
Meridianin D analogues possess antibiofilm activity against Mycobacterium smegmatis†
Sara M.
Brackett‡
a,
Karlie E.
Cox‡
a,
Samantha L.
Barlock
a,
William M.
Huggins
b,
David F.
Ackart
c,
Randall J.
Bassaraba
c,
Roberta J.
Melander
a and
Christian
Melander
 *a
*a
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: cmelande@nd.edu
bDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695, USA
cDepartment of Microbiology, Immunology, and Pathology, Colorado State University, Fort Collins, CO 80523, USA
First published on 16th December 2019
Abstract
The formation of bacterial biofilms significantly decreases the efficacy of antibiotic treatments. Herein, we've investigated the antibiofilm properties of the natural product meridianin D and a library of analogues against Mycobacterium smegmatis. As a result, we discovered several analogues that both inhibit and disperse M. smegmatis biofilms.
Introduction
The continued emergence of antibiotic resistance is one of the greatest global health threats.1 Multidrug resistant bacterial infections are increasing in frequency, and require the use of more aggressive antibiotic therapies to eradicate.2 While the continued development of novel antibiotics is crucial, alternative strategies are also needed, such as the development of adjuvants that target bacterial pathways responsible for antibiotic tolerance or resistance.3 One way in which bacteria avoid the effects of antibiotic treatment, is the formation of biofilms.4 Biofilms are an assembly of surface associated bacteria that are encased in an extracellular polymeric substance (EPS).5 Bacteria in this state have an increased ability to tolerate antibiotic treatment due to several factors including the protection provided by the EPS, and increased gene transfer within the biofilm.6 Biofilm-associated bacteria are upwards of 1000-fold more tolerant to antibiotics when compared to planktonic (free-floating) bacteria, which often renders antibiotic treatment ineffective.4Biofilms are believed to play an important role in the persistence of Mycobacterium tuberculosis infections.7M. tuberculosis is the causative agent of tuberculosis (TB) and is the leading cause of death from a single infectious agent.8 TB is primarily an infection of the lungs with symptoms including chest pain, cough, fatigue, weight loss, and fever.9 In 2016, there were an estimated 1.6 million TB-related deaths worldwide, and it is estimated that one-third of the global population has been exposed to M. tuberculosis.8 Current treatment for TB lasts a minimum of six months and involves antibiotic combination therapy with isoniazid, rifampin, and pyrazinamide for two months followed by isoniazid and rifampin for four months to eliminate persistent TB reservoirs and prevent disease relapse. Therapy for the treatment of multi-drug resistant TB is even more extensive, lasting upwards of two years. This extended antibiotic therapy regimen often leads to patient non-compliance because of drug toxicity and drug availability.10 It has been shown that M. tuberculosis forms biofilm-like entities on cellular debris in vitro, which are more tolerant to treatment with isoniazid.11 It has been hypothesized that such communities in vivo are a source of persistent TB reservoirs.7 Small molecules that can target these biofilm-like communities of mycobacteria could potentially be used as adjuvants in combination with conventional TB antibiotics to decrease treatment time, leading to increased patient compliance and limiting the development of resistant TB.
Natural products have historically been rich sources of molecules with diverse biological activities. The meridianin compounds are an example of structurally related marine natural products with a wide array of biological activities including antitumor activity, kinase inhibition, and antimalarial activity (Fig. 1).12–14
 | ||
| Fig. 1 Structure of lead compound meridianin D and halogenated phenazine compounds. | ||
Select meridianin derivatives have also been reported to exhibit antimicrobial activity against M. tuberculosis.15 Recently, our group has established that compounds from our library of meridianin D analogues possess antibiofilm activity against methicillin-resistant Staphylococcus aureus (MRSA).16 Also, Garrison et al. noted that halogenated phenazines (Fig. 1) exhibit antibiofilm activity against MRSA and display low micromolar minimal inhibitory concentrations (MIC) against M. tuberculosis.17,18 With this literature precedent, we posited that our meridianin D analogues would exhibit antibiofilm activity against mycobacteria.
Mycobacterium smegmatis is commonly used as a surrogate bacterium for M. tuberculosis. M. smegmatis is a non-pathogenic, faster growing mycobacterium and shares over 2000 genetic homologs with M. tuberculosis.19 Previously, we have shown that molecules with antibiofilm properties against M. smegmatis also exhibited antibiofilm properties against M. tuberculosis.11 Therefore, we began by determining the biofilm inhibition and dispersion activity of meridianin D (1) and subsequently a library of meridianin D analogues against M. smegmatis. We then analyzed the ability of our analogues to potentiate the activity of a series of β-lactam antibiotics. β-Lactams are the most prescribed antimicrobial agents and can disrupt cell wall synthesis in both Gram-positive and Gram-negative bacteria, making them an advantageous treatment option. However, mycobacteria are intrinsically resistant to β-lactams. Previously, we've shown that other small molecule adjuvants can be effective in reversing this resistance in mycobacteria.20 The initial structure–activity relationship (SAR) data obtained from these screens prompted us to synthesize further derivatives to obtain a more complete understanding of the activity within the library.
Results and discussion
For this study, we began by testing meridianin D (1) for antibiofilm and β-lactam potentiation activity against M. smegmatis. The initial screening of this compound focused on determining the ability to disperse pre-formed M. smegmatis biofilms and inhibit the formation of biofilms as assessed using a crystal violet reporter assay.5 Meridianin D (1) displayed moderate dispersion activity, with an EC50 value of 51.0 ± 1.3 μM, but more potent biofilm inhibition activity with an IC50 value of 21.5 ± 1.6 μM. Here, the EC50 value represents the concentration at which 50% of a pre-formed biofilm is dispersed in comparison to an untreated control. The IC50 value represents the concentration necessary to inhibit 50% of biofilm formation in comparison to an untreated control. We next analyzed the antibiotic activity of the compound alone, and its ability to potentiate a wide array of commonly prescribed β-lactam antibiotics: cefotaxime, oxacillin, cefoxitin, ceftazidime and ampicillin. We first measured the MIC of 1, which was determined to be 200 μM. When analyzing the β-lactam potentiation capability, the compound was tested at lower than one-third of the MIC, or 60 μM, to avoid inherent toxicity. At 60 μM, 1 did not lower the MIC of any of the five antibiotics tested.After determining that meridianin D (1) possessed limited biofilm dispersion activity and no β-lactam potentiation activity, we chose to focus our screening on our library of previously synthesized meridianin D analogues16 (compounds 2–31, Fig. 2) to see if a trend could be observed between structural changes and activity. Several of these analogues displayed increased activity against MRSA when compared to meridianin D so we hypothesized that we would see improved activity against M. smegmatis as well.
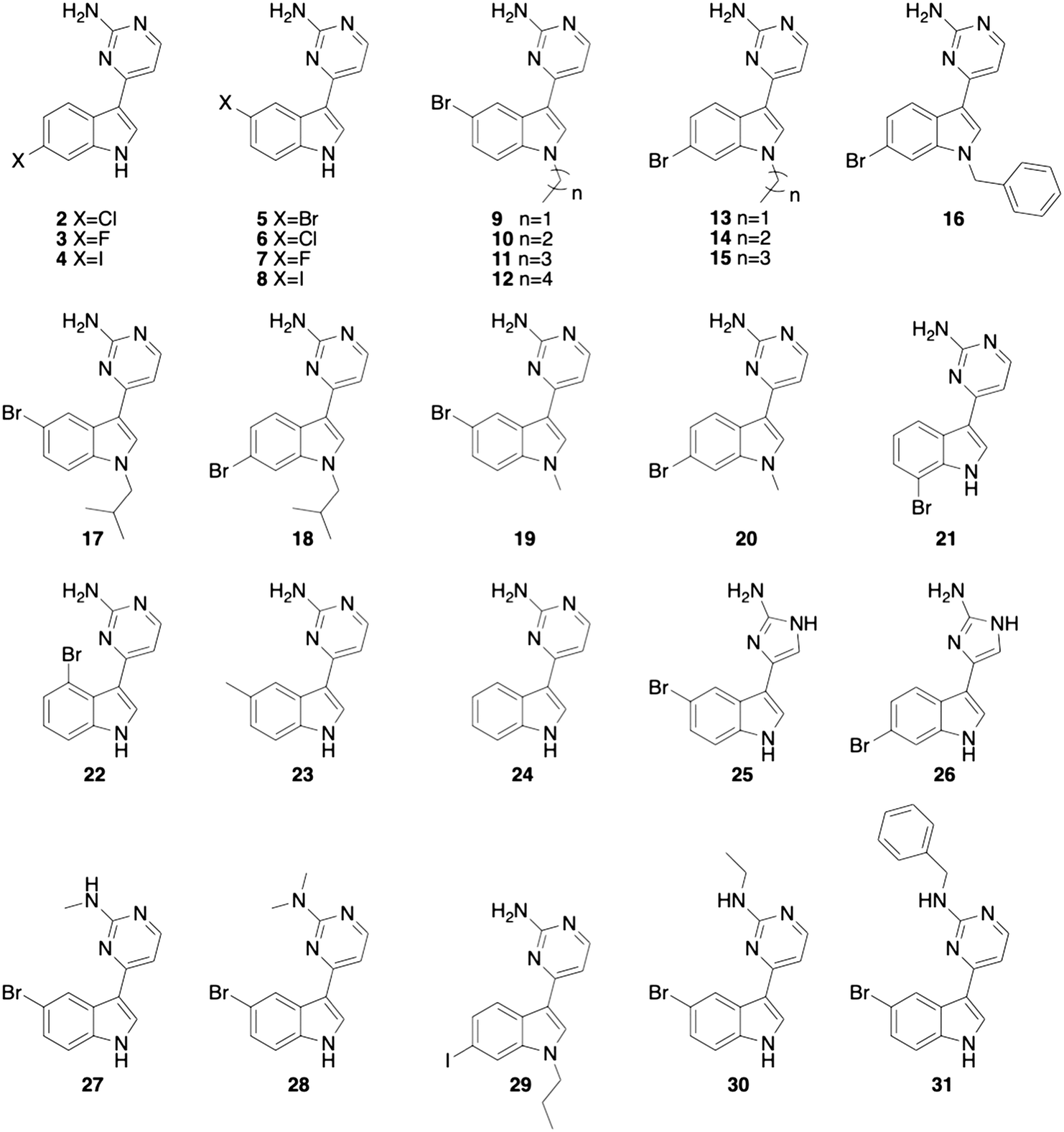 | ||
| Fig. 2 Previously synthesized meridianin analogue library.16 | ||
The 30 analogues were first screened for biofilm inhibition and biofilm dispersion activity at an upper concentration of 50 μM. Analogues that displayed activity below this threshold where then subjected to a dose–response study to determine EC50 and IC50 values. Of the compounds screened, seven analogues exhibited EC50 values less than 50 μM and 25 displayed IC50 values less than 50 μM (Table 1).
| Compound | IC50 (μM) | EC50 (μM) |
|---|---|---|
| 1 | 21.5 ± 1.6 | 51.0 ± 1.3 |
| 2 | 26.6 ± 1.5 | >50 |
| 3 | 21.6 ± 3.4 | >50 |
| 4 | 13.2 ± 1.8 | 29.4 ± 0.9 |
| 5 | 22.7 ± 1.2 | >50 |
| 6 | 18.3 ± 0.7 | >50 |
| 7 | 16.0 ± 2.3 | >50 |
| 8 | 13.1 ± 1.5 | 28.1 ± 3.7 |
| 9 | 37.1 ± 3.8 | >50 |
| 10 | 15.1 ± 1.0 | >50 |
| 11 | 22.7 ± 2.9 | >50 |
| 12 | >50 | >50 |
| 13 | 39.1 ± 3.6 | >50 |
| 14 | 34.5 ± 1.4 | >50 |
| 15 | >50 | >50 |
| 16 | 27.3 ± 3.5 | >50 |
| 17 | 14.9 ± 2.4 | >50 |
| 18 | 50.4 ± 3.4 | >50 |
| 19 | 27.8 ± 1.8 | >50 |
| 20 | 26.7 ± 1.7 | >50 |
| 21 | 26.0 ± 3.0 | >50 |
| 22 | >50 | >50 |
| 23 | 15.7 ± 0.5 | >50 |
| 24 | 29.5 ± 3.2 | >50 |
| 25 | 9.5 ± 0.6 | >50 |
| 26 | 7.5 ± 0.3 | 27.9 ± 3.2 |
| 27 | 25.5 ± 0.9 | 45.0 ± 3.9 |
| 28 | 14.3 ± 0.4 | 35.7 ± 1.3 |
| 29 | >50 | >50 |
| 30 | 14.2 ± 0.4 | 39.3 ± 2.4 |
| 31 | 6.9 ± 0.8 | 28.3 ± 2.3 |
From this data, it was obvious that most of the analogues did not possess potent EC50 values, as was similarly seen with the original natural product, meridianin D (1). There were however, twelve derivatives with lower IC50 values than compound 1 (IC50 21.5 ± 1.6 μM) and seven with EC50 values less than 50 μM. Substitution of the exocyclic nitrogen on the 2-aminopyrimidine moiety resulted in biofilm dispersal activity. The unsubstituted derivative 5 has an EC50 value greater than 50 μM while the substituted analogues 27, 28, 30, and 31 can disperse preformed M. smegmatis biofilms. One of the most active analogues that possessed both inhibition and dispersion capabilities is compound 26, which has an EC50 of 27.9 ± 3.2 μM and an IC50 of 7.5 ± 0.3 μM. Interestingly, 26 differs from meridianin D (1) only by the incorporation of a 2-aminoimidazole (2-AI) moiety in place of the 2-aminopyrimidine group, and we have previously identified a variety of 2-AI containing molecules that display both antibiofilm activity and the ability to potentiate several cephalosporin and penicillin antibiotics against planktonic M. smegmatis.21
Since 2-AI containing compounds have shown β-lactam potentiation activity in the past and some of the analogues improved upon the antibiofilm activity seen with meridianin D, we chose to screen all the analogues against the same five antibiotics meridianin D was screened with. As for meridianin D, the MIC was determined for all 30 analogues. Most of the analogues had an MIC of ≥200 μM, and were therefore tested at 60 μM, except for 25 and 26, which exhibited an MIC of 100 μM and were therefore tested at 30 μM. Potentiation activity was minimal across all five antibiotics and all analogues, with the largest reduction in MIC being eight-fold with both cefotaxime and ceftazidime for 19. Due to the minimal activity, no obvious trend between structure and activity could be determined (Tables 2 and S1†).
| Compound | Concentration tested (μM) | Cefotaxime MIC (μg mL−1) | Oxacillin MIC (μg mL−1) | Cefoxitin MIC (μg mL−1) | Ceftazidime MIC (μg mL−1) | Ampicillin MIC (μg mL−1) |
|---|---|---|---|---|---|---|
| 128 | 512 | 16 | 512 | 128 | ||
| 4 | 60 | 128 (0) | 512 (0) | 16 (0) | 512 (0) | 128 (0) |
| 8 | 60 | 64 (2) | 512 (0) | 16 (0) | 512 (0) | 128 (0) |
| 19 | 60 | 16 (8) | 256 (2) | 8 (2) | 64 (8) | 32 (4) |
| 26 | 30 | 128 (0) | 512 (0) | 16 (0) | 128 (4) | 128 (0) |
| 27 | 60 | 64 (2) | 256 (2) | 16 (0) | 512 (0) | 64 (2) |
| 28 | 60 | 128 (0) | 512 (0) | 16 (0) | 512 (0) | 64 (2) |
| 30 | 60 | 128 (0) | 512 (0) | 16 (0) | 512 (0) | 64 (2) |
| 31 | 60 | 64 (2) | 256 (2) | 16 (0) | 512 (0) | 64 (2) |
Since there was minimal β-lactam potentiation activity, we decided to look for activity trends within the antibiofilm activity. One obvious trend observed for the 2-AI containing derivatives was that when the bromine was moved from the 5-position on the indole, 25, to the 6-position in compound 26, the antibiofilm activity increased. This trend was reversed for the analogues containing a 2-aminopyrimidine moiety in place of the 2-AI. In the pyrimidine containing compounds, the presence of halogens at the 5-position of the indole imparted increased activity as compared to halogenation at the 6-position. Additionally, we noted that activity with the pyrimidine containing derivatives varied depending on the identity of the halogen at the 5-postition. The trend indicated that bromine containing analogues were the least active followed by chlorine, then fluorine and then iodine. With this in mind, we chose to perform a small SAR study with the 2-AI containing analogues since there were only a small number of analogues containing the 2-AI subunit in the initial library, and 26 was one of the most active antibiofilm analogues.
Following the synthetic procedure outlined by our group previously,16 the 5-chloro and fluoro analogues and the 6-chloro analogue were synthesized (Scheme 1). Attempts were made to synthesize the 6-fluoro analogue and the non-halogenated analogue, but the desired products appeared to lack the stability necessary for biological testing, so no further synthetic attempts were made. No attempts were made to access the corresponding iodinated derivatives given the moderate activity of 4, 8, and 29.
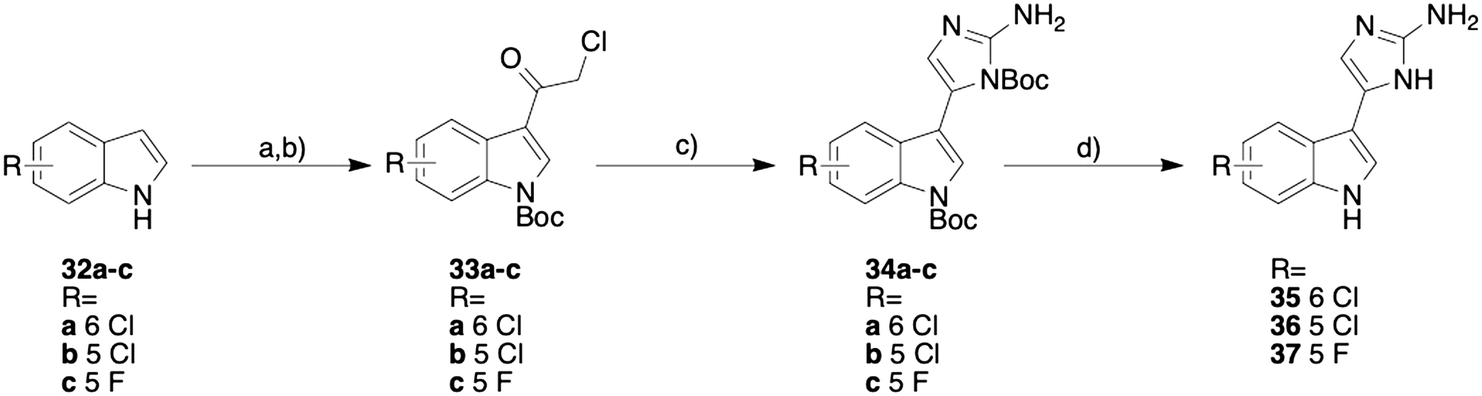 | ||
| Scheme 1 Synthesis of the 2-AI meridianin D analogues. a) 1) chloroacetyl chloride, toluene, 60 °C, 2 h; 2) MeOH, H2O, room temperature, 1 h; b) Boc anhydride, DMAP, THF, room temperature, 4 h; c) N-Boc guanidine, sodium iodide, DMF, room temperature, 48 h; d) 1) 30% TFA, DCM, 0 °C warming to room temperature, 16 h; 2) MeOH/HCl. | ||
The synthesis was initiated by acylating the desired indole (32a–c) with chloroacetyl chloride. The corresponding acylated indole was purified by recrystallizing with ethanol and was immediately Boc protected using Boc anhydride. The Boc protected indole (33a–c) was then subjected to a Boc guanidine cyclization and then a subsequent Boc deprotection with trifluoroacetic acid (TFA). The resulting product was then converted from the TFA salt to the HCl salt using hydrochloric acid and methanol. This synthetic procedure yielded the three new analogues (compounds 35–37, Scheme 1).
The resulting analogues were tested for their antibiofilm activity in the same manner as the initial library (Table 3). As predicted from the activity exhibited by 26, the 6-substituted analogue, 35, again showed an EC50 value less than 50 μM at 33.7 ± 2.4 μM. Additionally, this analogue exhibited good inhibition activity with an IC50 value of 10.3 ± 1.3 μM. Although the newly synthesized analogue exhibited good activity, it did not show an increase in activity compared to the lead compound 26. When analyzing the other derivatives, the analogues with the halogen at the 5-position (36 and 37) exhibited no biofilm dispersion activity, with EC50 values greater than 50 μM. 36 was slightly less active than the brominated analogue with regards to biofilm inhibition, exhibiting an IC50 of 16.0 ± 0.7 μM and the fluorinated derivative 37 was substantially less active, with an IC50 of 40.2 ± 2.4 μM.
| Compound | IC50 (μM) | EC50 (μM) |
|---|---|---|
| 35 | 10.3 ± 1.3 | 33.7 ± 2.4 |
| 36 | 16.0 ± 0.7 | >50 |
| 37 | 40.2 ± 2.4 | >50 |
Lastly, we tested the toxicity of the lead meridianin analogues against eukaryotic red blood cells. This was measured by quantifying hemolysis of meridianin D (1) and lead compounds 26 and 31 as well as inactive compound 15. At 500 μM (highest concentration tested), 1, 26, 31, and 15 caused 13%, 14%, 32% and 73% lysis respectively compared to 1% Triton-X. At 200 μM, 1, 26, 31, and 15 effected 8%, 7%, 9%, and 56% lysis respectively. Both active analogues 26 and 31 were relatively non-toxic while the inactive derivative 15, which contains a long alkyl chain, resulted in the greatest hemolytic activity.
In conclusion, we have described several meridianin D analogues that improve upon the biofilm inhibition and dispersion activity exhibited by the natural product. The most active analogue contained a 2-AI moiety in place of the 2-aminopyrimidine substituent. With this, we chose to synthesize additional 2-AI containing analogues with varying halogen substitutions. This SAR study showed that the trend of activity displayed by the 2-AI containing compounds was opposite to the trend displayed by the pyrimidine containing derivatives. For the 2-AI containing compounds, substitution at the 6-position led to both biofilm inhibition and dispersion activity while the 5-substituted analogues only displayed biofilm inhibition activity. Alternatively, the analogues containing a pyrimidine moiety displayed greater activity if the halogen was in the 5-position instead of the 6-position. Additionally, for the 2-AI containing analogues activity was greatest for bromine containing derivatives, then diminished for chlorine containing compounds and further diminished for fluorine containing compounds. For the pyrimidine containing analogues, activity was greatest for iodine-containing derivatives, reduced for fluorine, reduced further for chlorine, and the least active analogues contained bromine as the halogen substitution. Although these molecules exhibit promising antibiofilm activity, none of them were able to notably potentiate a range of β-lactam antibiotics. Future studies will entail testing lead compounds for their ability to potentiate antibiotics against M. tuberculosis biofilms.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the National Institutes of Health (AI106733) for support.References
- D. J. Payne, Microbiology. Desperately seeking new antibiotics, Science, 2008, 321(5896), 1644–1645 CrossRef CAS PubMed.
- H. C. Neu, The crisis in antibiotic-resistance, Science, 1992, 257(5073), 1064–1073 CrossRef CAS PubMed.
- C. A. Arias and B. E. Murray, Antibiotic-resistant bugs in the 21st century--a clinical super-challenge, N. Engl. J. Med., 2009, 360(5), 439–443 CrossRef CAS PubMed.
- K. Lewis, Riddle of biofilm resistance, Antimicrob. Agents Chemother., 2001, 45(4), 999–1007 CrossRef CAS.
- T. F. Mah and G. A. O'Toole, Mechanisms of biofilm resistance to antimicrobial agents, Trends Microbiol., 2001, 9(1), 34–39 CrossRef CAS.
- R. M. Donlan and J. W. Costerton, Biofilms: survival mechanisms of clinically relevant microorganisms, Clin. Microbiol. Rev., 2002, 15(2), 167–193 CrossRef CAS.
- I. M. Orme, A new unifying theory of the pathogenesis of tuberculosis, Tuberculosis, 2014, 94(1), 8–14 CrossRef CAS PubMed.
- Organization, W. H. Global Tuberculosis Report Search PubMed.
- L. Rook, Tuberculosis in Priority Medicines for Europe and the World “A Public Health Approach to Innovation”, 2013 Search PubMed.
- C. R. Horsburgh Jr., C. E. Barry 3rd and C. Lange, Treatment of Tuberculosis, N. Engl. J. Med., 2015, 373(22), 2149–2160 CrossRef PubMed.
- D. F. Ackart, E. A. Lindsey, B. K. Podell, R. J. Melander, R. J. Basaraba and C. Melander, Reversal of Mycobacterium tuberculosis phenotypic drug resistance by 2-aminoimidazole-based small molecules, Pathog. Dis., 2014, 70(3), 370–378 CrossRef CAS.
- M. A. A. Radwan and M. El-Sherbiny, Synthesis and antitumor activity of indolylpyrimidines: Marine natural product meridianin D analogues, Bioorg. Med. Chem., 2007, 15(3), 1206–1211 CrossRef CAS PubMed.
- S. B. Bharate, R. R. Yadav, S. I. Khan, B. L. Tekwani, M. R. Jacob, I. A. Khan and R. A. Vishwakarma, Meridianin G and its analogs as antimalarial agents, MedChemComm, 2013, 4(6), 1042–1048 RSC.
- M. Gompel, M. Leost, E. B. D. Joffe, L. Puricelli, L. H. Franco, J. Palermo and L. Meijer, Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum, Bioorg. Med. Chem. Lett., 2004, 14(7), 1703–1707 CrossRef CAS PubMed.
- R. R. Yadav, S. I. Khan, S. Singh, I. A. Khan, R. A. Vishwakarma and S. B. Bharate, Synthesis, antimalarial and antitubercular activities of meridianin derivatives, Eur. J. Med. Chem., 2015, 98, 160–169 CrossRef CAS PubMed.
- W. M. Huggins, W. T. Barker, J. T. Baker, N. A. Hahn, R. J. Melander and C. Melander, Meridianin D Analogues Display Antibiofilm Activity against MRSA and Increase Colistin Efficacy in Gram-Negative Bacteria, ACS Med. Chem. Lett., 2018, 9(7), 702–707 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, F. Bai, M. Ukhanova, V. Mai, S. Jin, H. Luesch and R. W. Huigens 3rd, Halogenated Phenazines that Potently Eradicate Biofilms, MRSA Persister Cells in Non-Biofilm Cultures, and Mycobacterium tuberculosis, Angew. Chem., Int. Ed., 2015, 54(49), 14819–14823 CrossRef CAS PubMed.
- A. T. Garrison, Y. Abouelhassan, D. Kallifidas, H. Tan, Y. S. Kim, S. Jin, H. Luesch and R. W. Huigens 3rd, An Efficient Buchwald-Hartwig/Reductive Cyclization for the Scaffold Diversification of Halogenated Phenazines: Potent Antibacterial Targeting, Biofilm Eradication, and Prodrug Exploration, J. Med. Chem., 2018, 61(9), 3962–3983 CrossRef CAS.
- M. Altaf, C. H. Miller, D. S. Bellows and R. O'Toole, Evaluation of the Mycobacterium smegmatis and BCG models for the discovery of Mycobacterium tuberculosis inhibitors, Tuberculosis, 2010, 90(6), 333–337 CrossRef CAS PubMed.
- T. V. Nguyen, M. S. Blackledge, E. A. Lindsey, B. M. Minrovic, D. F. Ackart, A. B. Jeon, A. Obregon-Henao, R. J. Melander, R. J. Basaraba and C. Melander, The Discovery of 2-Aminobenzimidazoles That Sensitize Mycobacterium smegmatis and M. tuberculosis to beta-Lactam Antibiotics in a Pattern Distinct from beta-Lactamase Inhibitors, Angew. Chem., Int. Ed., 2017, 56(14), 3940–3944 CrossRef CAS PubMed.
- T. V. Nguyen, B. M. Minrovic, R. J. Melander and C. Melander, Identification of Anti-Mycobacterial Biofilm Agents Based on the 2-Aminoimidazole Scaffold, ChemMedChem, 2019, 14(9), 927–937 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00466a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |