
Broad-spectrum antitumor properties of Withaferin A: a proteomic perspective
Martin
Dom
 ,
Wim
Vanden Berghe
and
Xaveer
Van Ostade
*
,
Wim
Vanden Berghe
and
Xaveer
Van Ostade
*
Laboratory of Protein Chemistry, Proteomics and Epigenetic Signalling (PPES), Department of Biomedical Sciences, University of Antwerp (UA), Universiteitsplein 1, 2610 Wilrijk, Belgium. E-mail: xaveervanostade@uantwerpen.be; Tel: +3232562319
First published on 16th December 2019
Abstract
The multifunctional antitumor properties of Withaferin A (WA), the manifold studied bioactive compound of the plant Withania somnifera, have been well established in many different in vitro and in vivo cancer models. This undoubtedly has led to a much better insight in the underlying mechanisms of WAs broad antitumor activity range, but also raises additional challenging questions on how all these antitumor properties could be explained on a molecular level. Therefore, a lot of effort was made to characterize the cellular WA target proteins, since these binding events will lead and initiate the observed downstream effects. Based on a proteomic perspective, this review provides novel insights in the molecular chain of events by which WA potentially exercises its antitumor activities. We illustrate that WA triggers multiple cellular stress pathways such as the NRF2-mediated oxidative stress response, the heat shock response and protein translation events and at the same time inhibits these cellular protection mechanisms, driving stressed cancer cells towards a fatal state of collapse. If cancer cells manage to restore homeostasis and survive, a stress-independent WA antitumor response comes into play. These include the known inhibition of cytoskeleton proteins, NFκB pathway inhibition and cell cycle inhibition, among others. This review therefore provides a comprehensive overview which integrates the numerous WA–protein binding partners to formulate a general WA antitumor mechanism.
1 Introduction
The current understanding of the antitumor properties of Withaferin A (WA) is scattered throughout the oncology field. A variety of seemingly unrelated pathways has been identified to explain its activity, suggesting promiscuous activities for this molecule. Furthermore, the plethora of experimental treatments described for Withania Somnifera extracts or its major bioactive constituent WA in different cancer cell types or in vivo models, makes it very hard to get a clear picture of its therapeutic mechanism of action against tumors.Most reports usually focus on a few WA target proteins, while it is recognized that WA is a multifunctional antitumor compound targeting tens to hundreds of proteins.1,2 For instance, the deficiency of vimentin, a well-known cancer target of WA, does not completely abrogate the biological activity of WA and suggests additional WA targets.3 This immediately demonstrates the experimental Achilles' heel of multifunctional compounds like WA which act through modulation of an intracellular network rather than single target proteins. The functional validation of WA targets by gene specific knockdown experiments typically fail to abrogate WA effects due to redundancy of the signaling network which bypasses the defect by promoting WA binding to alternative targets.
In addition, most studies describe general WA antitumor effects based on the measurements of secondary effects like ROS generation or cell cycle inhibition. Such observations are very useful in describing the biological effects of WA, but provide less information on the primary molecular targets of WA for further optimization of target-based cancer drug development.
For this review, we reasoned that, based on the above concerns, it is more straightforward to characterize the pleiotropic antitumor effects of WA based on its very first action: binding to the target proteins. WA-target proteins were extracted from literature as well as results obtained via chemoproteomic approaches.4 Information from differential proteomics expression studies following WA treatment4 will be used to complement and support our conclusions. To our knowledge, this is the first comprehensive overview which integrates the numerous WA–protein binding partners and differential protein expression profiles to formulate a general WA antitumor mechanism.
2 WA-responsive cellular stress pathways
Mammalian cells utilize a wide range of biochemical pathways and systems as they seek to maintain homeostasis, repair or replace damaged cellular components, carry out organ-specific functions and respond to environmental signals or stress.5 Both electrophiles and reactive oxygen species (ROS) are such stress signals for a cell and it is well described that WA holds electrophilic stress inducing6 and ROS inducing7–10 properties. The electrophilic stress inducing properties of WA result from the electrophilic nature and the preference to react with nucleophilic groups: i.e. WA is a Michael addition electron acceptor.1The incomplete reduction of molecular oxygen (O2) produces a superoxide radical (˙O2−) which is the precursor of most other reactive oxygen species,11–13 including hydrogen peroxide (H2O2) and hydroxyl radicals (˙OH):
| (O2 + e− → ˙O2−) |
Although WA is well-known as a “ROS-inducing” compound, we found no evidence that WA directly promotes ROS production. In contrast, increased ROS in response to WA may be an indirect consequence of WA-specific inhibition of ROS eliminating pathways. Fig. 1 describes our hypothesis of electrophilic stress inducing and ROS accumulating effects of WA based on recently described dual stress-modulating concepts.18 Treatment of WA and binding to Keap1 (ref. 19) results in elevated NRF2 protein levels that in turn regulate the expression of antioxidant proteins protecting the cell against oxidative damage.20 Meanwhile, WA targets numerous proteins from the anti-stress pathway, thereby augmenting the basal ROS levels being a byproduct of the aerobic metabolism.21 The augmented basal ROS levels in turn further stimulate antioxidant pathways and the resulting imbalanced ROS/cytoprotection levels will eventually determine the cancer cells fate: live or die. Indeed, various ROS-dependent mechanisms have been proposed to explain WA-induced cell death via apoptosis,22 paraptosis9 or ferroptosis.8
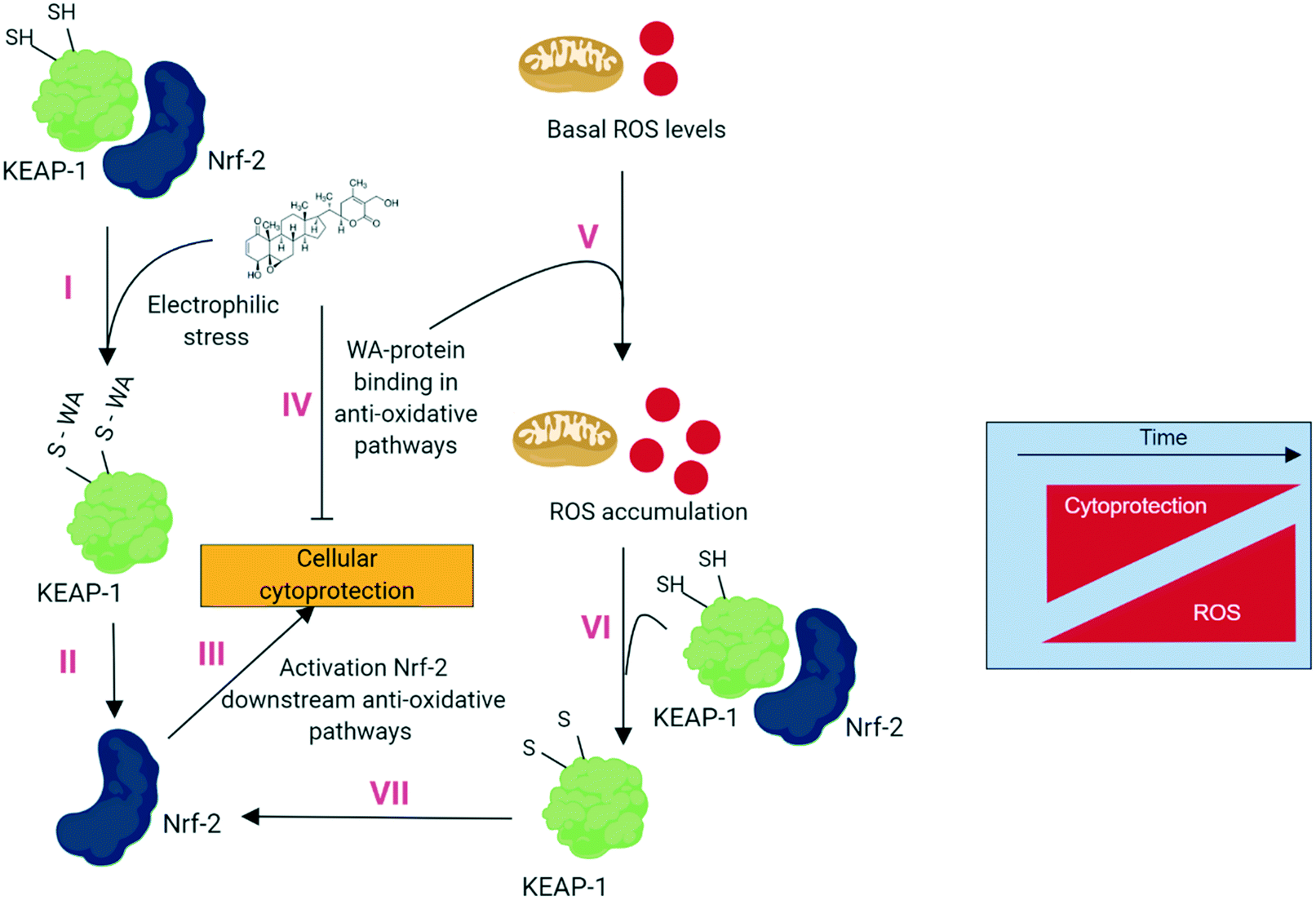 | ||
| Fig. 1 Overview of the dual action antitumor mechanism of WA: inducing anti-stress pathways and at the same time inhibiting them. (I) WA treatment results in electrophilic stress and binding of WA to Keap1 results in (II) Nrf-2 activation and translocation. (III) Activation of Nrf-2 downstream anti-oxidative pathways that are prone to (IV) WA–protein binding and inhibition. As result WA induces and inhibits the anti-oxidative pathways simultaneously. (V) Low, basal, ROS levels further accumulate due to inhibition of anti-oxidative pathways. (VI) ROS accumulation triggers Keap1 and (VII) further activates Nrf-2. The resulting imbalanced cytoprotection/ROS levels will eventually determine cellular fate: live or die. | ||
In the following subchapters we describe this dual action hypothesis on the basis of protein upregulation in different anti-oxidative stress pathways (= activation) and the known binding events of WA and proteins of these anti-oxidative stress pathways (= inhibition).
2.1 The NRF2-mediated oxidative stress response
Cells have numerous mechanisms by which they counteract stress response of oxidative insults. A pivotal role herein is provided by the ‘NRF2-mediated Oxidative Stress Response’ pathway. In the presence of oxidative stress, cytosolic Nrf2 uncouples from the oxidative stress sensor Keap1 (Keap1) and is translocated to the nucleus where it binds to anti-oxidant response elements (ARE) and subsequently induces stress-protective genes thereby trying to restore cellular homeostasis levels (Fig. 2).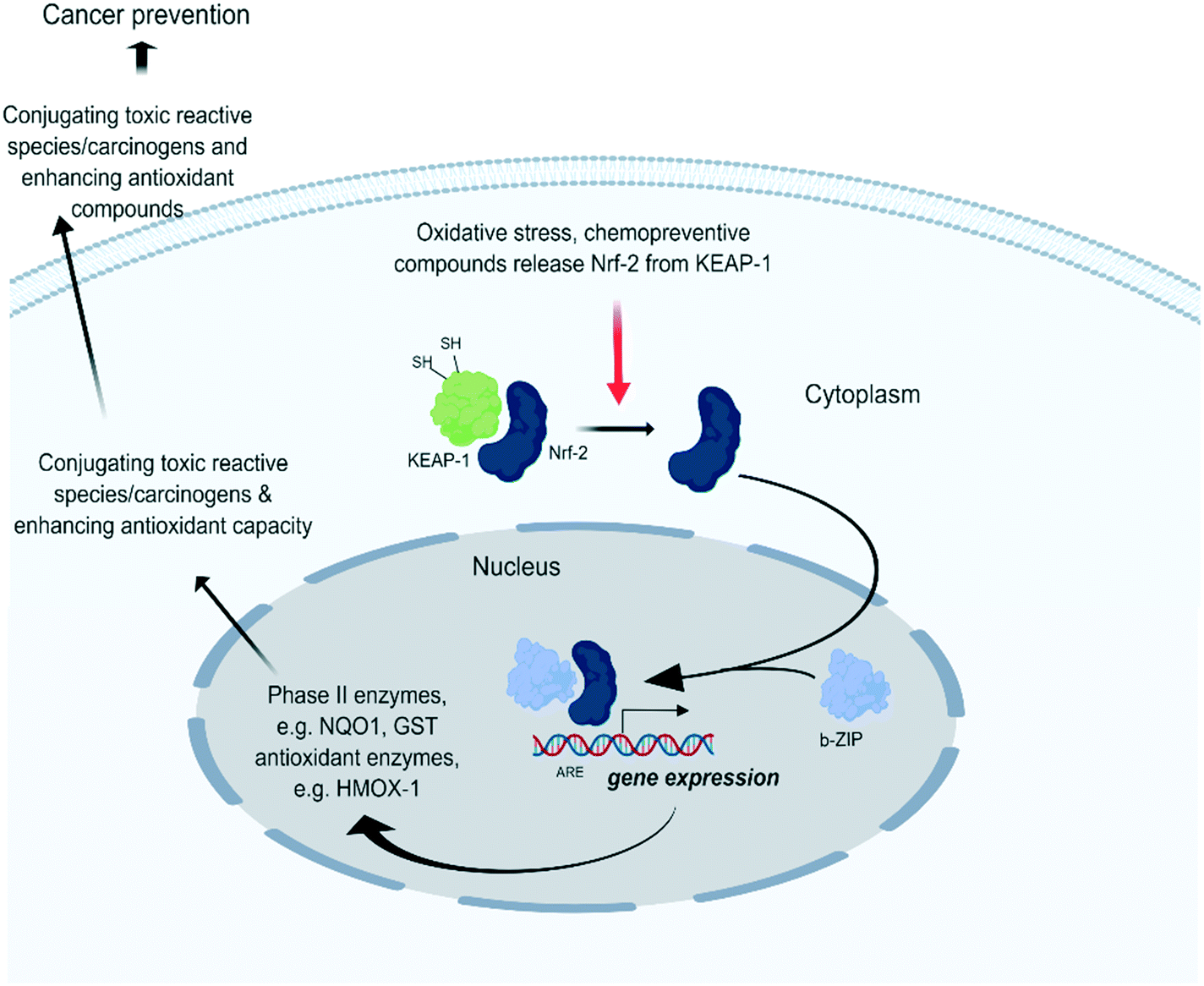 | ||
| Fig. 2 Under quiescent conditions, binding of Kelch-like ECH-associated protein 1 (Keap1) with nuclear factor erythroid-derived 2-like 2 (Nrf2) facilitates the sustained ubiquitination and proteasome degradation of Nrf2.23 Upon oxidative stress, Keap1 becomes inactivated through binding of its cysteine thiols with electrophilic groups. This releases Keap1 from Nrf2 which allows Nrf2 translocation to the nucleus. There it forms heterodimers with small Maf proteins containing a leucine zipper (bZIP) domain and binds with the ARE of target genes such as NAD(P)H dehydrogenase [quinone] 1 (NQO1) and heme-oxygenase 1 (HMOX1) which initiate stress homeostasis mechanisms. | ||
Recently, we identified Keap1 as a direct target of WA in endothelial cells.19 We found that WA binds to Keap1 independently of Nrf2 and binds the Cys151 in the BTB domain and 4 other cysteine sites located in the C-terminal Kelch domain. This led us to assume that the WA activation of Nrf2 is mediated via disruption of the Keap1-Nrf2 complex.
Proteins related to oxidative stress response that are differentially upregulated upon WA treatment:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Heme oxygenase 1 | HMOX1_HUMAN | Anti-oxidant enzyme | 4, 25 |
| CDGSH iron sulfur domain 1 | CISD1_HUMAN | Sensor of oxidative signals | 4 |
| Aldose reductase | ALDR_HUMAN | NADPH-dependent oxidoreductase | 4, 25 |
| Sepiapterin reductase | SPRE_HUMAN | Mediator of chemical redox cycling | 4 |
However, some of these stress response proteins eventually fail to perform their function following WA treatment because of covalent WA-binding, as has already been reported for inhibition of peroxiredoxin 1 (PRDX1) activity.26
WA target protein related to the oxidative stress response:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Peroxiredoxin 1 | PRDX1_HUMAN | Dual functioning thiol-specific peroxidase and molecular chaperon | 26 |
PRDX1 is a dual-functioning protein that serves both an antioxidant enzyme activity27 and a molecular chaperone activity,28 depending on its oligomeric state.29 As a homodimer, PRDX1 functions as a thiol-specific peroxidase that catalyzes the reduction of hydrogen peroxide. On the other hand, homodecamers, or higher-order oligomers function as molecular chaperone, preventing client proteins from stress-induced aggregation. Although binding of WA is only described to inhibit PRDX1 chaperone activity and not affecting peroxidase activity in kidney cells,26 it would be very interesting to further explore the WA effects on different PRDX1 oligomeric states and activities since the type of oligomerization is redox state dependent.30
Another way for WA to inhibit the stress response pathway is by downregulation of oxidative stress response pathways:
Proteins related to oxidative stress response that are differentially downregulated upon WA treatment:
This has been demonstrated for glutathione peroxidase 1 (GPX1), an anti-oxidant enzyme which reduces hydrogen peroxide to water.31 Along the same line, GPX4 protein levels were found to drop upon covalent cysteine binding of WA which can be rescued upon mutation of cysteine residues in Gpx4 which abrogate WA binding.8 This suggest that WA triggers excessive oxidative damage and cancer cell death via a dual mechanism, involving elevated basal ROS levels as well as disrupting the cytoprotective Keap1/Nrf2 pathway.
Proteins related to the ubiquitin proteasome system that are differentially upregulated upon WA treatment:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Proteasome subunit beta type-1 | PSB1_HUMAN | 20S core particle | 4 |
| Proteasome subunit alpha type-2 | PSA2_HUMAN | 20S core particle | 4 |
| Proteasome activator complex subunit-4 | PSME4_HUMAN | 20S core particle | 4 |
| 26S proteasome regulatory subunit 10B | PRS10_HUMAN | 19S regulatory particle | 4 |
| Ubiquitin carboxyl-terminal hydrolase 24 | UBP24_HUMAN | De-ubiquitinating enzyme | 4 |
Intriguingly, ubiquitin carboxyl-terminal hydrolase 24 (USP24), a deubiquitinating enzyme, is required for cellular tumor antigen p53 (TP53) stabilization under both stressed- and unstressed conditions by deubiquitinating TP53.35 Since it is nearly impossible for a cell to become cancerous unless it inactivates the p53 network,36 the upregulation of USP24 and the subsequent TP53 stabilization could explain the observed TP53 accumulation and TP53-dependent apoptosis upon WA treatment.37
However, similar to inactivation of various oxidative stress responsive proteins upon covalent WA binding, various components of the ubiquitin–proteasome system also become inactivated and/or degraded shortly after their transcriptional upregulation. The following WA-target proteins from the ubiquitin–proteasome system seem to lose their function upon WA treatment:
WA target proteins related to the ubiquitin–proteasome system:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Proteasome subunit beta type-10 | PSB10_HUMAN | Proteasome subunit involved in antigen processing | 4 |
| Proteasome subunit beta type-5 | PSB5_HUMAN | Chymotrypsin-like activity of the 20S proteasome | 38 |
| AAA+ chaperone p97 | TERA_HUMAN | ATPase activity | 39 |
| Ubiquitin carboxyl-terminal hydrolase 14 | USP14_HUMAN | Deubiquitinating enzyme | 4 |
| Ubiquitin carboxyl-terminal hydrolase isozyme L5 | UCHL5_HUMAN | Deubiquitinating enzyme | 4 |
Impairment of the chymotrypsin-like catalytic activity of the proteasomal 20S core particle (CP) and associated accumulation of ubiquitinated proteins has been explained by WA binding with the proteasome subunit beta type-5 (PSMB5), as suggested by in silico docking.38 Surprisingly, proteasome activity assays in our lab demonstrated no inhibition of any catalytic 20S CP activity following WA treatment, although this may still be the case in other experimental cell models (e.g. other WA treatment regimes, other types of cancer cells, etc.). Recently, 20S CP-independent WA proteasome inhibiting mechanisms have also been suggested. For example, treating HEK293 cells with different WA analogues targeting the AAA+ chaperone p97 (VCP) protein resulted in the accumulation of poly-ubiquitinated proteins in HEK293 cells, a phenomenon which was also observed upon treatment with CB-5083, a selective inhibitor of VCP.39
In addition, the deubiquitinating (DUB) enzymes ubiquitin carboxyl-terminal hydrolase 14 (USP14) and ubiquitin carboxyl-terminal hydrolase isozyme L5 (UCHL5) are also protein targets of WA.4 Hence, blocking the activity of these proteins may also result in inhibition of proteasome degradation as was shown in our lab by the specific accumulation of Lys48 poly-ubiquitin chains on proteins from WA-treated cells. These observations suggest that the WA antitumor mode of action occurs via 20S core proteasome dependent and 20S core proteasome independent mechanisms, thereby holding promising potential in avoiding multi-drug resistance.40
The upregulated proteasome associated proteins serve as a cellular rescue towards homeostasis and the binding of WA to the cytoprotective proteasome associated proteins undermines this homeostasis. Indeed, downregulation/inhibition of a limited number of proteasome components will always dominate over upregulation of other proteasome components because the complex lacks essential components and stays dysfunctional, even when other components are highly upregulated. This could explain the seemingly the contradictory observations, i.e. simultaneous up and downregulation of different proteasome associated proteins.
Proteins related to the autophagy pathway and differentially upregulated upon WA treatment:
Here again, WA compromises the autophagy pathway by direct binding and subsequent inactivation of several essential autophagy modulating proteins (Fig. 3):
WA target proteins related to the autophagy pathway:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Histone deacetylase 6 | HDAC6_HUMAN | Deacetylation of lysine residues of histones and adapter protein | 4 |
| Phospholipase A-2-activating protein | PLAP_HUMAN | Autophagy cofactor | 4 |
| WD repeat domain phosphoinositide-interacting protein 2 | WIPI2_HUMAN | Formation of pre-autophagosome structures | 4 |
| SNARE-associated protein Snapin | SNAPN_HUMAN | Required for biogenesis of lysosome-related organelles | 4 |
| Tubulin beta chain | TBB1_HUMAN | Major constituent of microtubules | 4, 43 |
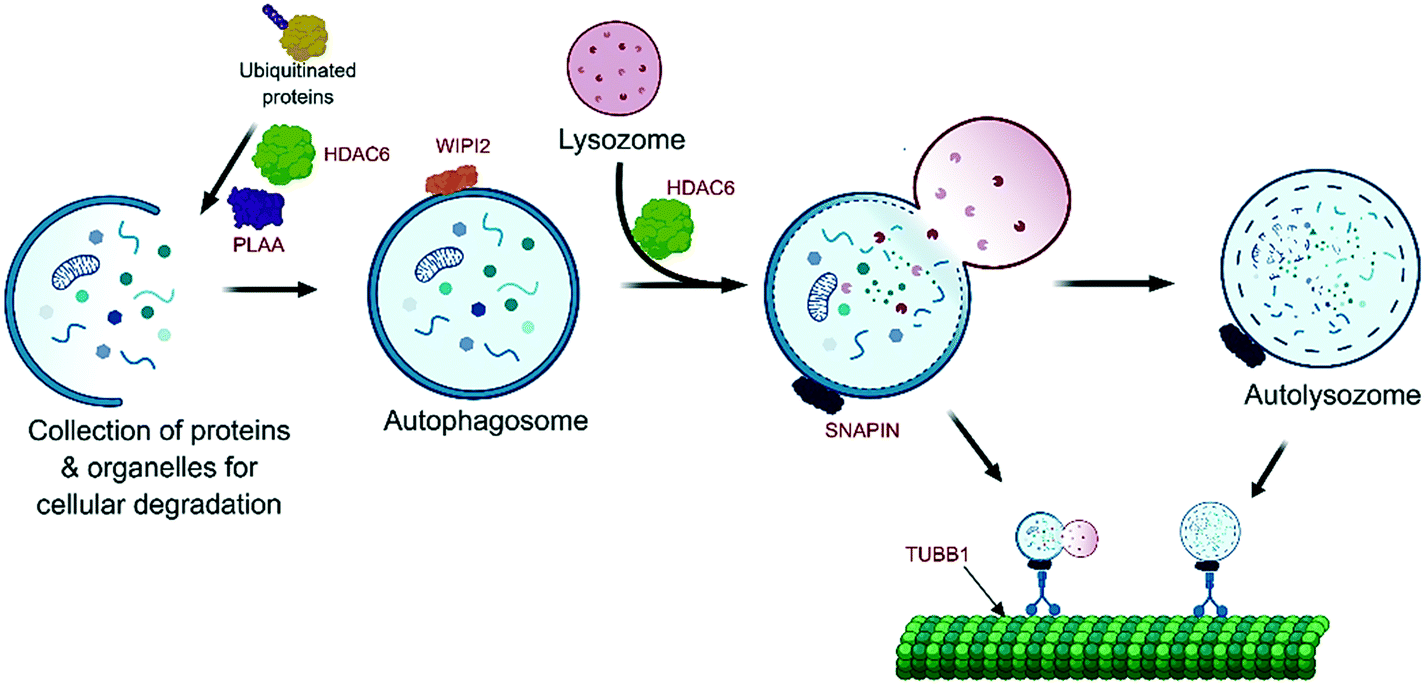 | ||
| Fig. 3 Overview of the autophagy pathway and its main components. Within each step of the formation of the autolysosome, WA targets the 5 proteins (gene names HDAC6, PLAA, WIPI2, SNAPIN and TUBB), suggesting serious disruption of the autophagy process with subsequent cellular impotency to deal with the WA-induced cellular stress. Marked in red are the WA-target proteins (gene names). | ||
For example, WA targets histone deacetylase 6 (HDAC6), an adapter protein that recognizes poly-ubiquitinated misfolded proteins and targets them to the aggresomes.44 Interestingly, clinical combination therapies exist where the selective HDAC6 inhibitor WT161 and the selective proteasome inhibitor bortezomib were used together. This combination treatment was effective in bortezomib resistant multiple myeloma cells and in the presence of bone marrow stromal cells45 that have been shown to mediate multiple myeloma (MM) cell drug resistance. This confirms that HDAC6 is a promising target protein for novel combination treatments of MM cancers.45
Additional targeting of phospholipase A-2-activating protein (PLAA), a cofactor in autophagy, was also demonstrated.4 PLAA is necessary to remove Lys48 linked ubiquitin chains to promote autophagosome formation and subsequent autophagy of ruptured lysosomes.46 The recent observation in our lab of accumulation of Lys48 poly-ubiquitinated target proteins following WA treatment could be the result of inhibition of USP14 and UCHL5, associated with the proteasome 19S regulatory article (RP), as well as the inhibition of the PLAA cofactor responsible for autophagosome formation.
Furthermore the WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) was identified as a potential WA target.4 WIPI2 is crucial in the formation of pre-autophagosomal structures and their maturation into mature autophagosomes, hence its inhibition prevents autophagy.47
Also, SNARE-associated protein Snapin (SNAPIN) was identified as a potential WA target.4 SNAPIN regulates lysosome positioning,48 a crucial aspect since the fusion of autophagosome and lysosomes will result in the actual degradation in the autolysosome.49 Interestingly this positioning and autolysosome formation is dependent on the microtubular network.50 Since WA induces decreased tubulin polymerization and binds directly to tubulin beta (TUBB)51 or tubulin in breast cancer cells,9,43 the formation of autolysosomes could be impaired at multiple stages of the autophagy pathway.
Finally, downregulation of annexin A4 (ANXA4), a calcium/phospholipid-binding protein that promotes membrane fusion, could be observed upon WA treatment.4 The impairment of autophagy membrane fusion processes also contributes to the inhibition of autophagy pathway. These results suggests again that WA elicits excessive stress by triggering the autophagy pathway and meanwhile inhibiting the rescue proteins that must ensure proper autophagy function.
Proteins related to the autophagy pathway and differentially downregulated upon WA treatment:
2.2 Heat shock response
Cells possess several mechanisms and processes to maintain proteostasis, i.e. protein quality control depending on the balance of protein degradation and re-synthesis. At one end of the spectrum, the proteasome and autophagy as well as other lysosome-dependent systems function in the degradation of dysfunctional proteins. At the other end, heat shock proteins modulate protein folding and repair.52 Heat shock factor-1 transcription factor (HSF1) is a stress-inducible and DNA-binding transcription factor responsible for the transcriptional activation of the heat shock response elements (HSE, Fig. 4). Under normal cellular conditions, HSF1 resides in the cytoplasm as a monomer, but upon stress induction, HSF1 becomes activated and trimerizes, allowing nuclear translocation and binding to its HSE.53 This leads to the expression of a large class of molecular chaperones, including heat shock proteins (HSPs), that protect cells from cellular insults54 such as accumulation of misfolded proteins in the endoplasmic reticulum (ER).55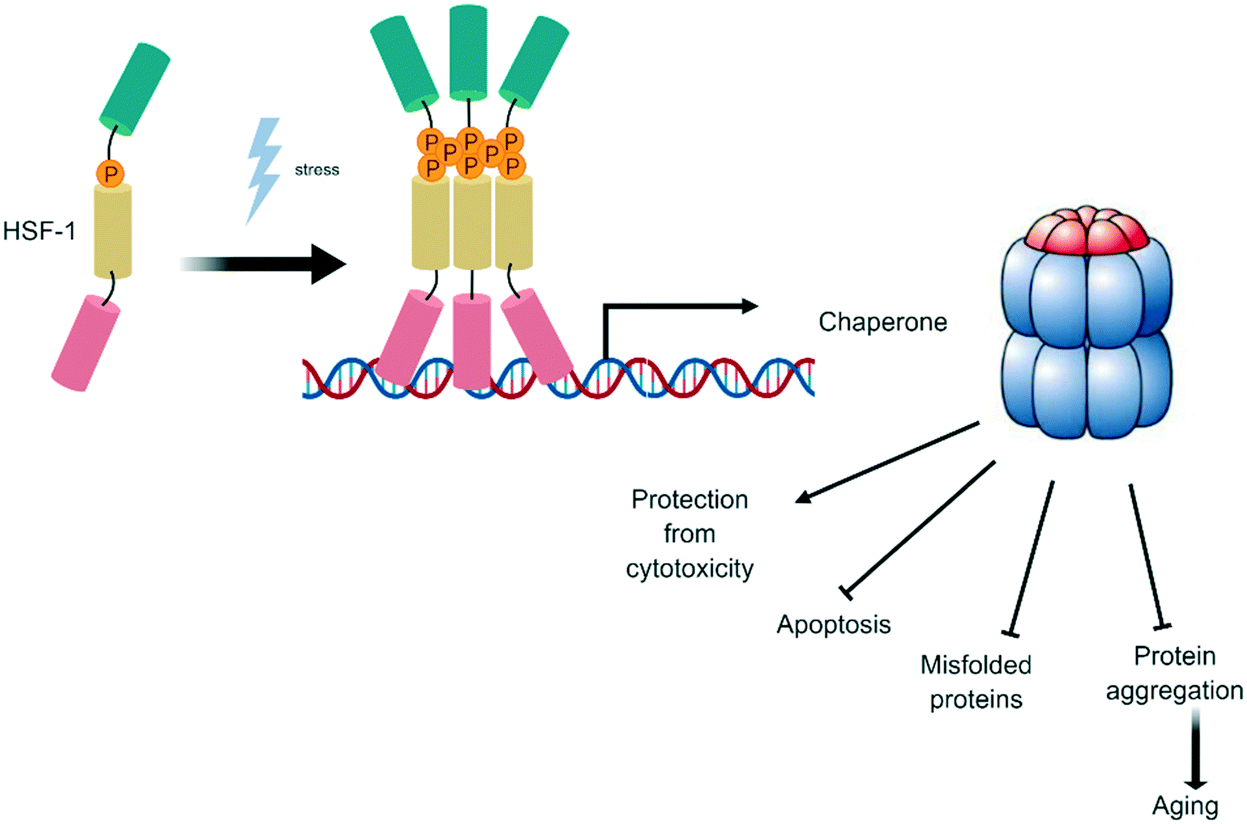 | ||
| Fig. 4 Heat shock factor-1 transcription factor (HSF1): a stress-inducible and DNA-binding transcription factor. | ||
WA was initially identified as a promising antitumor compound based on a HSF1 reporter assay, showing very strong ER-stress inducing properties.56 Remarkably, HSF1 is indicated as potential WA binding target and is able to downregulate HSF-1 controlled genes HSP13, DJC10 and FKBP4 (ref. 4) (see also below). Therefore, it is possible that additional factors also contribute in fine tuning the HSF1-dependent ER-stress response, although binding of WA to HSF-1 as such may also selectively modulate HSF1-dependent gene regulation.
The process of protein folding ensures that proteins have the correct 3D structure since they are produced as unfolded polypeptides with random coiling. Cancer cells are characterized by high proliferation rates, implicating protein synthesis and folding must be performed at high speed. Hence, molecular chaperones that aid in protein folding are crucial for cancer survival making them an attractive antitumor target.57 We defined three functional categories of proteins that are affected by WA treatment: (1) protein chaperone, (2) enzymes (isomerase and reductase activities) helping protein folding and (3) ER associated degradation.
Proteins related to the protein chaperone functioning and that are differentially upregulated upon WA treatment:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Protein unc-45 homolog A | UN45A_HUMAN | Co-chaperone of heat shock protein 90 | 4 |
| Heat shock 70 kDa protein 1A | HS71A_HUMAN | Molecular chaperone essential in protein quality control | 4 |
| Heat shock 70 kDa protein 1B | HS71B_HUMAN | Molecular chaperone essential in protein quality control | 4 |
The protein unc 45 homolog (UNC45A) is a co-chaperone of HSP90 and is required for proper protein folding. Its observed upregulation upon WA treatment is likely a consequence of the induced WA stress, and hence a signal that the cancer cells are trying to insure homeostasis. Similarly, WA is inducing Heat shock 70 kDa protein 1A/1B (HSPA1A and HSPA1B), two major orchestrators of the cellular stress response.58 Members of the HSP70 family control all aspects of cellular proteostasis such as nascent protein chain folding, protein import into organelles, recovering of proteins from aggregation, and assembly of multi-protein complexes.59
Also here, WA seems to play a paradoxal role in the heat shock response by simultaneously increasing HSP-chaperone levels as well as disrupting enzymatic protein folding and degradation salvage pathways.
WA target proteins related to the protein chaperone functioning:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Heat shock protein HSP 90-alpha/beta | HS90A_HUMAN | Maturation and structural maintenance of proteins | 60, 61 |
| HS90B_HUMAN | |||
| DnaJ homolog subfamily A member 2 | DNJA2_HUMAN | Co-chaperone of HSP70 that stimulates ATP hydrolysis | 4 |
| BAG family molecular chaperone regulator 2 | BAG2_HUMAN | Co-chaperone of HSP70 | 4 |
| ER membrane protein complex – subunit 9 | EMC9_HUMAN | Chaperone complex that is relatively recently discovered | 4 |
The binding of WA with heat shock protein HSP 90-α/β (HSP90AA1/HSP90AB1) was already demonstrated in vitro as well as in vivo with pancreatic cancer xenografts60,61 and likely results in impaired protein folding.
DnaJ homolog subfamily A member 2 (DNAJA2) has an essential role in the folding mechanism of HSP70, a heat shock protein strongly upregulated by heat stress and toxic chemicals. A DNAJA2 mutant (DJA2-Δm2) lacking the region between the zinc finger motifs of DNAJA2 is incapable of releasing substrates during its transfer to HSP70 proteins.62 It would be interesting to determine whether binding of WA affects the same region, thereby inhibiting proper DNAJA2 co-activity with HSP70.
Moreover, BAG family molecular chaperone regulator 2 (BAG2) is another co-chaperone of HSP70 that is potentially inhibited by WA. Intriguingly, cancer cells treated with the proteasome inhibitor MG132 showed induction of BAG2 (ref. 63) and this effect was more pronounced in cells sensitive to proteasome inhibitors. Indeed, preventing BAG2 upregulation inhibited the cytotoxicity induced by MG132 and a similar reasoning can be used for WA since it is known for its proteasome inhibiting properties (see section 2.1.2). The explicit mechanisms of BAG2 gene transcriptional activation by proteasome inhibitors and BAG2 promotion of apoptosis still remains to be elucidated.
The role of the WA target protein EMC chaperone complex and its EMC9 subunit remains largely unknown.64 Three EMC9 subunits (KIAA0090, TMEM111 and TTC35) suggest a close link between this complex and ER-associated degradation (ERAD) components implicated in ubiquitin recognition and protein dislocation.65
Finally, in addition to inhibition of chaperone proteins, WA also downregulates heat shock 70 kDa protein 13 (HSPA13), another chaperone protein:
Protein related to the protein chaperone pathway that is differentially downregulated upon WA treatment:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Heat shock 70 kDa protein 13 | HSP13_HUMAN | Role in the processing of cytosolic and secretory proteins | 4 |
Heat shock 70 kDa protein 13 (HSPA13) is a member of the heat shock protein 70 family that plays a crucial role in the processing of cytosolic and secretory proteins, as well as in the removal of denatured or incorrectly folded proteins.59 Interestingly, the upregulation of several HSPs, including HSPA13 in tumor tissues is associated with poor outcomes from early-stage elated hepatocellular carcinoma66 Hence, downregulation of HSPA13 will most likely result in reduced removal of incorrectly folded proteins and prevent stressed cancer cells to insure homeostasis, again illustrating the dual stress response effects of WA.
Whereas HSPs upregulation by WA initially promotes a cytoprotective response, subsequent binding of WA to HSPs undermines this homeostasis and will ultimately trigger cell death.
WA target proteins related to the isomerase and reductase enzymatic activity:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Peptidyl-prolyl cis–trans isomerase D | PPID_HUMAN | Co-chaperone of HSP90 complexes | 4 |
| Peptidyl-prolyl isomerase domain and WD repeat-containing protein 1 | PPWD1_HUMAN | cis–trans isomerization of proline imidic peptide bonds | 4 |
| Peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 | PIN1_HUMAN | Phosphorylation-specific prolyl isomerase | 68 |
Peptidyl-prolyl cis–trans isomerase D (PPID) is a crucial co-chaperone of HSP90 complexes since it accelerates the folding of proteins. Interestingly, upregulation of PPID is associated with anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma (ALK+ ALCL). Treatment with PPID siRNA resulted in the significant reduced viability of ALK+ ALCL cancer cell lines,69 suggesting that WA binding to PPID could also result in decreased cancer cell viability.
Peptidylprolyl isomerase domain and WD repeat-containing protein 1 (PPWD1) is described as part of the spliceosome,70 a nuclear multi-protein structure that cancer cells exploit to exhibit remarkable transcriptome alterations partly by adopting cancer-specific splicing isoforms.71 This suggest WA has potential as splicing disruptor, opening a complete new application field for WA.
Peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 (PIN1) acts as a molecular switch in multiple cellular processes, including cell cycle arrest.72 Molecular docking followed by mass spectrometry indicated covalent interaction of WA with Cys 113 of Pin1, suggesting a crucial role of WA-Pin1 binding in mitotic arrest.68
Furthermore, WA dependent decrease of peptidyl-prolyl cis–trans isomerase FKBP4 (FKBP4) and the disulfide reductase DJC10 (DNAJC10) was observed.
Proteins related to the isomerase and reductase enzymatic activities and that are differentially downregulated upon WA treatment:
The prolyl isomerase FKBP4 forms complexes in mitochondria with the glucocorticoid receptor and the Hsp90/Hsp70-based chaperone hetero-complex.73 Interestingly, FKBP4 overexpression protects cells against oxidative stress, whereas FKBP4 knockdown makes them more sensitive to injury. This suggests that FKBP4 is a major mitochondrial factor and may be related to anti-apoptotic mechanisms triggered during the stress response.73 Hence, downregulation of FKBP4 upon WA treatment will most likely result in reduced protection against oxidative stress.
DNAJC10 is a disulfide reductase that cleaves the disulfide bonds of misfolded proteins, thereby contributing to efficient retro-translocation into the cytosol.74 Downregulation of DNAJC10 upon WA treatment will therefore most likely impact the efficient removal of misfolded proteins and consequently prevent stressed cancer cells to insure homeostasis.
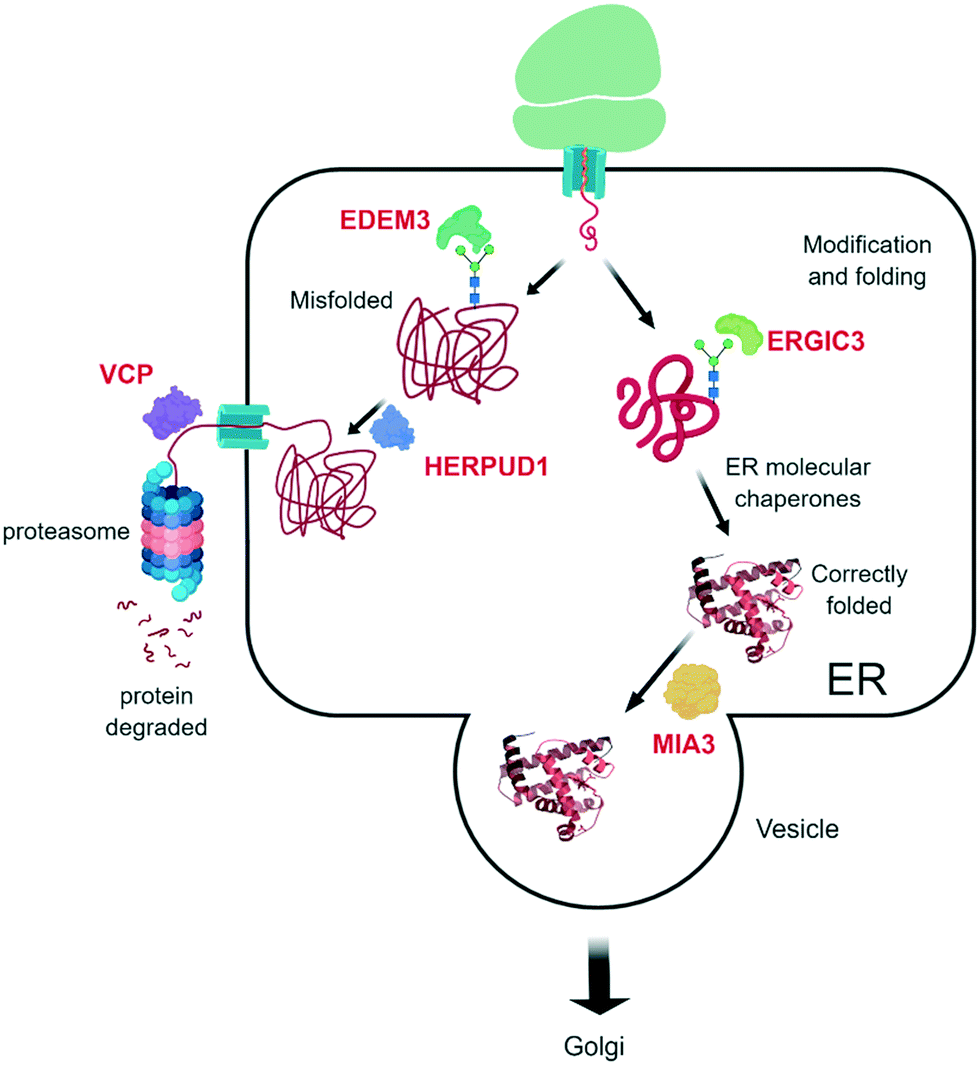 | ||
| Fig. 5 Overview of ERAD. Newly synthesized proteins are guided towards the Golgi structures in case of properly folding. Misfolded proteins are recognized by glycan processing and marked for proteasome degradation. Marked in red are the WA-target proteins (gene names). | ||
Five ERAD associated proteins were identified as WA target proteins:
WA target proteins related to ERAD functioning:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| ER degradation-enhancing alpha-mannosidase-like protein 3 | EDEM3_HUMAN | Catalyzes mannose trimming from Man8GlcNAc2 to Man7GlcNAc2 | 4 |
| AAA+ chaperone p97 (VCP) | TERA_HUMAN | ATPase activity | 39 |
| Endoplasmic reticulum–Golgi intermediate compartment protein 3 | ERGI_HUMAN | Transport between endoplasmic reticulum and Golgi | 4 |
| Homocysteine inducible ER protein with ubiquitin like domain 1 | HERP1_HUMAN | Involved in ubiquitin-dependent degradation of misfolded endoplasmic reticulum proteins | 4 |
| Transport and Golgi organization protein 1 homolog | TGO1_HUMAN | Involved in the export process exported from the endoplasmic reticulum | 4 |
Through the binding of WA with ER degradation-enhancing alpha-mannosidase-like protein 3 (EDEM3), WA potentially inhibits the removal of terminally misfolded proteins, thereby hampering the proper stress response. To further remove misfolded proteins to the UPS, a translocation between the ER and the cytosol, where the UPS is located, must happen. This is mediated by VCP and its ATPase activity,76 and different WA analogues have already demonstrated their VCP inhibiting effects.39 Finally, the terminally misfolded proteins are degraded by the 26S proteasome, a system that is also inhibited by WA (see above).
Also the homocysteine inducible ER protein with ubiquitin like domain 1 (HERPUD1), an ERAD component, was identified as a potential WA target. HERPUD1 is responsible for protein repair/removal via the chaperone stress response.77 Therefore, impairment of its function will likely lead to accumulation of misfolded proteins and consequently cell death.
Endoplasmic reticulum–Golgi intermediate compartment protein 3 (ERGIC3) is also identified as a WA target.4 ERGIC3 is possibly involved in the protein transport between endoplasmic reticulum and Golgi and is associated with poor clinical outcome in lung cancer patients. Knockdown of ERGIC3 led to ER stress-induced autophagy cell death and suppression of proliferation of the A549 human lung cancer cell line.78
Finally, the transport and Golgi organization protein 1 homolog (MIA3) is targeted by WA.4 MIA3 is critical for the organization of ER exit sites since its depletion disassembles COPII components that are part of a vesicle coat protein, as well as membrane-bound ER-resident complexes, resulting in fewer functional ER exit sites and delayed transit of folded proteins.79 Binding of WA to MIA3 could therefore explain the suggested accumulation of misfolded proteins and the subsequent cell death. Altogether, the multiple WA target proteins in the ER degradation pathway and other stress pathways make it extremely hard for cancer cells to escape its antitumor action and/or develop therapy resistance.
2.3 Protein translation
During the WA-modulated acute cellular stress response, protein translation must be controlled to prevent accumulation of newly synthesized misfolded proteins.80 WA also has broad effects on the protein translation machinery like upregulation of the eukaryotic translation initiation factor 2A (EIF2A):Protein related to the protein translation process and that is differentially upregulated upon WA treatment:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Eukaryotic translation initiation factor 2A | EIF2A_HUMAN | Translation initiation factor of specific mRNA's | 4 |
Interestingly, EIF2A is required for translation under stress conditions and the knockdown of eIF2A almost completely inhibited proliferation of cells under stress but not under normal conditions.81 Similarly, WA-induced cellular stress initially tries to maintain translation by increasing EIF2A expression. However, subsequent binding of WA to eukaryotic translation initiation factors like IF5A1 and IF4B will finally ultimately trigger cell death.
WA target proteins related to protein translation process:
Eukaryotic translation initiation factor 5A-1 (eIF5A1) is essential for sustained proliferation of mammalian cells and is intimately involved in malignant cell transformation.82 Indeed, eIF5A1 is widely overexpressed in cancer cells and is termed ‘the eIF5A regulon’, referring to the eIF5A1 control of a translational network of cancer driving genes in cervical cancer, thereby contributing to cervical cancer cell proliferation.83 As a consequence, eIF5A1 targeting and inactivation is explored in numerous cancer types, including leukemia,84 pancreatic cancer85 and hepatocellular carcinoma.86
Eukaryotic translation initiation factor 4B (eIF4B) is also essential for cellular survival and proliferation and is regulated by proto-oncogenic signaling pathways.87 Reducing eIF4B expression alone is sufficient to decrease synthesis of proteins associated with enhanced tumor cell survival, including DAXX, BCL2 and ERCC5.88 Moreover, eIF4B is described as a convergent target and critical effector of oncogenic Pim and PI3K/Akt/mTOR signaling pathways, making it a promising therapeutic target in leukemia tumors.89
Finally, the downregulation of eukaryotic peptide chain release factor subunit 1 (ETF1) and eukaryotic translation initiation factor 3 subunit M (EIF3M) was demonstrated:
Proteins related to the protein translation process and that are differentially downregulated upon WA treatment:
Eukaryotic peptide chain release factor subunit 1 (ETF1) is required for stop codon recognition and translational termination. Eukaryotic translation initiation factor 3 subunit M (EIF3M), a component of the eukaryotic translation initiation factor 3 complex, was confirmed to be highly expressed in human cancer cell lines and colon cancer patient tissues. By expression silencing with EIF3M-specific small interfering RNA, EIF3M was shown to influence cell proliferation, cell cycle progression and cell death in the human colon cancer cell line HCT-116.90
2.4 The dual action of WA cellular stress pathways
The higher metabolism of cancer cells will lead to higher oxidative radical production and consequently to increased oxidative stress. Further increasing this stress level by WA treatment in combination with blocking the corresponding stress protective pathways may cause an exponential buildup of stress beyond its maximum tolerance. Eventually, the WA-induced stress may drive a cancer cell beyond its limits of homeostasis such that cellular functions may collapse upon overload of metabolic stress.Above a certain threshold ROS-triggered mitochondrial permeability transition may occur, resulting in the release of cytochrome c and other pro-apoptotic proteins, which can trigger caspase activation and apoptosis.91,92 Alternatively, excessive ROS levels may exceed homeostatic capacity of cytoprotective redox responses (Nrf2, KEAP1, HMOX1, GPX proteins) and result in irreversible lipid peroxidation damage and ferroptosis.8 Because of the lower basal metabolic rate in normal cells (compared to cancer cells), these cells are more tolerant for WA-induced stress since homeostatic limits are not reached (i.e. oxidative stress below threshold), thereby explaining the selective cytotoxic effect of WA towards cancer cells. The existence of such a threshold could also explain the frequently observed dose-dependent effects of WA: whereas sub-toxic concentrations of WA may promote a homeostatic cytoprotective Nrf2 response to moderate electrophilic oxidative stress, cancer cytotoxic concentrations of WA may trigger a vicious circle of ROS production, which ultimately will result in cellular collapse and cancer cell death.
Based on our chemoproteomic approach, it becomes clear that WA binds to multiple proteins targeting different protein signaling networks. To our opinion, the synergistic polypharmaceutical effects in response to WA make it difficult to simplify its anti-cancer mechanism to one or a few “primary” protein targets (Fig. 6).
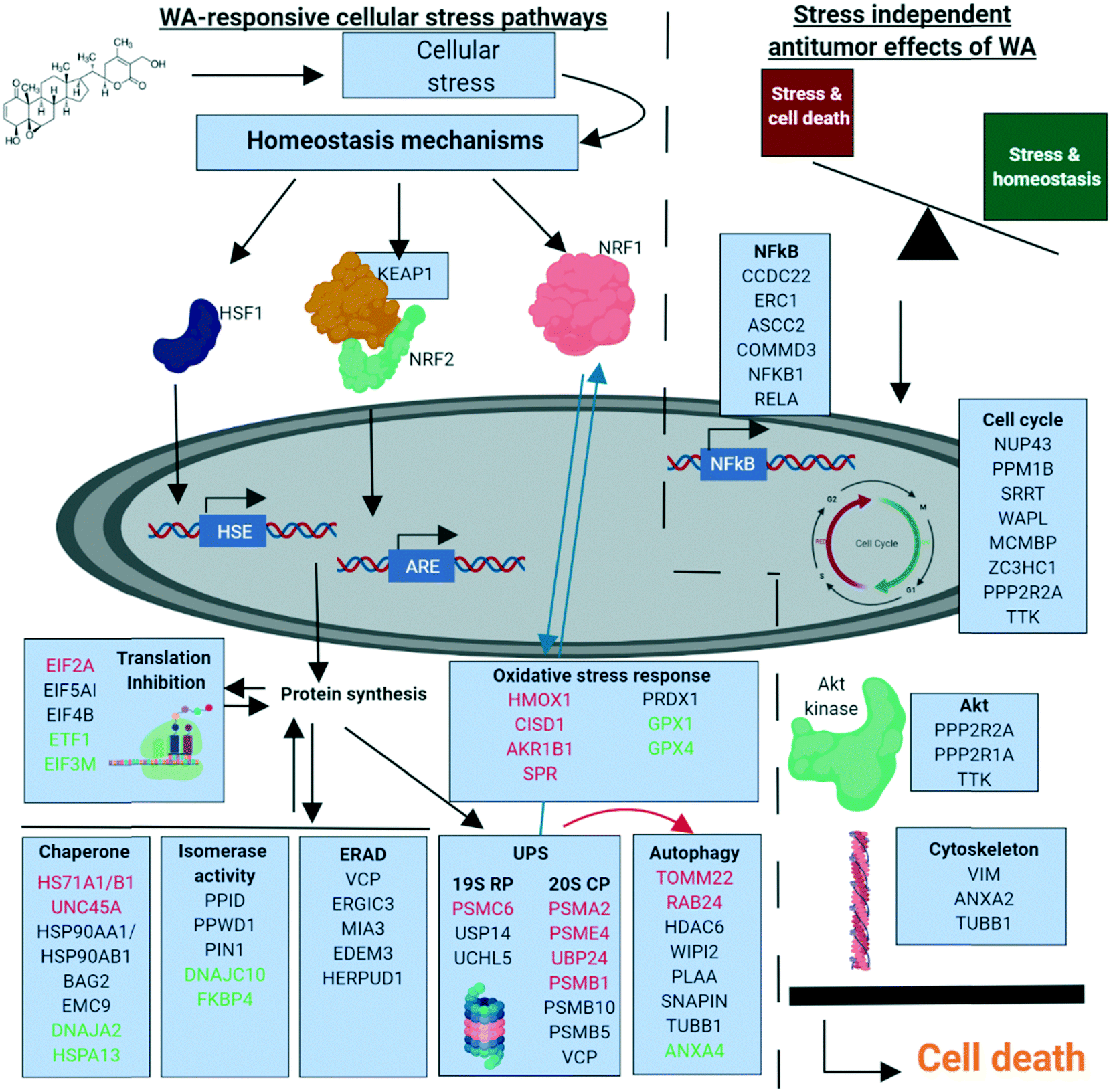 | ||
| Fig. 6 Overview of the proposed WA antitumor mechanism based on stress dependent (left) and – independent (right) effects. All proteins are represented by their gene names. Black = WA target; red = upregulated protein expression upon WA treatment; green = downregulated protein expression upon WA treatment. RP: regulatory particle. CP: core particle. | ||
3 Stress independent antitumor effects of WA
So far we described the quick stress response induced by WA in cancer cells. The eventual cellular fate, live or death, is dose dependent and co-determined by the basal stress levels and the efficiency of stress reduction by salvage pathways. However, we hypothesize that in sub-cytotoxic conditions, WA can still elicit slow therapeutic antitumor effects of WA which are only measurable after hours to days of incubation (in contrast to the quick acute effects observed minutes to a few hours after WA incubation). In this subchapter, we therefore aim to explain these effects on the basis of newly identified WA target proteins based on proteomic strategies. For global reviews on already established targets and secondary antitumor mechanisms of WA, we refer to.1,2 Below, the four most widely described antitumor effects are considered.3.1 Disruption of cytoskeleton functions
Probably the best characterized antitumor effects of WA are assigned to its cytoskeleton binding properties. Vimentin, annexin A2 and β-tubulin are well established WA targets:WA target proteins related to the cytoskeleton:
Vimentin (VIM) is responsible for supporting and anchoring organelles in the cytosol through attachment of nucleus, endoplasmic reticulum and mitochondria. As a result of the binding of WA, vimentin assembly is prevented, consequently inhibiting intermediate filament organization and subsequent loss of cell stability and eventual viability.108 Many subsequent effects of the WA–vimentin binding have been described and include phosphorylation events, cell cycle arrest and migration and invasiveness events.
Annexin A2 (ANXA2) supports diverse cellular function such as endocytosis, adhesion and migration. It is a calcium-dependent phospholipid binding protein that is aberrantly expressed in a wide range of cancers, participating in tumor cell adhesion, -proliferation and metastasis.109–111 The covalent binding of WA to the annexin A2 core domain results in the decrease of actin polymerization and subsequent limited migratory and invasive capabilities of the cancer cells.106
β-Tubulin (TUBB1) is the third WA target protein that is involved in maintaining cytoskeleton architecture. Together with α-tubulin, it is a major component of the microtubules, a hollow cylindrical structure critical for cell cycle progression (mitosis), cellular movement (cilia and flagella) and intracellular trafficking.112 WA–β-tubulin binding occurs at Cys303 of β-tubulin, as shown by mass spectrometry and in silico docking.43 Additional, downregulation of β-tubulin upon WA treatment was also observed in the same study. This resulted in reduced cell stability and eventually cell death. Hence, targeting the cytoskeleton architecture, especially important in cancer metastasis, undermine the mechano-structural integrity of the cancer cells and lead to cell death.113
3.2 Disruption of cell cycle functions
Oscillations in oxygen consumption, energy metabolism and redox state are intimately integrated with cell cycle progression,114 with S- to M phase transition occurring only during the reductive phase of metabolism and G1 in the oxidative phase (Fig. 7).115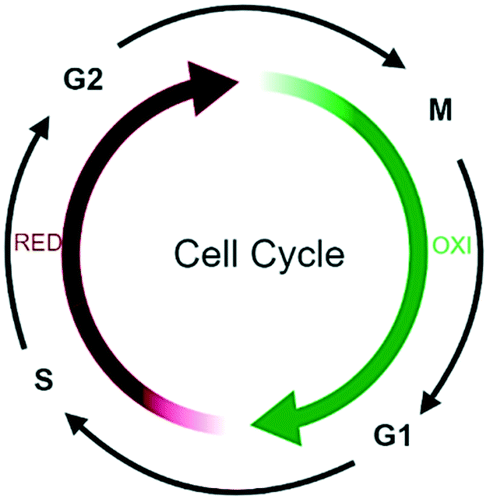 | ||
| Fig. 7 Schematic overview of the coupling between redox state of the metabolism and the cell cycle. | ||
For example, in G1, ROS stimulate mitogenic pathways that control the activity of cyclin-dependent kinases (CDKs) and phosphorylation of the retinoblastoma protein (pRB), thereby regulating S-phase entry.116
The G2/M transition is subjected to strict quality control by several checkpoint pathways and many reactive compounds that induce oxidative stress also cause cell cycle arrest in G2/M. For example, primary epithelial cultures deficient in the Nrf2 transcription factor accumulate oxidative damage and arrest in G2/M.117 A bioinformatics strategy used to identify cell cycle proteins with ROS sensitive motifs, identified 92 redox sensitive cell cycle related proteins from which 70% of these function in G2/M cell cycle regulation.118 Many downstream proteins of the Nrf2 transcription factor are influenced by WA and possibly cause an exponential increase in oxygen radicals upon WA treatment (see above). This implicates that the observed cell cycle inhibitory effects of WA treatment are potentially due to the increase of ROS concentrations by WA.
Many studies on the antitumor properties of WA observed cell cycle arrest at the G2/M and G1/S transition upon WA treatment,119–122 but the corresponding molecular targets remained unidentified. Eight WA target proteins were identified:
WA target proteins related to the cell cycle:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Dual specificity protein kinase TTK | TTK_HUMAN | Kinase essential for chromosome alignment | 4 |
| Nucleoporin Nup43 | NUP43_HUMAN | Involved in kinetochore microtubule attachment, mitotic progression and chromosome segregation | 4 |
| Protein phosphatase 1B | PPM1B_HUMAN | Phosphatase | 4 |
| Serrate RNA effector molecule homolog | SRRT_HUMAN | Mediator between the cap-binding complex (CBC) and the primary microRNAs (miRNAs) processing machinery during cell proliferation | 4 |
| Wings apart-like protein homolog | WAPL_HUMAN | Regulator of sister chromatid cohesion in mitosis | 4 |
| Nuclear-interacting partner of ALK | NIPA_HUMAN | Controls entering mitotic phase | 4 |
| Mini-chromosome maintenance complex-binding protein | MCMBP_HUMAN | Associated component of the MCM complex that acts as a regulator of DNA replication | 4 |
| Serine/threonine-protein phosphatase 2A 55 kDa regulatory subunit B, alpha isoform | 2ABA_HUMAN | Phosphatase. The B regulatory subunit modulates substrate selectivity and catalytic activity | 4 |
Dual specificity protein kinase TTK (TTK) is a kinase essential for chromosome alignment at the centromere and the mitotic checkpoint. It regulates Cdc25 proteins in order to maintain checkpoint control.123 The binding of WA to TKK could therefore explain the observed Cdc25 downregulation and accompanied mitotic arrest upon WA treatment in ovarian carcinoma.124
Nucleoporin Nup43 (NUP43), a component of the Nup107-160 sub-complex of the nuclear pore complex (NPC), is a complex required for the assembly of a functional NPC, normal kinetochore microtubule attachment, mitotic progression and chromosome segregation.125 The binding of WA to NUP43 and subsequent inhibition could therefore explain the observed G2/M attenuation upon WA treatment as a result of impaired kinetochore formation linked to impaired Spindle Assembly Checkpoint modulation.120,126
Protein phosphatase 1B (PPM1B) is an enzyme with broad specificity. For example, PPM1B dephosphorylates CDK2 and subsequently activates it. Binding of WA to PPM1B could therefor result in continued phosphorylation of CDK2, its continued inhibition and subsequent G1/S attenuation.93
Serrate RNA effector molecule homolog (SRRT) acts as a mediator between the cap-binding complex (CBC) and the primary microRNAs (miRNAs) processing machinery during cell proliferation. SRRT contributes to the stability and delivery of capped primary miRNA transcripts to the primary miRNA processing complex, thereby playing a role in RNA-mediated gene silencing (RNAi). SRRT is involved in cell cycle progression at the S-phase and is crucial in regulating transcription. Binding of WA and SRRT could suggest the cell cycle is halted at the S-phase due to the binding inhibition, like observed upon WA treatment.127
Wings apart-like protein homolog (WAPL) is involved in both sister chromatid cohesion during interphase and sister-chromatid resolution during early stages of mitosis. Binding of WA and anticipated inhibition of WAPL could therefore explain the observed G2/M arrest upon WA treatment as a result of mitotic delay by blocking spindle assembly checkpoint function in colorectal cancer.120
Nuclear-interacting partner of ALK (NIPA) is an essential protein that controls mitotic entry by mediating ubiquitination and subsequent degradation of the cell cycle regulatory protein cyclin B1 (CCNB1).128 NIPA inactivation by WA binding could therefore result in nuclear accumulation of CCNB1 in the interphase and premature mitotic entry, thereby deregulating the entire cell cycle.
Mini-chromosome maintenance complex-binding protein (MCMBP) is an associated component of the MCM complex that acts as a regulator of DNA replication. It binds to the MCM complex during the late S-phase and promotes the disassembly of the MCM complex from chromatin, thereby acting as a key regulator of pre-replication complex unloading from replicated DNA. WA-binding and inhibition of MCMBP could explain the observed halted G2/M entry upon WA treatment of leukemia cell lines in a does dependent manner.121
The phosphothreonine preference of serine/threonine-protein phosphatase 2A 55 kDa regulatory subunit B, alpha isoform (PPP2A2R) provides an essential regulatory element of mitotic exit.129 The B regulatory subunit modulates substrate selectivity and catalytic activity, and also might direct the localization of the catalytic enzyme to a particular subcellular compartment. WA binding and inhibition of PPP2R2A could therefore explain the observed halted mitotic phase upon WA treatment in breast cancer cell lines.122
Although most studies report G2/M cell cycle arrest upon WA treatment, some studies also found WA-induced G0/G1 phase arrest.93 Most presumably, context dependent factors such as the used cancer cell model, WA concentration and cell culture differences in cell cycle synchronization may explain variations in the observed cell cycle effects.
On the other hand, since it is shown that WA targets the activity of Hsp90,38,61,130 it is hypothesized that WA treatment results in the decreased expression of the Hsp90 client proteins, including Cdk1 and Cdk4. This will affect different phases of the cell cycle, but does not have a significant effect on non-client proteins of Hsp90 including Cdk2 or cyclin A1. Noteworthy, many client proteins of Hsp90 are involved in different stages of the cell cycle and may be unique to different tumor models, thus explaining the observed differences.130
3.3 Disruption of kinase functions
Protein kinase B (PKB), also known as Akt, is a serine/threonine-specific protein kinase that plays a key role in multiple cellular processes such as glucose metabolism, cell proliferation, transcription, cell migration and apoptosis. Many studies on the antitumor properties of WA observed Akt inhibition in the PI3K/AKT/mTOR pathway,131 a pathway that is a crucial regulator (e.g. proliferation) in cancer. However the corresponding molecular targets remained unidentified. Three upstream WA target proteins related to Akt regulation, which were also involved in cell cycle regulation (see above), were identified.WA target proteins related to the Akt pathway:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| TTK protein kinase | TTK_HUMAN | Involved in chromosome alignment | 4 |
| Serine/threonine-protein phosphatase 2A 55 kDa regulatory subunit B alpha isoform | 2ABA_HUMAN | Phosphatase. The B regulatory subunit modulates substrate selectivity and catalytic activity | 4 |
| Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform | 2AAA_HUMAN | Phosphatase. The B regulatory subunit modulates substrate selectivity and catalytic activity | 132 |
TTK is essential for chromosome alignment and functions as a cell cycle regulator.133,134Via direct CDCA8 phosphorylation, TTK enhance AURKB activity at the centromere, and for the mitotic checkpoint. Besides its function as a cell cycle regulator, TTK also activates Akt.135 Assuming that WA binding results in TTK inhibition, this in turn could decrease AURKB136 and Akt kinase activities as has been observed.137
PPP2R2A is also described to sustain the hyperactivity of multiple oncogenic signaling pathways, including Akt and ERK.138 The observed binding of WA to PPP2R2A therefore suggests an alternative mode of action for the observed Akt downregulation upon WA treatment.137 Also PPP2R1A is described as WA target. Binding of WA to the Cys 377 is suggested to lead to PPP2A activation and subsequent inhibition of Akt signaling and breast cancer cell proliferation.132
3.4 Disruption of NFκB signaling pathways
The NFκB pathway is intensively studied as it proves a key pathway dysregulated in many types of cancer.139,140 Today, many antitumor properties of WA are attributed to its NFκB inhibiting effects, including covalent Cys179 binding of WA and inhibition of the IKK2 kinase responsible for IκB phosphorylation and NFκB activation.141,142 This downregulates gene transcription of various downstream target genes (i.e. IL6, IL8, MCP1, etc.) involved in inflammation. Interestingly, various additional WA target proteins involved in the NFκB pathway, were identified, thereby further supporting WA's role as a poly-pharmaceutical NFκB inhibitor.WA target proteins related to the NFκB signaling:
| Protein name | Uniprot accession | Function | Ref. |
|---|---|---|---|
| Coiled-coil domain-containing protein 22 | CCD22_HUMAN | Promoting IκB ubiquitination | 4 |
| Activating signal co-integrator 1 complex subunit 2 | ASCC2_HUMAN | Trans-activator of NFκB | 4 |
| ELKS/Rab6-interacting/CAST family member 1 | RB6I2_HUMAN | Regulatory subunit of IKK | 4 |
| COMM domain-containing protein 3 | COMD3_HUMAN | Downregulates activation of NFκB | 4 |
| Transcription factor p65 (RelA) | TF65_HUMAN | Transcription factor | 143 |
| Nuclear factor NFκB p105 subunit (p50) | NFκB1_HUMAN | DNA binding | 4, 144 |
Coiled-coil domain-containing protein 22 (CCDC22) promotes ubiquitination of IκB and its subsequent proteasomal degradation leading to NFκB activation.145 WA binding to CCD22 and subsequent CCD22 inactivation may therefore prevent NFκB activation.
Activating signal co-integrator 1 complex subunit 2 (ASCC2) is another promising WA target. It is described to enhance NFκB transactivation, but also that of other crucial transcription factors like activator protein 1 (AP-1) which regulates gene expression in response to a variety of stimuli, including cytokines, growth factors, stress and bacterial and viral infections thereby controlling a number of cellular processes including differentiation, proliferation and apoptosis,146 all of which could be inhibited after binding of WA to ASCC2, leading to a high level of cell dysregulation.
Interestingly, we also identified ELKS/Rab6-interacting/CAST family member 1 (ERC1) as a potential WA target protein.4 ERC1 is a regulatory subunit of the IKK complex which recruits IKK1 to the IKK2 complex.147 The binding and ERC1 inhibition supports the hypothesis that WA inhibits NFκB activity through blocking the formation of active IKK complexes.148 Also NFκB subunits RelA (p65) and p50 were suggested to bind WA.143,144
Finally, not all identified potential WA target proteins may contribute to the observed NFκB inhibition. For example, COMM domain-containing protein 3 (COMMD3) downregulates the activity of NFκB,149 hence it remains unclear how WA binding of this protein affects cellular NFκB activity.
4 Conclusion
Although various antitumor properties of WA have already been reported, it has been difficult do discriminate direct and indirect WA effects. We hereby propose that WA triggers two waves of antitumor responses: (I) a quick cytoprotective oxidative stress response, followed by elimination of stress salvage pathways which may lead to a quick cellular collapse and (II) a slow response during which WA targets multiple cancer hallmarks such as inhibition of cell cycle progression, suppression of metastatic motility through cytoskeletal disruption and anti- inflammatory signaling via NFκB pathway inhibition and Akt downregulation. The early phase directly kills cancer cells through excessive production of reactive oxygen radicals and lipid peroxidation damage, beyond the point of no return, due to failure of the cellular repair systems. The later phase kills cancer cells indirectly through suppression of several essential cellular cancer cell proliferation and survival pathways and processes.To our knowledge, this is the first proteomic perspective on WA-specific cancer therapeutic effects obtained across manifold experimental setups. Various pre-clinical studies in vivo support its pleiotropic effects to overcome therapy resistance.150 Nano-targeted delivery systems or prodrug formulations may further improve its antitumor efficacy and/or selectivity.151 Dose limiting toxicity, safety and pharmacokinetics have been recently evaluated in one clinical phase I study in osteosarcoma patients receiving a standardized root extract of the plant (Withania somnifera) containing 4.5% of WA (w/w). WA formulation was well tolerated, but showed low oral bioavailability and warrants further studies with improved formulations.152
The molecular roadmap described here underscoring the broad antitumor action of WA, may help us in the rational selection of other (FDA approved) antitumor compounds from which combination with WA can be predicted to synergistically boost their clinical efficacy. In addition, design of new antitumor targets on the basis of the proposed WA antitumor target network becomes possible and may hold an exciting potential for development of novel antitumor treatments.
Conflicts of interest
The authors declare no potential conflicts of interest.Acknowledgements
This research was supported by FWO grants G059713N and G079614N.References
- W. Vanden Berghe, L. Sabbe, M. Kaileh, G. Haegeman and K. Heyninck, Molecular insight in the multifunctional activities of Withaferin A, Biochem. Pharmacol., 2012, 84, 1282–1291, DOI:10.1016/j.bcp.2012.08.027.
- A. R. Vyas and S. V. Singh, Molecular targets and mechanisms of cancer prevention and treatment by withaferin a, a naturally occurring steroidal lactone, AAPS J., 2014, 16, 1–10, DOI:10.1208/s12248-013-9531-1.
- P. Bargagna-Mohan, A. Hamza, Y. E. Kim, Y. Khuan, N. Mor-Vaknin, N. Wendschlag, J. Liu, R. M. Evans, D. M. Markovitz, C. G. Zhan, K. B. Kim and R. Mohan, The Tumor Inhibitor and Antiangiogenic Agent Withaferin A Targets the Intermediate Filament Protein Vimentin, Chem. Biol., 2007, 14, 623–634, DOI:10.1016/j.chembiol.2007.04.010.
- M. Dom, F. Offner, W. Vanden Berghe and X. Van Ostade, Proteomic characterization of Withaferin A-targeted protein networks for the treatment of monoclonal myeloma gammopathies, J. Proteomics, 2018, 179, 17–29, DOI:10.1016/j.jprot.2018.02.013.
- K. A. Smith, G. B. Waypa and P. T. Schumacker, Redox signaling during hypoxia in mammalian cells, Redox Biol., 2017, 13, 228–234, DOI:10.1016/j.redox.2017.05.020.
- H.-W. Chang, R.-N. Li, H.-R. Wang, J.-R. Liu, J.-Y. Tang, H.-W. Huang, Y.-H. Chan and C.-Y. Yen, Withaferin A Induces Oxidative Stress-Mediated Apoptosis and DNA Damage in Oral Cancer Cells, Front. Physiol., 2017, 8, 634, DOI:10.3389/fphys.2017.00634.
- F. Malik, A. Kumar, S. Bhushan, S. Khan, A. Bhatia, K. A. Suri, G. N. Qazi and J. Singh, Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic cell death of human myeloid leukemia HL-60 cells by a dietary compound withaferin A with concomitant protection by N-acetyl cysteine, Apoptosis, 2007, 12, 2115–2133, DOI:10.1007/s10495-007-0129-x.
- B. Hassannia, B. Wiernicki, I. Ingold, F. Qu, S. Van Herck, Y. Y. Tyurina, H. Bayir, B. A. Abhari, J. P. F. Angeli, S. M. Choi, E. Meul, K. Heyninck, K. Declerck, C. S. Chirumamilla, M. Lahtela-Kakkonen, G. Van Camp, D. V. Krysko, P. G. Ekert, S. Fulda, B. G. De Geest, M. Conrad, V. E. Kagan, W. Vanden Berghe, P. Vandenabeele and T. Vanden Berghe, Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma, J. Clin. Invest., 2018, 128, 3341–3355, DOI:10.1172/JCI99032.
- K. Ghosh, S. De, S. Das, S. Mukherjee and S. Sengupta Bandyopadhyay, Withaferin A Induces ROS-Mediated Paraptosis in Human Breast Cancer Cell-Lines MCF-7 and MDA-MB-231, PLoS One, 2016, 11, e0168488, DOI:10.1371/journal.pone.0168488.
- M. H. Raza, S. Siraj, A. Arshad, U. Waheed, F. Aldakheel, S. Alduraywish and M. Arshad, ROS-modulated therapeutic approaches in cancer treatment, J. Cancer Res. Clin. Oncol., 2017, 143(9), 1789–1809, DOI:10.1007/s00432-017-2464-9.
- D. P. Jones, Redefining oxidative stress, Antioxid. Redox Signaling, 2006, 8, 1865–1879, DOI:10.1089/ars.2006.8.1865.
- J. F. Woolley, J. Stanicka and T. G. Cotter, Recent advances in reactive oxygen species measurement in biological systems, Trends Biochem. Sci., 2013, 38, 556–565, DOI:10.1016/j.tibs.2013.08.009.
- T. P. A. Devasagayam, J. C. Tilak, K. K. Boloor, K. S. Sane, S. S. Ghaskadbi and R. D. Lele, Free radicals and antioxidants in human health: current status and future prospects, J. Assoc. Physicians India, 2004, 52, 794–804 CAS.
- J. N. Moloney and T. G. Cotter, ROS signalling in the biology of cancer, Semin. Cell Dev. Biol., 2017, 1–15, DOI:10.1016/j.semcdb.2017.05.023.
- X. Liu, L. Chen, T. Liang, X.-D. Tian, Y. Liu and T. Zhang, Withaferin A induces mitochondrial-dependent apoptosis in non-small cell lung cancer cells via generation of reactive oxygen species, J. BUON, 2017, 22, 244–250 Search PubMed.
- A.-X. Li, M. Sun and X. Li, Withaferin-A induces apoptosis in osteosarcoma U2OS cell line via generation of ROS and disruption of mitochondrial membrane potential, Riv. Eur. Sci. Med. Farmacol., 2017, 21, 1368–1374, DOI:10.4103/0973-1296.211042.
- E. Mayola, C. Gallerne, D. D. Esposti, C. Martel, S. Pervaiz, L. Larue, B. Debuire, A. Lemoine, C. Brenner and C. Lemaire, Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2, Apoptosis, 2011, 16, 1014–1027, DOI:10.1007/s10495-011-0625-x.
- J.-Y. Tang, A. A. Farooqi, F. Ou-Yang, M.-F. Hou, H.-W. Huang, H.-R. Wang, K.-T. Li, S. Fayyaz, C.-W. Shu and H.-W. Chang, Oxidative stress-modulating drugs have preferential anticancer effects - involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration, Semin. Cancer Biol., 2019, 58, 109–117, DOI:10.1016/j.semcancer.2018.08.010.
- K. Heyninck, L. Sabbe, C. S. Chirumamilla, K. Szarc Vel Szic, P. Vander Veken, K. J. A. Lemmens, M. Lahtela-Kakkonen, S. Naulaerts, K. Op De Beeck, K. Laukens, G. Van Camp, A. R. Weseler, A. Bast, G. R. M. M. Haenen, G. Haegeman and W. Vanden Berghe, Withaferin A induces heme oxygenase (HO-1) expression in endothelial cells via activation of the Keap1/Nrf2 pathway, Biochem. Pharmacol., 2016, 109, 48–61, DOI:10.1016/j.bcp.2016.03.026.
- Y. Huang, W. Li, Z. Su and A.-N. T. Kong, The complexity of the Nrf2 pathway: beyond the antioxidant response, J. Nutr. Biochem., 2015, 26, 1401–1413, DOI:10.1016/j.jnutbio.2015.08.001.
- R. Mittler, ROS Are Good, Trends Plant Sci., 2017, 22, 11–19, DOI:10.1016/j.tplants.2016.08.002.
- E.-R. Hahm, M. B. Moura, E. E. Kelley, B. Van Houten, S. Shiva and S. V. Singh, Withaferin A-Induced Apoptosis in Human Breast Cancer Cells Is Mediated by Reactive Oxygen Species, PLoS One, 2011, 6, e23354, DOI:10.1371/journal.pone.0023354.
- D. D. Zhang and M. Hannink, Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress, Mol. Cell. Biol., 2003, 23, 8137–8151 CrossRef CAS PubMed.
- S. S. Cao and R. J. Kaufman, Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease, Antioxid. Redox Signaling, 2014, 21, 396–413, DOI:10.1089/ars.2014.5851.
- M. Narayan, K. W. Seeley and U. K. Jinwal, Identification and quantitative analysis of cellular proteins affected by treatment with withaferin a using a SILAC-based proteomics approach, J. Ethnopharmacol., 2015, 175, 86–92, DOI:10.1016/j.jep.2015.09.024.
- Q. Zhao, Y. Ding, Z. Deng, O.-Y. Lee, P. Gao, P. Chen, R. J. Rose, H. Zhao, Z. Zhang, X.-P. Tao, A. J. R. Heck, R. Kao and D. Yang, Natural products triptolide, celastrol, and withaferin A inhibit the chaperone activity of peroxiredoxin I †Electronic supplementary information (ESI) available: Chemical synthesis and characterization of triptolide probes; Fig. S1–S10; Table S1–S3. See D, Chem. Sci., 2015, 6, 4124–4130, 10.1039/c5sc00633c.
- H. A. Woo, H. Z. Chae, S. C. Hwang, K.-S. Yang, S. W. Kang, K. Kim and S. G. Rhee, Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation, Science, 2003, 300, 653–656, DOI:10.1126/science.1080273.
- F. Saccoccia, P. Di Micco, G. Boumis, M. Brunori, I. Koutris, A. E. Miele, V. Morea, P. Sriratana, D. L. Williams, A. Bellelli and F. Angelucci, Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin, Structure, 2012, 20, 429–439, DOI:10.1016/j.str.2012.01.004.
- H. H. Jang, K. O. Lee, Y. H. Chi, B. G. Jung, S. K. Park, J. H. Park, J. R. Lee, S. S. Lee, J. C. Moon, J. W. Yun, Y. O. Choi, W. Y. Kim, J. S. Kang, G.-W. Cheong, D.-J. Yun, S. G. Rhee, M. J. Cho and S. Y. Lee, Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function, Cell, 2004, 117, 625–635, DOI:10.1016/j.cell.2004.05.002.
- A. Hall, K. Nelson, L. B. Poole and P. A. Karplus, Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins, Antioxid. Redox Signaling, 2011, 15, 795–815, DOI:10.1089/ars.2010.3624.
- E. Lubos, J. Loscalzo and D. E. Handy, Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities, Antioxid. Redox Signaling, 2011, 15, 1957–1997, DOI:10.1089/ars.2010.3586.
- M.-K. Kwak, N. Wakabayashi, J. L. Greenlaw, M. Yamamoto and T. W. Kensler, Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway, Mol. Cell. Biol., 2003, 23, 8786–8794, DOI:10.1128/MCB.23.23.8786-8794.2003.
- Z. Sha and A. L. Goldberg, Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97, Curr. Biol., 2014, 24, 1573–1583, DOI:10.1016/j.cub.2014.06.004.
- S. Khan, A. W. Rammeloo and J. J. Heikkila, Withaferin A Induces Proteasome Inhibition, Endoplasmic Reticulum Stress, the Heat Shock Response and Acquisition of Thermotolerance, PLoS One, 2012, 7(11), e50547, DOI:10.1371/journal.pone.0050547.
- L. Zhang, L. Nemzow, H. Chen, A. Lubin, X. Rong, Z. Sun, T. K. Harris and F. Gong, The Deubiquitinating Enzyme USP24 Is a Regulator of the UV Damage Response, Cell Rep., 2015, 10, 140–147, DOI:10.1016/j.celrep.2014.12.024.
- P. A. J. Muller and K. H. Vousden, p53 mutations in cancer, Nat. Cell Biol., 2013, 15, 2–8, DOI:10.1038/ncb2641.
- R. Munagala, H. Kausar, C. Munjal and R. C. Gupta, Withaferin a induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells, Carcinogenesis, 2011, 32, 1697–1705, DOI:10.1093/carcin/bgr192.
- A. Grover, A. Shandilya, V. S. Bisaria and D. Sundar, Probing the anticancer mechanism of prospective herbal drug Withaferin A on mammals: a case study on human and bovine proteasomes, BMC Genomics, 2010, 11(Suppl 4), S15, DOI:10.1186/1471-2164-11-S4-S15.
- S. Tao, J. Tillotson, E. M. K. Wijeratne, Y. Xu, M. Kang, T. Wu, E. C. Lau, C. Mesa, D. J. Mason, R. V. Brown, J. J. La Clair, A. A. L. Gunatilaka, D. D. Zhang and E. Chapman, Withaferin A Analogs That Target the AAA+ Chaperone p97, ACS Chem. Biol., 2015, 10, 1916–1924, DOI:10.1021/acschembio.5b00367.
- P. Borst, Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what?, Open Biol., 2012, 2, 120066, DOI:10.1098/rsob.120066.
- N. Mizushima and M. Komatsu, Autophagy: renovation of cells and tissues, Cell, 2011, 147, 728–741, DOI:10.1016/j.cell.2011.10.026.
- N. Muniraj, S. Siddharth, A. Nagalingam, A. Walker, J. Woo, B. Gyorffy, E. Gabrielson, N. K. Saxena and D. Sharma, Withaferin A inhibits lysosomal activity to block autophagic flux and induces apoptosis via energetic impairment in breast cancer cells, Carcinogenesis, 2019 DOI:10.1093/carcin/bgz015 , [Epub ahead of print].
- M. L. Antony, J. Lee, E. R. Hahm, S. H. Kim, A. I. Marcus, V. Kumari, X. Ji, Z. Yang, C. L. Vowell, P. Wipf, G. T. Uechi, N. A. Yates, G. Romero, S. N. Sarkar and S. V. Singh, Growth arrest by the antitumor steroidal lactone withaferin a in human breast cancer cells is associated with down-regulation and covalent binding at cysteine 303 of β-tubulin, J. Biol. Chem., 2014, 289, 1852–1865, DOI:10.1074/jbc.M113.496844.
- J. A. Olzmann, L. Li, M. V. Chudaev, J. Chen, F. A. Perez, R. D. Palmiter and L.-S. Chin, Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6, J. Cell Biol., 2007, 178, 1025–1038, DOI:10.1083/jcb.200611128.
- T. Hideshima, J. Qi, R. M. Paranal, W. Tang, E. Greenberg, N. West, M. E. Colling, G. Estiu, R. Mazitschek, J. A. Perry, H. Ohguchi, F. Cottini, N. Mimura, G. Görgün, Y.-T. Tai, P. G. Richardson, R. D. Carrasco, O. Wiest, S. L. Schreiber, K. C. Anderson and J. E. Bradner, Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13162–13167, DOI:10.1073/pnas.1608067113.
- C. Papadopoulos, P. Kirchner, M. Bug, D. Grum, L. Koerver, N. Schulze, R. Poehler, A. Dressler, S. Fengler, K. Arhzaouy, V. Lux, M. Ehrmann, C. C. Weihl and H. Meyer, VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy, EMBO J., 2017, 36, 135–150, DOI:10.15252/embj.201695148.
- H. E. J. Polson, J. de Lartigue, D. J. Rigden, M. Reedijk, S. Urbe, M. J. Clague and S. A. Tooze, Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation, Autophagy, 2010, 6, 506–522, DOI:10.4161/auto.6.4.11863.
- J. Pu, C. Schindler, R. Jia, M. Jarnik, P. Backlund and J. S. Bonifacino, BORC, a multisubunit complex that regulates lysosome positioning, Dev. Cell, 2015, 33, 176–188, DOI:10.1016/j.devcel.2015.02.011.
- D. Glick, S. Barth and K. F. Macleod, Autophagy: cellular and molecular mechanisms, J. Pathol., 2010, 221, 3–12, DOI:10.1002/path.2697.
- R. Mackeh, D. Perdiz, S. Lorin, P. Codogno and C. Pous, Autophagy and microtubules - new story, old players, J. Cell Sci., 2013, 126, 1071–1080, DOI:10.1242/jcs.115626.
- I. Monastyrska, E. Rieter, D. J. Klionsky and F. Reggiori, Multiple roles of the cytoskeleton in autophagy, Biol. Rev. Cambridge Philos. Soc., 2009, 84, 431–448, DOI:10.1111/j.1469-185X.2009.00082.x.
- K. Dokladny, O. B. Myers and P. L. Moseley, Heat shock response and autophagy--cooperation and control, Autophagy, 2015, 11, 200–213, DOI:10.1080/15548627.2015.1009776.
- A. V. Gómez, G. Córdova, R. Munita, G. E. Parada, Á. P. Barrios, G. I. Cancino, A. R. Álvarez and M. E. Andrés, Characterizing HSF1 Binding and Post-Translational Modifications of hsp70 Promoter in Cultured Cortical Neurons: Implications in the Heat-Shock Response, PLoS One, 2015, 10, e0129329, DOI:10.1371/journal.pone.0129329.
- A. Vihervaara and L. Sistonen, HSF1 at a glance, J. Cell Sci., 2014, 127, 261–266, DOI:10.1242/jcs.132605.
- Y. Liu and A. Chang, Heat shock response relieves ER stress, EMBO J., 2008, 27, 1049–1059, DOI:10.1038/emboj.2008.42.
- S. Santagata, Y. Xu, E. M. K. Wijeratne, R. Kontnik, C. Rooney, C. C. Perley, H. Kwon, J. Clardy, S. Kesari, L. Whitesell, S. Lindquist and A. A. L. Gunatilaka, Using the Heat-Shock Response To Discover Anticancer Compounds that Target Protein Homeostasis, ACS Chem. Biol., 2012, 7, 340–349, DOI:10.1021/cb200353m.
- K. Shen, D. W. Johnson, D. A. Vesey, M. A. McGuckin and G. C. Gobe, Role of the unfolded protein response in determining the fate of tumor cells and the promise of multi-targeted therapies, Cell Stress Chaperones, 2018, 23(3), 317–334, DOI:10.1007/s12192-017-0844-3.
- M. P. Mayer, Hsp70 chaperone dynamics and molecular mechanism, Trends Biochem. Sci., 2013, 38, 507–514, DOI:10.1016/j.tibs.2013.08.001.
- M. R. Fernandez-Fernandez and J. M. Valpuesta, Hsp70 chaperone: a master player in protein homeostasis, F1000Research, 2018, 7, F1000 Faculty Rev-1497, DOI:10.12688/f1000research.15528.1.
- S. Verma, S. Goyal, S. Jamal, A. Singh and A. Grover, Hsp90: Friends, clients and natural foes, Biochimie, 2016, 127, 227–240, DOI:10.1016/j.biochi.2016.05.018.
- Y. Yu, A. Hamza, T. Zhang, M. Gu, P. Zou, B. Newman, Y. Li, A. A. L. Gunatilaka, C. G. Zhan and D. Sun, Withaferin A targets heat shock protein 90 in pancreatic cancer cells, Biochem. Pharmacol., 2010, 79, 542–551, DOI:10.1016/j.bcp.2009.09.017.
- I. Baaklini, M. J. H. Wong, C. Hantouche, Y. Patel, A. Shrier and J. C. Young, The DNAJA2 substrate release mechanism is essential for chaperone-mediated folding, J. Biol. Chem., 2012, 287, 41939–41954, DOI:10.1074/jbc.M112.413278.
- H.-Q. Wang, H.-Y. Zhang, F.-J. Hao, X. Meng, Y. Guan and Z.-X. Du, Induction of BAG2 protein during proteasome inhibitor-induced apoptosis in thyroid carcinoma cells, Br. J. Pharmacol., 2008, 155, 655–660, DOI:10.1038/bjp.2008.302.
- M. C. Jonikas, S. R. Collins, V. Denic, E. Oh, E. M. Quan, V. Schmid, J. Weibezahn, B. Schwappach, P. Walter, J. S. Weissman and M. Schuldiner, Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum, Science, 2009, 323, 1693–1697, DOI:10.1126/science.1167983.
- J. C. Christianson, J. A. Olzmann, T. A. Shaler, M. E. Sowa, E. J. Bennett, C. M. Richter, R. E. Tyler, E. J. Greenblatt, J. W. Harper and R. R. Kopito, Defining human ERAD networks through an integrative mapping strategy, Nat. Cell Biol., 2012, 14, 93–105, DOI:10.1038/ncb2383.
- Z. Yang, L. Zhuang, P. Szatmary, L. Wen, H. Sun, Y. Lu, Q. Xu and X. Chen, Upregulation of heat shock proteins (HSPA12A, HSP90B1, HSPA4, HSPA5 and HSPA6) in tumour tissues is associated with poor outcomes from HBV-related early-stage hepatocellular carcinoma, Int. J. Med. Sci., 2015, 12, 256–263, DOI:10.7150/ijms.10735.
- B. M. Dunyak and J. E. Gestwicki, Peptidyl-Proline Isomerases (PPIases): Targets for Natural Products and Natural Product-Inspired Compounds, J. Med. Chem., 2016, 59, 9622–9644, DOI:10.1021/acs.jmedchem.6b00411.
- S. K. Samanta, J. Lee, E.-R. Hahm and S. V. Singh, Peptidyl-prolyl cis/trans isomerase Pin1 regulates withaferin A-mediated cell cycle arrest in human breast cancer cells, Mol. Carcinog., 2018, 57, 936–946, DOI:10.1002/mc.22814.
- J. D. Pearson, Z. Mohammed, J. T. C. Bacani, R. Lai and R. J. Ingham, The heat shock protein-90 co-chaperone, Cyclophilin 40, promotes ALK-positive, anaplastic large cell lymphoma viability and its expression is regulated by the NPM-ALK oncoprotein, BMC Cancer, 2012, 12, 229, DOI:10.1186/1471-2407-12-229.
- Z. Zhou, L. J. Licklider, S. P. Gygi and R. Reed, Comprehensive proteomic analysis of the human spliceosome, Nature, 2002, 419, 182–185, DOI:10.1038/nature01031.
- E. El Marabti and I. Younis, The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer, Front. Mol. Biosci., 2018, 5, 80, DOI:10.3389/fmolb.2018.00080.
- C.-W. Cheng and E. Tse, PIN1 in Cell Cycle Control and Cancer, Front. Pharmacol., 2018, 9, 1367, DOI:10.3389/fphar.2018.01367.
- L. I. Gallo, M. Lagadari, G. Piwien-Pilipuk and M. D. Galigniana, The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress, J. Biol. Chem., 2011, 286, 30152–30160, DOI:10.1074/jbc.M111.256610.
- R. Ushioda, J. Hoseki, K. Araki, G. Jansen, D. Y. Thomas and K. Nagata, ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER, Science, 2008, 321, 569–572, DOI:10.1126/science.1159293.
- N. Hosokawa, Y. Kamiya, D. Kamiya, K. Kato and K. Nagata, Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans, J. Biol. Chem., 2009, 284, 17061–17068, DOI:10.1074/jbc.M809725200.
- P. P. Shah and L. J. Beverly, Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress, Oncotarget, 2015, 6, 17725–17737, DOI:10.18632/oncotarget.3918.
- A. Schulze, S. Standera, E. Buerger, M. Kikkert, S. van Voorden, E. Wiertz, F. Koning, P.-M. Kloetzel and M. Seeger, The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway, J. Mol. Biol., 2005, 354, 1021–1027, DOI:10.1016/j.jmb.2005.10.020.
- S.-H. Hong, S.-H. Chang, K.-C. Cho, S. Kim, S. Park, A. Y. Lee, H.-L. Jiang, H.-J. Kim, S. Lee, K.-N. Yu, H. W. Seo, C. Chae, K. P. Kim, J. Park and M.-H. Cho, Endoplasmic reticulum-Golgi intermediate compartment protein 3 knockdown suppresses lung cancer through endoplasmic reticulum stress-induced autophagy, Oncotarget, 2016, 7, 65335–65347, DOI:10.18632/oncotarget.11678.
- M. Maeda, T. Katada and K. Saito, TANGO1 recruits Sec16 to coordinately organize ER exit sites for efficient secretion, J. Cell Biol., 2017, 216, 1731–1743, DOI:10.1083/jcb.201703084.
- Y. Gonskikh and N. Polacek, Alterations of the translation apparatus during aging and stress response, Mech. Ageing Dev., 2017, 168, 30–36, DOI:10.1016/j.mad.2017.04.003.
- O. S. Kwon, S. An, E. Kim, J. Yu, K. Y. Hong, J. S. Lee and S. K. Jang, An mRNA-specific tRNAi carrier eIF2A plays a pivotal role in cell proliferation under stress conditions: stress-resistant translation of c-Src mRNA is mediated by eIF2A, Nucleic Acids Res., 2017, 45, 296–310, DOI:10.1093/nar/gkw1117.
- P. M. J. Clement, H. E. Johansson, E. C. Wolff and M. H. Park, Differential expression of eIF5A-1 and eIF5A-2 in human cancer cells, FEBS J., 2006, 273, 1102–1114, DOI:10.1111/j.1742-4658.2006.05135.x.
- E. Memin, M. Hoque, M. R. Jain, D. S. Heller, H. Li, B. Cracchiolo, H. M. Hanauske-Abel, T. Pe'ery and M. B. Mathews, Blocking eIF5A modification in cervical cancer cells alters the expression of cancer-related genes and suppresses cell proliferation, Cancer Res., 2014, 74, 552–562, DOI:10.1158/0008-5472.CAN-13-0474.
- S. Balabanov, A. Gontarewicz, P. Ziegler, U. Hartmann, W. Kammer, M. Copland, U. Brassat, M. Priemer, I. Hauber, T. Wilhelm, G. Schwarz, L. Kanz, C. Bokemeyer, J. Hauber, T. L. Holyoake, A. Nordheim and T. H. Brummendorf, Hypusination of eukaryotic initiation factor 5A (eIF5A): a novel therapeutic target in BCR-ABL-positive leukemias identified by a proteomics approach, Blood, 2007, 109, 1701–1711, DOI:10.1182/blood-2005-03-037648.
- K. Fujimura, T. Wright, J. Strnadel, S. Kaushal, C. Metildi, A. M. Lowy, M. Bouvet, J. A. Kelber and R. L. Klemke, A hypusine-eIF5A-PEAK1 switch regulates the pathogenesis of pancreatic cancer, Cancer Res., 2014, 74, 6671–6681, DOI:10.1158/0008-5472.CAN-14-1031.
- N. P. Lee, F. H. Tsang, F. H. Shek, M. Mao, H. Dai, C. Zhang, S. Dong, X.-Y. Guan, R. T. P. Poon and J. M. Luk, Prognostic significance and therapeutic potential of eukaryotic translation initiation factor 5A (eIF5A) in hepatocellular carcinoma, Int. J. Cancer, 2010, 127, 968–976, DOI:10.1002/ijc.25100.
- D. Shahbazian, A. Parsyan, E. Petroulakis, J. Hershey and N. Sonenberg, eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways, Cell Cycle, 2010, 9, 4106–4109, DOI:10.4161/cc.9.20.13630.
- E. Horvilleur, T. Sbarrato, K. Hill, R. V. Spriggs, M. Screen, P. J. Goodrem, K. Sawicka, L. C. Chaplin, C. Touriol, G. Packham, K. N. Potter, S. Dirnhofer, A. Tzankov, M. J. S. Dyer, M. Bushell, M. MacFarlane and A. E. Willis, A role for eukaryotic initiation factor 4B overexpression in the pathogenesis of diffuse large B-cell lymphoma, Leukemia, 2014, 28, 1092–1102, DOI:10.1038/leu.2013.295.
- K. Chen, J. Yang, J. Li, X. Wang, Y. Chen, S. Huang and J.-L. Chen, eIF4B is a convergent target and critical effector of oncogenic Pim and PI3K/Akt/mTOR signaling pathways in Abl transformants, Oncotarget, 2016, 7, 10073–10089, DOI:10.18632/oncotarget.7164.
- S.-H. Goh, S.-H. Hong, S.-H. Hong, B.-C. Lee, M.-H. Ju, J.-S. Jeong, Y.-R. Cho, I.-H. Kim and Y.-S. Lee, eIF3m expression influences the regulation of tumorigenesis-related genes in human colon cancer, Oncogene, 2011, 30, 398–409, DOI:10.1038/onc.2010.422.
- M. Ott, V. Gogvadze, S. Orrenius and B. Zhivotovsky, Mitochondria, oxidative stress and cell death, Apoptosis, 2007, 12, 913–922, DOI:10.1007/s10495-007-0756-2.
- S. Orrenius, V. Gogvadze and B. Zhivotovsky, Mitochondrial oxidative stress: implications for cell death, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 143–183, DOI:10.1146/annurev.pharmtox.47.120505.105122.
- P. Bargagna-Mohan, R. R. Paranthan, A. Hamza, N. Dimova, B. Trucchi, C. Srinivasan, G. I. Elliott, C. G. Zhan, D. L. Lau, H. Zhu, K. Kasahara, M. Inagaki, F. Cambi and R. Mohan, Withaferin A targets intermediate filaments glial fibrillary acidic protein and vimentin in a model of retinal gliosis, J. Biol. Chem., 2010, 285, 7657–7669, DOI:10.1074/jbc.M109.093765.
- P. Bargagna-Mohan, S. P. Deokule, K. Thompson, J. Wizeman, C. Srinivasan, S. Vooturi, U. B. Kompella and R. Mohan, Withaferin A Effectively Targets Soluble Vimentin in the Glaucoma Filtration Surgical Model of Fibrosis, PLoS One, 2013, 8(5), e63881, DOI:10.1371/journal.pone.0063881.
- P. Bargagna-Mohan, R. R. Paranthan, A. Hamza, C. G. Zhan, D. M. Lee, K. B. Kim, D. L. Lau, C. Srinivasan, K. Nakayama, K. I. Nakayama, H. Herrmann and R. Mohan, Corneal antifibrotic switch identified in genetic and pharmacological deficiency of vimentin, J. Biol. Chem., 2012, 287, 989–1006, DOI:10.1074/jbc.M111.297150.
- G. Lahat, Q. S. Zhu, K. L. Huang, S. Wang, S. Bolshakov, J. Liu, K. Torres, R. R. Langley, A. J. Lazar, M. C. Hung and D. Lev, Vimentin is a novel anti-cancer therapeutic target; insights from In Vitro and In Vivo mice xenograft studies, PLoS One, 2010, 5(4), e10105, DOI:10.1371/journal.pone.0010105.
- S. A. Maxwell, E. M. Cherry and K. J. Bayless, Akt, 14-3-3ζ, and vimentin mediate a drug-resistant invasive phenotype in diffuse large B-cell lymphoma, Leuk. Lymphoma, 2011, 52, 849–864, DOI:10.3109/10428194.2010.551793.
- J. T. Thaiparambil, L. Bender, T. Ganesh, E. Kline, P. Patel, Y. Liu, M. Tighiouart, P. M. Vertino, R. D. Harvey, A. Garcia and A. I. Marcus, Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation, Int. J. Cancer, 2011, 129, 2744–2755, DOI:10.1002/ijc.25938.
- J. Lee, E. R. Hahm, A. I. Marcus and S. V. Singh, Withaferin A inhibits experimental epithelial-mesenchymal transition in MCF-10A cells and suppresses vimentin protein level in vivo in breast tumors, Mol. Carcinog., 2015, 54(6), 417–429, DOI:10.1002/mc.22110.
- B. Grin, S. Mahammad, T. Wedig, M. M. Cleland, L. Tsai, H. Herrmann and R. D. Goldman, Withaferin A alters intermediate filament organization, cell shape and behavior, PLoS One, 2012, 7, 1–13, DOI:10.1371/journal.pone.0039065.
- A. A. Challa, M. Vukmirovic, J. Blackmon and B. Stefanovic, Withaferin-A reduces type I collagen expression in vitro and inhibits development of myocardial fibrosis in vivo, PLoS One, 2012, 7(8), e42989, DOI:10.1371/journal.pone.0042989.
- E. Gladilin, P. Gonzalez and R. Eils, Dissecting the contribution of actin and vimentin intermediate filaments to mechanical phenotype of suspended cells using high-throughput deformability measurements and computational modeling, J. Biomech., 2014, 47, 2598–2605, DOI:10.1016/j.jbiomech.2014.05.020.
- Y. Miyatake, N. Sheehy, S. Ikeshita, W. W. Hall and M. Kasahara, Anchorage-dependent multicellular aggregate formation induces CD44 high cancer stem cell-like ATL cells in an NF-κB- and vimentin-dependent manner, Cancer Lett., 2015, 357, 355–363, DOI:10.1016/j.canlet.2014.11.055.
- Y. Nishikawa, D. Okuzaki, K. Fukushima, S. Mukai, S. Ohno, Y. Ozaki, N. Yabuta and H. Nojima, Withaferin A induces cell death selectively in androgen-independent prostate cancer cells but not in normal fibroblast cells, PLoS One, 2015, 10, 1–20, DOI:10.1371/journal.pone.0134137.
- O. Biskou, V. Casanova, K. M. Hooper, S. Kemp, G. P. Wright, J. Satsangi, P. G. Barlow and C. Stevens, The type III intermediate filament vimentin regulates organelle distribution and modulates autophagy, PLoS One, 2019, 14, e0209665, DOI:10.1371/journal.pone.0209665.
- R. R. Falsey, M. T. Marron, G. M. K. B. Gunaherath, N. Shirahatti, D. Mahadevan, A. A. L. Gunatilaka and L. Whitesell, Actin microfilament aggregation induced by withaferin A is mediated by annexin II, Nat. Chem. Biol., 2006, 2, 33–38, DOI:10.1038/nchembio755.
- G. Ozorowski, C. M. Ryan, J. P. Whitelegge and H. Luecke, Withaferin A binds covalently to the N-terminal domain of annexin A2, Biol. Chem., 2012, 393, 1151–1163, DOI:10.1515/hsz-2012-0184.
- J. Zhao, L. Zhang, X. Dong, L. Liu, L. Huo and H. Chen, High Expression of Vimentin is Associated With Progression and a Poor Outcome in Glioblastoma, Appl. Immunohistochem. Mol. Morphol., 2018, 26(5), 337–344, DOI:10.1097/PAI.0000000000000420.
- H. Bao, M. Jiang, M. Zhu, F. Sheng, J. Ruan and C. Ruan, Overexpression of Annexin II affects the proliferation, apoptosis, invasion and production of proangiogenic factors in multiple myeloma, Internet J. Hematol., 2009, 90, 177–185, DOI:10.1007/s12185-009-0356-8.
- N. A. Lokman, M. P. Ween, M. K. Oehler and C. Ricciardelli, The role of annexin A2 in tumorigenesis and cancer progression, Cancer Microenviron., 2011, 4, 199–208, DOI:10.1007/s12307-011-0064-9.
- X. Liu, D. Ma, X. Jing, B. Wang, W. Yang and W. Qiu, Overexpression of ANXA2 predicts adverse outcomes of patients with malignant tumors: a systematic review and meta-analysis, Med. Oncol., 2015, 32, 392, DOI:10.1007/s12032-014-0392-y.
- K. J. Verhey and J. Gaertig, The tubulin code, Cell Cycle, 2007, 6, 2152–2160, DOI:10.4161/cc.6.17.4633.
- C. M. Fife, J. A. McCarroll and M. Kavallaris, Movers and shakers: cell cytoskeleton in cancer metastasis, Br. J. Pharmacol., 2014, 171, 5507–5523, DOI:10.1111/bph.12704.
- W. C. Burhans and N. H. Heintz, The cell cycle is a redox cycle: Linking phase-specific targets to cell fate, Free Radical Biol. Med., 2009, 47, 1282–1293, DOI:10.1016/j.freeradbiomed.2009.05.026.
- J. Chiu and I. W. Dawes, Redox control of cell proliferation, Trends Cell Biol., 2017, 22, 592–601, DOI:10.1016/j.tcb.2012.08.002.
- C. J. Sherr and J. M. Roberts, Living with or without cyclins and cyclin-dependent kinases, Genes Dev., 2004, 18, 2699–2711, DOI:10.1101/gad.1256504.
- N. M. Reddy, S. R. Kleeberger, J. H. Bream, P. G. Fallon, T. W. Kensler, M. Yamamoto and S. P. Reddy, Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling, Oncogene, 2008, 27, 5821–5832, DOI:10.1038/onc.2008.188.
- J. E. Conour, W. V. Graham and H. R. Gaskins, A combined in vitro/bioinformatic investigation of redox regulatory mechanisms governing cell cycle progression, Physiol. Genomics, 2004, 18, 196–205, DOI:10.1152/physiolgenomics.00058.2004.
- G. Kim, T. Kim, E. Hwang, K. Chang, J. Hong and J. Park, Withaferin A inhibits the proliferation of gastric cancer cells by inducing G2/M cell cycle arrest and apoptosis, Oncol. Lett., 2017, 14, 416–422, DOI:10.3892/ol.2017.6169.
- T. Das, K. S. Roy, T. Chakrabarti, S. Mukhopadhyay and S. Roychoudhury, Withaferin A modulates the Spindle Assembly Checkpoint by degradation of Mad2-Cdc20 complex in colorectal cancer cell lines, Biochem. Pharmacol., 2014, 91, 31–39, DOI:10.1016/j.bcp.2014.06.022.
- S. Okamoto, T. Tsujioka, S. Ichiro Suemori, J. Ichiro Kida, T. Kondo, Y. Tohyama and K. Tohyama, Withaferin A suppresses the growth of myelodysplasia and leukemia cell lines by inhibiting cell cycle progression, Cancer Sci., 2016, 107, 1302–1314, DOI:10.1111/cas.12988.
- S. D. Stan, Y. Zeng and S. V. Singh, Ayurvedic medicine constituent withaferin a causes G2 and M phase cell cycle arrest in human breast cancer cells, Nutr. Cancer, 2008, 60(Suppl 1), 51–60, DOI:10.1080/01635580802381477.
- J.-H. Wei, Y.-F. Chou, Y.-H. Ou, Y.-H. Yeh, S.-W. Tyan, T.-P. Sun, C.-Y. Shen and S.-Y. Shieh, TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68, J. Biol. Chem., 2005, 280, 7748–7757, DOI:10.1074/jbc.M410152200.
- X. Zhang, A. K. Samadi, K. F. Roby, B. Timmermann and M. S. Cohen, Inhibition of cell growth and induction of apoptosis in ovarian carcinoma cell lines CaOV3 and SKOV3 by natural withanolide Withaferin A, Gynecol. Oncol., 2012, 124, 606–612, DOI:10.1016/j.ygyno.2011.11.044.
- M. Zuccolo, A. Alves, V. Galy, S. Bolhy, E. Formstecher, V. Racine, J.-B. Sibarita, T. Fukagawa, R. Shiekhattar, T. Yen and V. Doye, The human Nup107-160 nuclear pore subcomplex contributes to proper kinetochore functions, EMBO J., 2007, 26, 1853–1864, DOI:10.1038/sj.emboj.7601642.
- I. Mossaid and B. Fahrenkrog, Complex Commingling: Nucleoporins and the Spindle Assembly Checkpoint, Cell, 2015, 4, 706–725, DOI:10.3390/cells4040706.
- L.-H. Shi, X.-J. Wu, J.-S. Liu and Y.-B. Gao, Withaferin A activates stress signalling proteins in high risk acute lymphoblastic leukemia, Int. J. Clin. Exp. Pathol., 2015, 8, 15652–15660 Search PubMed.
- N. R. Brown, E. D. Lowe, E. Petri, V. Skamnaki, R. Antrobus and L. N. Johnson, Cyclin B and cyclin A confer different substrate recognition properties on CDK2, Cell Cycle, 2007, 6, 1350–1359, DOI:10.4161/cc.6.11.4278.
- A. Kurimchak and X. Grana, PP2A: more than a reset switch to activate pRB proteins during the cell cycle and in response to signaling cues, Cell Cycle, 2015, 14, 18–30, DOI:10.1080/15384101.2015.1056614.
- M. K. McKenna, B. W. Gachuki, S. S. Alhakeem, K. N. Oben, V. M. Rangnekar, R. C. Gupta and S. Bondada, Anti-cancer activity of withaferin A in B-cell lymphoma, Cancer Biol. Ther., 2015, 16, 1088–1098, DOI:10.1080/15384047.2015.1046651.
- A. K. Samadi, S. M. Cohen, R. Mukerji, V. Chaguturu, X. Zhang, B. N. Timmermann, M. S. Cohen and E. A. Person, Natural withanolide withaferin A induces apoptosis in uveal melanoma cells by suppression of Akt and c-MET activation, Tumor Biol., 2012, 33, 1179–1189, DOI:10.1007/s13277-012-0363-x.
- E. A. Grossman, C. C. Ward, J. N. Spradlin, L. A. Bateman, T. R. Huffman, D. K. Miyamoto, J. I. Kleinman and D. K. Nomura, Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products, Cell Chem. Biol., 2017, 1–23, DOI:10.1016/j.chembiol.2017.08.013.
- X. Liu and M. Winey, The MPS1 family of protein kinases, Annu. Rev. Biochem., 2012, 81, 561–585, DOI:10.1146/annurev-biochem-061611-090435.
- K. L. Thu, J. Silvester, M. J. Elliott, W. Ba-Alawi, M. H. Duncan, A. C. Elia, A. S. Mer, P. Smirnov, Z. Safikhani, B. Haibe-Kains, T. W. Mak and D. W. Cescon, Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E1570–E1577, DOI:10.1073/pnas.1719577115.
- X. Liu, W. Liao, Q. Yuan, Y. Ou and J. Huang, TTK activates Akt and promotes proliferation and migration of hepatocellular carcinoma cells, Oncotarget, 2015, 6, 34309–34320, DOI:10.18632/oncotarget.5295.
- A. Koch, A. Maia, A. Janssen and R. H. Medema, Molecular basis underlying resistance to Mps1/TTK inhibitors, Oncogene, 2015, 35, 2518, DOI:10.1038/onc.2015.319.
- D. H. Lee, I. H. Lim, E. G. Sung, J. Y. Kim, I. H. Song, Y. K. Park and T. J. Lee, Withaferin A inhibits matrix metalloproteinase-9 activity by suppressing the Akt signaling pathway, Oncol. Rep., 2013, 30, 933–938, DOI:10.3892/or.2013.2487.
- A. L. Hein, P. Seshacharyulu, S. Rachagani, Y. M. Sheinin, M. M. Ouellette, M. P. Ponnusamy, M. C. Mumby, S. K. Batra and Y. Yan, PR55alpha Subunit of Protein Phosphatase 2A Supports the Tumorigenic and Metastatic Potential of Pancreatic Cancer Cells by Sustaining Hyperactive Oncogenic Signaling, Cancer Res., 2016, 76, 2243–2253, DOI:10.1158/0008-5472.CAN-15-2119.
- V. Baud and M. Karin, Is NF-[kappa]B a good target for cancer therapy? Hopes and pitfalls, Nat. Rev. Drug Discovery, 2009, 8, 33–40, DOI:10.1038/nrd2781.
- L. M. Staudt, Oncogenic activation of NF-kappaB, Cold Spring Harbor Perspect. Biol., 2010, 2, a000109, DOI:10.1101/cshperspect.a000109.
- M. Kaileh, W. Vanden Berghe, A. Heyerick, J. Horion, J. Piette, C. Libert, D. De Keukeleire, T. Essawi and G. Haegeman, Withaferin A strongly elicits IκB kinase β hyperphosphorylation concomitant with potent inhibition of its kinase activity, J. Biol. Chem., 2007, 282, 4253–4264, DOI:10.1074/jbc.M606728200.
- K. Heyninck, M. Lahtela-kakkonen, P. Van Der Veken, G. Haegeman and W. Vanden, Withaferin A inhibits NF-kappaB activation by targeting cysteine 179 in IKK b, Biochem. Pharmacol., 2014, 91(4), 501–509, DOI:10.1016/j.bcp.2014.08.004.
- S. Ashkenazi, A. Plotnikov, A. Bahat, E. Ben-Zeev, S. Warszawski and R. Dikstein, A Novel Allosteric Mechanism of NF-κB Dimerization and DNA Binding Targeted by an Anti-Inflammatory Drug, Mol. Cell. Biol., 2016, 36, 1237–1247, DOI:10.1128/MCB.00895-15.
- L. Gambhir, R. Checker, D. Sharma, M. Thoh, A. Patil, M. Degani, V. Gota and S. K. Sandur, Thiol dependent NF-κB suppression and inhibition of T-cell mediated adaptive immune responses by a naturally occurring steroidal lactone Withaferin A, Toxicol. Appl. Pharmacol., 2015, 289, 297–312, DOI:10.1016/j.taap.2015.09.014.
- P. Starokadomskyy, N. Gluck, H. Li, B. Chen, M. Wallis, G. N. Maine, X. Mao, I. W. Zaidi, M. Y. Hein, F. J. McDonald, S. Lenzner, A. Zecha, H.-H. Ropers, A. W. Kuss, J. McGaughran, J. Gecz and E. Burstein, CCDC22 deficiency in humans blunts activation of proinflammatory NF-kappaB signaling, J. Clin. Invest., 2013, 123, 2244–2256, DOI:10.1172/JCI66466.
- J. Hess, P. Angel and M. Schorpp-Kistner, AP-1 subunits: quarrel and harmony among siblings, J. Cell Sci., 2004, 117, 5965–5973, DOI:10.1242/jcs.01589.
- J. L. Ducut Sigala, V. Bottero, D. B. Young, A. Shevchenko, F. Mercurio and I. M. Verma, Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit, Science, 2004, 304, 1963–1967, DOI:10.1126/science.1098387.
- A. Grover, A. Shandilya, A. Punetha, V. S. Bisaria and D. Sundar, Inhibition of the NEMO/IKKβ association complex formation, a novel mechanism associated with the NF-κB activation suppression by Withania somnifera's key metabolite withaferin A, BMC Genomics, 2010, 11(Suppl 4), S25, DOI:10.1186/1471-2164-11-S4-S25.
- E. Burstein, J. E. Hoberg, A. S. Wilkinson, J. M. Rumble, R. A. Csomos, C. M. Komarck, G. N. Maine, J. C. Wilkinson, M. W. Mayo and C. S. Duckett, COMMD proteins, a novel family of structural and functional homologs of MURR1, J. Biol. Chem., 2005, 280, 22222–22232, DOI:10.1074/jbc.M501928200.
- B. Hassannia, E. Logie, P. Vandenabeele, T. Vanden Berghe and W. Vanden Berghe, Withaferin A: From ayurvedic folk medicine to preclinical anti-cancer drug, Biochem. Pharmacol., 2019, S0006-2952(19)30292-8, DOI:10.1016/j.bcp.2019.08.004.
- A. H. Kyakulaga, F. Aqil, R. Munagala and R. C. Gupta, Withaferin A inhibits Epithelial to Mesenchymal Transition in Non-Small Cell Lung Cancer Cells, Sci. Rep., 2018, 8, 15737, DOI:10.1038/s41598-018-34018-1.
- N. Pires, V. Gota, A. Gulia, L. Hingorani, M. Agarwal and A. Puri, Safety and Pharmacokinetics of Withaferin-A in advanced stage high grade Osteosarcoma: A phase I trial, J. Ayurveda Integr. Med., 2019, S0975-9476(18)30789-7, DOI:10.1016/j.jaim.2018.12.008.
| This journal is © The Royal Society of Chemistry 2020 |