
Targeting prenylation inhibition through the mevalonate pathway
Pimyupa
Manaswiyoungkul
b,
Elvin D.
de Araujo
a and
Patrick T.
Gunning
 *ab
*ab
aDepartment of Chemical and Physical Sciences, University of Toronto Mississauga, 3359 Mississauga Rd N., Mississauga, Ontario L5L 1C6, Canada. E-mail: patrick.gunning@utoronto.ca
bDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
First published on 23rd December 2019
Abstract
Protein prenylation is a critical mediator in several diseases including cancer and acquired immunodeficiency syndrome (AIDS). Therapeutic intervention has focused primarily on directly targeting the prenyltransferase enzymes, FTase and GGTase I and II. To date, several drugs have advanced to clinical trials and while promising, they have yet to gain approval in a medical setting due to off-target effects and compensatory mechanisms activated by the body which results in drug resistance. While the development of dual inhibitors has mitigated undesirable side effects, potency remains sub-optimal for clinical development. An alternative approach involves antagonizing the upstream mevalonate pathway enzymes, FPPS and GGPPS, which mediate prenylation as well as cholesterol synthesis. The development of these inhibitors presents novel opportunities for dual inhibition of cancer-driven prenylation as well as cholesterol accumulation. Herein, we highlight progress towards the development of inhibitors against the prenylation machinery.
Introduction
Post-translational lipidation extends the functional and structural diversity of the eukaryotic proteome through the covalent attachment of a non-peptidic hydrophobic moiety. Collectively termed lipidation, lipid modifications can include N-myristoylation,1 palmitoylation,2 glycosylphosphatidylinositol (GPI) anchor addition,3 cholesterol attachment,3 and prenylation.4 In all cases, covalent attachment of a lipid to a protein dramatically alters the physical chemical properties and activity of a protein as well as its cellular distribution.5 This process facilitates the interaction of several proteins with cellular membranes and subsequently mediates multiple cell signalling pathways.6In this review, we will focus on small molecule efforts to inhibit protein prenylation. Prenylation involves the thiolinkage of either a farnesyl (C15) or geranylgeranyl (C20) isoprenoid. The isoprenoids utilized by the prenyltransferases are initially generated from acetyl-coenzyme A (CoA) through the multistep mevalonate pathway.7 (Fig. 1) Following generation of the isoprenoid, prenylation is carried out by one of three distinct prenyltransferases: farnesyl transferase (FTase), geranylgeranyl transferase I (GGTase I), RabGGTase (GGTase II), and the recently identified GGTase III.8,9 FTase and GGTase I are heterodimeric proteins that recognize a consensus sequence at the C-terminus of target substrates, referred to as the CAAX box, where C refers to Cys and A is an aliphatic residue. Although both proteins share an identical α subunit, the identity of X is most commonly a Leu or Glu for GGTase I recognition and a Met, Ser, Gln, Ala, or Cys for FTase recognition.8 The GGTase II recognition motif is at the C-terminus of the target protein, but differs from the other two prenyltransferases as it is more variable in sequence (XXCC, XCXC, CCXX, CCXXX or CXXX).10,11 The recently identified GGTase III differs drastically from its counterparts, and consists of a unique α subunit, referred to as prenyltransferase α subunit repeat-containing protein 1 (PTAR1), and shares the same β subunit as GGTase II. GGTase III recruits the additional protein, F-box/LRR-repeat protein 2 (FBXL2), through the LRR domain of F-box protein by the α subunit and its target through CAAX recognition motifs via the β subunit.9
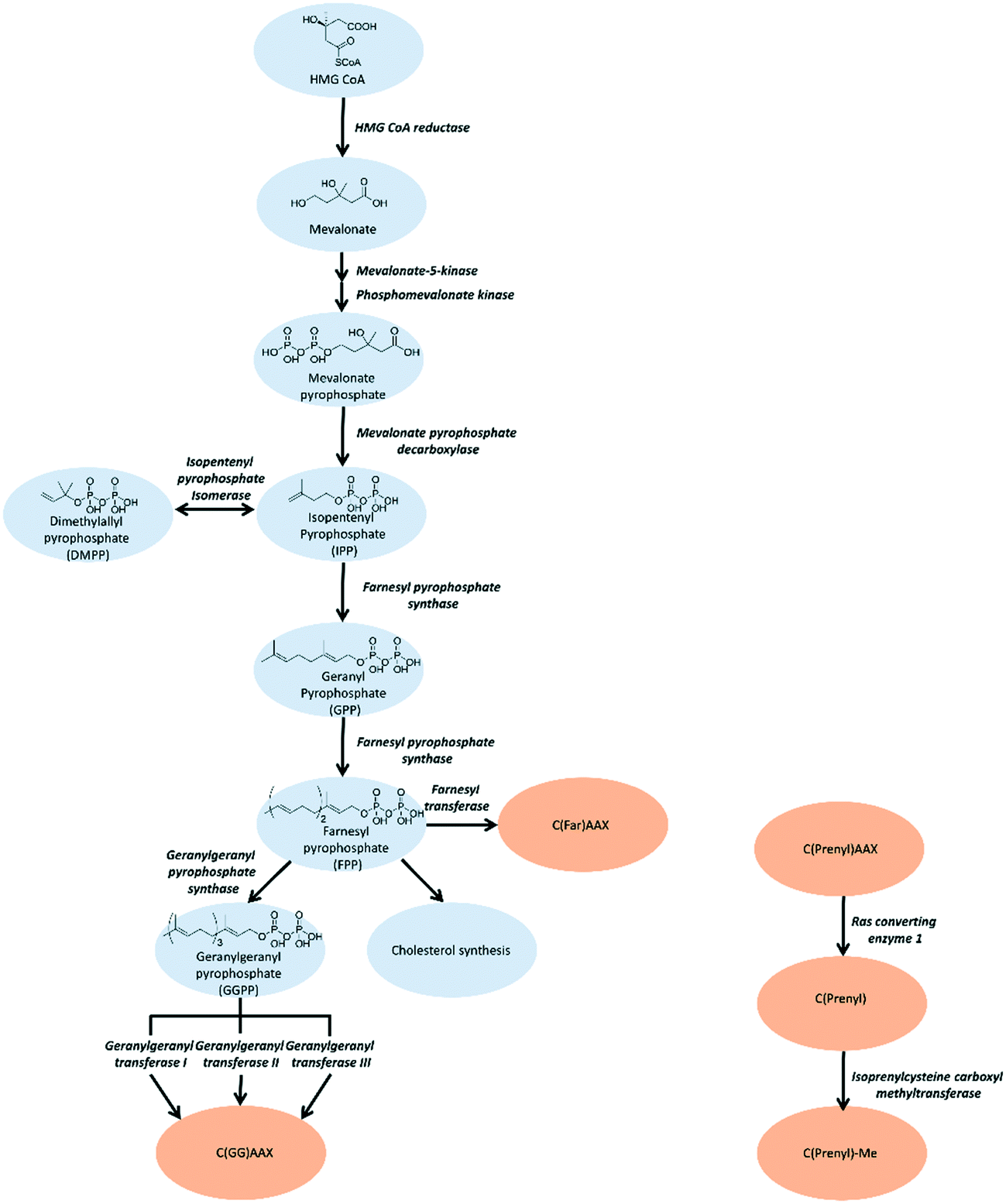 | ||
| Fig. 1 To target the isoprenoid synthesis in this pathway, inhibition strategies are predominantly aimed towards FPPS, GGPPS, and prenyltransferases. Alternative approaches involve targeting the post-prenylation processing enzymes, Rce1 and ICMT. | ||
Following prenylation by FTase and GGTase I, the –AAX residues are cleaved by Ras-converting enzyme 1 (Rce1), a protein-specific endoprotease of the endoplasmic reticulum.8,12 The resulting free carboxyl group of the prenylated Cys is methylated by the methyl transferase, isoprenylcysteine carboxyl methyltransferase (Icmt).13 Dysregulation of the post-isoprenylation process as seen in Rce1 and Icmt deficiency can lead to protein mislocalization and detrimental effects in cell growth and viability.13–15 Substrates of GGTase II are doubly geranylgeranylated and often remain uncleaved although methylation has been observed in specific cases.11,16 GGTase III recognizes a similar CAAX sequence as GGTase I, suggesting similar post-prenylation processing to its targets as well.9
Other post-translational modifications to the human proteome are of therapeutic interest, including phosphorylation (30–75% proteome),17,18 acetylation (80–90% proteome),19 and ubiquitination (∼60% proteome).20 To date, 30 clinical drugs target phosphorylation through inhibition of the kinases and only 2 target the ubiquitin system.21 By comparison, prenylation modifies 0.5–2% of the proteome.22,23 However, given that oncogenic proteins, Ras1 and Ras-like proteins, are prenylation substrates, there was significant interest in developing prenylation-targeting pharmacologic agents. Moreover, early preclinical success against prenylation pathways further propagated interest.
The most well-known prenylated substrates are Ras subfamily members, important signalling cascade initiators that play a major role in tumorigenesis. K-RAS, the most frequently mutated form of RAS, is found to be present in 90% of patients with pancreatic cancer.24,25 The activity of K-RAS greatly affects downstream proteins including RALA and RALB in enhancing oncogenesis.26 In addition to cancer, abnormal prenylation levels, resulting from a surplus of the isoprenoid pool, may be indicative of a variety of disorders, including cardiac hypertrophy,27 neuronal apoptosis,28 and a plethora of metabolic conditions.29
Current therapeutic strategies for targeting prenylation include inhibiting the three different prenylation enzymes directly, as well as indirect inhibition of these same enzymes by targeting the mevalonate pathway, which disrupts the intracellular concentrations of the isoprene pool. Herein, we will review the advances in inhibiting the mevalonate pathway enzymes and prenylation precursors as well as the three prenylation enzymes. More complex therapeutic methods are geared towards hijacking the lipidation machinery to repurpose it towards delivering a specific therapeutic to a designated site.
The mevalonate pathway
The mevalonate pathway, also known as the isoprenoid pathway, or the hydroxymethylglutaryl-CoA (HMG-CoA) reductase pathway, produces isoprenoids that are essential for various cellular functions such as cholesterol synthesis and growth control in eukaryotes, archaea, and some bacteria.30,31 This pathway exclusively converts acetyl-Coenzyme A (acetyl-CoA) to isopentenyl pyrophosphate (IPP). IPP is utilized for synthesis of farnesyl pyrophosphate (FPP) that branches into either squalene synthesis for cholesterol production or geranylgeranyl pyrophosphate (GGPP) for prenylation. Depletion of IPP can be replenished through isomerization of dimethylallyl pyrophosphate (DMPP).32 (Fig. 1) Strategies for inhibition of cholesterol synthesis have generally focused on upstream enzymes, such HMG CoA reductase, whereas prenylation inhibition is targeted towards the downstream enzymes of the mevalonate pathway.Upstream mevalonate pathway enzymes
The first steps of the mevalonate pathway are focused on generating DMPP from acetyl CoA. HMG CoA synthase 1 (HMGCS1) is the first enzyme in the mevalonate pathway which catalyzes the condensation reaction of acetyl CoA and acetoacetyl CoA into HMG CoA.33 The molecule is reduced to mevalonate by HMG CoA reductase (HMGCR) through a four-electron reductive deacylation which is the rate limiting step in the pathway.30,34,35 Statins reversibly bind the active site of HMGCR and inhibit cholesterol synthesis for treatment of cardiovascular diseases such as hypertension and hypercholesterolemia.34,36 The statins have been comprehensively reviewed and are published elsewhere.37–39 Mevalonate kinase (MK) catalyzes the transfer of the γ-phosphoryl group of ATP onto the C5 hydroxyl oxygen of mevalonic acid, resulting in the production of mevalonate-5-phosphate(MP).40–42 Progress has been made towards the development of MK inhibitors, particularly for use as antibacterial agents.43 Phosphomevalonate kinase (PMK) transfers the γ-phosphoryl group from ATP onto MP, yielding 5-diphosphomevalonante (MPP).44,45 Despite discovery of lead compounds for PMK inhibitor development, to the best of our knowledge no compounds have demonstrated drug-like efficacy to progress clinically.46 Mevalonate pyrophosphate decarboxylase (MPDC) catalyzes the ATP-dependent decarboxylation of MPP to form IPP.47 Despite the promising MPDC drug candidate, 6-fluoromevalonatepyrophosphate, discovered in 1985, its successors failed to yield higher efficacy compared to the parent, and no MPDC-targeting drugs have been approved to date.48–50Farnesyl pyrophosphate synthase inhibitor
Farnesyl pyrophosphate synthase (FPPS) is the most attractive upstream target for inhibition of prenylation. This enzyme catalyzes the sequential condensation of IPP with DMPP to form a molecule of geranyl pyrophosphate (GPP) which further reacts with a second molecule of IPP to form farnesyl pyrophosphate (FPP).51,52 The molecular mechanism of catalysis has been well characterized through crystallography and is described as a concerted three-step reaction involving ionization, condensation, and elimination steps.53,54 Initially, DMPP is held at the active site (often referred to as S1, A1 or the allylic site) containing three octahedral coordinated Mg2+ ions and a series of Asp motifs that assist in stabilizing the pyrophosphate (PPi) tail. These interactions help stabilize the release of PPi and formation of the DMPP carbocation. The hydrophobic portion of the DMPP substrate stretches deeper into the active site where it can also reach the opposing dimer. IPP binding occurs at the S2 (often referred to as I or homoallylic) site and is directed by salt bridges with the C-terminal tail and PPi, which positions the double bond of IPP for nucleophilic attack on the carbocation.54,55 Binding of IPP, triggers ordering of the C-terminal tail, which sterically closes the active site. The PPi bound in the Mg2+ subpocket acts as a catalytic base to abstract a proton in an elimination reaction. The condensation reaction is repeated to yield FPP.56 Adjacent to the active site, there is a S3 (or allosteric) site which inhibits activity of FPPS when occupied. A subpocket below the S3 allosteric site, termed the S4 site, has also been suggested as a potential site for FPPS inhibitors (Fig. 2).57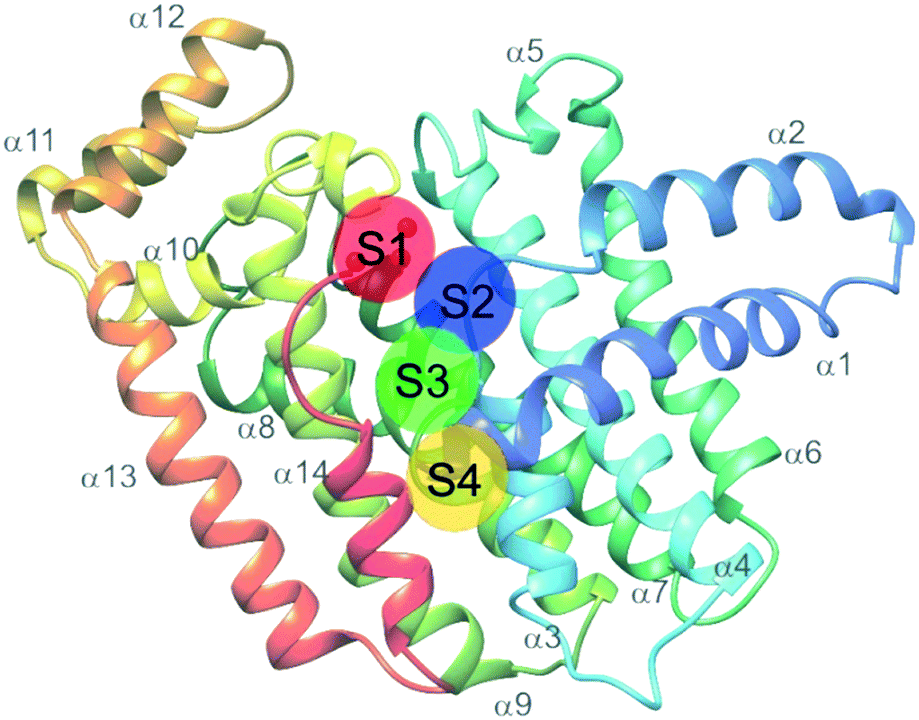 | ||
| Fig. 2 Structure of FFPS with possible small molecule binding sites. S1 represents the allylic subpocket; S2 depicts the homoallylic/IPP subpocket; S3 illustrates the allosteric pocket; S4 portrays a plausible binding groove on the protein surface. | ||
Bisphosphonates
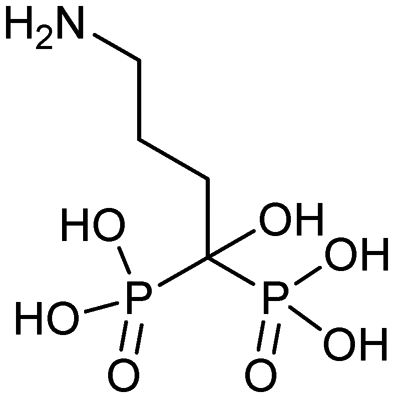
Currently, clinically available FPPS inhibitor structures focus around a bisphosphonate (BP) moiety that targets the FPPS active site, predominantly through mimicking PPi of the allylic substrates within the S1 site. However, BPs are unusual scaffolds due to their hydrophilicity and poor oral bioavailability (<1–5% for most BPs).58,59 Furthermore, BPs have a high propensity for Ca2+ ion chelation and rapidly target the skeletal system. As such, BP-class drugs are commonly used to treat osteoporosis and hypercalcemia through inhibition of bone resorption.60,61 The use of nitrogen-containing BPs (NBPs), such as pamidronate and alendronate, was also found to successfully inhibit cholesterol biosynthesis in a mouse macrophage cell line, J774.62 However, these molecules showed limited activity against squalene synthase which suggested a target further upstream in the mevalonate pathway, later confirmed to be FPPS.51 There are 7 FDA approved NBPs which include pamidronate, risedronate, alendronate, zoledronate, etidronate, tiludronate, and ibandronate (Table 1).63,64 An interesting correlation between in vitro FPPS inhibition potency and in vivo anti-resorptive potency was revealed. Upon treatment, the compound prevented leaching of hydroxyapatite, Mg2+, Na+, and HCO3− into the blood stream.65 Through inhibition of bone resorption, NBPs deprive tumor cells of bone-derived factors required for growth resulting in anti-tumor activity.| Name | Structure | Approval date (company) | Treatment |
|---|---|---|---|
| Alendronate (Fosamax) |
|
11/24/1999 (Merck Research Laboratories) | Osteogenesis imperfecta in pediatric patients (age >4) |
| Bone manifestations of Gaucher disease | |||
| Paget's disease | |||
| Glucocorticoid-induced osteoporosis | |||
| Osteoporosis | |||
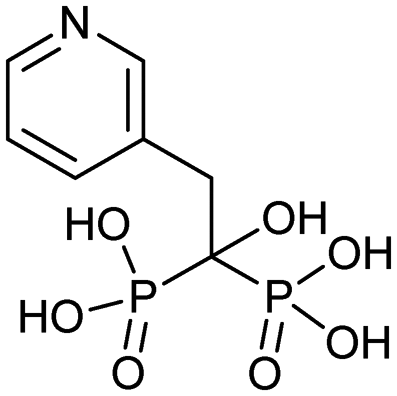
| Risedronate (Actonel) |
|
3/27/1998 (Procter and Gamble Pharmaceuticals) | Osteogenesis imperfecta |
| Ibandronate (Boniva) |
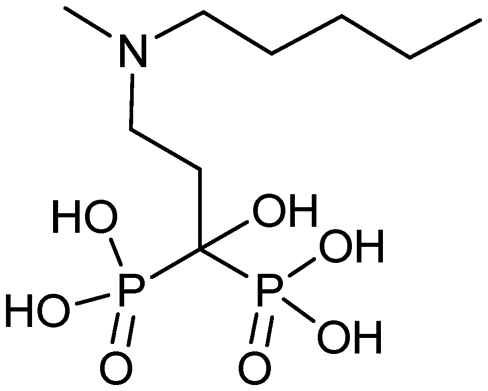
|
5/16/2003 (Hoffmann-La Roche Inc.) | Osteoporosis in post-menopausal women |
| Pamidronate (Aredia) |
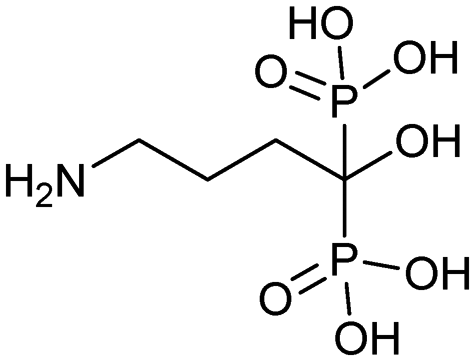
|
9/22/1998 (Novartis) | Osteoporosis |
| Hypercalcemia | |||
| Etidronate (Didronel) |
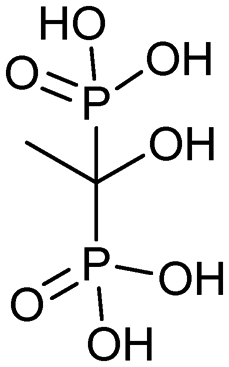
|
4/20/1987 (MGI Pharma Inc.) | Osteoporosis |
| Paget's disease | |||
| Tiludronate (Skelid) |
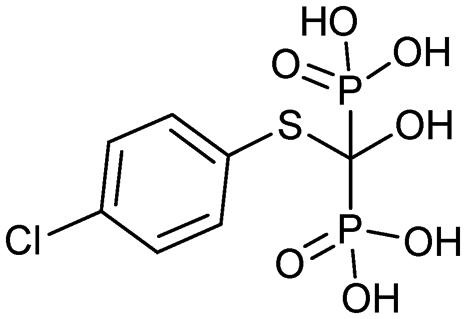
|
3/7/1997 (Sanofi Aventis US) | Paget's disease |
| Zoledronate (Zometa, Reclast) |
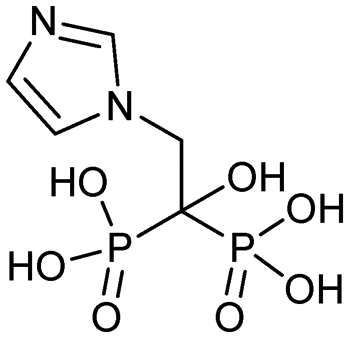
|
2/22/2002 (Novartis) | Tumor-induced hypercalcemia |
| Hypercalcemia of malignancy |
The binding mechanism of NBPs to FPPS has been resolved through crystallography and biophysical studies.66 NBP binding at the S1 site is competitive with DMPP and initially reversible. Sequestering of IPP at the S2 site results in a ternary complex and subsequent closing of the C-terminal tail, potentially leading to irreversible inhibition.67
NBPs induce tumor cell apoptosis in cellulo, when used as single agents or in combination with chemotherapeutic agents.68–70In vivo, several studies provide evidence that NBPs can inhibit the growth of soft tissue tumors and visceral metastases in animals.71–73 However, in some metastatic settings, the treatment involving NBPs and anticancer therapeutics have shown delay in skeletal morbidity associated with bone metastasis but have yet to provide benefit in overall patient survival.74,75 Molecular dynamics simulations and crystallographic studies revealed the importance of residues, Thr201, Gln240 and Lys200, in the binding of NBPs (risedronate and zoledronate) which is similar to the binding mode of DMPP. However, mutagenesis studies revealed that mutation of Thr201 does not reduce NBP affinity, suggesting additional sites of interaction. Further investigation revealed Tyr204 as a critical residue for NBP binding, which also participates in orienting Gln96 and Arg60 for IPP binding. Mutation of Tyr204 reduces FPPS activity which can be partially recovered at lower pH conditions, demonstrating the role of the hydroxyl group in substrate binding.76 These studies validated the role of the S1 and S2 sites in NBP binding.
The wealth of structural data on FPPS coupled with chemical diversity that can be accommodated by the R2 site on NBPs, has triggered the emergence of compound libraries with improved lipophilicity. Despite the development of several NBP-based inhibitor libraries, improved potency over the FDA approved zoledronate (IC50 = 28 nM, in vitro) has not been achieved.77 Nonetheless, significant advances in BP inhibitors have been made in recent years to expand our understanding of FPPS inhibition. Many of these have focused on manipulating the affinity of BPs for bone or re-targeting inhibitors towards the homoallylic or allosteric sites.
Modulating affinity of BPs for the skeletal system
The high affinity of BPs for the skeletal system has elicited specific safety concerns, such as drug accumulation in the jaw resulting in osteronecrosis.78 Thus, lowering bone affinity would assist in a more uniform drug distribution. Replacement of the hydroxyl moiety with fluorine substantially reduces BP Ca2+-chelating properties. Binding studies of 1-fluoro-2-(imidazo-[1,2alpha]pyridin-3-yl)-ethyl-bisphosphonate (1), revealed equivalent inhibition of FPPS to currently used NBPs (IC50 = 2.6 nM), while demonstrating reduced skeletal binding. However, it should be noted that lowering compound 1 dosage resulted in no significant differences in inhibitory activity between control and treated groups.79 Contrastingly, growth inhibition of osteoclasts (bone cells responsible for bone resorption) has been exploited by BPs to initiate localized release of H2S. This was accomplished through the fusion of aryl-isothiocyanate with alendronate into DM-22 (2). Although specific binding against FPPS was not examined, reduced FPPS binding affinity was expected based on lower cytotoxicity in cells treated with 2, compared to treatment with the parent inhibitor, alendronate, and well as known structural properties of the protein.80 The addition of the bulky aryl-isothiocyanate moiety alters the coordination between the NH2 and Thr201, suggesting a rationale for the reduced affinity of 2 compared to the parent.81 These compounds propose new strategies in tailoring the affinity of BPs to bone.80An alternative approach for manipulating NBP bone affinity, involves encapsulating zoledronate within a liposome, which bypasses direct chemical modification of the backbone. Liposome-encapsulated zoledronate containing phosphatidylcholine from egg yolk/cholesterol/1,2-diacyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol)-2000], referred to as Lipozol, was shown to mask many of the off-target effects and improve bioavailability. Lipozol treatment significantly reduced mechanical hypersensitivity of mice with spared nerve injury (SNI), compared to zoledronate treatment alone. Moreover, in SNI afflicted mice, fluorescence-activated cell sorting analysis (FACS) of tissue samples indicated that Lipozol demonstrated bioavailability in the spinal cord and brain. These studies highlight progression in FPPS inhibition that is independent of bone resorption properties.82
Thienopyrimidine bisphosphonate and monophosphate derivatives
A theme central to several efforts aimed at improving NBP inhibitors involves increasing drug lipophilicity without compromising potency. Thienopyrimidine (Th) derivatives (3) represent one of the most successful NBP classes, exhibiting low nM activity against FPPS. The addition of long alkyl groups significantly increases the hydrophobicity whilst maintaining high affinity for hydroxyapatite (HAP). Similar to NBPs, co-crystal structures of FPPS-thienopyrimidine bisphosphonate (ThBP) derivatives in the presence of Mg2+ revealed interactions at the active site as well as displacement of Thr201 and Lys200 by the steric bulk of Th.63 As shown by differential scanning fluorimetry (DSF), binding of ThBP prevents IPP from interacting at the S2 site likely due to steric clashes between the Th core and the prenyl side chain. However, FPPS still adopts a closed conformation resulting in a similar irreversible inhibition observed with NBPs.63Modification of ThBP to Th-monophosphate (ThMP) (4) resulted in allosteric site selectivity with only a modest loss of potency. In contrast, both binding and biological potency were abolished in the monophosphate analogs of NBPs.51 In the absence of Mg2+, ThMPs are proposed to adopt an alternative upside down binding pose in the allosteric site. However, The physiological relevance of the Mg2+-free binding mode remains unclear.83 ThMPs have been shown to alter intracellular p-Tau levels in human immortalized neurons, providing a tool for the interrogation of the biochemical link between prenylation and the accumulation of p-Tau in neurons.83
The effects of monophosphate (MP) on allosteric-site targeting were investigated on the clinically-used drug, AMP397 (5). Although a HAP binding assay assessing 5 showed 18-fold weaker affinity when compared to zoledronate, these strategies inspired the replacement of carboxylic acid functionalities with MPs on allosteric-targeting inhibitors. This was accomplished for S3 site-targeting compounds of a benzoindole (6a) (IC50 = 0.2–0.4 μM), salicylic acid (6b) (IC50 = 0.02–0.52 μM), and quinolone (6c) (IC50 = 0.04–1.0 μM) derivatives of allosteric inhibitors. Despite substitution of the carboxylate functionality with MP, bone affinity was insufficient for efficient osteoclasts-targeting. However, increasing the linker length between the MP and the allosteric scaffold modestly increased bone affinity.84
Future of NBPs
While NBPs dominate the FPPS inhibitor landscape, inhibition strategies have shifted from targeting the S1 site to the allosteric pockets. NBP re-purposing, as with zoledronate (breast cancer and glioblastoma cell proliferation), has been gaining traction with inhibitors registering IC50 values in the 1–50 μM range.85 These treatments result in autophagy but can be reversed by treating cells with geranylgeraniol. Similar results were also obtained in treated cervical cancer cell lines, where key markers of autophagy, ATG5, beclin1, and LC3-II were monitored.86 These orthogonal effects highlight the importance of exploring FDA approved drugs across oncology indications, and to explore combinatorial therapies that can lead to lower dosing regimens. Finally, FPPS inhibitors are increasingly explored as anti-parasitic agents targeting Plasmodium or Trypanosoma. This work has been reviewed elsewhere.87Additional targeting of the S1 and S2 sites
Binding of non-BPs to FPPS has also been shown with N6-isopentenyladenosine (7), an adenosine derivative.88 Compound 7 exhibits anti-tumoral effects as well as FPPS inhibition through occupancy of the S1 site via the sugar core through to the S2 isoprenoid and S3 allosteric pocket by the isopentenyl group. Inverse virtual screening identified FPPS as a potential target of 7 from a panel of 296 (+50 blank) PDB structures. Saturation transfer difference (STD) NMR confirmed binding of 7 to FPPS which was not dependent on the presence of IPP.89 These studies highlight the efficacy of novel computational strategies to identify and characterize FPPS inhibitors.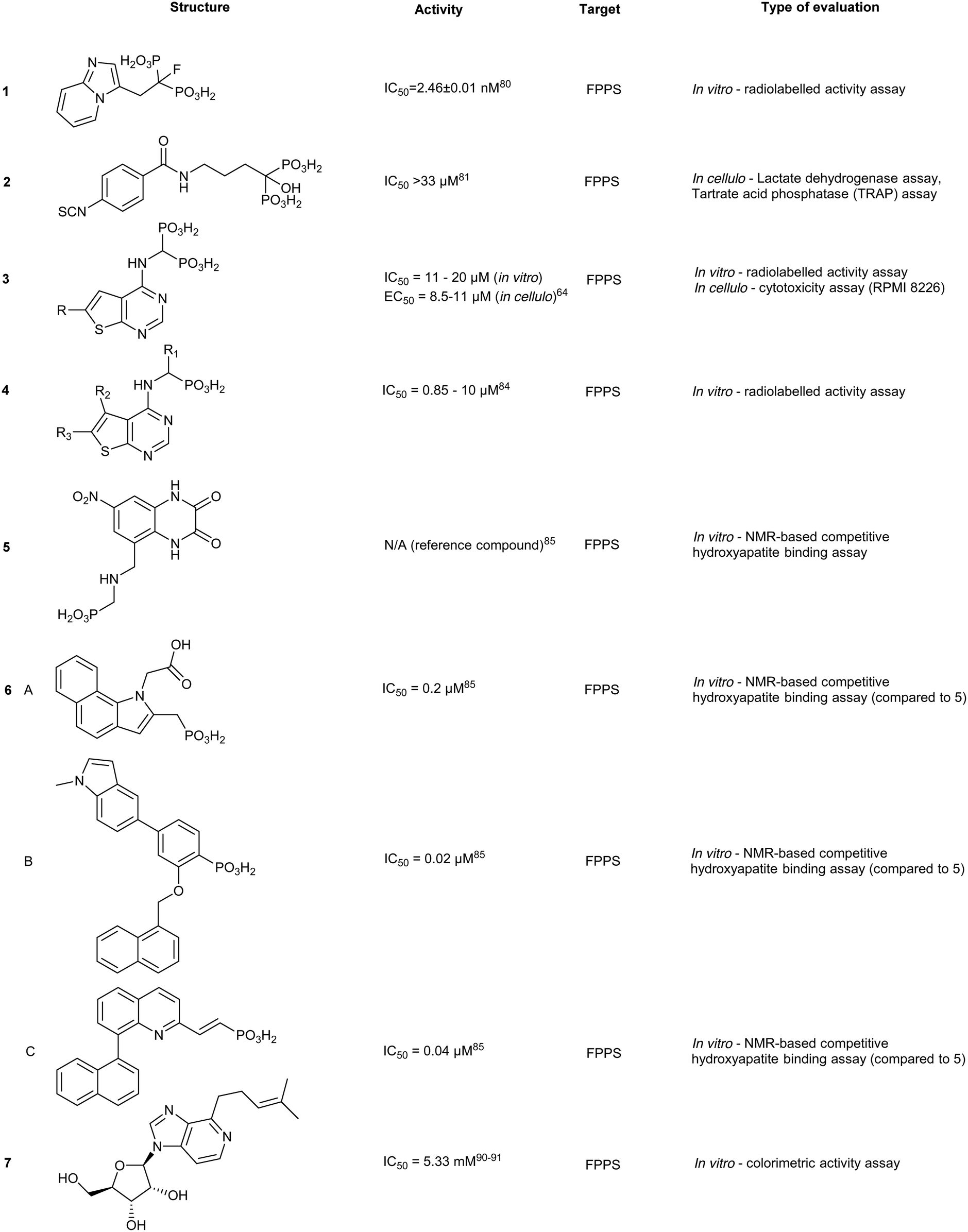
Further studies on benzyl modification of the N6 position of the adenosine, yielded compound CM223 (8) (Kd ∼ 200 μM, compared to Kd (7) ∼ 1 mM, STD-NMR). The improved binding affinity was suggested to arise from the increase in stabilization of the H-bond interaction with Asp257 and Lys214 and π–π stacking between Phe113 and Tyr218. Regardless of binding mechanism, the compounds exhibited cellular cytotoxicity IC50 ∼ 1000 fold lower than in vitro activity IC50. This suggests additional in cellulo targets that may contribute to the observed cellular apoptosis, including the signal transducer and activator of transcription 3 (STAT3) protein pathway, as STAT3 phosphorylation was also shown to be inhibited upon treatment with 10 μM of 8 in patient-derived glioma cell lines, GBM37 and GBM50.90
Targeting FPPS outside S1 and S2
The allosteric site of FPPS has been targeted, although the biological relevance of this site was only speculated to be involved in negative feedback inhibition. Initial allosteric inhibitors were identified by Novartis with nanomolar interactions with FPPS.91 Since these initial discoveries, the popularity of targeting the allosteric site has increased, as shown with previously discussed ThBPs and ThMPs.Crystallographic studies have shown FPP interactions at the allosteric site and are postulated to result in feedback inhibition of FPPS via a conformational change that locks the protein in an open conformation that prevents additional substrate conversion to FPP. The α-phosphate of FPP participates in H-bonding with Asn59 as well as electrostatic interactions with Phe239 at the entrance of the allosteric sub-pocket. The β-phosphate forms an ionic interaction with Lys57 and Arg60. Furthermore, the prenyl tail forms van der Waals interactions with the interior cavity of the pocket. These interactions result in conformational changes that likely impede binding of DMPP to FPPS and result in enzyme inhibition.92
The identification of FPP as an allosteric inhibitor provides an alternative strategy in FPPS inhibition. From an 850![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-compound library screen, a salicylic acid derivative (9a), and a quinolone derivative (10a) were discovered with an IC50 of 54 μM and 1.2 μM in the scintillation proximity assay and LC-MS high-throughput (HTS) assay, respectively. Crystallography of the complex revealed interactions between 9a and Asn59 and Phe239, similar to the allosteric interactions of FPP with FPPS with additional side-on interactions to the aromatic ring of Phe239. The naphthyl group of 9a sits in a hydrophobic pocket where it participates in an amide-π stacking interaction with Asn59. Further optimization of compound 9a yielded 9b and 9c with IC50 values of 38 nM and 17 nM, respectively. Despite impressive IC50 values, poor cellular permeability was observed, likely due to the carboxylic acid substituent. Crystal structures of compound 10a bound to FPPS revealed an analogous binding pose, with H-bond interactions present between the quinolone nitrogen and Asn59. The increase in inhibitory activity was suggested to arise from improved hydrophilic interactions between R1/R2 group and the enzymatic allosteric site. An SAR study of 10a yielded 10b and 10c with IC50 values of 24 nM and 37 nM, respectively.93
000-compound library screen, a salicylic acid derivative (9a), and a quinolone derivative (10a) were discovered with an IC50 of 54 μM and 1.2 μM in the scintillation proximity assay and LC-MS high-throughput (HTS) assay, respectively. Crystallography of the complex revealed interactions between 9a and Asn59 and Phe239, similar to the allosteric interactions of FPP with FPPS with additional side-on interactions to the aromatic ring of Phe239. The naphthyl group of 9a sits in a hydrophobic pocket where it participates in an amide-π stacking interaction with Asn59. Further optimization of compound 9a yielded 9b and 9c with IC50 values of 38 nM and 17 nM, respectively. Despite impressive IC50 values, poor cellular permeability was observed, likely due to the carboxylic acid substituent. Crystal structures of compound 10a bound to FPPS revealed an analogous binding pose, with H-bond interactions present between the quinolone nitrogen and Asn59. The increase in inhibitory activity was suggested to arise from improved hydrophilic interactions between R1/R2 group and the enzymatic allosteric site. An SAR study of 10a yielded 10b and 10c with IC50 values of 24 nM and 37 nM, respectively.93
Additional allosteric inhibitors have been identified, such as a diamidine-based compound, N-[3-(4,5-dihydro-1H-imidazol-2-yl)phenyl]-4-[4-[[3-(4,5-dihydro-1H-imidazol-2-yl) phenyl]carbamoyl]phenyl]benzamide dihydrochloride (11) (hFPPS, IC50 = 1.8 μM). This molecule is unique in that it occupies both the S2 and S3 binding sites and stretches deeper into the allosteric site, termed S4. These allosteric inhibitors represent an alternative approach to inhibiting FPPS without the use of a non-drug-like phosphate moiety.57
Geranylgeranyl pyrophosphate synthase inhibitor
GGPPS catalyzes condensation of FPP and IPP to form geranylgeranyl pyrophosphate (GGPP),94 similar to its upstream enzyme, FPPS. However, inhibition of GGPPS results in GGTase inhibition through the depletion of cellular GGPP level. This approach is postulated to be more selective for GGTase targets as opposed to FPPS-targeting, which depletes cellular FPP utilized in both prenylation and cholesterol synthesis. As a result, fewer off-target effects should be observed with a GGPPS targeting drug.95Similar to FPPS, the catalytic mechanism involves an ionization–condensation–elimination reaction. The allylic site contains acidic residues that coordinate three Mg2+ ions and a hydrophobic pocket that extends deep into the GGPPS cavity. The homoallylic site also contains basic residues to stabilize the PPi group and is lined with hydrophobic residues to accommodate the lipophilic IPP tail. As previously discussed, NBPs do not target GGPPS, likely due to the long hydrophobic channels present in the enzyme. Inhibition of GGPPS was originally achieved with the so-called “V-shaped” BPs that possess long isoprene derivatives on the R1 and R2 position.94,96,97
A co-crystallized structure of GGPPS complexed with GGPP identified an allosteric pocket. The binding pose showed the PPi of GGPP at the allylic site and the hydrophobic tail stretching beyond the active site.94 As with FPPS, the natural product is suspected to result in feedback inhibition. Other GGPPS inhibitors have been co-crystalized in yeast homologs with similar conformations observed, where the hydrophilic head group occupies the allylic site with the hydrophobic tail positioned in the allosteric site.98,99 Here, we highlight notable advancements in GGPPS inhibitor development.
Bisphosphonates
GGPPS inhibitors also possess the BP scaffold. Initial efforts to develop GGPPS inhibitors yielded digeranyl bisphosphonate (DGBP) (12) (IC50 = 260 nM, in vitro), which can simultaneously occupy the FPP binding cavity and the GGPP product subpocket.100 Wills et al. have developed novel triazole bisphosphonate (TBP) inhibitors of GGPPS from DGBP. A triazole moiety was predicted to optimally link the hydrophobic digeranyl group and the BP moiety to mimic the length of the natural substrate, GGPP. Crystallographic data indicated that DGBP activity can be attributed to the V-shape. This allows one hydrophobic chain to simultaneously block FPP from entering the pocket and GGPP exiting from the active site. All TBPs were found to offer selective inhibitory activity against GGPPS compared to the related enzyme, FPPS, in an in vitro enzymatic assay. However, in cellulo experiments showed reduced potency, likely due to poor cell permeability.101 Studies involving modifications to the V-shaped motif showed that combinations of DGBPs and TBPs improved inhibition 40-fold compared to treatment with the geranyl isomer alone.102 The homoneryl triazole (HNT) (13) exhibited higher cellular geranylgeranylation than homogeranyl triazole (HGT). (14) An increasing trend in inhibitory activity was observed as the concentration of HGT to HNT was increased from 1:3, 1:1 and 3:1. The highest potency was observed with mixtures of homogeranyl/homoneryl (3:1) TBP (IC50 of 45 nM).101 Molecular docking of HNT and HGT to the GGPP and FPP site suggested that both isomers have preferential occupancy to each of the two sites with HNT favoring the FPP binding sites and HGT favoring the GGPP binding sites. The requirement for specific concentrations of both isomers may be correlated to changes in the multimeric state of GGPPS due to cooperative binding between HNT and HGT.103,104
Despite favourable GGPPS inhibitory observed, synthesis of a cell permeable prodrug version of 14 was deemed problematic due to acidity of proton at the α position. Thus, development of α-methylated isoprenoid triazole BP yielded 15 which demonstrated an IC50 comparable to its parent (IC50 = 86 ± 22 nM) and 693-fold selectivity for GGPPS over FPPS.106 Additional studies revealed that 14 was capable of inhibiting geranylgeranylation and inducing apoptosis in cellular models in 7 pancreatic ductal adenocarcinoma (PDAC) cell lines.107
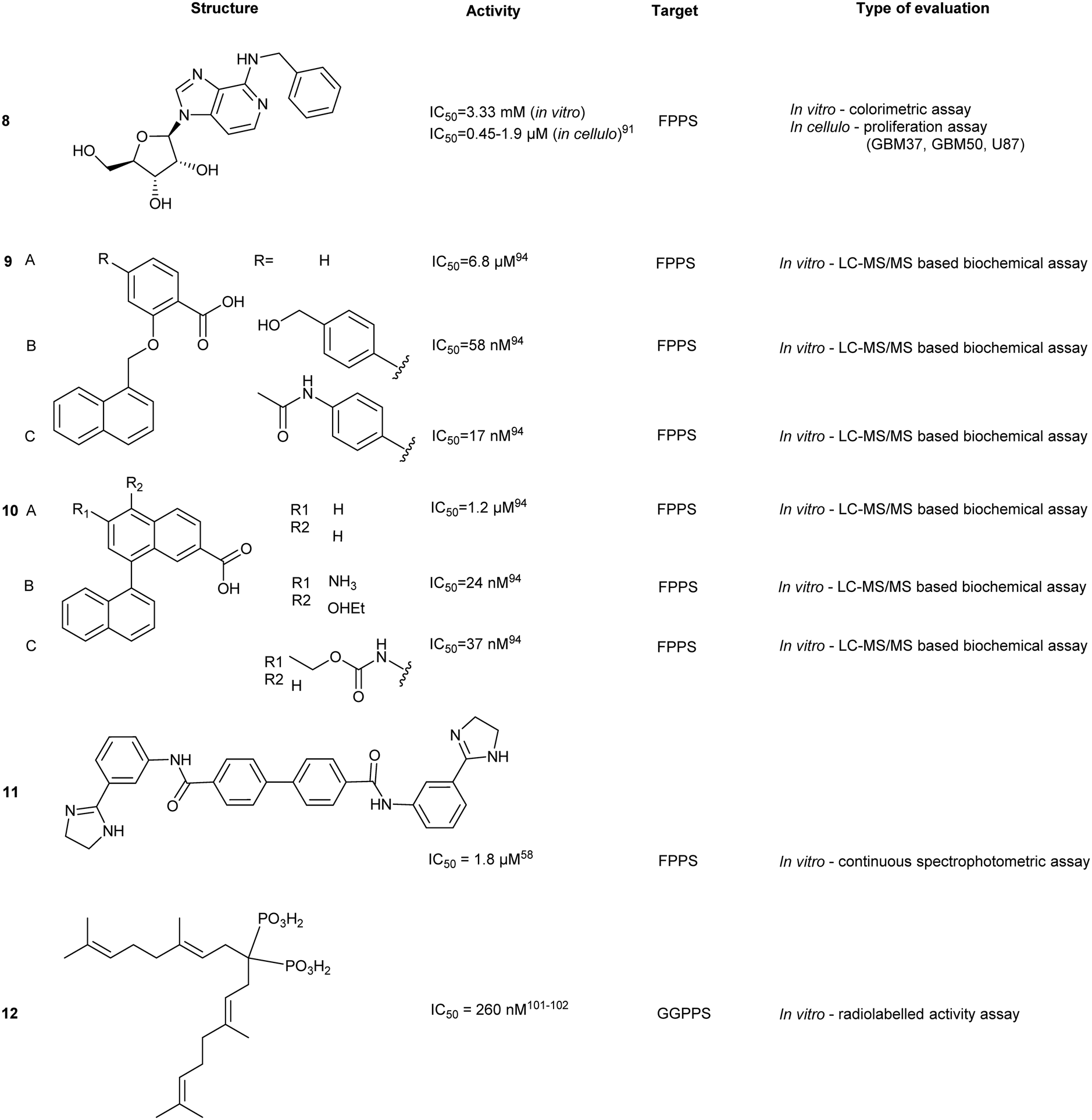
Modification of triazole to thienopyrimidine on BP-based compounds results in ThBP GGPPS inhibitors (16, 17). Crystallographic studies of GGPPS with ThBP revealed a binding mechanism reliant on electrostatic interactions between the BP and Asp-rich motifs to compete with the PPi tail of FPP. Moreover, the extension of the thienopyrimidine moiety into the IPP binding site contributed towards the observed increase in potency. Cell viability assays in multiple myeloma (MM) cell line, RPMI-8226, showed inhibition of cell proliferation and was concomitant with inhibition of GGPPS. Administration of 16 in a Vk*MYC transgenic MM mouse model (3 mg kg−1 per day, 14 days) validated down regulation of Rap1A geranylgeranylation, as well as reduction of M-proteins, a disease burden indicator in the serum. This study highlighted GGPPS as a potential therapeutic target in MM cancer.105
Non-bisphosphonates
The development of non-BP GGPPS inhibitors allows for greater specificity and reduces off-target FPPS inhibition. This can result from lack of PPi-tunel targeting interactions and utilization of steric hindrance from the GGPP-FPP isoprenyl size difference to prevent FPPS binding. A library of >100000 compounds was docked against the allosteric site of GGPPS. The top 1% of compounds were used to create rules to guide inhibitor structure selection based on a site-moiety map. These rules were based on charge distribution within the molecule and bond angles and were computationally validated using known GGPPS inhibitors. Stratification of inhibitors based on these rules yielded 10 compounds, with top hits (17 and 18). Although the observed binding affinities were relatively weak, these molecules provided a potential starting point for hit-to-lead optimization of a non-BP molecule.106
Benzoic acids
Similar to FPPS, advances in GGPPS inhibitors originated from anti-parasitic agents targeting malaria-carrying Plasmodium vivax. Inhibition has focused around blocking GGPP synthesis through the enzyme GGPPS. However, pvGGPPS is structurally similar to human FPPS in comparison to human GGPPS, offering the basis for the preliminary design of pvGGPPS inhibitors. Since pvGGPPS is only weakly inhibited by BPs, benzoic acid derivatives were investigated for enzyme inhibition.107 Novel lipophilic benzoic acids were tested against pvGGPPS in vitro with the most potent compound, 19, exhibiting an IC50 of 50 μM. Co-crystals revealed the benzoic acid occupies the IPP/GGPP binding sites forming electrostatic interactions with Arg135, Arg136, and Lys301. Additionally, molecular dynamic simulations have predicted stabilization of loops which control accessibility to the binding pocket, similar to the C-terminal tail of FPPS. Regardless of the weak affinity observed, in silico approaches to rationale-designed drug offer an alternative method in lead compound identification.108Farnesyl transferase
Early characterization of farnesyl transferase (FTase) revealed its involvement in the activation of the oncogenic Ras signalling pathway and subsequently triggered multiple inhibitor development programs.109 Despite promising in vitro data, poor in vivo profiles, attributed to a compensatory upregulation of GGTase, precluded clinical development.110,111 However, select molecules still retain their efficacy against cancer, parasites, and progeria.There are four key pockets in the FTase protein active site, including the lipid binding site, substrate binding site, metal binding site, and exit cavity. The catalytic mechanism is initiated by FPP binding at the lipid binding site (Kd = 2 nM)112 followed by substrate binding to the CAAX pocket. The CAAX containing substrate binds in an extended conformation, with the Cys residue coordinating to the active site Zn2+. The Cys residue reacts with FPP forming a thioester linkage to the isoprenoid. A second FPP molecule binds to FTase and repositions the lipidated C-terminal CAAX in the ‘exit groove’. Product release is kinetically slow, thus limiting the rate in the entire FTase mechanism.113–116
Based on the mechanism of action, different FTase inhibitors have been broadly classified as CAAX-targeting (peptidomimetics) or lipid binding (FPP-mimicking analogues). An overview on the evolution of FTIs is given by Ochocki and Distefano.11 The authors attribute the lack of FPP-based drugs to the high affinity of the enzyme for its natural substrate, the likelihood for off-target effects, and the necessary incorporation of non-drug-like hydrophilic head groups. The use of CAAX mimicking motifs is the conventional strategy as various potent compounds have been developed with high affinities to FTase. The majority of these CAAX-mimicking compounds have transitioned into non-peptidics due to lack of cell permeability, hydrolysation via peptidases, and greater chemical diversity.117 While FTase inhibition has seen a tremendous increase in both basic research and clinical studies, toxicity and poor efficacy in vivo have hampered clinical development. Here we will highlight the recent advancements in FTI.
Zn2+ dependent inhibitor coordination at the CAAX site
An attractive molecular target for thiol-based CAAX peptidomimetics is the functionally critical Zn2+ metal centre. However, this approach is complicated by metal-binding moieties which non-specifically interact with metalloproteins and disarm their catalytic activity, leading to over-estimation of in vitro or in cellulo activities. As such, several identified compounds also require additional off-target screening.Structure-based screening and virtual library optimization resulted in a class of pentanedioic acid derivatives (20) with observed potency against FTase (IC50 = 0.0029–7.1 μM, in vitro). Although this class of compounds interacts with the bound FPP, the mechanism of action is principally based on COOH-mediated coordination of the Zn2+ centre. Esterification of the carboxylate or replacement with a sulphonamide leads to a loss of activity. SAR studies on 20 revealed the importance of sterics in the binding pocket. Removal of the para-chloro- from the phenyl ring resulted in a 35-fold drop in activity whereas substitution with a larger halogen, Br, resulted in greater potency. At the opposite end of the molecule, substitution with hydrophilic groups was observed to strongly reduce binding, as a result of exclusion from the hydrophobic pocket.118
Simultaneous targeting of both the Zn2+ centre and the FPP binding cavity was explored to develop a multi-site binding inhibitor. SN1, a dithiol compound that inhibits Zn2+ metalloenzymes, was fused with FPP to give compound 21, as well as other FTase targeting compounds such as napthylene (22). Docking studies predicted that the first thiol group binds to the Zn2+ and Tyr361 and the secondary thiol interacts with Arg202. The farnesyl moiety occupies the FPP binding pocket hindering substrate entry. Previous studies into dual targeting of the FPP binding site and the active site have been plagued by challenges of inhibitor solubility as well as outcompeting the high affinity natural substrate, FPP. In vitro assays revealed an IC50 of 0.9 μM for 21 against human FTase. Compound 21 also inhibited the proliferation of pancreatic cell line AsPC-1 expressing the cancer-driving K-Ras mutant.119
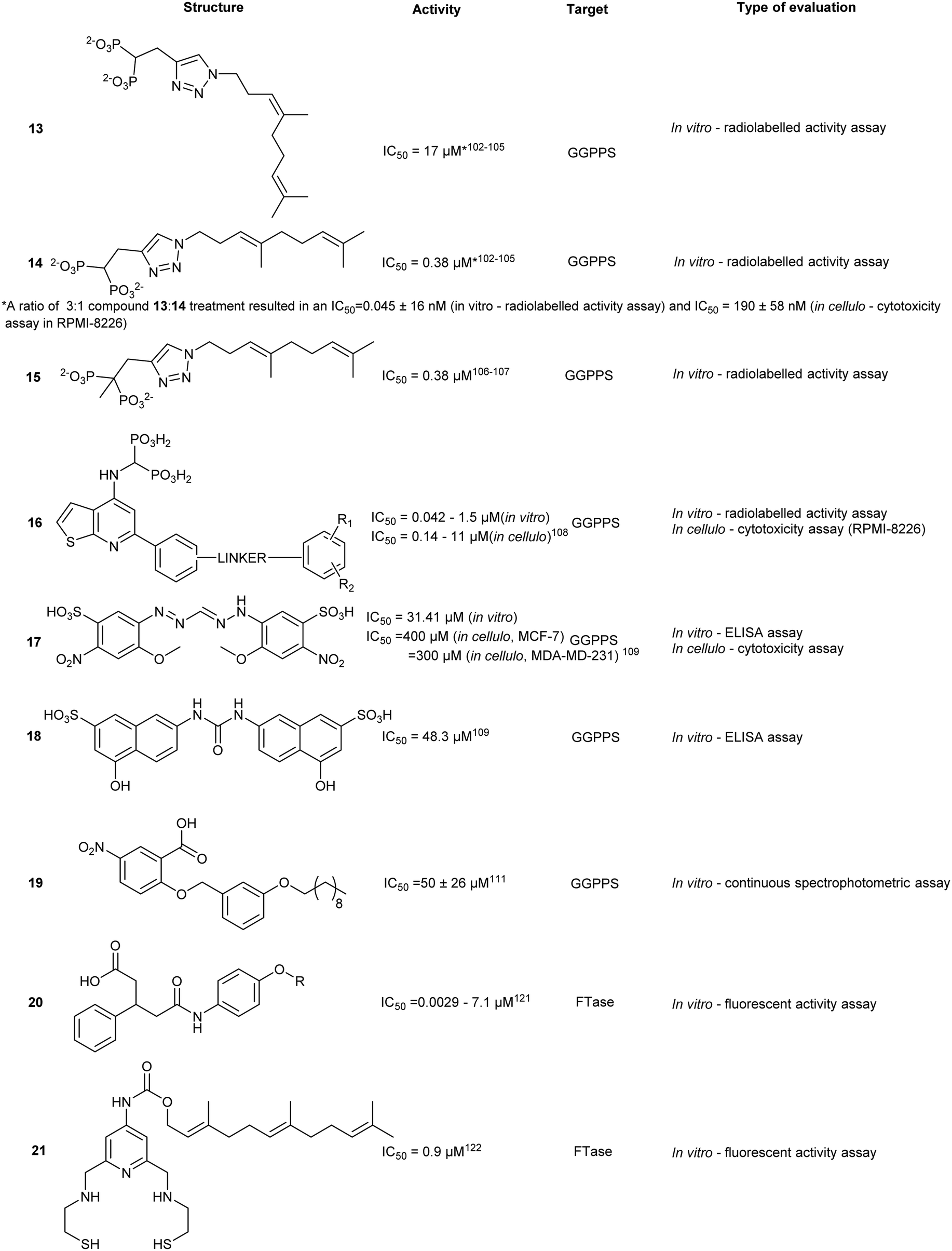
Pyroglutamic derivatives
Fluorescence-based activity assays identified pyroglutamic acid derivative, 23, as a single digit nanomolar FTase inhibitor. Different binding modes have been predicted using computational modelling, but in all cases, the compounds remain in close proximity to FPP and Zn2+. Although proximal to the Zn2+, they are not predicted to chelate the metal center, which was confirmed by UV-vis spectroscopy.120Thiazole derivatives
Computational predictions of previously generated FTI-2148 (24) suggests interactions of the aliphatic spacers from the CAAX motif with the FTase hydrophobic pocket. Molecular docking studies based on 24 proposed interactions between 2-o-tolylbenzoyl and the hydrophobic cavity of FTase. The substitution of the tolyl with a bulky aromatic system (25) was hypothesized to provide additional interactions and increase inhibitory potential. However, the most potent compound appears to be the non-modified system. This indicates the importance of inhibitor positioning within the active site through increasing protein-drug contacts.121N-Ylides
Different functionalities were designed to explore the potential of the secondary aliphatic CAAX site. A series of N-ylides 26 were discovered to inhibit with potencies in the μM range. Binding was shown to be dependent on three substituents. Firstly, the pyridine ring and the phenothiazine moiety were found to stabilize the compound within the active site by engaging in π-stacking interactions with two Tyr residues. Secondly, Zn2+ chelation was achieved by interaction with the cyano ethylacetate moiety. Finally, the addition of a para-chloro/bromo on the phenyl ring provided increased inhibitory activity against FTase (in vitro). Despite well-defined interaction motifs, FTase inhibitory activity of the SAR library was not observed to be lower than low μM range.122Indolizine derivatives
Indolizin-3-yl(phenyl)methanone (27) exhibited single digit μM potency against FTase. An SAR study around 27 revealed the biological importance of halogens at the p-benzyl position.123 While replacement of the amide with an ester afforded an increase in potency (11.4 to 1.3 μM) the effects were not as drastic as those seen in the phenothiazine class for the analogous modification.A follow-up study was conducted to investigate the effects of the conjugation of chalcone, a compound class known to exhibit anti-cancer properties, as a linkage between indolizine and phenothiazine derivatives. An SAR study around the indolizine–chalcone–phenothiazine core revealed several compounds with strong inhibitory activity with the most active compound 28 possessing an acrylamide moiety with IC50 of 9 nM (hFTase). These findings highlight the development of novel scaffolds for FTI activity and instigate further investigation of linking pharmacologically active moieties together to improve biological inhibitory activity.124 The acrylamide moiety also suggests the possibility of a covalent targeted approach, on the basis that target selectivity can be maintained.
High-throughput screening
Through virtual screening, Yu et al. investigated approximately 260000 compounds with diverse scaffolds against FTase where hits were clustered according to their structural redundancy and similarity. The screen identified 22 promising compounds that were tested against FTase and human breast cancer cell lines (MCF-7). The most potent compound (29), showed IC50 values of 48.9 nM and 14.28 μM, respectively. Docking of 29 suggested key interactions with the allosteric subpocket and effective occupancy of the product exit cleft.125
IMB-1406 (30) was identified by Jin et al. from a virtual screen of over 900 compounds upon docking against FTase. Each molecular docking pose obtained from the screen was compared against a known ethylenediamine-based FTase inhibitor (FTI) (31). Cytotoxicity was evaluated in 4 cancer cell lines where 30 was demonstrated to be equipotent (IC50 = 6.92–8.99 μM) with FDA approved inhibitor, sunitinib (IC50 = 7.60–10.36 μM). Moreover, compound 30 was shown to induce apoptosis through S-phase arrest within the cell cycle.126 In spite of the cytotoxic potency, experimental validation against the direct target, FTase, was not reported. Thus, the observed potency may be due to an accumulation of effects include the inhibition of FTase and off-target toxicity and thereby only suggesting 29 as a possible FTase inhibitor without further evidential support.
Chaetomellic acid A (32a) isolated from microbial coelomycete, Chaetomella acutiseta, was identified as an FTI (IC50 = 55 nM, hFTase) from screening of a natural product library. Compound (32a) spontaneously cyclizes upon in vitro synthesis (32b), and is hydrolyzed at physiological pH to yield the dicarboxylate anion (32c). Notably, inhibition of FTase by 32c is non-competitive to the native CAAX-acceptor peptide but is highly competitive towards the isoprenoid, FPP.127 Bellesia et al. identified 32d (IC50 = 190 nM, rat FTase) which possesses a higher potency compared to 32a (IC50 = 910 nM, rat FTase). Analysis of the SAR data revealed a direct correlation between increasing hydrocarbon tail length and inhibitory activity. Molecular docking of 32d predicts binding to the active site in the same orientation as FPP. The aliphatic tail occupies the hydrophobic pocket while forming van der Waals contacts with the second aliphatic residue of the CAAX peptide.128
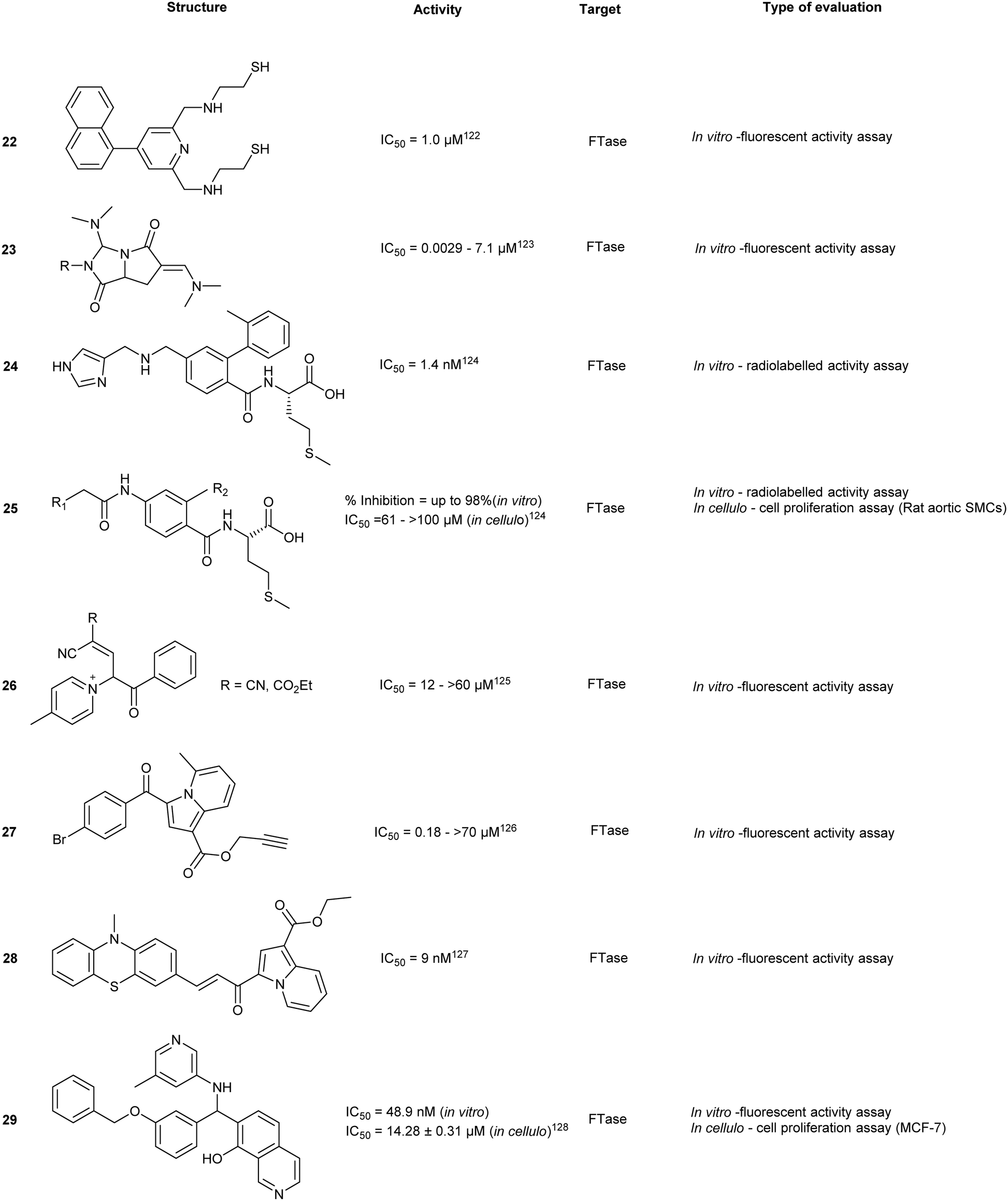
Tecomaquinone I (33) was identified as a potent inhibitor from a focused natural product library screen against hFTase (IC50 = 65 nM), Trypanosoma brucei FTase (IC50 = 112 nM) and Plasmodium falciparum FTase (IC50 > 22 μM). The majority of 32 analogues prepared revealed selective inhibitory profiles for hFTase and TbFTase while showing minimal activity against PfFTase, with 33 exhibiting the lowest selectivity. Contrary to the findings with 32a and its derivatives, increasing the aliphatic chain length of 33 did not confer increased bioactivity.129
The future of FTIs
Modest efficacy/potencies coupled with elevated toxicity profiles, necessitate further exploration in FTI development, whether it be identification of a new lead scaffold or repurposing of known FTI clinical candidates. For instance, tipifarnib, a highly potent FTI, has been reinvestigated against other cancers such as juvenile myelomonocytic leukemia (JMML) following rejection by the FDA for the treatment of acute myeloid leukemia.130 Due to promising data in numerous phase II trials, tipifarnib remains under investigation in both single agent and combinatorial therapy for indications that it can excel in such as solid tumors and hematologic malignancies.134 Other strategies for FTI improvement have involved nanoparticle conjugation of FTIs in an attempt to minimize off-target effects.131 Finally, targeting species-specific FTase isoforms can assist in selective treatments with anticancer (hFTase), anti-malaria (PfFTase) and antibacterial (TbFtase) therapies as shown with 33.129Geranylgeranyl transferase inhibitors
Since inhibition of FTase has been associated with a compensatory GGTase I upregulation, the development of selective GGTase (specifically GGTase I) inhibitors is particularly popular. There are three isoforms of GGTase, GGTase I, GGTase II (also referred to as RabGGTase), and GGTase III. The structures and reaction mechanisms of GGTase I are highly similar to FTase, whereas GGTase II is unique in that substrate specificity is achieved through complexation with REP (Rab escort protein). REP complexes with a Rab protein and results in a REP-Rab-GGTase II ternary complex.132 As with other prenyltransferases, dissociation of the prenylated (Rab-REP) substrate is initiated by GGPP binding. GGTase III shares the same β subunit as RabGGTase while recognizing similar CAAX sequence as GGTase I of FBXL2 substrate receptor subunits of SKP1-CUL1-F-box protein (SCF) ubiquitin ligase complexes.133 The PTAR1 α subunit of GGTase III specifically recruits FBXL2 for geranylgeranylation via interaction at its conserved N-terminal extension (NTE). Through evidence of GGTase III – FBXL2 complexation, the fourth prenyltransferase family member which can geranylgeranylate ubiquitin ligases is now revealed.9While limited structural differences in GGTase-I and GGTase-II favor pan-isoform inhibitors, current strategies have focused on improving selectivity between GGTases.
GGTase-I
Tetrahydropyridine derivatives
A small molecule library, created by phosphine-catalyzed annulation reactions, was screened to reveal two novel classes of GGTI, dihydropyrrole (P5-H6) (34) and tetrahydropyridine (P3-E5) (35). Both of these compounds demonstrated selectivity for GGTaseI over RabGGtase (>160-fold and >20-fold, respectively) in a radiolabelled in vitro activity assay.134 Mechanistic studies revealed these scaffolds operate through competitive displacement of the target protein substrate, but are non-competitive towards the GGPP site. The lead tetrahydropyridine, P3-E5 compound (35), was further modified with a L-Leu methylester in place of the free carboxylic acid to yield, P61-E7 (36).135 This compound was shown to have improved GGTase inhibition, demonstrated by accumulation of cytosolic RalA and RhoA as well as increased potency in Jurkat cell lines (IC50 = 20 μM, 35; IC50 = 3.5 μM, 36). Despite the high selectivity observed in vitro, GGTase-I inhibition in vivo has not been achieved.136RabGGTase
Phosphonocarboxylate
Phosphonocarboxylates (PCs) are derived from bisphosphonates (BPs) and involve the replacement of a phosphate group with a carboxylic acid. PC derivatives of risedronic acid (3-(3-pyridyl)-2-hydroxy-2-phosphonopropanoic acid) (37) and mindronic acid (2-hydroxy-3-(imidazo[1,2-a]pyridin-3-yl)-2-phosphonopropanoic acid) (38) were found to be inhibitors of RabGGTase.137,138 Compound 38 exhibited a 25-fold increased potency as compared to the + enantiomer of (37).139 An SAR study around the effects of the α-OH position showed less potency compared to the lead compound 38. Molecular docking studies suggested binding proximal to the first geranylgeranylated Cys residue. Compound binding is predicted to be mediated by multiple interactions including H-bonding, salt bridges and van der Waals interactions of the imidazo[1,2-a]pyridine heterocycle in the hydrophobic pocket.140 Cell-based FRET studies revealed a significant reduction in membrane localization of dual geranylgeranylated Rab proteins but not of mono-geranylgeranylated substrates. This suggested that the mechanism of inhibition occurs through the second geranylgeranylation event.137N-Oxide derivatives of 37 were found to lower the inhibitory activity against Rab GGTase to 1.8 mM from the parent compound (0.7 mM), supporting the author's hypothesis that the inclusion of uncharged substituents or bulk may increase the compound's potency.141 Inhibitory activity was identified for PC with GGTase II but not against FPPS/GGPPS, due to the presence of a chiral center at the α-carbon. However, substitution of the α-OH with an alkyl chain restores activity against FPPS/GGPPS with a strong correlation to hydrophobicity and chain length.142Psoromic acid
Screening >10000 compounds for activity against GGTase II identified, psoromic acid, (39) a natural product derived from lichen (IC50 = 1.4 μM, in vitro fluorometric assay) in a gel-based assay, analogues of 39 selectively inhibited GGTase II over related prenyltransferases, FTase and GGTase I. A fluorometric assay revealed 39-analogues compete for the IPP binding site and covalently bind to the N-terminus of GGTase II (α subunit) through formation of a Schiff base with the aldehyde functionality of 39. Crystallographic analysis of the 39-GGTaseII complex suggested that the compound binding mode exploits an auto-regulatory function unique to GGTase II. In the apo-state, activity is regulated by an N-terminal adjacent His residue which coordinates the Zn2+ ion. Displacement of the N-terminus, as a result of 39-covalent binding, allows for further interactions of 39 at the active site and occupancy of the lipid substrate binding site. Despite the molecule's interesting in vitro mode of inhibition, no inhibitory activity was detected in HeLa cells up to a concentration of 300 μM. This may be a consequence of low cell membrane permeability of 39 as shown by PAMPA assays. These studies do however provide a novel mechanism of action for RabGGTase inhibition.143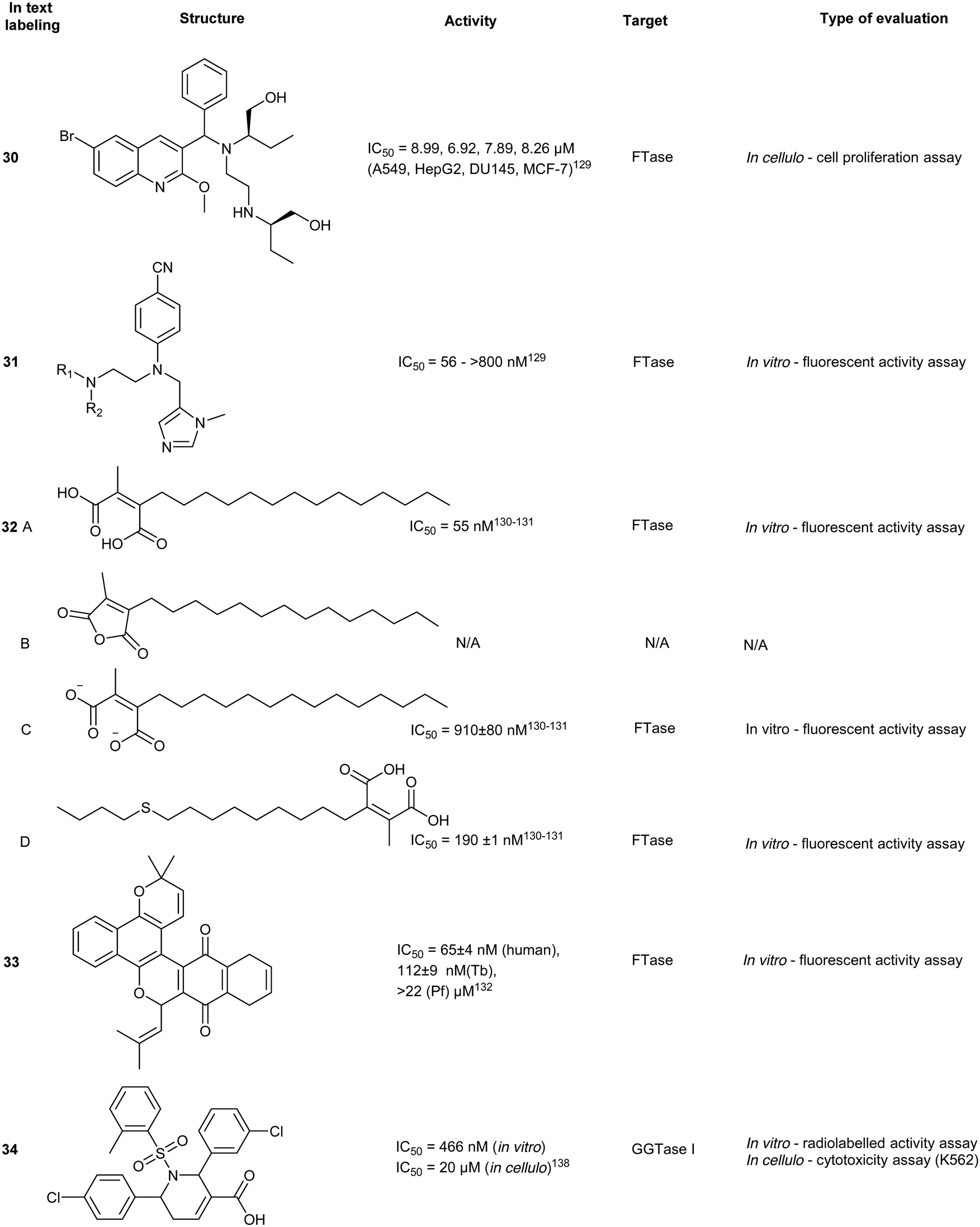
GGTase III
GGTase III was recently identified as a prenylation enzyme, and thus there have been limited targeted efforts to generate enzyme-specific inhibitors. However, this discovery presents a unique opportunity for the inhibition of ubiquitin ligase prenylation via the accessory protein, FBXL2 which prevents the recruitment of the target proteins.152,157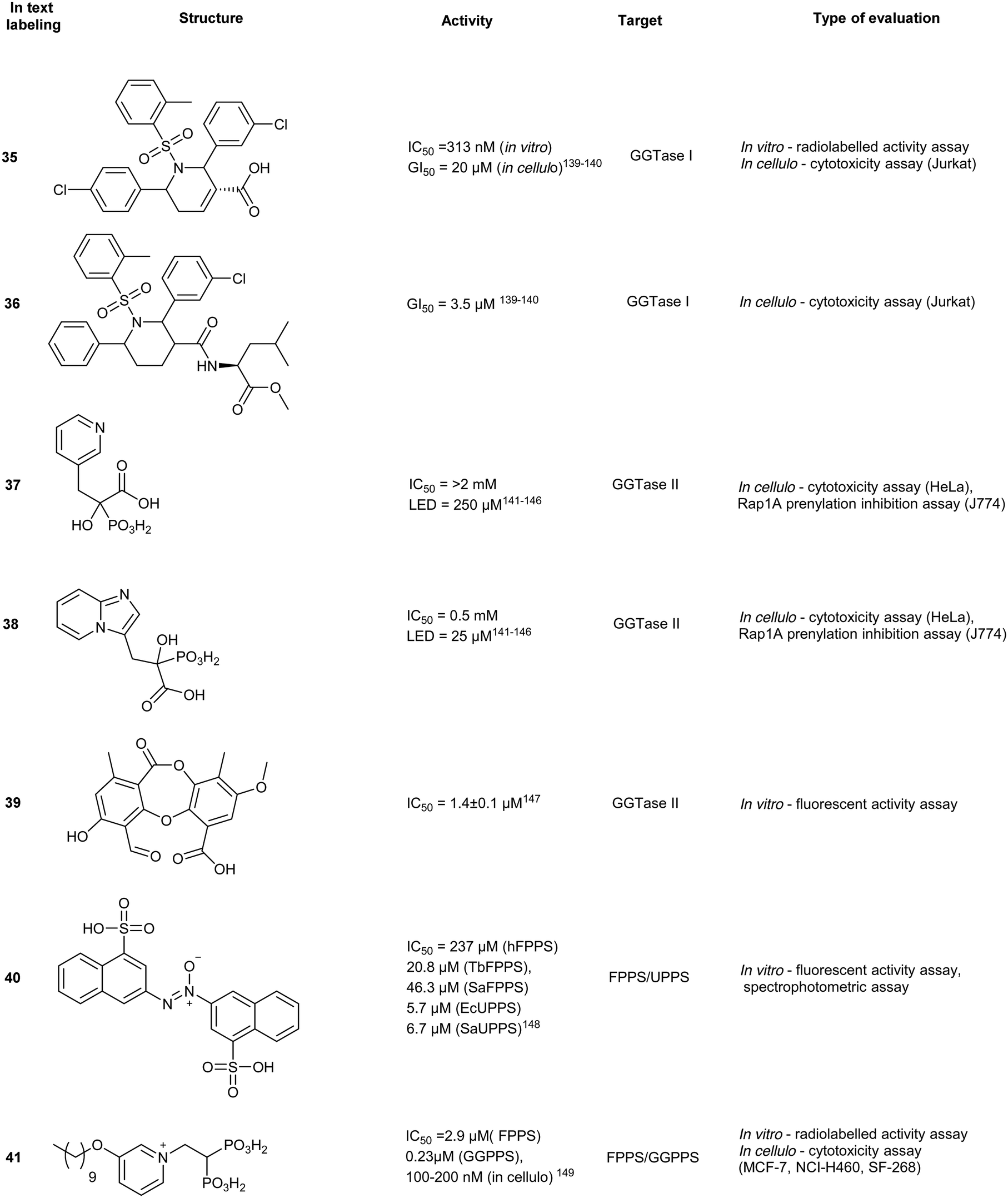
Dual inhibitors
An alternative and arguably more effective strategy for inhibiting prenylation involves simultaneous targeting of FPPS and GPPS or FTase/GTase, with dual inhibitors. The overall strategy involves potently reducing Ras prenylation while exploiting the structural similarities of the prenylsynthase/transferase enzymes. Importantly, this approach seeks to circumvent the challenges of traditional BP-based inhibitors that accumulate in the skeletal system.FPPS/UPPS inhibitors
The use of non-BPs as FPPS drugs is an attractive inhibition strategy since they are less vulnerable to accumulation in calcified tissues. Through molecular docking, 23 compounds were predicted as possible inhibitors of Trypanosoma brucei FPPS (a carrier of Chagas disease). In vitro assays revealed compound 40, as the most potent hit against various bacterial and human FPPS isoforms (IC50 = 5.7 to 237 μM). Binding interactions were predicted to be driven by sulfonate–Mg2+ coordination within the active site, similar to the phosphate–Mg2+ coordination pose observed by traditional BPs. In addition to pan-FPPS isoform inhibitory activity, (40) was observed to possess therapeutic effects against undecaprenyl diphosphate synthase (UPPS), an enzyme essential for bacterial cell growth.144 However, further evaluation of antibacterial activity as well as cellular cytotoxicity is necessary to conclude the efficacy of this class of compounds as a dual inhibition agents of FPPS/UPPS.Lipophilic bisphosphonates
Various FPPS-targeting BPs demonstrated off-target binding for GGPPS due to structural similarity of the two enzymes. Given these selectivity challenges, dual FPPS/GGPPS inhibitors were explored. Several BPs were engineered with positively charged groups adjacent to the BP-pharmacophore for increasing FPPS inhibitory activity. Additionally, incorporation of large hydrophobic motifs in the scaffold results in reduction of bone uptake and drug bioavailability. Cationic BPs such as 41, show higher inhibitory profiles (IC50 = 100–200 nM) compared to established BPs, such as zoledronate and pamidronate (IC50 = 15–140 μM), possibly due to the increase in cell permeability. Large hydrophobic BPs, such as 42, also demonstrated greater growth inhibition with reversibility was observed upon the addition of 20 μM geranylgeranylhydroxide (GGOH). In vivo experiments with lipophilic BPs, performed in murine xenograft models with SK-ES-1 sarcoma cells, elucidated greater tumour reduction than zoledronate. The combined effects of electrostatics and hydrophobicity in lipophilic-BP treatments improved a traditional class of compounds furthering their utility as therapeutic agents.145 From the demonstrated potential of lipophilic bisphosphonates, the top hits from this class of compounds were investigated for the treatment of KRAS-driven lung cancer in a combination treatment with rapamycin. Substantial tumour suppression was observed by 43, a lipophilic zoledronate analogue, in mouse syngeneic orthotopic graft models. However, the development of ascites at the injection site was observed resulting in the discontinuation of the study suggesting that 43, in the current delivery system, also acts an irritant.146 Further research also revealed efficacy against activated hepatic stellate cells, which are responsible for liver fibrogenesis.147Isoprenoid analogues
Synthetic isoprenoid analogues mimic the natural ligand of prenyltransferases which leads to high selectivity for both GGTase and FTase. However, administration of synthetic isoprenoids is challenging due to high lipophilicity and metabolic instability of the PPi moiety. To improve the drug-like attributes, a pro-drug approach was employed using mimicry of pathway intermediates by the replacement of PPi with a hydroxyl group. The synthetic isoprenoids, anilinogeraniol (44) and anilinofarnesol (45) behave as the natural products, farnesol or geranylgeraniol, and are converted to their PPi analogues. The resulting anilinoprenylpyrophosphate is transferred to the target substrate by the prenyltransferase without translocating to the membrane.148,149 Chen et al. have investigated the utility of the synthetic isoprenoids as prodrugs to inhibit breast cancer invasion in a 3D in cellulo assay and cell cytotoxicity assay in the MDA-MB-231 (breast cancer) and MCF10A (non-malignant) cell lines. Compound 44, a GGTase substrate, appears to demonstrate higher potency compared to compound 45, a FTase substrate, in the inhibition of the metastatic cell line. This effect originates from the longer carbon chain required for GGTase recognition of 45, which may play a larger role in breast cancer invasion, demonstrating the applicability of hijacking the prenyltransferase activity to modify targets with a non-membrane directing functionality.150Bivalent inhibitors
Disruption of transient protein–protein interactions (PPI) can impact signalling cascades utilized by various regulatory pathways. Although the mechanism of action for PPIs is highly variable between different systems and challenging to investigate, a recent study on PPI within the mitogen activated protein kinase (MAPK) family revealed the importance of the electrostatic surface potential. Disruption of the surface charges can dramatically alter PPI. A similar approach was adopted for inhibition of prenyltransferases to block their interactions with cancer-driving K-Ras4B. The positively charged poly-Lys C-terminus of K-Ras4B was found to interact with the acidic surfaces of FTase and GGTase. Machida et al. developed a bivalent inhibitor mimicking K-Ras4B containing positively charged poly-Lys and the tetrapeptide CVIM (46) to block both the active site and acidic surface of the prenyltransferases, inhibiting transient PPI and thus, the prenylation of K-Ras4B by both FTase (Ki = 5 ± 1 nM) and GGTase I (Ki = 344 ± 86 nM), which demonstrates the utility of bivalent inhibitors. However, due to the size of 46, the compound has limited cell permeability, halting further evaluations of the compound series.151To improve cell permeability of bivalent inhibitors, Tsubamoto et al. explored the utilization of C3-alkylated guanidine moiety and FTI-249 to improve upon the previous peptide-based scaffold (46). The guanidinium cation interacts with the lipid phosphate head and disrupts membrane curvature thereby allowing the C3-alkyl side chain to insert into the membrane and increase membrane penetration. Alternatively, FTI-249 was developed as a CVIM peptidomimetic which possesses FTase selectivity. Upon comprehensive SAR development, the FTI-249-based core was substituted with a biphenyl-based peptidomimetic, FTI-276 to improve potency of the scaffold. Despite increased inhibitory activity against FTase, GGTase I inhibitory activity remains moderate (Ki = 710 ± 2 nM). Regardless, a methyl ester derivative 47 was able to induce G1 arrest and cytosolic delocalization of K-Ras and its c-Raf association, revealing potential therapeutic strategy for K-Ras targeting through dual inhibition of both the active site and acidic surfaces.156
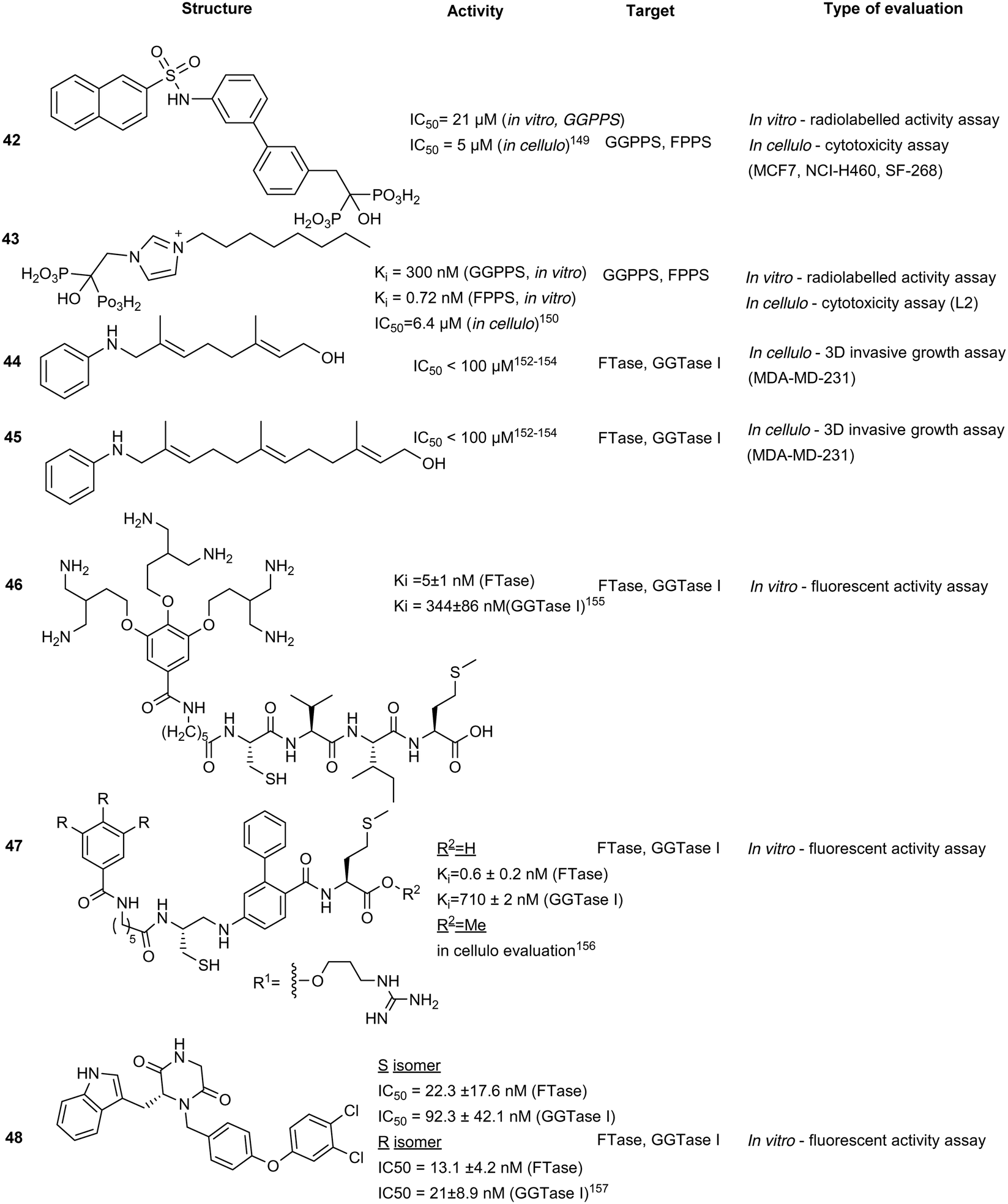
Piperazinedione analogues
Piperazinedione analogues, derived from the CAAX recognition sequence, inhibit the action of both FTase and GGTase I. Compound 48 was found to be the most potent inhibitor from an SAR study with an in vitro IC50 of 13.1 nM and 21 nM against FTase and GGTase I, respectively. Based on kinetic studies, compound 48 was predicted to interact with both the isoprenoid and CAAX binding cavities and adopts a U-shaped conformation according to in silico docking.153 This strategy improves on the traditional peptidomimetic approach by incorporating the isoprenoid binding region to facilitate dual inhibitory activity.Conclusions
Development of mevalonate pathway inhibitors to halt prenylation of oncogenic drivers and reducing cholesterol production has been advancing for decades. Potent and selective molecules have progressed into clinical trials, although results for FPPS and GGPPS inhibitors as well as for the prenyltransferases have been modest. Collectively, screening technology developments have led to new methods of lead compound discovery from novel scaffolds to existing natural products. However, inhibitor-based therapeutic strategies have suffered from similar disadvantages such as limited bioavailability, clearance rate, and active secondary metabolites. A possible alternative approach in the therapeutic use of the upregulated prenylation pathway in cancer driving cells involves hijacking of the system to target other vital proteins required for the cell survival. Previous attempts have shown the utility of the lipid covalent attachment onto protein targets for immobilization which prevents organelle translocation.154,155 This can also prevent the formation of protein–protein interactions necessary for functional activity. Protein membrane anchorage (PMA) has also been proposed as a viable strategy for targeting transcription factors in cancer cells in order to sequester them to the membrane and away from the site of activity.158–160 Focusing on a more traditional inhibitor-based route of inhibition of prenylation, the development of a FTase/GGTase dual inhibitor is still relatively unexplored. Regardless of therapeutic strategy, evaluation of prenylation inhibition requires a collective biophysical and biochemical approach to understand the mechanisms involved in direct target engagement. Due to the possibility of incorporating false hits arising from off-target toxicity in cell-based proliferation and viability assays, utilization of orthogonal methods such as in vitro activity or binding assays should be considered an essential step to avoid false-positive molecules. Other modern techniques such as cellular thermal shift assay (CETSA) and activity-based protein profiling (ABPP) are also critical to validate lead molecules and avoid unnecessary effort with off-target molecules or false-positives bearing pan-assay interference (PAINs) scaffolds.161,162In summary, targeting prenylation through the mevalonate pathway can be achieved through multiple routes but currently remains a significant challenge. The direct inhibition of the upstream enzymes, FPPS and GGPPS, are conventionally targeted by active site-binding NBPs. Optimization of NBPs yielded improved scaffolds including ThBPs and ThMPs which increased biological efficacy and reduced off-target effects compared to the parent, NBPs. A more direct approach to inhibiting prenylation is through targeting the prenyltransferases, FTase and GGTase I/II/III. However, inhibition of FTase alone is insufficient due to similarities arising from the recognition CAAX motif between FTase and GGTase I which can result in compensatory activity. Moreover, introduction of potent FTase and GGTase I inhibitors have led to detrimental cytotoxic effects as prenylation is required for cell viability. Due to the major difference in recognition sequences between the GGTase I and II, the two enzymes are not used interchangeably within the cellular system. This has pushed the development of GGTase II as a therapeutic target. GGTase II not only possesses a more specific recognition sequence, its targets are usually oncogenic drivers, namely clinically-relevant Rab protein. The combination of structural guided inhibitor design and multimodal treatment strategies offer potential in developing candidates for targeting prenyl transferases in cancer as well as a plethora of other diseases due to branching points in the mevalonate pathway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. T. G is supported by research grants from NSERC (RGPIN-2014-05767), CIHR (MOP-130424, MOP-137036), Canada Research Chair (950-232042), Canadian Cancer Society (703963), Canadian Breast Cancer Foundation (705456) and infrastructure grants from CFI (33536) and the Ontario Research Fund (34876).Notes and references
- D. D. O. Martin, E. Beauchamp and L. G. Berthiaume, Biochimie, 2011, 93, 18–31 CrossRef CAS PubMed.
- M. E. Linder and R. J. Deschenes, Nat. Rev. Mol. Cell Biol., 2007, 8, 74–84 CrossRef CAS PubMed.
- D. J. Siegwart, K. A. Whitehead, L. Nuhn, G. Sahay, H. Cheng, S. Jiang, M. Ma, A. Lytton-Jean, A. Vegas, P. Fenton, C. G. Levins, K. T. Love, H. Lee, C. Cortez, S. P. Collins, Y. F. Li, J. Jang, W. Querbes, C. Zurenko, T. Novobrantseva, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12996–13001 CrossRef CAS PubMed.
- M. Wang and P. J. Casey, Nat. Rev. Mol. Cell Biol., 2016, 17, 110–122 CrossRef CAS PubMed.
- M. D. Resh, Curr. Biol., 2013, 23, R431R435 CrossRef PubMed.
- M. Avadisian and P. T. Gunning, Mol. BioSyst., 2013, 9, 2179–2788 RSC.
- G. P. Hooff, W. G. Wood, W. E. Müller and G. P. Eckert, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2010, 1801, 896–905 CrossRef CAS PubMed.
- J. Gao, J. Liao and G. Y. Yang, Am. J. Transl. Res., 2009, 1, 312–325 Search PubMed.
- S. Kuchay, H. Wang, A. Marzio, K. Jain, H. Homer, N. Fehrenbacher, M. R. Philips, N. Zheng and M. Pagano, Nat. Struct. Mol. Biol., 2019, 26, 628–636 CrossRef CAS PubMed.
- M. A. Hast and L. S. Beese, J. Biol. Chem., 2008, 283, 31933–31940 CrossRef CAS PubMed.
- J. D. Ochocki and M. D. Distefano, MedChemComm, 2013, 4, 476 RSC.
- E. Kim, P. Ambroziak, J. C. Otto, B. Taylor, M. Ashby, K. Shannon, P. J. Casey and S. G. Young, J. Biol. Chem., 1999, 274, 8383–8390 CrossRef CAS PubMed.
- M. O. Bergo, G. K. Leung, P. Ambroziak, J. C. Otto, P. J. Casey, A. Q. Gomes, M. C. Seabra and S. G. Young, J. Biol. Chem., 2001, 276, 5841–5845 CrossRef CAS PubMed.
- I. Manolaridis, K. Kulkarni, R. B. Dodd, S. Ogasawara, Z. Zhang, G. Bineva, N. O'Reilly, S. J. Hanrahan, A. J. Thompson, N. Cronin, S. Iwata and D. Barford, Nature, 2013, 504, 301–305 CrossRef CAS PubMed.
- D. R. Tobergte and S. Curtis, Encyclopedia of Signaling Molecules, 2013, vol. 53 Search PubMed.
- E. M. Novak and E. M. Rego, Physiopathogenesis of Hematological Cancer, Bentham Science Publishers, 2012 Search PubMed.
- K. Sharma, R. C. J. D'Souza, S. Tyanova, C. Schaab, J. Wiśniewski, J. Cox and M. Mann, Cell Rep., 2014, 8, 1583–1594 CrossRef CAS PubMed.
- F. Wolschin, S. Wienkoop and W. Weckwerth, Proteomics, 2005, 5, 4389–4397 CrossRef CAS PubMed.
- Y. Liu and Y. Lin, Genomics, Proteomics Bioinf., 2004, 2, 253–255 CrossRef CAS.
- P. Mertins, J. W. Qiao, J. Patel, N. D. Udeshi, K. R. Clauser, D. R. Mani, M. W. Burgess, M. A. Gillette, J. D. Jaffe and S. A. Carr, Nat. Methods, 2013, 10, 634–637 CrossRef CAS PubMed.
- S.-L. Paiva, S. R. da Silva, E. D. de Araujo and P. T. Gunning, J. Med. Chem., 2017, 61, 405–421 CrossRef PubMed.
- C. Banfi, M. Brioschi, S. Barcella, R. Wait, S. Begum, S. Galli, A. Rizzi and E. Tremoli, Proteomics, 2009, 9, 1344–1352 CrossRef CAS PubMed.
- W. W. Epstein, D. Lever, L. M. Leining, E. Bruenger and H. C. Rilling, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9668–9670 CrossRef CAS PubMed.
- D. Zeitouni, Y. Pylayeva-Gupta, C. J. Der and K. L. Bryant, Cancers, 2016, 8, 45 CrossRef PubMed.
- S. Eser, A. Schnieke, G. Schneider and D. Saur, Br. J. Cancer, 2014, 111, 817–822 CrossRef CAS PubMed.
- B. Győrffy, I. Stelniec-Klotz, C. Sigler, K. Kasack, T. Redmer, Y. Qian and R. Schäfer, Oncotarget, 2015, 6, 13334–13346 CrossRef PubMed.
- L. Chang, J. Zhang, Y. H. Tseng, C. Q. Xie, J. Ilany, J. C. Brüning, Z. Sun, X. Zhu, T. Cui, K. A. Youker, Q. Yang, S. M. Day, C. R. Kahn and Y. E. Chen, Circulation, 2007, 116, 2976–2983 CrossRef CAS PubMed.
- S. Gao, J. Mo, L. Chen, Y. Wang, X. Mao, Y. Shi, X. Zhang, R. Yu and X. Zhou, Neuropharmacology, 2016, 103, 44–56 CrossRef CAS PubMed.
- N. Xu, N. Shen, X. X. Wang, S. Jiang, B. Xue and C. J. Li, Sci. China: Life Sci., 2015, 58, 328–335 CrossRef CAS PubMed.
- J. L. Goldstein and M. S. Brown, Nature, 1990, 343, 425–430 CrossRef CAS PubMed.
- I. Buhaescu and H. Izzedine, Clin. Biochem., 2007, 40, 575–584 CrossRef CAS PubMed.
- V. J. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman and J. D. Keasling, Nat. Biotechnol., 2003, 21, 796–802 CrossRef CAS PubMed.
- E. S. Istvan, M. Palnitkar, S. K. Buchanan and J. Deisenhofer, EMBO J., 2000, 19, 819–830 CrossRef CAS PubMed.
- R. W. Sarver, E. Bills, G. Bolton, L. D. Bratton, N. L. Caspers, J. B. Dunbar, M. S. Harris, R. H. Hutchings, R. M. Kennedy, S. D. Larsen, A. Pavlovsky, J. A. Pfefferkorn and G. Bainbridge, J. Med. Chem., 2008, 51, 3804–3813 CrossRef CAS PubMed.
- E. S. Istvan, Science, 2001, 292, 1160–1164 CrossRef CAS PubMed.
- C. Stancu and A. Sima, J. Cell. Mol. Med., 2001, 5, 378–387 CrossRef CAS PubMed.
- H. V. Ganga, H. B. Slim and P. D. Thompson, Am. Heart J., 2014, 168, 6–15 CrossRef CAS PubMed.
- R. K. Krishna, O. Issa, D. Saha, F. Y. B. Macedo, B. Correal and O. Santana, Pulm. Pharmacol. Ther., 2015, 30, 134–140 CrossRef CAS PubMed.
- M. Needham and F. L. Mastaglia, Neuromuscular Disord., 2014, 24, 4–15 CrossRef CAS PubMed.
- M. K. Akula, M. X. Ibrahim, E. G. Ivarsson, O. M. Khan, I. T. Kumar, M. Erlandsson, C. Karlsson, X. Xu, M. Brisslert, C. Brakebusch, D. Wang, M. Bokarewa, V. I. Sayin and M. O. Bergo, Nat. Commun., 2019, 10, 1–13 CrossRef PubMed.
- B. L. Schafer, R. W. Bishop, V. J. Kratunis, S. S. Kalinowski, S. T. Mosley, K. M. Gibson and R. D. Tanaka, J. Biol. Chem., 1992, 267, 13229–13238 CAS.
- D. Potter and H. M. Miziorko, J. Biol. Chem., 1997, 272, 25449–25454 CrossRef CAS PubMed.
- Z. Fu, N. E. Voynova, T. J. Herdendorf, H. M. Miziorko and J. J. P. Kim, Biochemistry, 2008, 47, 3715–3724 CrossRef CAS PubMed.
- M. Gharehbeglou, G. Arjmand, M. R. Haeri and M. Khazeni, Cholesterol, 2015, 147601 Search PubMed.
- D. Pilloff, K. Dabovic, M. J. Romanowski, J. B. Bonanno, M. Doherty, S. K. Burley and T. S. Leyh, J. Biol. Chem., 2003, 278, 4510–4515 CrossRef CAS PubMed.
- S. Hogenboom, J. J. Tuyp, M. Espeel, J. Koster, R. J. Wanders and H. R. Waterham, J. Lipid Res., 2004, 45, 697–705 CrossRef CAS PubMed.
- P. Boonsri, T. S. Neumann, A. L. Olson, S. Cai, T. J. Herdendorf, H. M. Miziorko, S. Hannongbua and D. S. Sem, Biochem. Biophys. Res. Commun., 2013, 430, 313–319 CrossRef CAS PubMed.
- N. E. Voynova, Z. Fu, K. P. Battaile, T. J. Herdendorf, J. J. P. Kim and H. M. Miziorko, Arch. Biochem. Biophys., 2008, 480, 58–67 CrossRef CAS PubMed.
- J. F. Nave, H. d'Orchymont, J. B. Ducep, F. Piriou and M. J. Jung, Biochem. J., 1985, 227, 247–254 CrossRef CAS PubMed.
- Y. Qiu and D. Li, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 1080–1087 CrossRef CAS PubMed.
- Y. Qiu and D. Li, Org. Lett., 2006, 8, 1013–1016 CrossRef CAS PubMed.
- J. E. Dunford, A. A. Kwaasi, M. J. Rogers, B. L. Barnett, F. H. Ebetino, R. G. G. Russell, U. Oppermann and K. L. Kavanagh, J. Med. Chem., 2008, 51, 2187–2195 CrossRef CAS PubMed.
- M. K. Dhar, A. Koul and S. Kaul, New Biotechnol., 2013, 30, 114–123 CrossRef CAS PubMed.
- S. Chang, T. Ko, A. P.-C. Chen, A. H.-J. Wang and P.-H. Liang, Protein Sci., 2004, 13, 971–978 CrossRef CAS PubMed.
- P. H. Liang, T. P. Ko and A. H. J. Wang, Eur. J. Biochem., 2002, 269, 3339–3354 CrossRef CAS PubMed.
- D. J. Hosfield, Y. Zhang, D. R. Dougan, A. Broun, L. W. Tari, R. V. Swanson and J. Finn, J. Biol. Chem., 2004, 279, 8526–8529 CrossRef CAS PubMed.
- S. B. Gabelli, J. S. McLellan, A. Montalvetti, E. Oldfield, R. Docampo and L. M. Amzel, Proteins, 2006, 62, 80–88 CrossRef CAS PubMed.
- Y. L. Liu, R. Cao, Y. Wang and E. Oldfield, ACS Med. Chem. Lett., 2015, 6, 349–354 CrossRef CAS PubMed.
- B. J. Gertz, S. D. Holland, W. F. Kline, B. K. Matuszewski, A. Freeman, H. Quan, K. C. Lasseter, J. C. Mucklow and A. G. Porras, Clin. Pharmacol. Ther., 1995, 58, 288–298 CrossRef CAS PubMed.
- A. Ezra and G. Golomb, Adv. Drug Delivery Rev., 2000, 42, 175–195 CrossRef CAS PubMed.
- S. Cremers and S. Papapoulos, Bone, 2011, 49, 42–49 CrossRef CAS PubMed.
- J. R. Berenson, Semin. Oncol., 2002, 29, 12–18 CrossRef CAS PubMed.
- D. Amin, S. A. Cornell, S. K. Gustafson, S. J. Needle, J. W. Ullrich, G. E. Bilder and M. H. Perrone, J. Lipid Res., 1992, 33, 1657–1663 CAS.
- C. Y. Leung, J. Park, J. W. De Schutter, M. Sebag, A. M. Berghuis and Y. S. Tsantrizos, J. Med. Chem., 2013, 56, 7939–7950 CrossRef CAS PubMed.
- Y. S. Lin, J. Park, J. W. De Schutter, X. F. Huang, A. M. Berghuis, M. Sebag and Y. S. Tsantrizos, J. Med. Chem., 2012, 55, 3201–3215 CrossRef CAS PubMed.
- E. van Beek, E. Pieterman, L. Cohen, C. Löwik and S. Papapoulos, Biochem. Biophys. Res. Commun., 1999, 255, 491–494 CrossRef CAS PubMed.
- K. L. Kavanagh, K. Guo, J. E. Dunford, X. Wu, S. Knapp, F. H. Ebetino, M. J. Rogers, R. G. G. Russell and U. Oppermann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7829–7834 CrossRef CAS PubMed.
- J. Mao, S. Mukherjee, Y. Zhang, R. Cao, J. M. Sanders, Y. Song, Y. Zhang, G. A. Meints, Y. G. Gao, D. Mukkamala, M. P. Hudock and E. Oldfield, J. Am. Chem. Soc., 2006, 128, 14485–14497 CrossRef CAS PubMed.
- D. R. Budman and A. Calabro, Oncology, 2006, 70, 147–153 CrossRef CAS PubMed.
- A. Guenther, S. Gordon, M. Tiemann, R. Burger, F. Bakker, J. R. Green, W. Baum, A. J. Roelofs, M. J. Rogers and M. Gramatzki, Int. J. Cancer, 2010, 126, 239–246 CrossRef CAS PubMed.
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef CAS PubMed.
- T. Hiraga, P. J. Williams, A. Ueda, D. Tamura and T. Yoneda, Clin. Cancer Res., 2004, 10, 4559–4567 CrossRef CAS PubMed.
- F. Daubiné, C. Le Gall, J. Gasser, J. Green and P. Clézardin, J. Natl. Cancer Inst., 2007, 99, 322–330 CrossRef PubMed.
- A. T. Stopeck, A. Lipton, J.-J. Body, G. G. Steger, K. Tonkin, R. H. de Boer, M. Lichinitser, Y. Fujiwara, D. A. Yardley, M. Viniegra, M. Fan, Q. Jiang, R. Dansey, S. Jun and A. Braun, J. Clin. Oncol., 2010, 28, 5132–5139 CrossRef CAS PubMed.
- A. Lipton, R. L. Theriault, G. N. Hortobagyi, J. Simeone, R. D. Knight, K. Mellars, D. J. Reitsma, M. Heffernan and J. J. Seaman, Cancer, 2000, 88, 1082–1090 CrossRef CAS.
- I. J. Diel, J. J. Body, M. R. Lichinitser, E. D. Kreuser, W. Dornoff, V. A. Gorbunova, M. Budde and B. Bergström, Eur. J. Cancer, 2004, 40, 1704–1712 CrossRef CAS PubMed.
- M. K. Tsoumpra, J. R. Muniz, B. L. Barnett, A. A. Kwaasi, E. S. Pilka, K. L. Kavanagh, A. Evdokimov, R. L. Walter, F. Von Delft, F. H. Ebetino, U. Oppermann, R. G. G. Russell and J. E. Dunford, Bone, 2015, 81, 478–486 CrossRef CAS.
- R. Coleman, R. Cook, V. Hirsh, P. Major and A. Lipton, Cancer, 2011, 117, 11–23 CrossRef CAS PubMed.
- S. L. Ruggiero, Ann. N. Y. Acad. Sci., 2011, 1218, 38–46 CrossRef CAS PubMed.
- M. A. Lawson, F. H. Ebetino, A. Mazur, A. D. Chantry, J. Paton-Hough, H. R. Evans, D. Lath, M. K. Tsoumpra, M. W. Lundy, R. L. M. Dobson, M. Quijano, A. A. Kwaasi, J. E. Dunford, X. Duan, J. T. Triffitt, G. Jeans and R. G. G. Russell, J. Bone Miner. Res., 2017, 32, 1860–1869 CrossRef CAS PubMed.
- S. Rapposelli, L. Gambari, M. Digiacomo, V. Citi, G. Lisignoli, C. Manferdini, V. Calderone and F. Grassi, Sci. Rep., 2017, 7, 11940 CrossRef PubMed.
- J. M. Rondeau, F. Bitsch, E. Bourgier, M. Geiser, R. Hemmig, M. Kroemer, S. Lehmann, P. Ramage, S. Rieffel, A. Strauss, J. R. Green and W. Jahnke, ChemMedChem, 2006, 1, 267–273 CrossRef CAS PubMed.
- M. Caraglia, L. Luongo, G. Salzano, S. Zappavigna, M. Marra, F. Guida, S. Lusa, C. Giordano, V. De Novellis, F. Rossi, A. Abbruzzese Saccardi, G. De Rosa and S. Maione, Mol. Pharmaceutics, 2013, 10, 1111–1118 CrossRef CAS PubMed.
- J. Park, C. Y. Leung, A. N. Matralis, C. M. Lacbay, M. Tsakos, G. Fernandez De Troconiz, A. M. Berghuis and Y. S. Tsantrizos, J. Med. Chem., 2017, 60, 2119–2134 CrossRef CAS PubMed.
- W. Jahnke, G. Bold, A. L. Marzinzik, S. Ofner, X. Pell, S. Cotesta, E. Bourgier, S. Lehmann, C. Henry, R. Hemmig, F. Stauffer, J. C. D. Hartwieg, J. R. Green and J. M. Rondeau, Angew. Chem., Int. Ed., 2015, 54, 14575–14579 CrossRef CAS PubMed.
- I. S. Woo, S. Y. Eun, H. J. Kim, E. S. Kang, H. J. Kim, J. H. Lee, K. C. Chang, J. H. Kim, S. C. Hong and H. G. Seo, Neurosci. Lett., 2010, 474, 115–120 CrossRef CAS PubMed.
- I.-T. Wang, S.-C. Chou and Y.-C. Lin, Tumour Biol., 2014, 35, 11913–11920 CrossRef CAS PubMed.
- D. Fernández, R. Ramis, J. Ortega-Castro, R. Casasnovas, B. Vilanova and J. Frau, J. Comput.-Aided Mol. Des., 2017, 31, 675–688 CrossRef PubMed.
- C. Laezza, M. Notarnicola, M. G. Caruso, C. Messa, M. Macchia, S. Bertini, F. Minutolo, G. Portella, L. Fiorentino, S. Stingo and M. Bifulco, FASEB J., 2006, 20, 412–418 CrossRef CAS PubMed.
- M. Scrima, G. Lauro, M. Grimaldi, S. Di Marino, A. Tosco, P. Picardi, P. Gazzerro, R. Riccio, E. Novellino, M. Bifulco, G. Bifulco and A. M. D'Ursi, J. Med. Chem., 2014, 57, 7798–7803 CrossRef CAS PubMed.
- E. Ciaglia, M. Grimaldi, M. Abate, M. Scrima, M. Rodriquez, C. Laezza, R. Ranieri, S. Pisanti, P. Ciuffreda, C. Manera, P. Gazzerro, A. M. D'Ursi and M. Bifulco, Br. J. Pharmacol., 2017, 174, 2287–2301 CrossRef CAS.
- W. Jahnke, J.-M. Rondeau, S. Cotesta, A. Marzinzik, X. Pellé, M. Geiser, A. Strauss, M. Götte, F. Bitsch, R. Hemmig, C. Henry, S. Lehmann, J. F. Glickman, T. P. Roddy, S. J. Stout and J. R. Green, Nat. Chem. Biol., 2010, 6, 660–666 CrossRef CAS PubMed.
- J. Park, M. Zielinski, A. Magder, Y. S. Tsantrizos and A. M. Berghuis, Nat. Commun., 2017, 8, 14132 CrossRef CAS PubMed.
- A. L. Marzinzik, R. Amstutz, G. Bold, E. Bourgier, S. Cotesta, J. F. Glickman, M. Götte, C. Henry, S. Lehmann, J. C. D. Hartwieg, S. Ofner, X. Pellé, T. P. Roddy, J. M. Rondeau, F. Stauffer, S. J. Stout, A. Widmer, J. Zimmermann, T. Zoller and W. Jahnke, ChemMedChem, 2015, 10, 1884–1891 CrossRef CAS PubMed.
- K. L. Kavanagh, J. E. Dunford, G. Bunkoczi, R. G. G. Russell and U. Oppermann, J. Biol. Chem., 2006, 281, 22004–22012 CrossRef CAS PubMed.
- N. Ullah, M. Mansha and P. J. Casey, Curr. Cancer Drug Targets, 2016, 16, 563–571 CrossRef CAS PubMed.
- X. Zhou, J. E. Reilly, K. A. Loerch, R. J. Hohl and D. F. Wiemer, Beilstein J. Org. Chem., 2014, 10, 1645–1650 CrossRef PubMed.
- C. K. M. Chen, M. P. Hudock, Y. Zhang, R. T. Guo, R. Cao, H. N. Joo, P. H. Liang, T. P. Ko, T. H. Chang, S. C. Chang, Y. Song, J. Axelson, A. Kumar, A. H. J. Wang and E. Oldfield, J. Med. Chem., 2008, 51, 5594–5607 CrossRef CAS.
- S. Vandermoten, É. Haubruge and M. Cusson, Cell. Mol. Life Sci., 2009, 66, 3685–3695 CrossRef CAS.
- R.-T. Guo, R. Cao, P.-H. Liang, T.-P. Ko, T.-H. Chang, M. P. Hudock, W.-Y. Jeng, C. K.-M. Chen, Y. Zhang, Y. Song, C.-J. Kuo, F. Yin, E. Oldfield and A. H.-J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10022–10027 CrossRef CAS PubMed.
- B. M. Wasko, A. Dudakovic and R. J. Hohl, J. Pharmacol. Exp. Ther., 2011, 337, 540–546 CrossRef CAS PubMed.
- V. S. Wills, C. Allen, S. A. Holstein and D. F. Wiemer, ACS Med. Chem. Lett., 2015, 6, 1195–1198 CrossRef CAS.
- A. J. Wiemer, H. Tong, K. M. Swanson and R. J. Hohl, Biochem. Biophys. Res. Commun., 2007, 353, 921–925 CrossRef CAS PubMed.
- C. Allen, S. Kortagere, H. Tong, R. A. Matthiesen, J. I. Metzger, D. F. Wiemer and S. A. Holstein, Mol. Pharmacol., 2017, 91, 229–236 CrossRef CAS PubMed.
- X. Zhou, S. D. Ferree, V. S. Wills, E. J. Born, H. Tong, D. F. Wiemer and S. A. Holstein, Bioorg. Med. Chem., 2014, 22, 2791–2798 CrossRef CAS.
- R. A. Matthiesen, M. L. Varney, P. C. Xu, A. S. Rier, D. F. Wiemer and S. A. Holstein, Bioorg. Med. Chem., 2015, 6, 1195–1198 Search PubMed.
- L. Haney, M. L. Varney, Y. S. Chhonker, S. Shin, K. Mehla, A. J. Crawford, H. J. Smith, L. M. Smith, D. J. Murry, M. A. Hollingsworth and S. A. Holstein, Oncogene, 2019, 38, 5308–5320 CrossRef PubMed.
- C. M. Lacbay, D. D. Waller, J. Park, M. G. Palou, F. Vincent, X. F. Huang, V. Ta, A. M. Berghuis, M. Sebag and Y. S. Tsantrizos, J. Med. Chem., 2018, 61(15), 6904–6917 CrossRef CAS.
- S. Chen, S. Lin, S. Lin, P. Liang and J. Yang, J. Chem. Inf. Model., 2013, 53, 2299–2311 CrossRef CAS PubMed.
- D. Mukkamala, H. N. Joo, L. M. Cass, T. K. Chang and E. Oldfield, J. Med. Chem., 2008, 51, 7827–7833 CrossRef CAS.
- C. G. Ricci, Y. L. Liu, Y. Zhang, Y. Wang, W. Zhu, E. Oldfield and J. A. McCammon, Biochemistry, 2016, 55, 5180–5190 CrossRef PubMed.
- J. E. Buss and J. C. Marsters, Chem. Biol., 1995, 2, 787–791 CrossRef CAS PubMed.
- E. A. Zverina, C. L. Lamphear, E. N. Wright and C. A. Fierke, Curr. Opin. Chem. Biol., 2012, 16, 544–552 CrossRef CAS.
- A. D. Cox and C. J. Der, Curr. Opin. Pharmacol., 2002, 2, 388–393 CrossRef CAS.
- M. Schlitzer, R. Ortmann and M. Altenkämper, Drug Design of Zinc-Enzyme Inhibitors: Functional, Structural, and Disease Applications, John Wiley & Sons, 2009, pp. 813–857 Search PubMed.
- S. B. Long, P. J. Casey and L. S. Beese, Nature, 2002, 419, 645–650 CrossRef CAS.
- S. B. Long, P. J. Casey and L. S. Beese, Biochemistry, 1998, 37, 9612–9618 CrossRef CAS.
- J. L. Hougland, K. A. Hicks, H. L. Hartman, R. A. Kelly, T. J. Watt and C. A. Fierke, J. Mol. Biol., 2010, 395, 176–190 CrossRef CAS PubMed.
- J. L. Hougland, C. L. Lamphear, S. A. Scott, R. A. Gibbs and C. A. Fierke, Biochemistry, 2009, 48, 1691–1701 CrossRef CAS PubMed.
- M. Shen, P. Pan, Y. Li, D. Li, H. Yu and T. Hou, Drug Discovery Today, 2015, 20, 267–276 CrossRef CAS PubMed.
- L. Yang, W. Liu, H. Mei, Y. Zhang, X. Yu, Y. Xu, H. Li, J. Huang, Z. Zhao, J.-L. Kraus, T. J. D. Jørgensen, P. M. Alzari, S. Neimanis, M. Engel, R. M. Biondi, M. S. Kies, V. Papadimitrakopoulou, F. V. Fossella, P. Kirschmeier, W. R. Bishop, W. K. Hong and R. L. Smith, Med. Chem. Commun., 2015, 6, 671–676 RSC.
- A. Tanaka, M. O. Radwan, A. Hamasaki, A. Ejima, E. Obata, R. Koga, H. Tateishi, Y. Okamoto, M. Fujita, M. Nakao, K. Umezawa, F. Tamanoi and M. Otsuka, Bioorg. Med. Chem. Lett., 2017, 27, 3862–3866 CrossRef CAS PubMed.
- G. Homerin, E. Lipka, B. Rigo, A. Farce, J. Dubois and A. Ghinet, Org. Biomol. Chem., 2017, 15, 8110–8118 RSC.
- V. Straniero, M. Pallavicini, G. Chiodini, P. Ruggeri, L. Fumagalli, C. Bolchi, A. Corsini, N. Ferri, C. Ricci and E. Valoti, Bioorg. Med. Chem. Lett., 2014, 24, 2924–2927 CrossRef CAS PubMed.
- C.-M. Abuhaie, A. Ghinet, A. Farce, J. Dubois, B. Rigo and E. Bîcu, Bioorg. Med. Chem. Lett., 2013, 23, 5887–5892 CrossRef CAS PubMed.
- C. Dumea, D. Belei, A. Ghinet, J. Dubois, A. Farce and E. Bîcu, Bioorg. Med. Chem. Lett., 2014, 24, 5777–5781 CrossRef CAS PubMed.
- I.-M. Moise, A. Ghinet, D. Belei, J. Dubois, A. Farce and E. Bîcu, Bioorg. Med. Chem. Lett., 2016, 26, 3730–3734 CrossRef CAS PubMed.
- X. Yu, X. Zhao, L. Zhu, C. Zou, X. Liu, Z. Zhao, J. Huang and H. Li, MedChemComm, 2013, 4, 962 RSC.
- Y. Jin, L. Li, Z. Yang, M. Liu, H. Guo and W. Shen, Oncotarget, 2017, 8, 24635–24643 Search PubMed.
- J. B. Gibbs, D. L. Pompliano, S. D. Mosser, E. Rands, R. B. Lingham, S. B. Singh, E. M. Scolnick, N. E. Kohl and A. Oliff, J. Biol. Chem., 1993, 268, 7617–7620 CAS.
- F. Bellesia, S. R. Choi, F. Felluga, G. Fiscaletti, F. Ghelfi, M. C. Menziani, A. F. Parsons, C. D. Poulter, F. Roncaglia, M. Sabbatini and D. Spinelli, Bioorg. Med. Chem., 2013, 21, 348–358 CrossRef CAS PubMed.
- M. M. Cadelis, M. L. Bourguet-Kondracki, J. Dubois, A. Valentin, D. Barker and B. R. Copp, Bioorg. Med. Chem., 2016, 24, 3102–3107 CrossRef CAS PubMed.
- E. Stieglitz, A. F. Ward, R. B. Gerbing, T. A. Alonzo, R. J. Arceci, Y. L. Liu, P. D. Emanuel, B. C. Widemann, J. W. Cheng, N. Jayaprakash, F. M. Balis, R. P. Castleberry, N. J. Bunin, M. L. Loh and T. M. Cooper, Pediatr. Blood Cancer, 2015, 629–636 CrossRef CAS PubMed.
- N. M. G. M. Appels, Oncologist, 2005, 10, 565–578 CrossRef PubMed.
- S. Ghasemi, S. Ghanbarzadeh, S. Mozaffari and S. Davaran, Pharm. Ind., 2015, 77(6), 920–926 CAS.
- F. L. Zhang and P. J. Casey, Annu. Rev. Biochem., 1996, 65, 241–269 CrossRef CAS.
- J. R. Skaar, J. K. Pagan and M. Pagano, Nat. Rev. Mol. Cell Biol., 2013, 14, 369 CrossRef CAS PubMed.
- M. Watanabe, H. D. G. Fiji, L. Guo, L. Chan, S. S. Kinderman, D. J. Slamon, O. Kwon and F. Tamanoi, J. Biol. Chem., 2008, 283, 9571–9579 CrossRef CAS PubMed.
- L. N. Chan, H. D. G. Fiji, M. Watanabe, O. Kwon and F. Tamanoi, PLoS One, 2011, 6, e26135 CrossRef CAS PubMed.
- F. Tamanoi and J. Lu, in The Enzymes, 2013, vol. 34, pp. 181–200 Search PubMed.
- F. Coxon, Ł. Joachimiak, A. K. Najumudeen, G. Breen, J. Gmach, C. Oetken-Lindholm, R. Way, J. Dunford, D. Abankwa and K. M. Błazewska, Eur. J. Med. Chem., 2014, 84, 77–89 CrossRef CAS PubMed.
- C. E. McKenna, B. A. Kashemirov, K. M. Błazewska, I. Mallard-Favier, C. A. Stewart, J. Rojas, M. W. Lundy, F. H. Ebetino, R. A. Baron, J. E. Dunford, M. L. Kirsten, M. C. Seabra, J. L. Bala, M. S. Marma, M. J. Rogers and F. P. Coxon, J. Med. Chem., 2010, 53, 3454–3464 CrossRef CAS PubMed.
- R. A. Baron, R. Tavaré, A. C. Figueiredo, K. M. Błazewska, B. A. Kashemirov, C. E. McKenna, F. H. Ebetino, A. Taylor, M. J. Rogers, F. P. Coxon and M. C. Seabra, J. Biol. Chem., 2009, 284, 6861–6868 CrossRef CAS PubMed.
- K. M. Błaewska, F. Ni, R. Haiges, B. A. Kashemirov, F. P. Coxon, C. A. Stewart, R. Baron, M. J. Rogers, M. C. Seabra, F. H. Ebetino and C. E. McKenna, Eur. J. Med. Chem., 2011, 46, 4820–4826 CrossRef PubMed.
- X. Zhou, E. J. Born, C. Allen, S. A. Holstein and D. F. Wiemer, Bioorg. Med. Chem. Lett., 2015, 25, 2331–2334 CrossRef CAS PubMed.
- P. Merino, L. Maiuolo, I. Delso, V. Algieri and A. De, RSC Adv., 2017, 7, 10947–10967 RSC.
- C. Deraeve, Z. Guo, R. S. Bon, W. Blankenfeldt, R. DiLucrezia, A. Wolf, S. Menninger, E. A. Stigter, S. Wetzel, A. Choidas, K. Alexandrov, H. Waldmann, R. S. Goody and Y. W. Wu, J. Am. Chem. Soc., 2012, 134, 7384–7391 CrossRef CAS.
- J. D. Durrant, R. Cao, A. A. Gorfe, W. Zhu, J. Li, A. Sankovsky, E. Oldfield and J. A. Mccammon, Chem. Biol. Drug Des., 2011, 78, 323–332 CrossRef CAS.
- Y. Zhang, R. Cao, F. Yin, M. P. Hudock, R. T. Guo, K. Krysiak, S. Mukherjee, Y. G. Gao, H. Robinson, Y. Song, J. H. No, K. Bergan, A. Leon, L. Cass, A. Goddard, T. K. Chang, F. Y. Lin, E. Van Beek, S. Papapoulos, A. H. J. Wang, T. Kubo, M. Ochi, D. Mukkamala and E. Oldfield, J. Am. Chem. Soc., 2009, 131, 5153–5162 CrossRef CAS PubMed.
- Y. Xia, Y. L. Liu, Y. Xie, W. Zhu, F. Guerra, S. Shen, N. Yeddula, W. Fischer, W. Low, X. Zhou, Y. Zhang, E. Oldfield and I. M. Verma, Sci. Transl. Med., 2014, 6, 263ra161 CrossRef PubMed.
- X. Zhou, Y. Gu, H. Xiao, N. Kang, Y. Xie, G. Zhang, Y. Shi, X. Hu, E. Oldfield, X. Zhang and Y. Zhang, Front. Immunol., 2017, 8, 1381 CrossRef PubMed.
- K. A. H. Chehade, D. A. Andres, H. Morimoto and H. P. Spielmann, J. Org. Chem., 2000, 65, 3027–3033 CrossRef CAS PubMed.
- J. M. Troutman, K. A. H. Chehade, K. Kiegiel, D. A. Andres and H. P. Spielmann, Bioorg. Med. Chem. Lett., 2004, 14, 4979–4982 CrossRef CAS PubMed.
- M. Chen, T. Knifley, T. Subramanian, H. P. Spielmann and K. L. O'Connor, PLoS One, 2014, 9, e89892 CrossRef PubMed.
- S. Machida, N. Kato, K. Harada and J. Ohkanda, J. Am. Chem. Soc., 2011, 133, 958–963 CrossRef CAS PubMed.
- M. Tsubamoto, T. K. Le, M. Li, T. Watanabe, C. Matsumi, P. Parvatkar, H. Fujii, N. Kato, J. Sun and J. Ohkanda, Chem. – Eur. J., 2019, 25, 13531–13536 CrossRef CAS PubMed.
- Y. Qiao, J. Gao, Y. Qiu, L. Wu, F. Guo, K. Kam-Wing Lo and D. Li, Eur. J. Med. Chem., 2011, 46, 2264–2273 CrossRef CAS PubMed.
- M. Avadisian, S. Fletcher, B. Liu, W. Zhao, P. Yue, D. Badali, W. Xu, A. D. Schimmer, J. Turkson, C. C. Gradinaru and P. T. Gunning, Angew. Chem., Int. Ed., 2011, 50, 6248–6253 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341 CrossRef CAS PubMed.
- M. Avadisian, P. T. Gunning, D. Ludwig, C. Ungermann, R. S. Goody, A. Rak, M. Geyer, N. I. Tarasova, J. S. McMurray, E. Chen, A. Moscona, M. Porotto, E. L. Nelson, P. R. Sista and A. Dusek, Mol. BioSyst., 2013, 9, 2179 RSC.
- M. Schürmann, P. Janning, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2016, 23, 435–441 CrossRef PubMed.
- E. M. Storck, J. Morales-Sanfrutos, R. A. Serwa, N. Panyain, T. Lanyon-Hogg, T. Tolmachova, L. N. Ventimiglia, J. Martin-Serrano, M. C. Seabra, B. Wojciak-Stothard and E. W. Tate, Nat. Chem., 2019, 11, 552–561 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |