
Insights into structural and physicochemical properties required for β-hematin inhibition of privileged triarylimidazoles
Clinton G. L.
Veale
 *a,
Janeeka
Jayram
a,
Shivani
Naidoo
a,
Dustin
Laming
b,
Tarryn
Swart
b,
Tania
Olivier
c,
Matthew P.
Akerman
a,
Katherine A.
de Villiers
c,
Heinrich C.
Hoppe
b and
Vineet
Jeena
*a
*a,
Janeeka
Jayram
a,
Shivani
Naidoo
a,
Dustin
Laming
b,
Tarryn
Swart
b,
Tania
Olivier
c,
Matthew P.
Akerman
a,
Katherine A.
de Villiers
c,
Heinrich C.
Hoppe
b and
Vineet
Jeena
*a
aSchool of Chemistry and Physics, Pietermaritzburg Campus, University of KwaZulu-Natal, Private Bag X01, Scottsville, 3209, South Africa
bDepartment of Biochemistry and Microbiology, Rhodes University, Grahamstown, 6140, South Africa
cDepartment of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1, Matieland, 7602, South Africa. E-mail: VealeC@ukzn.ac.za; JeenaV1@ukzn.ac.za
First published on 16th December 2019
Abstract
In this study, we investigated a series of triarylimidazoles, in an effort to elucidate critical SAR information pertaining to their anti-plasmodial and β-hematin inhibitory activity. Our results showed that in addition to the positional effects of ring substitution, subtle changes to lipophilicity and imidazole ionisability were important factors in SAR interpretation. Finally, in silico adsorption analysis indicated that these compounds exert their effect by inhibiting β-hematin crystal growth at the fast growing 001 face.
Introduction
In 2017, malarial infections from species of the Plasmodium genus affected in excess of 200 million people living in 90 countries situated in tropical regions of the world, resulting in approximately 435![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.1 While multiple orthogonal strategies have considerably reduced the incidence of malarial transmission,2 this progress is continuously undermined by the pernicious emergence of resistance to front-line chemotherapeutics.3 Efforts to combat this problematic trend have tended to focus either on the identification, validation and subsequent inhibition of new biological pathways, which would circumvent resistance pathways, or through phenotypic screening programmes which aim to identify unique anti-plasmodial chemotypes, which possibly inhibit new valid biological targets.4,5
000 deaths.1 While multiple orthogonal strategies have considerably reduced the incidence of malarial transmission,2 this progress is continuously undermined by the pernicious emergence of resistance to front-line chemotherapeutics.3 Efforts to combat this problematic trend have tended to focus either on the identification, validation and subsequent inhibition of new biological pathways, which would circumvent resistance pathways, or through phenotypic screening programmes which aim to identify unique anti-plasmodial chemotypes, which possibly inhibit new valid biological targets.4,5
While widespread resistance has rendered chloroquine obsolete in modern treatment regimens, it remains a compound of interest in anti-malarial medicinal chemistry research. This is due in-part, to the discovery that its mechanism of action involves the inhibition of insoluble hemozoin crystal formation within the plasmodial digestive vacuole.6 Hemozoin formation is an essential detoxification pathway which sequesters liberated heme following haemoglobin digestion.7 Critically, this pathway is non-enzymatic, and is therefore not subject to the same selective pressures of proteomic targets. Resistance is more commonly as a result of insufficient drug accumulation within the digestive vacuole, following mutations in transmembrane transport proteins;8–10 therefore this target remains relevant in contemporary anti-malarial drug discovery.

