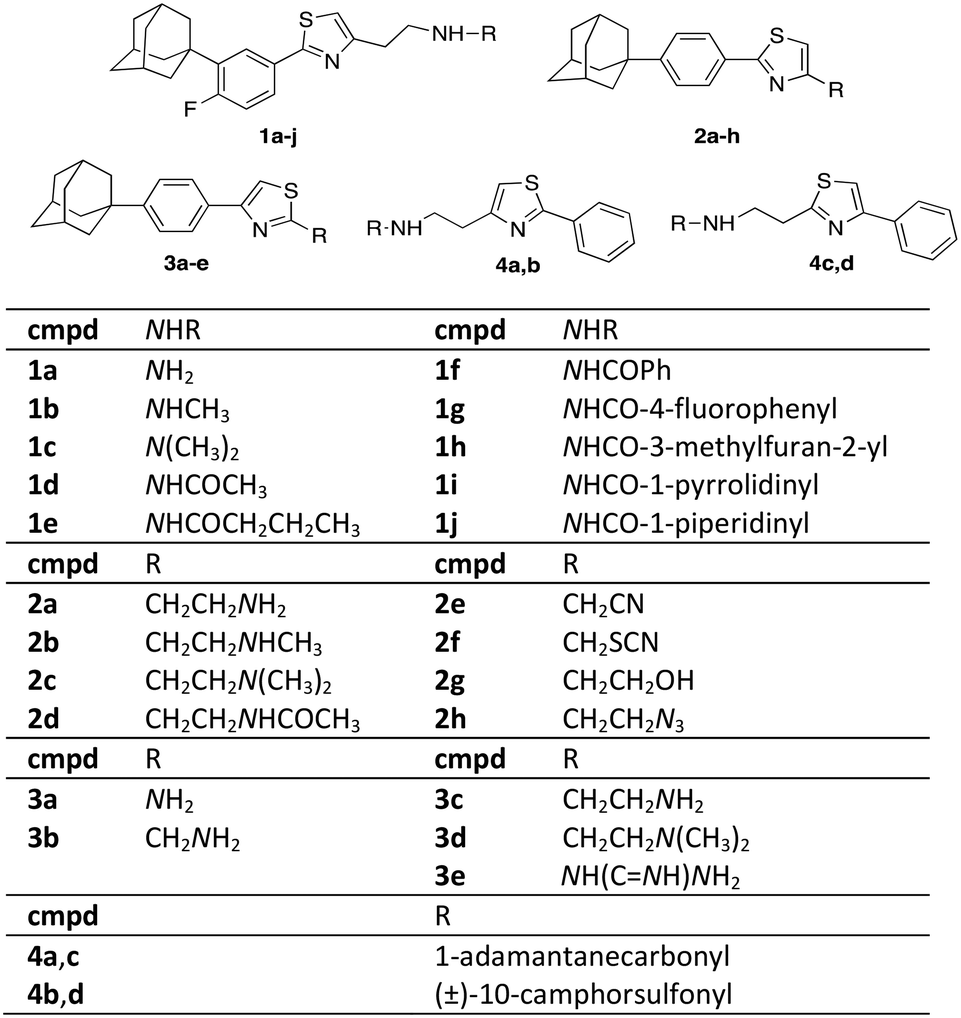 | ||
| Fig. 2 Novel thiazole derivatives 1a–j, 2a–h, 3a–e, 4a–d. | ||
2-Phenylthiazol-4-ethylamines 1a–d and 2a–d share the same structural features, apart from the relative position of the 1-adamantyl core and the addition of a fluoro-substituent in series 1. Fluorine alters the biophysical and chemical properties, such as lipophilicity, acidity, as well as the reactivity and conformation of the substituted derivatives.23 In 2018, 18 out of the 38 small drug molecules, that were approved by the FDA, contain a fluorine atom.24,25 Derivatives 3 differ in the thiazole moiety compared to adducts 1 and 2. The 2,4-substituents of the thiazoles 2a, c have their positions switched in derivatives 3c, d. The functionalization of the amino-end of congeners 1 involves various amide (aromatic and non-aromatic) and urea substituents. In adducts 2, the polar heads were translocated to the functional end of the general type scaffold IV. The length of the side chain of derivatives 2e, 2g and 3e was kept at the distance of three atoms (2C and 1N and vice versa), which in the derivatives I, II and III was found to be the optimal length for enhanced trypanocidal potency.6–8 The length of the R group is different in adducts 2f, h and 3a, b. 2-Aminothiazole (adduct 3a), is a frequent-hitting fragment in biophysical binding assays.26 Moreover, an analogous thiazole guanidinium system of derivative 3e has been used as a substitute for other aromatic rings improving biological activity.27
The relative position of the adamantane cage, the phenyl ring and the thiazole moiety was altered in derivatives 4a, c. Compounds 4a, c bear the same thiazole ring substituents as derivatives 2 and 3. Additionally, the adamantane core was replaced in the camphor skeleton in adducts 4b, 4. The latter molecules are sulfonamides in alignment with the scaffolds of compounds III.7
Results and discussion
Synthesis
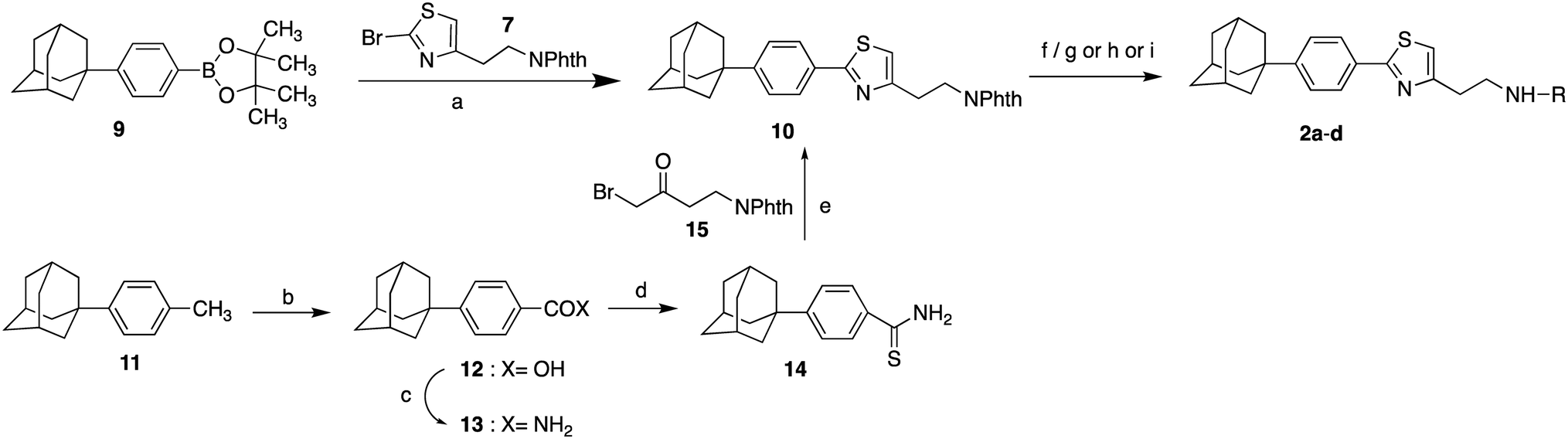
The 4-substituted-2[3-(adamant-1-yl)-4-fluorophenyl]thiazoles 1a–j were synthesized as shown in Scheme 1. As starting material, for the synthesis of thiazoles 1a–j, the (3-adamant-1-yl)-(4-fluorophenyl)boronic acid (6) was used. The reported method for the preparation of the boronic acid 6 (ref. 28) has been modified, by changing the reaction times. The synthetic route involved a Suzuki–Miyaura palladium-catalyzed coupling between the boronic acid 6 and the 2-thiazole bromide 7 (ref. 6) to provide the phthalimide protected adamantane derivative 8. The hydrazinolysis of phthalimide derivative 8 led to the deprotected parent compound 2-{2-[3-(adamant-1-yl)-4-fluorophenyl]thiazol-4-yl}ethan-1-amine (1a), which was subsequently methylated, dimethylated,29 acylated and carbamoylated to deliver adducts 1b–j, via the procedures shown in Scheme 1. | ||
| Scheme 1 Reagents and conditions: (a) i. n-BuLi, THF, −78 °C, 20 min, ii. (i-PrO)3B, r.t. 18 h, 90%; (b) Pd(PPh3)4, Na2CO3, anh. toluene, 80 °C, 18 h, 80%; (c) H2NNH2.H2O, EtOH, reflux, 1 h; (d) i. ClCOOEt, NEt3, THF, r.t. 18 h, ii. LiAlH4, THF, reflux 4 h, iii. EtOH, H2O, NaOH 10%, 0 °C, 88% from 8; (e) MeONa, CH3COOH, HCHO 38% in H2O, NaBH3CN, MeOH, r.t. 18 h, 75% from 8. (f) Benzoyl chloride (1f: 96% from 8) or 4-fluorobenzoyl chloride (1g: 93% from 8) or 1-piperidinecarbonyl chloride (1j: 36% from 8) or 1-pyrrolidinecarbonyl chloride (1i: 46% from 8), Et3N, EtOAc, r.t. 18 h; (g) acetic anhydride (1d: 84% from 8) or butyric anhydride (1e: 92% for 8), Et3N, EtOAc, r.t. 48 h; (h) 3-methyl-furoic acid, EDCI, HOBt, DMAP, DMF/DCM, 45 °C, 18 h, 29% from 8. | ||
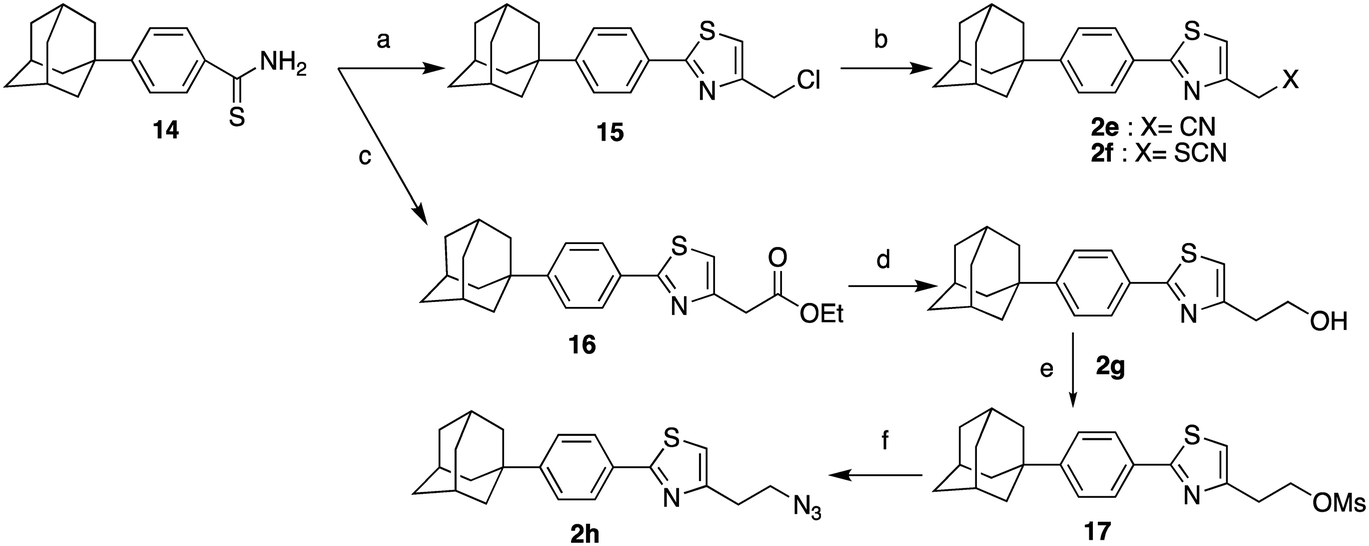
The synthesis of the 4-substituted-2[4-(adamant-1-yl)phenyl]thiazoles 2a–d was realized following two synthetic pathways, as illustrated in Scheme 2. The key-compound for the preparation of the thiazoles 2a–h, the 2-{2-[(2-(adamant-1-yl)phenyl)thiazol-4-yl]ethyl}isoindoline-1,3-dione (10) was obtained via two different synthetic routes. The first involves a Suzuki–Miyaura30 palladium-catalysed coupling between the 4,4,5,5-tetramethyl-2-[4-(adamant-1-yl)phenyl]-2H-1,3,2-dioxaborolane (9)31 and the 2-thiazole bromide 7, which led to the protected precursor 10. The second synthetic approach, towards the thiazole adduct 10, was based on the Hantzsch condensation32 of thiobenzamide 14 with the α-bromoketone 15.6 Our lab has previously published the preparation of the 4-(adamant-1-yl)benzoic acid (12),31 which was now obtained by a different transition metal ion catalyzed oxidation of 1-(4-tolyl)adamantane (11).33 The reported method of oxidation34 was modified as the reaction mixture was bubbled with oxygen gas and heated at 105 °C for 6 h. The benzoic acid 12 was subsequently converted to the corresponding benzamide 13 and the thiobenzamide 14. Comparing the two methods, the first involves 5 steps (17% total yield), while the second 6 steps (25% total yield), a more facile work-up and cheaper reagents. The parent thiazole 2a was methylated, dimethylated and acetylated to the respective congeners 2b–d, as previously shown.
 | ||
| Scheme 2 Reagents and conditions: (a) Pd(PPh3)4, Na2CO3, anh. toluene, 80 °C, 18 h, 87%; (b) O2, NaBr, Mn(OAc)2, Co(OAc)2, AcOH/dioxane, 90 °C, 6 h, 89%; (c) i. SOCl2, 65 °C, 45 min, ii. aq. NH3, 25%, r.t. 1 h, 96%; (d) Lawesson's reagent, dioxane, 110 °C, 2 h, 50%; (e) i-PrOH, autoclave, 120 °C, 18 h, 65%; (f) H2NNH2·H2O, EtOH, reflux 1 h; (g) acetic anhydride, Et3N, EtOAc, r.t. 48 h, 53% from 10; (h) i. ClCOOEt, NEt3, THF, r.t. 18 h, ii. LiAlH4, THF, reflux 4 h, iii. EtOH, H2O, NaOH 10%, 0 °C, 54% from 10; (i) MeONa, CH3COOH, HCHO 38% in H2O, NaBH3CN, MeOH, r.t. 18 h, 85% from 10. | ||
The functionalized thiazoles 2e–h were obtained by the route shown in Scheme 3. The thiobenzamide 14 was condensed with 1,3-dichloroacetone and 4-chloroacetoacetate, under Hantzsch reaction conditions, to give the chloromethylthiazole 15 and the thiazolethyl acetate 16, respectively. Treatment of the choromethylthiazole 15 with KCN or KSCN led to the respective cyanide 2e and the thiocyanide 2f. The thiazolethyl acetate 16 was reduced to the corresponding alcohol 2g, which was then converted to the azide 2h, via activation of the methanesulphonyl derivative 17.
 | ||
| Scheme 3 Reagents and conditions: (a) 1,3-dichloroacetone, acetone, reflux, 18 h, 75%; (b) KCN, anh. DMF, 60 °C, 36 h (2e: 41%) or KSCN, EtOH, 45 °C, 18 h (2f: 67%); (c) 4-chloroacetoacetate, i-PrOH, autoclave, 120 °C, 18 h, 92%; (d) i. LiAlH4, THF, r.t. 2 h, ii. EtOH, H2O, NaOH 10%, 0 °C, 80%; (e) MsCl, Et3N, DCM, 0 °C then r.t. 18 h, 95%; (f) NaN3, anh. DMF, 60 °C, 2 h, 65%. | ||
The synthesis of the 2-substituted-4-{4-(adamant-1-yl)phenyl}thiazoles 3a–d and the guanidyl derivative 3e, is shown in Scheme 4. (1-Phenyl)adamantane (18)4 was acylated under Friedel–Crafts reaction conditions36 to deliver the corresponding α-bromoketone 19, which via a Hantzsch condensation with the appropriate reagent, thiourea,37 thioamides 20,3821 (ref. 7) and guanylthiourea provided the desired thiazoles 3a–c and 3e, respectively. The dimethylthiazole 3d was prepared from the parent thiazole 3c, as shown before.
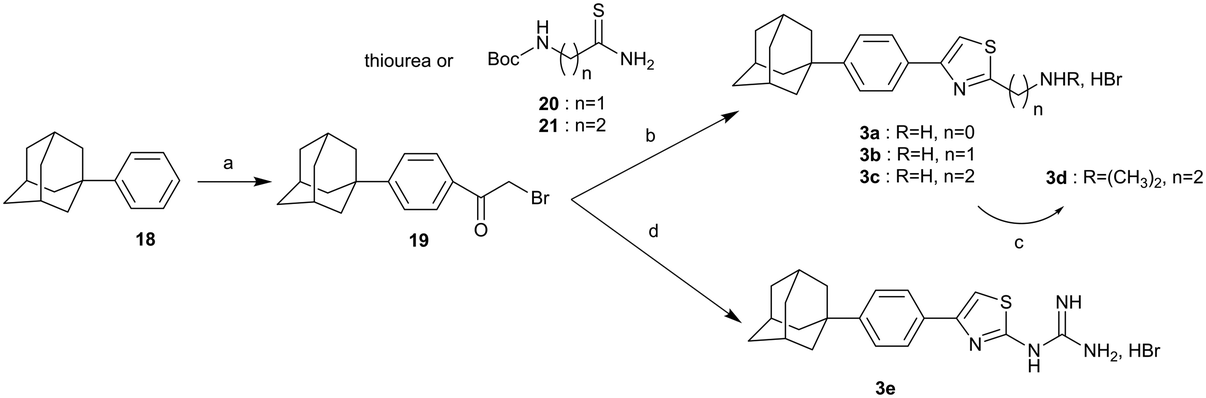 | ||
| Scheme 4 Reagents and conditions: (a) BrCOCH2Br, AlCl3, DCM, −10 °C then r.t. 18 h, 57%; (b) appropriate thiobenzamide 20 (3b: 36%), 21 (3c: 85%), i-PrOH, autoclave, 120 °C 18 h or thiourea (3a: 88%), EtOH, reflux, 18 h; (c) MeONa, CH3COOH, HCHO 38% in H2O, NaBH3CN, MeOH, r.t. 18 h, 85%; (d) guanylthiourea, EtOH, reflux, 18 h, 89%. | ||
The 1-adamantylcarbonylamides 4a, c and the (±)-10-camphorsulfonyl amides 4b, d were obtained upon coupling the commercially available 1-adamantylcarboxylic acid and (±)-10-camphorsulfonyl chloride with the 2-phenylthiazol-4-ethylamine (22)6 and the 4-phenylthiazol-2-ethylamine (23),7 respectively. The acid reacted in the presence of the coupling reagent HBTU, while the chlorides reacted without the aid of any activating reagent (Scheme 5).
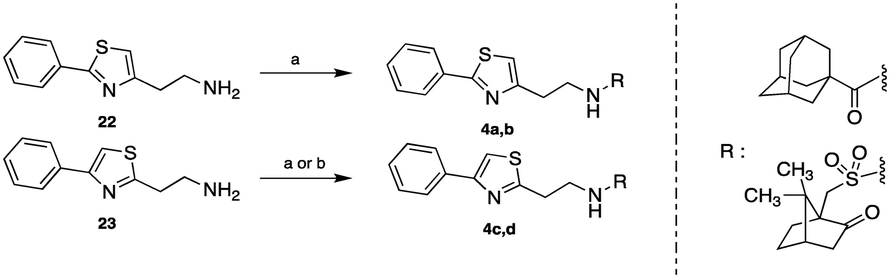 | ||
| Scheme 5 Reagents and conditions: (a) appropriate chloride NEt3, DCM or THF, r.t. 18 h, (4a: 49%, 4b: 50%, 4d: 81%); (b) 1-adamantanecarboxylic acid HBTU, DIPEA, DCM/DMF, r.t. 24 h, 94%. | ||
Pharmacology
The 27 new thiazole derivatives were tested for their activity against the bloodstream form Trypanosoma brucei and the results are shown in Table 1.| Cmpd | T. brucei IC50a (μM) | T. brucei IC90a (μM) | L6 cells IC50a (μM) | S.I.b |
|---|---|---|---|---|
| a IC50 and IC90; concentration that inhibits growth by 50% and 90%. b S.I.; selectivity index, the ratio of IC50 values obtained with L6 cells and T. brucei respectively. | ||||
| 1a | 0.42 ± 0.01 | 0.56 ± 0.01 | 1.05 ± 0.23 | 2.5 |
| 1b | 0.90 ± 0.01 | 1.10 ± 0.01 | 2.01 ± 0.32 | 2.2 |
| 1c | 0.79 ± 0.02 | 1.03 ± 0.01 | 1.53 ± 0.09 | 1.9 |
| 1d | 15.3 ± 0.2 | 18.1 ± 0.2 | — | — |
| 1e | >25 | — | — | — |
| 1f | >20 | — | — | — |
| 1g | >20 | — | — | — |
| 1h | >20 | — | — | — |
| 1i | >20 | — | — | — |
| 1j | 10.7 ± 0.3 | 12.6 ± 0.2 | <10.30 | <1 |
| 2a | 0.80 ± 0.03 | 1.17 ± 0.01 | 4.08 ± 0.15 | 5.1 |
| 2b | 0.59 ± 0.02 | 0.79 ± 0.01 | 0.96 ± 0.26 | 1.6 |
| 2c | 1.27 ± 0.07 | 1.60 ± 0.22 | — | — |
| 2d | >20 | — | — | — |
| 2e | >20 | — | — | — |
| 2f | ∼10 | — | — | — |
| 2g | ∼10 | — | — | — |
| 2h | >20 | — | — | — |
| 3a | 22.5 ± 0.6 | 30.5 ± 4.5 | 13.8 ± 1.6 | <1 |
| 3b | 9.76 ± 0.77 | 12.8 ± 0.2 | 12.6 ± 0.9 | <1 |
| 3c | 2.74 ± 0.29 | 4.40 ± 0.07 | 4.16 ± 0.24 | 1.5 |
| 3d | 1.41 ± 0.09 | 3.58 ± 0.05 | 3.19 ± 0.25 | 2.3 |
| 3e | ∼10 | — | — | — |
| 4a | 12.2 ± 0.8 | 18.7 ± 0.4 | — | — |
| 4b | 20.6 ± 0.7 | 31.1 ± 0.3 | — | — |
| 4c | 9.82 ± 0.22 | 13.1 ± 0.2 | — | — |
| 4d | 23.8 ± 1.1 | 31.5 ± 0.6 | — | — |
It is apparent from the test results that the ethylamines 1a–c exhibit the highest activity among the new 2,4-disubstituted arylthiazoles. Bulkier substituents than the methyl group at the amino end have a negative impact on trypanocidal activity. Amido adducts (alkyl 1d and 1c, the aromatic 1f and 1g and the heteroaromatic 1h) and the ureido derivatives, 1i and 1j, have a non-significant activity. The same pattern is also observed in the 2 series, as compounds 2a–c are ca. 20 times more active than their acetamido congener 2d. Comparing series 1 and 2, it becomes apparent that the fluorine substitution has little positive effect on the activity. The dimethylamino isomeric thiazoles 2c and 3d present almost the same potency, while the nor-derivatives, the isomeric thiazoles 2a and 3c, show a substantial difference in potency. The 2-phenylthiazol-4-ethylamines 1a, c and 2a, c seem to be, in general, more potent than their isomeric 4-phenylthiazol-2-ethylamines 3c and 3d. The decrease of the length of the side chain does not enhance activity. Methanamine 3b bears two atoms (carbon and nitrogen) in its side chain and is twice as potent as the 2-aminothiazole 3a, which has only one nitrogen atom. The polar functionalization of the side chain did not improve the trypanocidal activity. The azido and cyano-tailored derivatives 2e and 2h are less potent, and the thiocyanate 2f, the ethanol 2g and the guanyl derivative 3e exhibit modest activity. The change in the relative position of the adamantane cage, the phenyl ring and the thiazole moiety, in adducts 4a and 4c, did not lead to activity enhancement. Last, the replacement of the adamantane skeleton by the camphorsulfonyl moiety in derivatives 4b and 4d has not led to antitrypanosomal enhancement. The 2,4-disubstituted arylthiazole adamantane derivatives, the ethylamines 1a–c and 2a–c, present a notable pharmacological profile, which merits further investigation in terms of activity and toxicity. These findings suggest that an aliphatic amine moiety at the side chain is mandatory to achieve notable trypanocidal activity. This amine group is positively charged at the cytosolic pH, which is not the case for all the other polar heads tested. The presence of this particular group might also enhance the cellular accumulation into the protozoa, as it is reported in the case of bacteria.39,40 Thus, the ethylamines 1a–c and 2a–c seem to exhibit promising trypanocidal properties, although further optimisation will be necessary to reduce their cytotoxicity and to develop a more drug-like profile.
Conclusions
In this work, we describe the synthesis of a new series of aromatic 2,4-disubstituted 1,3-thiazole analogues with trypanocidal potency. Among their congeners, the 2-phenylthiazol-4-ethylamines 1a–c and 2a–c presented the most significant trypanocidal activity against T. brucei. Analogues 1a and 2a exhibit antitrypanosomal activity in the range of IC50 = 0.42 μM and IC50 = 0.80 μM, respectively. Primary amine 2a is less potent than its congener 1a, but exhibits higher selectivity, which is a promising perspective for designing new trypanocidals in the future. Both of these classes of derivatives bear a lipophilic end, which consists of a 4-(1-adamantyl)phenyl or a 3-(1-adamantyl)phenyl moiety, a 1,3-thiazole ring and a functional end, which comprises of an alkylamine. The addition of the adamantane ring into the scaffold of the thiazole reference compounds6,7 has not improved their pharmacological profile, in terms of activity and toxicity. On the other hand, the new congeners exhibit promising trypanocidal properties that merit further investigation. These tailored-made structural modifications will be implemented in the future in the design of trypanocidal agents.Experimental part
Biology
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 166.6 (2-Cth), 162.8 (d, J = 251.9 Hz, 4-Car), 152.8 (4-Cth), 137.5 (d, J = 11.4 Hz, 3-Car), 132.9 (1′,6′-Car), 132.8 (3′,4′-Car), 129.2 (1-Car), 126.2 (d, J = 8.4 Hz, 2-Car), 125.1 (6-Car), 122.2 (2′,5′-Car) 117.5 (d, J = 25.4, 5-Car), 116.4 (5-Cth), 40.5 (2,8,9-Cad), 38.4 (NCH2), 36.2 (1-Cad), 36.0 (4,6,10-Cad), 28.8 (CH2), 28.2 (3,5,7-Cad). Anal. calcd for C29H27FN2O2S: C, 71.58; H, 5.59; N, 5.76 found C, 71.35; H, 5.41; N, 5.54.
O), 166.6 (2-Cth), 162.8 (d, J = 251.9 Hz, 4-Car), 152.8 (4-Cth), 137.5 (d, J = 11.4 Hz, 3-Car), 132.9 (1′,6′-Car), 132.8 (3′,4′-Car), 129.2 (1-Car), 126.2 (d, J = 8.4 Hz, 2-Car), 125.1 (6-Car), 122.2 (2′,5′-Car) 117.5 (d, J = 25.4, 5-Car), 116.4 (5-Cth), 40.5 (2,8,9-Cad), 38.4 (NCH2), 36.2 (1-Cad), 36.0 (4,6,10-Cad), 28.8 (CH2), 28.2 (3,5,7-Cad). Anal. calcd for C29H27FN2O2S: C, 71.58; H, 5.59; N, 5.76 found C, 71.35; H, 5.41; N, 5.54.
A solution of the crude amide (220 mg, 0.51 mmol) in anhydrous THF (5 mL) was added dropwise to a stirred suspension of LiAlH4 (100 mg, 2.52 mmol) in anhydrous THF (5 mL), under an argon atmosphere. The mixture was stirred at ambient temperature for 25 min and then refluxed for 4 h. Next, the reaction mixture was cooled in an ice bath, and ethanol, water and a NaOH (10%) solution were sequentially added. The resulting suspension was then filtered, the filtrate was evaporated in vacuo and the resulting residue was treated with water and a HCl (5%) solution. The aqueous phase was then washed with Et2O and solid Na2CO3 was added until pH = 10. The aqueous phase was then extracted with DCM and the combined organic phase was dried over Na2CO3 and the solvent evaporated in vacuo to afford compound 1b, as a viscous oil (170 mg, 88% from compound 8). M.p. (dihydrochloride): 221–223 °C (EtOH/Et2O). 1H NMR (400 MHz, DMSO-d6) δ 9.26 (br.s, 1H, NHth), 7.97 (bs, 2H, NH2), 7.86–7.76 (m, 2H, 2,6-Har), 7.53 (s, 1H, 5-Hth), 7.24 (dd, J = 12.8, 8.3 Hz, 1H, 5-Har), 3.24 (d, J = 4.7 Hz, 2H, NCH2), 3.17 (d, J = 7.5 Hz, 2H, CH2), 2.57 (t, J = 5.4 Hz, 3H, CH3), 2.07 (s, 3H, 3,5,7-Had), 2.02 (s, 6H, 2,8,9-Had), 1.74 (s, 6H, 4,6,10-Had). 13C NMR (150 MHz, DMSO-d6) δ 166.5 (2-Cth), 162.7 (d, J = 251.7 Hz, 4-Car), 152.7 (4-Cth), 137.4 (d, J = 11.1 Hz, 3-Car), 129.3 (1-Car), 126.0 (d, J = 10.2 Hz, 2-Car), 124.9 (d, J = 7.1 Hz, 6-Car), 117.4 (d, J = 26.5 Hz, 5-Car), 116.1 (5-Cth), 47.2 (NCH2), 40.4 (2,8,9-Cad), 36.2 (4,6,10-Cad), 36.0 (1-Cad), 32.4 (CH3), 28.1 (3,5,7-Cad), 27.4 (CH2). Anal. calcd for C22H31FCl2N2S: C, 58.59; H, 6.59; N, 6.32 found C, 58.71; H, 6.25; N, 6.09.
O), 166.1 (2-Cth), 164.5 (d, J = 349.7 Hz, 4-Car), 155.5 (4-Cth), 138.6 (d, J = 11.2 Hz, 3-Car), 129.6 (1-Car), 125.8 (d, J = 2.9 Hz, 6-Car), 125.6 (2-Car), 117.2 (d, J = 25.8 Hz, 5-Car), 114.2 (5-Cth), 41.1 (d, J = 3.5 Hz, 2,8,9-Cad), 39.1 (CH2N), 36.9 (4,6,10-Cad), 36.7 (1-Cad), 31.0 (CH2), 28.9 (3,5,7-Cad), 23.5 (CH3). M.p. (fumarate): 143-145 °C (MeOH/Et2O). Anal. calcd for C27H31FN2O5S: C, 63.03; H, 6.07; N, 5.44 found C, 62.89; H, 6.01; N, 5.21.
O), 166.1 (2-Cth), 164.5 (d, J = 349.7 Hz, 4-Car), 155.5 (4-Cth), 138.3 (d, J = 11.2 Hz, 3-Car), 129.6 (1-Car), 125.8 (d, J = 2.9 Hz, 6-Car), 125.6 (2-Car), 117.2 (d, J = 25.8 Hz, 5-Car), 114.3 (5-Cth), 41.1 (2,8,9-Cad), 39.2 (CH2N), 36.9 (4,6,10-Cad), 36.7 (1-Cad), 31.0 (CH2), 28.9 (3,5,7-Cad), 28.9 (CH2), 19.3 (CH2), 13.9 (CH3). M.p. (fumarate): 291–293 °C (MeOH/Et2O). Anal. calcd for C29H35FN2O5S: C, 64.19; H, 6.50; N, 5.16 found C, 64.38; H, 6.61; N, 5.33.
O), 164.5 (d, J = 349.7 Hz, 4-Car), 155.5 (4-Cth), 138.3 (d, J = 11.2 Hz, 3-Car), 131.3 (4′-Car), 129.6 (1-Car), 128.5 (3′,5′-Car), 127.0 (2′,6′-Car), 125.8 (d, J = 2.9 Hz, 6-Car), 125.6 (2-Car), 117.2 (d, J = 25.8 Hz, 5-Car), 114.3 (5-Cth), 41.1 (2,8,9-Cad), 39.2 (CH2N), 36.9 (4,6,10-Cad), 36.7 (1-Cad), 31.0 (CH2), 28.9 (3,5,7-Cad). M.p. (fumarate): 305 °C (dec) (MeOH/Et2O). Anal. calcd for C32H33FN2O5S: C, 66.65; H, 5.77; N, 4.86 found C, 65.39; H, 5.90; N, 4.72.
O), 164.5 (d, J = 349.7 Hz, 4-Car), 163.9 (d, J = 348.6 Hz, 4′-Car) 155.5 (4-Cth), 138.3 (d, J = 11.2 Hz, 3-Car), 130.1 (d, J = 3 Hz, 1′-Car), 129.6 (1-Car), 128.3 (d, J = 7 Hz, 2′,6′-Car), 125.8 (d, J = 2.9 Hz, 6-Car), 125.6 (2-Car), 117.2 (d, J = 25.8 Hz, 5-Car), 116.1 (d, J = 18 Hz, 3′,5′-Car) 114.3 (5-Cth), 41.1 (d, J = 3.5 Hz, 2,8,9-Cad), 39.2 (CH2N), 36.9 (4,6,10-Cad), 36.7 (1-Cad), 31.0 (CH2), 28.9 (3,5,7-Cad). M.p. (fumarate): 226–227 °C (MeOH/Et2O). Anal. calcd for C32H32F2N2O5S: C, 64.63; H, 5.42; N, 4.71 found C, 64.44; H, 5.73; N, 4.88.
O), 155.5 (4-Cth), 142.7 (4-Cfur), 141.1 (1-Cfur), 138.3 (d, J = 11.2 Hz, 3-Car), 129.6 (1-Car), 125.8 (d, J = 2.9 Hz, 6-Car), 125.6 (2-Car), 117.2 (d, J = 25.8 Hz, 5-Car), 115.6 (2-Cfur), 115.2 (5-Cth), 114.3 (3-Cfur), 41.1 (2,8,9-Cad), 39.2 (CH2N), 36.9 (4,6,10-Cad), 36.7 (1-Cad), 31.0 (CH2), 28.9 (3,5,7-Cad), 11.0 (CH3). M.p. (fumarate): 260 (dec) °C (MeOH/Et2O). Anal. Calcd for C31H32FN2O6S: C, 64.12; H, 5.73; N, 4.82 found C, 64.44; H, 5.54; N, 4.98.
O), 155.5 (4-Cth), 138.3 (d, J = 11.2 Hz, 3-Car), 129.6 (1-Car), 125.8 (d, J = 2.9 Hz, 6-Car), 125.6 (2-Car), 117.2 (d, J = 25.8 Hz, 5-Car), 114.3 (5-Cth), 45.4 (2,5-Cpy), 41.1 (d, J = 3.5 Hz, 2,8,9-Cad), 39.2 (CH2N), 36.9 (4,6,10-Cad), 36.7 (1-Cad), 31.0 (CH2), 28.9 (3,5,7-Cad), 25.6 (3,4-Cpy). M.p. (fumarate): 301 °C (dec) (MeOH/Et2O). Anal. calcd for C30H36FN3O5S: C, 63.25; H, 6.37; N, 7.38 found C, 63.09; H, 6.14; N, 7.19.
O), 152.8 (4-Cth), 137.5 (d, J = 11.4 Hz, 3-Car), 129.2 (1-Car), 126.2 (d, J = 8.4 Hz, 2-Car), 125.1 (6-Car), 117.5 (d, J = 25.4, 5-Car), 116.4 (5-Cth), 43.9 (2,6-Cpi), 40.5 (2,8,9-Cad), 38.4 (NCH2), 36.2 (1-Cad), 36.0 (4,6,10-Cad), 28.8 (CH2), 28.2 (3,5,7-Cad), 24.8 (3,5-Cpi), 23.5 (4-Cpi). Anal. calcd for C27H34FN3OS: C, 69.35; H, 7.33; N, 8.99 found C, 69.55; H, 7.39; N, 9.12.
S), 156.0 (1-Car), 136.4 (4-Car), 126.9 (2,6-Car), 125.1 (3,5-Car), 42.9 (2,8,9-Cad), 36.7 (4,6,10-Cad), 36.6 (1-Cad), 28.8 (3,5,7-Cad). C17H21NS: C, 75.23; H, 7.80 found C, 75.07; H, 7.99.
Method A. Phthalimide 10 was prepared in a similar way as for compound 8 using pinacolborane 9 (ref. 31) (300 mg, 0.88 mmol) and bromothiazole 7 (ref. 6) (250 mg, 0.73 mmol) as starting materials. Elution with 10–20% EtOAc in hexanes afforded compound 10 as a white solid (300 mg, 87%).
Method B. The bromoketone 15 (ref. 6) (200 mg, 0.66 mmol) was added to a stirred solution of the thiobenzamide 14 (180 mg, 0.66 mmol) in EtOH (4 mL) and the reaction mixture was refluxed overnight. The resulting suspension was filtered and the precipitate was washed with Et2O and dried over MgSO4 to afford compound 10, as a white solid (200 mg, 65%) M.p.: 213–214 °C. 1H NMR (600 MHz, CDCl3) δ 7.81 (dt, J = 6.8, 3.4 Hz, 2H, 2′,5′-Har), 7.73 (d, J = 8.4 Hz, 2H, 2,6-Har), 7.66 (dt, J = 6.8, 3.4 Hz, 2H, 3′,4′-Har), 7.34 (d, J = 8.4 Hz, 2H, 3,5-Har), 6.94 (s, 1H, 5-Hth), 4.10 (t, J = 7.1 Hz, 2H, NCH2), 3.21 (t, J = 7.1 Hz, 2H, CH2), 2.12 (br.s, 3H, 3,5,7-Had), 1.91 (br.s, 6H, 2,8,9-Had), 1.78 (dd, J = 25.4, 12.1 Hz, 6H, 4,6,10-Had). 13C NMR (150 MHz, CDCl3) δ 168.4 (C
O), 168.2 (2-Cth), 154.2 (4-Cth), 153.4 (1-Car), 133.9 (3′,4′-Car), 132.3 (1′,6′-Car), 131.1 (4-Car), 126.4 (2,6-Car), 125.4 (3,5-Car), 123.3 (2′,5′-Car), 114.2 (5-Cth), 43.1 (2,8,9-Cad), 37.7 (NCH2), 36.9 (4,6,10-Cad), 36.5 (1-Cad), 30.2 (CH2), 29.0 (3,5,7-Cad). C29H28N2O2S: C, 74.33; H, 6.02; N, 5.98 found C, 74.64; H, 6.12; N, 6.21.
O, fum), 153.2 (1-Car), 152.9 (4-Cth), 134.6 (C-fum), 130.5 (4-Car), 126.0 (2,6-Car), 125.6 (3,5-Car), 115.7 (5-Cth), 47.4 (NCH2), 42.4 (2,8,9-Cad), 36.1 (4,6,10-Cad), 36.0 (1-Cad), 32.5 (CH3), 28.3 (3,5,7-Cad), 27.6 (CH2). Anal. calcd for C30H36N2O8S: C, 61.63; H, 6.21; N, 4.79 found C, 61.47; H, 6.08; N, 5.01.
O, fum), 153.7 (4-Cth), 134.7 (1-Car), 134.6 (CH, fum), 130.9 (4-Car), 126.4 (2,6-Car), 126.0 (3,5-Car), 116.0 (5-Cth), 56.4 (CH2N), 43.0 (CH3), 42.9 (2,8,9-Cad), 36.6 (4,6,10-Cad), 36.5 (1-Cad), 28.7 (3,5,7-Cad), 27.0 (CH2). Anal. calcd for C31H38N2O8S: C, 62.19; H, 6.40; N, 4.68 found C, 62.31; H, 6.64; N, 4.14.
O), 7.84 (d, J = 8.2 Hz, 2H, 2,6-Har), 7.46 (d, J = 8.2 Hz, 2H, 3,5-Har), 7.34 (s, 1H, 5-Hth), 6.63 (s, 2H, CH-Fum), 3.39 (dt, J = 13.3, 6.7 Hz, 2H, NCH2), 2.88 (t, J = 7.2 Hz, 2H, CH2), 2.06 (s, 3H, 3,5,7-Had), 1.88 (s, 6H, 2,8,9-Had), 1.80 (s, 3H, CH3), 1.74 (br.s, 6H, 4,6,10-Had). 13C NMR (75 MHz, DMSO-d6) δ 169.1 (NCO), 166.5 (2-Cth), 166.0 (CO, fum), 155.2 (4-Cth), 153.0 (1-Car), 134.0 (CH-fum), 130.7 (4-Car), 125.9 (2,6-Car), 125.5 (3,5-Car), 114.6 (5-Cth), 42.4 (2,8,9-Cad), 38.2 (NCH2), 36.1 (4,6,10-Cad), 36.0 (1-Cad), 31.3 (CH2), 28.3 (3,5,7-Cad), 22.7 (CH3). Anal. calcd for C27H32N2O5S: C, 65.30; H, 6.50; N, 5.64 found C, 65.27; H, 6.33; N, 5.45.
O), 158.0 (1-Car), 129.0 (2,6-Cad), 127.5 (4-Cad), 125.6 (3,5-Car), 42.9 (2,8,9-Cad), 36.9 (1-Cad), 36.8 (3,6,10-Cad), 31.1 (CH2), 28.8 (3,5,7-Cad). Anal. calcd for C18H21BrO: C, 64.87; H, 6.35 found C, 64.63; H, 6.46.
O, fum), 151.0 (4-Cth), 134.5 (CH2fum), 131.4 (1-Car), 127.1 (4-Car), 125.9 (2,6-Car), 125.2 (3,5-Car), 114.5 (5-Cth), 42.6 (2,8,9-Cad), 40.8 (CH2N), 36.3 (4,6,10-Cad), 35.8 (1-Cad), 28.4 (3,5,7-Cad). Anal. calcd for C28H32N2O8S: C, 60.62; H, 5.79; N, 5.03 found C, 60.33; H, 6.02; N, 4.89.
O), 168.2 (2-Cth), 155.9 (4-Cth), 133.5 (1-Car), 130.0 (4-Car), 128.8 (2,6-Car), 126.3 (3,5-Car), 114.2 (5-Cth), 40.5 (CH2N), 39.3 (2,8,9-Cad), 38.8 (1-Cad), 36.5 (4,6–10-Cad), 30.7 (CH2), 28.1 (3,5,7-Cad). Anal. calcd for C22H26N2OS: C, 72.08; H, 7.15; N, 7.64 found C, 72.31; H, 7.09; N, 7.88.
O), 167.2 (2-Cth), 155.0 (4-Cth), 133.4 (1-Car), 130.7 (4-Car), 129.7 (2,6-Car), 126.6 (3,5-Car), 116.3 (5-Cth), 58.3 (2-Ccam), 48.2 (CH2), 48.0 (7-Ccam), 42.7 (6-Ccam), 42.5 (5-Ccam), 42.5 (CH2N), 32.2 (CH2), 26.7 (4-Ccam), 25.0 (3-Ccam), 19.8 (CH3), 19.7 (CH3). Anal. calcd for C21H27ClN2O3S: C, 55.43; H, 5.98; N, 6.16 found C, 55.21; H, 6.11; N, 6.02.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (10 mL), was added 1-adamantanecarboxylic acid (227 mg, 1.26 mmol), HBTU (478 mg, 1.26 mmol), and DIPEA (474 mg, 3.68 mmol) and the reaction mixture was stirred at ambient temperature under an argon atmosphere, overnight. The mixture was then partitioned between DCM and an aqueous solution of citric acid (10%) and the aqueous phase was extracted with DCM. The combined organic phase was then washed with water and brine, dried over MgSO4 and the solvent evaporated under reduced pressure. The resulting residue was purified with gradient column chromatography. Elution with 10% to 50% EtOAc in hexanes afforded compound 4a as a white solid (330 mg, 94%). M.p.: 107–108 °C. 1H NMR (400 MHz, CDCl3) δ 8.01–7.84 (m, 2H, 2,6-Har), 7.54–7.28 (m, 3,4,5-Har, 5-Hth), 6.92 (s, 1H, NH), 3.69 (dd, J = 11.8, 5.7 Hz, 3H, CH2N), 3.20 (t, J = 7.4 Hz, 3H, CH2), 2.02 (s, 3H, 3,5,7-Had), 1.87 (d, J = 2.3 Hz, 6H, 2,8,9-Had), 1.70 (q, J = 12.2 Hz, 6H, 4,6,10-Had).13C NMR (150 MHz, CDCl3) δ 177.9 (CO), 168.2 (2-Cth), 155.9 (4-Cth), 133.5 (1-Car), 130.0 (4-Car), 128.8 (2,6-Car), 126.3 (3,5-Car), 114.2 (5-Cth), 40.5 (CH2N), 39.3 (2,8,9-Cad), 38.8 (1-Cad), 36.5 (4,6–10-Cad), 30.7 (CH2), 28.1 (3,5,7-Cad). Anal. calcd for C22H26N2OS: C, 72.09; H, 7.15; N, 7.64 found C, 72.27; H, 7.23; N, 7.92.
O), 167.4 (2-Cth), 153.7 (4-Cth), 134.0 (1-Car), 128.7 (2,6-Car), 128.0 (4-Car), 126.0 (3,5-Car), 114.0 (5-Cth), 57.8 (2-Ccam), 47.9 (CH2S), 47.6 (7-Ccam), 42.3 (6-Ccam), 42.1 (5-Ccam), 42.0 (CH2N), 33.6 (CH2), 26.3 (4-Ccam), 24.5 (3-Ccam), 19.3 (CH3), 19.2 (CH3). Anal. calcd for C21H27ClN2O3S: C, 55.43; H, 5.98; N, 6.16 found C, 55.67; H, 5.77, N; 6.23.
:1 (10 mL), was added 1-adamantanecarboxylic acid (227 mg, 1.26 mmol), HBTU (478 mg, 1.26 mmol), and DIPEA (474 mg, 3.68 mmol) and the reaction mixture was stirred at ambient temperature under an argon atmosphere, overnight. The mixture was then partitioned between DCM and an aqueous solution of citric acid (10%) and the aqueous phase was extracted with DCM. The combined organic phase was then washed with water and brine, dried over MgSO4 and the solvent evaporated under reduced pressure. The resulting residue was purified with gradient column chromatography. Elution with 10% to 50% EtOAc in hexanes afforded compound 4a as a white solid (330 mg, 94%). M.p.: 107–108 °C. 1H NMR (400 MHz, CDCl3) δ 8.01–7.84 (m, 2H, 2,6-Har), 7.54–7.28 (m, 3,4,5-Har, 5-Hth), 6.92 (s, 1H, NH), 3.69 (dd, J = 11.8, 5.7 Hz, 3H, CH2N), 3.20 (t, J = 7.4 Hz, 3H, CH2), 2.02 (s, 3H, 3,5,7-Had), 1.87 (d, J = 2.3 Hz, 6H, 2,8,9-Had), 1.70 (q, J = 12.2 Hz, 6H, 4,6,10-Had).13C NMR (150 MHz, CDCl3) δ 177.9 (CO), 168.2 (2-Cth), 155.9 (4-Cth), 133.5 (1-Car), 130.0 (4-Car), 128.8 (2,6-Car), 126.3 (3,5-Car), 114.2 (5-Cth), 40.5 (CH2N), 39.3 (2,8,9-Cad), 38.8 (1-Cad), 36.5 (4,6–10-Cad), 30.7 (CH2), 28.1 (3,5,7-Cad). Anal. calcd for C22H26N2OS: C, 72.09; H, 7.15; N, 7.64 found C, 72.27; H, 7.23; N, 7.92.
O), 167.4 (2-Cth), 153.7 (4-Cth), 134.0 (1-Car), 128.7 (2,6-Car), 128.0 (4-Car), 126.0 (3,5-Car), 114.0 (5-Cth), 57.8 (2-Ccam), 47.9 (CH2S), 47.6 (7-Ccam), 42.3 (6-Ccam), 42.1 (5-Ccam), 42.0 (CH2N), 33.6 (CH2), 26.3 (4-Ccam), 24.5 (3-Ccam), 19.3 (CH3), 19.2 (CH3). Anal. calcd for C21H27ClN2O3S: C, 55.43; H, 5.98; N, 6.16 found C, 55.67; H, 5.77, N; 6.23.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. Dimitra Benaki (Division of Pharmaceutical Chemistry, Department of Pharmacy, School of Health Sciences, National and Kapodistrian University of Athens) and Dr. Kyriakos C. Prousis, (Institute of Chemical Biology, National Hellenic Research Found) for NMR experiments.Notes and references
- M. A. Sleigh, Protozoa and other protists, ed. E. Arnold, London, 2nd edn, 1989 Search PubMed.
- S. Aksoy, P. Buscher, M. Lehane, P. Solano and J. Van Den Abbeele, PLoS Neglected Trop. Dis., 2017, 11, e0005454 CrossRef PubMed.
- I. Beltran-Hortelano, S. Perez-Silanes and S. Galiano, Curr. Med. Chem., 2017, 24, 1066 CrossRef CAS PubMed.
- P. E. Cockram and T. K. Smith, J. Nat. Prod., 2018, 81, 2138 CrossRef CAS PubMed.
- S. P. S. Rao, M. P. Barrett, G. Dranoff, C. J. Faraday, C. R. Gimpelewicz, A. Hailu, C. L. Jones, J. M. Kelly, J. K. Lazdins-Helds, P. Mäser, J. Mengel, J. C. Mottram, C. E. Mowbray, D. L. Sacks, P. Scott, G. F. Späth, R. L. Tarleton, J. M. Spector and T. T. Diagana, ACS Infect. Dis., 2019, 5, 152 CrossRef CAS PubMed.
- S. Russell, R. Rahmani, A. J. Jones, H. L. Newson, K. Neilde, I. Cotillo, M. Rahmani Khajouei, L. Ferrins, S. Qureishi, N. Nguyen, M. S. Martinez-Martinez, D. F. Weaver, M. Kaiser, J. Riley, J. Thomas, M. De Rycker, K. D. Read, G. R. Flematti, E. Ryan, S. Tanghe, A. Rodriguez, S. A. Charman, A. Kessler, V. M. Avery, J. B. Baell and M. J. Piggott, J. Med. Chem., 2016, 59, 9686 CrossRef CAS PubMed.
- D. A. Patrick, T. Wenzler, S. Yang, P. T. Weiser, M. Z. Wang, R. Brun and R. R. Tidwell, Bioorg. Med. Chem., 2016, 24, 2451 CrossRef CAS PubMed.
- A. A. Rashad, A. J. Jones, V. M. Avery, J. Baell and P. A. Keller, ACS Med. Chem. Lett., 2014, 5, 496 CrossRef CAS PubMed.
- I. Papanastasiou, G. B. Foscolos, A. Tsotinis, J. Olah, J. Ovadi, S. R. Prathalingam and J. M. Kelly, Heterocycles, 2008, 75, 2043 CrossRef CAS.
- I. Papanastasiou, A. Tsotinis, G. B. Foscolos, S. R. Prathalingam and J. M. Kelly, J. Heterocycl. Chem., 2008, 45, 1401 CrossRef CAS.
- G. B. Foscolos, I. Papanastasiou and A. Tsotinis, Lett. Org. Chem., 2008, 5, 57 CrossRef.
- I. Papanastasiou, A. Tsotinis, N. Kolocouris, S. R. Prathalingam and J. M. Kelly, J. Med. Chem., 2008, 51, 1496 CrossRef CAS PubMed.
- I. Papanastasiou, A. Tsotinis, G. Zoidis, N. Kolocouris, S. R. Prathalingam and J. M. Kelly, ChemMedChem, 2009, 4, 1059 CrossRef CAS PubMed.
- I. Papanastasiou, K. C. Prousis, K. Georgikopoulou, T. Pavlidis, E. Scoulica, N. Kolocouris and T. Calogeropoulou, Bioorg. Med. Chem. Lett., 2010, 20, 5484 CrossRef CAS PubMed.
- A. Koperniku, I. Papanastasiou, G. B. Foscolos, A. Tsotinis, M. C. Taylor and J. M. Kelly, MedChemComm, 2013, 4, 856 RSC.
- A.-S. Foscolos, I. Papanastasiou, G. B. Foscolos, A. Tsotinis, T. F. Kellici, T. Mavromoustakos, M. C. Taylor and J. M. Kelly, MedChemComm, 2016, 7, 1229 RSC.
- I. Papanastasiou, G. Foscolos, B. A. Tsotinis and J. M. Kelly, in African Trypanosomiasis: Clinical Symptoms, Diagnosis and Treatment, ed. G. T. Hughes, Nova Science Publishers, NY, 2016, p. 63 Search PubMed.
- M.-O. Georgiadis, V. Kourbeli, V. Ioannidou, E. Karakitsios, I. Papanastasiou, A. Tsotinis, D. Komiotis, A. Vocat, S. T. Cole, M. C. Taylor and J. M. Kelly, Bioorg. Med. Chem. Lett., 2019, 29, 1278 CrossRef CAS PubMed.
- O. Karoutzou, D. Benaki, I. Papanastasiou, A. Vocat and S. T. Cole, ChemistrySelect, 2019, 4, 8727 CrossRef CAS.
- A. Papageorgiou, A.-S. Foscolos, I. P. Papanastasiou, M. Vlachou, A. Siamidi, A. Vocat, S. T. Cole, T. F. Kellici, T. Mavromoustakos and A. Tsotinis, Future Med. Chem., 2019, 11, 2779 CrossRef CAS.
- C. B. Scarim, D. H. Jornada, M. G. M. Machado, C. M. R. Ferreira, J. L. dos Santos and M. C. Chung, Eur. J. Med. Chem., 2019, 162, 378 CrossRef CAS PubMed.
- A. Foscolos, I. Papanastasiou, A. Tsotinis, M. C. Taylor and J. M. Kelly, ChemMedChem, 2019, 14, 1227 CrossRef CAS PubMed.
- S. Vukelić, J. Moschner, S. Huhmann, R. Fernandes, A. A. Berger and B. Koksch, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds, ed. H. Groult, F. Leroux and A. Tressaud, Elsevier, 2017, p. 427 Search PubMed.
- H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem. – Eur. J., 2019, 25, 1 CrossRef.
- B. G. de la Torre and F. Albericio, Molecules, 2019, 24, 809 CrossRef PubMed.
- S. M. Devine, M. D. Mulcair, C. O. Debono, E. W. W. Leung, J. W. M. Nissink, S. S. Lim, I. R. Chandrashekaran, M. Vazirani, B. Mohanty, J. S. Simpson, J. B. Baell, P. J. Scammells, R. S. Norton and M. J. Scanlon, J. Med. Chem., 2015, 58, 1205 CrossRef CAS PubMed.
- A. Flood, C. Trujillo, G. Sanchez-Sanz, B. Kelly, C. Muguruza, L. F. Callado and I. Rozas, Eur. J. Med. Chem., 2017, 138, 38 CrossRef CAS PubMed.
- M. I. Dawson, Z. Xia, T. Jiang, M. Ye, J. A. Fontana, L. Farhana, B. Patel, L. P. Xue, M. Bhuiyan, R. Pellicciari, A. Macchiarulo, R. Nuti, X.-K. Zhang, Y.-H. Han, L. Tautz, P. D. Hobbs, L. Jong, N. Waleh, W. Chao, G.-S. Feng, Y. Pang and Y. Su, J. Med. Chem., 2008, 51, 5650 CrossRef CAS PubMed.
- S. H. Pine and B. L. Sanchez, J. Org. Chem., 1971, 36, 829 CrossRef CAS.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS.
- A. Koperniku, A.-S. Foscolos, I. Papanastasiou, G. B. Foscolos, A. Tsotinis and D. Schols, Lett. Org. Chem., 2016, 13, 171 CrossRef CAS.
- Thiazole and its derivatives, ed. J. V. Metzger, Wiley, NY, 1979 Search PubMed.
- Z. I. Dehimat, A. Paşahan, D. Tebbani, S. Yaşar and İ. Özdemir, Tetrahedron, 2017, 73, 5940 CrossRef CAS.
- S. V. Krasnikov, T. A. Obuchova, O. A. Yasinskii and K. V. Balakin, Tetrahedron Lett., 2004, 45, 711 CrossRef CAS.
- P. Mosset and R. Grée, Synlett, 2013, 24, 1142 CrossRef CAS.
- F. W. Foss, T. P. Mathews, Y. Kharel, P. C. Kennedy, A. H. Snyder, M. D. Davis, K. R. Lynch and T. L. Macdonald, Bioorg. Med. Chem., 2009, 17, 6123 CrossRef CAS PubMed.
- O. Kouatly, Ph. Eleftheriou, A. Petrou, D. Hadjipavlou-Litina and A. Geronikaki, SAR QSAR Environ. Res., 2018, 29, 83 CrossRef CAS PubMed.
- K. Aihara, Y. Kano, S. Shiokawa, T. Sasaki, F. Setsu, Y. Sambongi, M. Ishii, K. Tohyama, T. Ida, A. Tamura, K. Atsumi and K. Iwamatsu, Bioorg. Med. Chem., 2003, 11, 3475 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299 CrossRef CAS PubMed.
- M. F. Richter and P. J. Hergenrother, Ann. N. Y. Acad. Sci., 2019, 1435, 18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00478e |
| This journal is © The Royal Society of Chemistry 2020 |