Through the development of a β-hematin model of hemozoin formation, there has been renewed interest in exploiting the quinoline scaffold for the development of hemozoin inhibitors,11–13 while several recent studies have sought to identify new chemotypes which target this pathway.14–16 Two separate target based screens of the Vanderbilt University Institute of Chemical Biology Library,17 and the MMV Malaria Box,18 identified numerous scaffolds, including several triarylimidazoles (1–9), whose encouraging β-hematin inhibition activity was translated into phenotypic inhibition of P. falciparum cultures. Furthermore, the variety of substituents and substitution patterns present on these compounds, including non-phenyl derivatives, suggests a wide scope for structure activity relationship (SAR) optimisation. Consequently, Wicht et al. sought to expand the SAR surrounding the seemingly privileged triarylimidazole scaffold.19 Due in part to the limited variety of starting materials to suit their synthetic procedure, they focussed mainly on the substitution pattern on the A-ring. Here they identified a small cohort of 13 compounds, 9 of which (10–18) possessed β-hematin inhibitory activity, superior to that of chloroquine, which in most instances translated into low μM anti-plasmodial activity in vitro. While limited, the SAR data from this study showed that the absence of phenyl rings or a lack of phenyl substitution, rendered the compounds practically inactive, while incorporation of multiple oxygen containing substituents on the A-ring, such as a 1,3-dimethoxy pattern (11), was beneficial for β-hematin inhibition. However, a β-hematin SAR pattern was not categorically discernible, with singly substituted hydroxyl- (12), nitro- (13) and amino- (14) containing compounds displaying similar activity. Noticeable increases in in vitro anti-plasmodial activity were observed upon the inclusion of an extra methoxy (15) or hydroxy (16) substituent on the A-ring, as well as methoxylation (17) or halogenation (18) on the B/C-ring.19 In a series of recent studies, we have sought to develop methodology toward the synthesis of triarylimidazoles which overcomes the limitations associated with starting material accessibility, thereby allowing us to explore a variety of ring substitution patterns on the A-, B- and C-rings.20–22 In so doing, we have produced a library of 27 variably decorated imidazoles (19–45), which we subjected to anti-plasmodial biological assessment in order to probe aspects pertaining to their SAR. Accordingly, we present our results in which we have identified several compounds with anti-plasmodial and β-hematin inhibitory activity, comparable to that obtained by Wicht et al.19 Furthermore, our results have allowed us to uncover certain physicochemical characteristics, which may be beneficial for further optimisation.
Results and discussion
As a preliminary insight into anti-plasmodial biological activity, we conducted a single concentration screen at 20 μM (Fig. 1). Compounds 19–29, which all feature a single substituent on the A-ring, as well as compounds 30 and 31, whose single substituent was on the B/C-ring, were generally inactive. However, of the para-substituted cohort (19–22), a methoxy substituent (19) was seemingly favoured. However, while an ortho-methoxy substituent (26) was inactive, the remaining ortho-substituted compounds (27–29) were marginally more active than their corresponding meta (23, 25) or para (20) substituted analogues. Incorporation of two para substituted phenyl rings (32–37) generally resulted in increased single concentration activity, with the exception of compounds 34 and 37, both of which featured either two chloride or bromide substituents respectively. While limited insight could be gleaned from analogues 38 and 39, the activity of di-brominated 40, particularly when compared to 34 and 37, indicates that rings B and C are regions where further SAR exploration could occur, which is a trend mirrored in previously identified β-hematin inhibitors 1–9. Replacement of the A-ring with a naphthalene (41), a furanyl (42) and non-aromatic cyclohexyl (43) containing analogues, were inactive. Incorporation of a 3-indolyl substituent (44) resulted in slightly improved activity, while the 3-pentanyl analogue (45) displayed particularly encouraging activity. Importantly, none of the compounds assayed here displayed significant mammalian cytotoxicity against a HeLa cell line at 20 μM (Fig. 1). Compounds 32, 33, 35, 36, 40 and 45, which inhibited P. falciparum growth below 10% in the single concentration screen, were taken forward for IC50 analysis (Table 1), and were all found to have moderate activity in line with that previously reported for triarylimidazoles.19
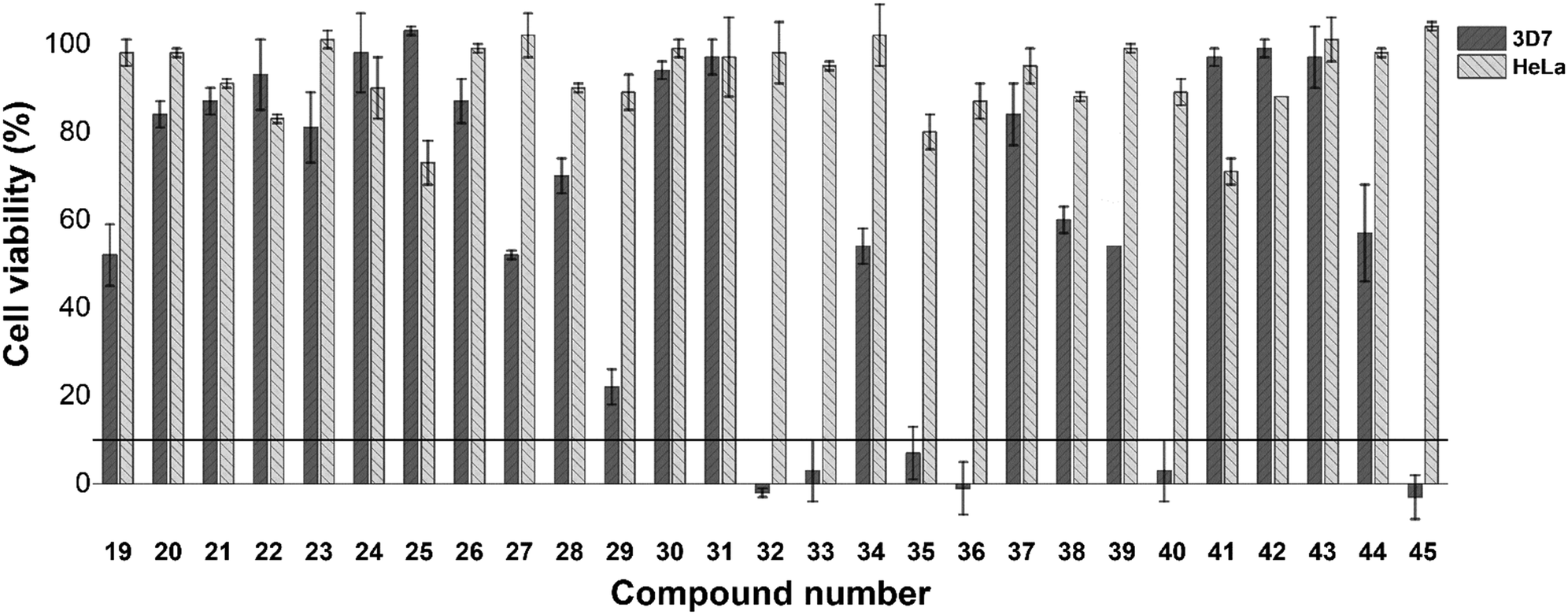 | ||
| Fig. 1 Cell viability (%) of P. falciparum (3D7 strain) and HeLa cells after incubation with compounds 19–45 at 20 μM for 48 hours. IC50 assessment was conducted for compounds, which inhibited cell viability below the 10% threshold against P. falciparum 3D7. In general, these compounds did not display significant HeLa cytotoxicity. | ||
| Compound no. | IC50 (μM) 3D7 | IC50 (μM) βH |
|---|---|---|
| IC50 values are the mean of experiments conducted in technical triplicate.a ND = not determined.b NA = not active. | ||
| 19 | NDa | 48.1 ± 11 |
| 20 | ND | 156 ± 14 |
| 21 | ND | NAb |
| 22 | ND | 489 ± 26 |
| 23 | ND | NA |
| 24 | ND | NA |
| 25 | ND | NA |
| 26 | ND | NA |
| 27 | ND | >500 |
| 28 | ND | >500 |
| 29 | ND | NA |
| 30 | ND | >500 |
| 31 | ND | 294 ± 32 |
| 32 | 11 | 20.5 ± 1.5 |
| 33 | 6.5 | 28.0 ± 4.1 |
| 34 | ND | 39.0 ± 1.5 |
| 35 | 8.0 | 28.8 ± 4.1 |
| 36 | 1.5 | 20.3 ± 4.1 |
| 37 | ND | 54.2 ± 11 |
| 38 | ND | >500 |
| 39 | ND | >500 |
| 40 | 13 | NA |
| 41 | ND | NA |
| 42 | ND | 401 ± 64 |
| 43 | ND | >500 |
| 44 | ND | 72.8 ± 6 |
| 45 | 11 | NA |
| Chloroquine | 0.010 | 16.9 ± 0.6 |
While the respective biological activities were not substantially different, the reduced activity of compound 40, which did not feature an A-ring substituent, and that of 45 which features a 3-pentanyl chain in place of the A-ring, strongly suggest that a para-substituted aromatic A-ring is beneficial to activity. While the most active compound in this assay (36) featured a para-methoxy substituent, comparison of 32, 33 and 35 indicate that a fluorine substituent may be tolerated in place of a methoxy substituent.
Following phenotypic biological assessment, all 28 compounds were investigated for their β-hematin inhibitory capacity (Table 1). Using the NP-40 detergent assay,23 nine compounds (21, 23–26, 29, 40, 41, 45) were found to be inactive, while a further seven compounds (27, 28, 30, 38, 39, 40, 43) inhibited β-hematin formation at IC50 values in excess of 500 μM, and were thus considered practically inactive. With the exception of compound 21, the A-ring of all of these compounds featured an unsubstituted, ortho or meta substituted phenyl ring, or alternatively a naphthalene (41), cylcohexanyl (43) or 3-pentanyl (45) substituent, and not a para substituent. The moderate anti-plasmodial activity of compounds 40 and 45, coupled to the absence of β-hematin inhibitory activity suggest that these compounds possibly exert their affect via an alternative biological pathway.
Compounds 20, 22, 31 and 42 were considered weak inhibitors of β-hematin formation (IC50 > 100 μM), while 19, 34, 37 and 44 were identified as comparatively moderate β-hematin inhibitors (IC50 > 30 μM), but encouragingly were within the range of clinically used hemozoin inhibiting antimalarial agents such as quinine.24 Similarly to the inactive chloride analogue 21, compound 22 features only a bromine on the A-ring, while 31 features only a B-ring bromide, suggesting that this structural motif in isolation is not sufficient for activity. In comparison to 21 and 22, the introduction of a methyl substituent (20) resulted in substantially improved activity, as in turn did a methoxy substituent (19) in the same position. Di-halogenated compounds 34 and 37, which combine the A/B-ring substitution patterns of 21, 22 and 31, showed improved activity over these mono-substituted analogues. While furanyl 43 was weakly active, the indole containing 44 displayed encouraging albeit moderate activity. Significantly, indole containing compound 7 has previously been identified as a potent β-hematin inhibitor,19 and these are the only current examples of activity being maintained when deviating from a substituted phenyl A-ring. Importantly, compounds 19, 20, 22, 31, 34, 37, 42 and 44 displayed no significant anti-plasmodial activity, which may be due to their weak to moderate β-hematin inhibitory activity as well as possible absorption and accumulation issues.
Compounds 32, 33, 35 and 36 were all found to be good inhibitors of β-hematin formation (IC50 < 30 μM). As was observed in the mono-substituted analogue (19), A-ring methoxylation imparts the greatest inhibitory activity amongst this library, with compounds 32 and 36 in particular, displaying activity comparable to that of chloroquine.
Mechanistically, there is still debate as to whether small molecules inhibit β-hematin crystallisation through direct complexation with heme subunits,25,26 or interaction with the crystal surface, to obstruct further growth.27 The β-hematin surface features four faces (001, 011, 010, 100), upon which crystal growth can propagate. The molecular topography of the fastest growing 001 face has led some to consider it the ideal region for inhibitor adsorption,28 while the slow growing and relatively featureless 100 face has also been proposed as a feasible region for inhibitor adsorption.29
Using a previously reported method,16 we conducted a series of in silico calculations to simulate the adsorption of compounds 19, 32, 36 and 44 in their unprotonated form onto the four faces of β-hematin. The conditions of the in vitro β-hematin assay are buffered to mimic the acidic pH (4.8) of the plasmodial digestive. Under these conditions, the weakly basic imidazole will exist in both its neutral and protonated states. Unfortunately, protonation of the imidazole in silico was found to be energetically unfavourable under our simulation conditions, resulting in pronounced molecular distortions, making analysis of the monoprotonated form unviable. The lowest adsorption energy for unprotonated 19, 32, 36 and 44 was calculated for interactions with the 001 face (Fig. 2).
 | ||
| Fig. 2 Plot of β-hematin IC50 values for compounds 19, 32, 36 and 44vs. calculated adsorption energy at the four faces of the β-hematin crystal. A statistically significant, linear correlation (r2 = 0.995, P-stat = 0.0023 ≪ 0.05) in the case of the 001 face indicates that this is the likely site of adsorption. | ||
However, a statistically-significant linear correlation between βH IC50 values and calculated adsorption energy was only observed for interactions at the 001 face, indicating that this is the likely surface for β-hematin inhibition. The corrugated 001 face features a series of grooves along the a-axis, where the pyrrole rings and alkyl side chains of the heme subunits forms a series of π-stacking regions and hydrophobic pockets, in which our compounds were predicted to adsorb. Structurally related compounds 19, 32 and 36 all orientated within these grooves in a similar manner, where the methoxylated A-ring, imidazole and B-ring positioned toward the crystal face, with the C-ring orientated out of the pocket (Fig. 3).
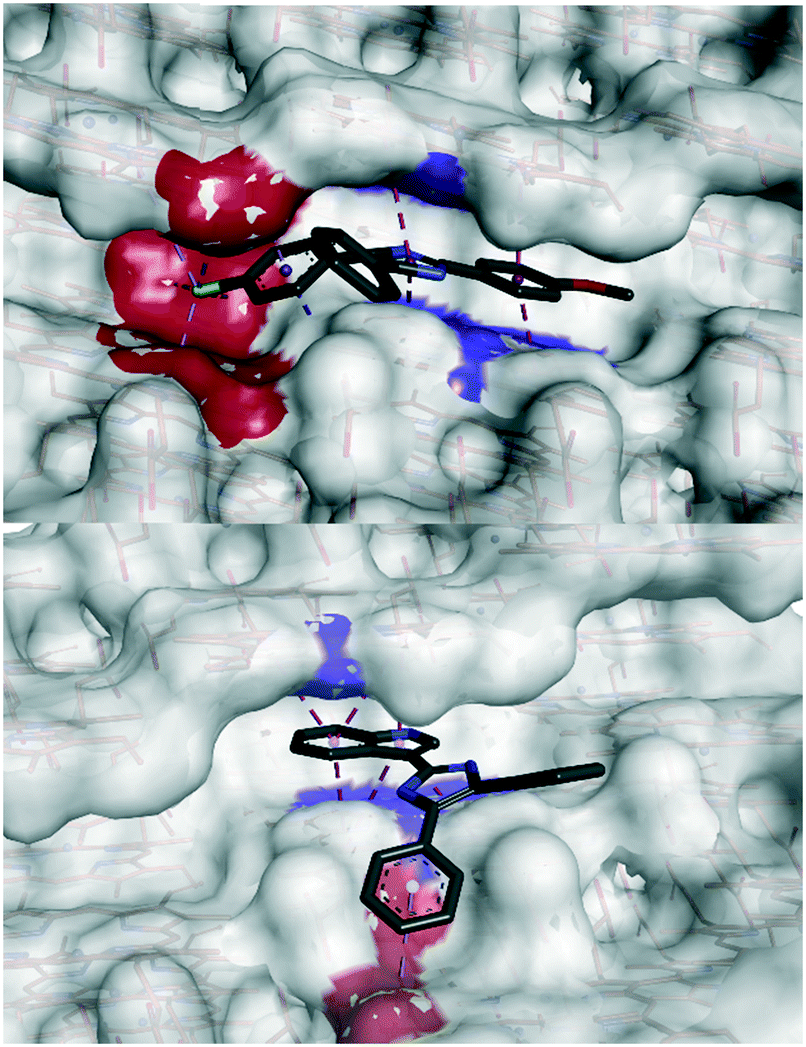 | ||
| Fig. 3 Docked poses of compounds 32 (top) and 44 (bottom) in the 001 face groove. Interacting aromatic residues are highlighted in blue and hydrophobic residues highlighted in red. The A-ring, imidazole and B-ring of compound 32 orientate toward the crystal face, forming π-stacking and hydrophobic interactions. Compound 44 occupies a slightly different region of the groove, with the indole ring intercalating inside the groove, and the B-ring interacting with the hydrophobic region outside the corrugated groove. | ||
The A-ring and imidazole were predicted to intercalate between two parallel heme rings via corresponding π-stacking interactions, while the B-ring orientated toward a hydrophobic pocket. This hydrophobic interaction was enhanced by the presence of a halogen. Compound 44 however, was predicted to occupy a different pose with the indole ring π-stacking between two heme rings, and the B-ring interacting hydrophobically with the step region outside of the groove. The capacity to occupy these different binding regions on the 001 surface offers interesting potential for elaboration, of this class of compounds, possibly analogous to that of compound 9.
As mentioned previously, SAR analysis indicated that para-substituted A- and B-rings were required for β-hematin inhibitory activity. The difference in activity profiles between dihalogenated compounds 34 and 37, to that of methoxylated compounds 32 and 36, indicated that not only does the position of a given substituent govern activity, but also the physicochemical properties imparted by said substituent. This idea has previously been explored by Wicht et al. who noted a linear correlation between biological activity and the number of oxygen-containing substituents.19 L'abbate et al. noted that the properties required for potent β-hematin inhibition need to be balanced with those required for cellular penetration such as lipophilicity.16
A plot of pIC50 (βH) vs. clogP (Fig. 4) revealed three distinct clusters of compounds. The first was the furanyl compound 42 in isolation (purple), the second series (black) were all compounds, which featured a single ring substituent in the para-position, with the exception of indole containing 44. The final cluster contained compounds with dual ring substituents with every compound in this cluster featuring an A-ring substituent in the para position. Compounds in this cluster for which phenotypic IC50 values were obtained are denoted in red, while compounds 34 and 37 (blue) were not sufficiently active in the single concentration screen to warrant anti-plasmodial dose-dependent analysis.
 | ||
| Fig. 4 Plot of pIC50 in the β-hematin assay vs. clogP. Compounds highlighted in black feature a single ring substituent, while those in blue (no anti-plasmodial IC50) and red (anti-plasmodial IC50) contain two substituents, with at least one appearing in the para position on the A-ring. Orange rings represent those compounds with methoxy substituents. Green numbers denote predicted substituent induced Δ-partial charge on the basic imidazole nitrogen comparted to unsubstituted triarylimidazole. | ||
Compounds highlighted with an orange ring are those compounds, which feature a methoxy substituent, which were the most active in their respective clusters. While the SAR results indicate that at least two substituents are required for reasonable biological activity, the resultant effect is typically an increase in clogP. However, within each cluster, the general trend indicates that β-hematin activity is improved with a reduction in clogP.
As mentioned previously, the acidic conditions of the β-hematin assay will increase the proportion of protonated imidazole species, the extent of which will be influenced by various ring substituents. The NBO partial charge on the protonatable imidazole nitrogen was calculated for each compound and subtracted from the value obtained for the unsubstituted triarylimidazole to derive the Δ-partial charge (Fig. 4). This value should be indicative of the ease of protonation relative to the unsubstituted species. An increase in magnitude of the Δ-partial charge shows that the compound of interest carries a more negative partial charge than the reference compound rendering protonation more favourable in acidic media.
These data indicate that the most active compounds in each cluster are also those, which carry the most negative partial charge, i.e. are most likely to be protonated at experimental pH. Ease of protonation of hemozoin inhibitors is commonly cited as an important factor in pH trapping within the digestive vacuole.30 Although this study does not show a definitive trend between ease of protonation and activity (since the β-hematin activity in these assays does not require permeation and trapping within the digestive vacuole) this observation does suggest that ionisability, in addition to other physical and structural properties, is a factor to consider in SAR optimisations. This is mainly with respect to direct heme interaction and not just digestive vacuole accumulation.
Conclusions
While the specific mechanism of how β-hematin inhibitors exert their effect is not yet resolved,31 the development of anti-malarial hemozoin inhibitors requires a delicate balance of properties required for cellular penetration, digestive vacuole accumulation and finally heme association. In this study, we describe the anti-plasmodial and β-hematin inhibitory activity of an expanded series of triarylimidazoles. Our results provide new insight into the structural features of triarylimidazoles required for phenotypic and β-hematin inhibitory activity. In silico calculations suggest that these compounds exert their effect at least in part, through adsorption to the fast growing 001 face of β-hematin. Furthermore, SAR analysis revealed that in addition to substituent position, substituent effects on lipophilicity and imidazole ionisability, play a subtle SAR role, and are worth considering in the development of potent β-hematin inhibitors, with corresponding anti-plasmodial activity.Experimental
Synthesis
The triarylimidazoles discussed have been reported previously using one of the methods described below. All compounds have been characterized using standard spectroscopic techniques and copies of the spectra are available in these manuscripts. Purity of all compounds were determined ≥95% by LC-MS.20–22Method 1
α-Methylene ketone (0.5 mmol), aldehyde (0.5 mmol), ammonium acetate (5.0 mmol) and copper chloride dihydrate (5 mol%) were mixed in a round bottom flask using dimethylformamide (1 mL) as a solvent. An oxygen balloon was attached and the resultant mixture heated at 50 °C for 24 hours. Thereafter, the mixture was cooled and ice water was added to form a precipitate, which was filtered and dried in an oven for 2 hours. The crude product was recrystallized to produce spectroscopically pure imidazoles.Method 2
α-Methylene ketone (1.0 mmol), molecular iodine (0.5 mmol) and DMSO (1 mL) were added to a round bottom flask and the mixture was heated to 100 °C for 2 hours. Thereafter, aldehyde (1.0 mmol), ammonium acetate (10 mmol) and ethanol (2 mL) were added and the mixture heated for an additional 2 hours. After cooling, a mixture of Na2S2O3/ice water was added to produce a precipitate, which was filtered, dried and recrystallized to produce spectroscopically pure imidazole.Method 3
A mixture of internal alkyne (0.5 mmol), molecular iodine (1.5 equiv.) and DMSO (1 mL) were heated at 130 °C for 1 hour. Thereafter, aldehyde (0.5 mmol), ammonium acetate (5.0 mmol) and ethanol (2 mL) were added and the mixture heated at 100 °C for 2 hours. Thereafter, a solution of 1% Na2S2O3 was added dropwise to produce a precipitate, which was filtered, dried and recrystallized to produce spectroscopically pure imidazole.Biological assessment
Single concentration and dose dependant anti-plasmodial assays against the 3D7 strain of P. falciparum, as well as cytotoxicity screening against HeLa cells, were conducted using previously reported methods.3 The β-hematin activity was assessed using a previously reported protocol.32In silico calculations
β-Hematin adsorption was calculated with the assistance of BIOVIA Materials Studio33 using a previously reported method.16 Compound structures were optimised and the NBO charges calculated using DFT methods at the B3LYP/6-311G(d,p) level of theory. A lack of negative frequency eigenvalues suggests the compounds are true minima on the global potential energy surface. Simulations were run using Gaussian 09W34 and input and output files prepared/analysed using GaussView 5.0.35Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of South Africa (NRF) CSUR under Grant No 116305 and Thuthuka under Grant No. - TTK180410319052. JJ, SN and TO were supported by postgraduate bursaries from the NRF. Anti-plasmodial assays (HCH and DL) were supported by the SA Medical Research Council. Centre for High Performance Computing (CHPC) is gratefully acknowledged for computational resources and Materials Studio license availability.References
- A. C. Cabrera, Drug Discovery Today, 2019, 24, 1304–1310 CrossRef PubMed.
- T. A. Tizifa, A. N. Kabaghe, R. S. McCann, H. van den Berg, M. Van Vugt and K. S. Phiri, Curr. Trop. Med. Rep., 2018, 5, 41–50 CrossRef PubMed.
- M. J. Lunga, R. L. Chisango, C. Weyers, M. Isaacs, D. Taylor, A. L. Edkins, S. D. Khanye, H. C. Hoppe and C. G. L. Veale, ChemMedChem, 2018, 13, 1353–1362 CrossRef CAS PubMed.
- M. Njoroge, N. M. Njuguna, P. Mutai, D. S. B. Ongarora, P. W. Smith and K. Chibale, Chem. Rev., 2014, 114, 11138–11163 CrossRef CAS PubMed.
- C. G. L. Veale, ChemMedChem, 2019, 14, 386–453 CrossRef CAS.
- T. J. Egan and H. M. Marques, in Coordination Chemistry Reviews, 1999, vol. 190–192, pp. 493–517 Search PubMed.
- J. M. Combrinck, T. E. Mabotha, K. K. Ncokazi, M. A. Ambele, D. Taylor, P. J. Smith, H. C. Hoppe and T. J. Egan, ACS Chem. Biol., 2013, 8, 133–137 CrossRef CAS PubMed.
- D. A. Fidock, T. Nomura, A. K. Talley, R. A. Cooper, S. M. Dzekunov, M. T. Ferdig, L. M. B. Ursos, A. Bir Singh Sidhu, B. Naudé, K. W. Deitsch, X. Z. Su, J. C. Wootton, P. D. Roepe and T. E. Wellems, Mol. Cell, 2000, 6, 861–871 CrossRef CAS PubMed.
- A. F. Cowman, D. Galatis and J. K. Thompson, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1143–1147 Search PubMed.
- S. R. Hawley, P. G. Bray, M. Mungthin, J. D. Atkinson, P. M. O'Neill and S. A. Ward, Antimicrob. Agents Chemother., 1998, 42, 682–686 CrossRef CAS PubMed.
- D. J. Sullivan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7483–7485 CrossRef CAS PubMed.
- M. C. Joshi, K. J. Wicht, D. Taylor, R. Hunter, P. J. Smith and T. J. Egan, Eur. J. Med. Chem., 2013, 69, 338–347 CrossRef CAS PubMed.
- W. J. Lu, K. J. Wicht, L. Wang, K. Imai, Z. W. Mei, M. Kaiser, I. E. T. El Sayed, T. J. Egan and T. Inokuchi, Eur. J. Med. Chem., 2013, 64, 498–511 CrossRef CAS PubMed.
- K. J. Wicht, J. M. Combrinck, P. J. Smith, R. Hunter and T. J. Egan, J. Med. Chem., 2016, 59, 6512–6530 CrossRef CAS PubMed.
- R. M. Young, M. R. Adendorff, A. D. Wright and M. T. Davies-Coleman, Eur. J. Med. Chem., 2015, 93, 373–380 CrossRef CAS PubMed.
- F. P. L'abbate, R. Müller, R. Openshaw, J. M. Combrinck, K. A. de Villiers, R. Hunter and T. J. Egan, Eur. J. Med. Chem., 2018, 159, 243–254 CrossRef PubMed.
- R. D. Sandlin, K. Y. Fong, K. J. Wicht, H. M. Carrell, T. J. Egan and D. W. Wright, Int. J. Parasitol.: Drugs Drug Resist., 2014, 4, 316–325 Search PubMed.
- K. Y. Fong, R. D. Sandlin and D. W. Wright, Int. J. Parasitol.: Drugs Drug Resist., 2015, 5, 84–91 Search PubMed.
- K. J. Wicht, J. M. Combrinck, P. J. Smith, R. Hunter and T. J. Egan, ACS Med. Chem. Lett., 2017, 8, 201–205 CrossRef CAS PubMed.
- J. Jayram and V. Jeena, Green Chem., 2017, 19, 5841–5845 RSC.
- J. Jayram and V. Jeena, RSC Adv., 2018, 8, 37557–37563 RSC.
- S. Naidoo and V. Jeena, Eur. J. Org. Chem., 2019, 2019, 1107–1113 CrossRef CAS.
- R. D. Sandlin, M. D. Carter, P. J. Lee, J. M. Auschwitz, S. E. Leed, J. D. Johnson and D. W. Wright, Antimicrob. Agents Chemother., 2011, 55, 3363–3369 CrossRef CAS PubMed.
- J. G. Woodland, R. Hunter, P. J. Smith and T. J. Egan, Org. Biomol. Chem., 2017, 15, 589–597 RSC.
- D. J. Sullivan, I. Y. Gluzman, D. G. Russell and D. E. Goldberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11865–11870 CrossRef CAS PubMed.
- T. J. Egan, J. Inorg. Biochem., 2006, 100, 916–926 CrossRef CAS.
- S. Pagola, P. W. Stephens, D. S. Bohle, A. D. Kosar and S. K. Madsen, Nature, 2000, 404, 307–310 CrossRef CAS.
- R. Buller, M. L. Peterson, Ö. Almarsson and L. Leiserowitz, Cryst. Growth Des., 2002, 2, 553–562 CrossRef CAS.
- K. N. Olafson, M. A. Ketchum, J. D. Rimer and P. G. Vekilov, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4946–4951 CrossRef CAS.
- C. H. Kaschula, T. J. Egan, R. Hunter, N. Basilico, S. Parapini, D. Taramelli, E. Pasini and D. Monti, J. Med. Chem., 2002, 45, 3531–3539 CrossRef CAS.
- S. M. Fitzroy, J. Gildenhuys, T. Olivier, N. O. Tshililo, D. Kuter and K. A. De Villiers, Langmuir, 2017, 33, 7529–7537 CrossRef CAS PubMed.
- M. D. Carter, V. V. Phelan, R. D. Sandlin, B. O. Bachmann and D. W. Wright, Comb. Chem. High Throughput Screening, 2010, 13, 285–292 CrossRef CAS PubMed.
- Accelrys Inc., BIOVIA Materials Studio.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, Semichem Inc., Shawnee Mission, Semichem Inc., KS, 2009 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |