
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperior anchoring effect of a Cu-benzenehexathial MOF as an aluminium–sulfur battery cathode host†
Preeti
Bhauriyal
 and
Biswarup
Pathak
*
and
Biswarup
Pathak
*
Discipline of Chemistry, Indian Institute of Technology (IIT) Indore, Indore, Madhya Pradesh 453552, India. E-mail: biswarup@iiti.ac.in
First published on 13th November 2020
Abstract
The poor reversibility and short lifespan of next generation Al–S batteries can be effectively overcome by opting for a cathode host able to trap sulfur and Al polysulfide species. In this work, with the help of density functional theory (DFT) calculations, a 2D Cu-benzenehexathial (Cu-BHT) metal organic framework (MOF) has been proposed as an excellent potential cathode host for Al–S batteries as compared to carbonaceous hosts such as graphene. The favourable electrical conductivity of the Cu-BHT MOF eliminates the insulating nature of most sulfur-based electrodes and is helpful in supporting the cathode charge/discharge processes. The results of a binding study show that while graphene has low binding energies (−0.76 to −0.88 eV) towards Al polysulfides, the Cu-BHT MOF exhibits improved binding to Al polysulfides (−1.11 to −3.56 eV) because of the presence of copper and sulfur, which results in increased charge transfer to the Al polysulfides. The synergic dual interactions based on the Al–S (of the sulfur host) bond and S (of the Al polysulfide)–Cu bond also lead to a uniform deposition of insoluble Al2S3 over the Cu-BHT surface, improving the utilization of the active cathode host. This leads to the incorporation of 45.81 wt% sulfur in the Cu-BHT monolayer host, higher than that of the experimentally reported S@HKUST-1-C cathode host (34 wt%) for Al–S batteries. Moreover, it is also observed that the Al polysulfide binding is much stronger than their interaction with the electrolytes, thus decreasing the overall dissolution of Al polysulfides into the electrolyte and improving the net cycling performance in Al–S batteries. With these desirable properties, it is expected that the Cu-BHT MOF can be used as a promising anchoring cathode host compared to graphene in Al–S batteries.
1. Introduction
The high abundance of aluminium (Al) in the earth's crust, the high volumetric (8.05 A h cm−3) and gravimetric capacity (2.98 A h g−1) associated with the 3e− redox reaction and its excellent safety make Al one of the most advantageous elements and Al–metal based batteries the future of leading energy storage systems.1–3 The use of ionic liquid-based electrolytes has increased the efficiency and stability of Al batteries via increasing reversible Al deposition/stripping with high Coulombic efficiency and thus has become the obvious choice for current Al batteries.4,5 These ionic liquid based Al batteries have been proposed to work via two distinctive reversible energy storage mechanisms depending upon the nature of the cathode materials: one is intercalation reaction and the other is conversion reaction. As the chloroaluminate ionic liquid electrolyte consists of Al ions in the form of both cation and anions, both Al3+ and AlCl4− can undergo intercalation in rocking chair type Al batteries6–16 and Al dual-ion batteries,17–30 respectively. The rocking chair Al batteries have been reported to exhibit high capacity (∼300 mA h g−1) but are limited by their small cycle life (<20 cycles), coulombic efficiency and cell voltage range (∼0.6 V), whereas Al dual-ion batteries can deliver higher voltages (∼2.0 V) with fast charge/discharge rates, but they have low storage capacity (<120 mA h g−1) and involve large volume expansion due to the involvement of a large sized anion,31 which can cause irreversible damage to the battery. On the other hand, the conversion reaction mechanism has been reported in the case of Ni- and Fe-based sulfides,32–35 which can hold higher storage capacities for the initial charge/discharge cycles, but they abruptly drop during long term usage due to destruction of the cathode lattice with continuous charge/discharge cycles.33 Also, low conductivity is a big obstacle in the application of these conversion based Al batteries. However, sulfur shows quite good advantages when used as a conversion cathode as it can undergo multi-electron transfer during electrochemical reactions to provide a high specific capacity of 1675 mA h g−1 and it is light-weight. Sulfur cathodes have been quite well-explored in Li–S,36–39 Na–S,40,41 and Mg–S42,43 batteries but the field of Al–S batteries is still very new. At present, limited numbers of studies44–49 have been reported for non-aqueous electrolyte-based Al–S batteries after the initial report of a successful demonstration of a primary Al–S battery in 2015.44 The involved charge/discharge processes were observed to follow the given reaction mechanism:| Anode: 2Al + 14AlCl4− ↔ 8Al2Cl7− + 6e− | (1) |
| Cathode: 8Al2Cl7− + 6e− + 3S ↔ Al2S3 + 14AlCl4− | (2) |
| Overall: 2Al + 3S ↔ Al2S3 | (3) |
In order to improve battery efficiency, studies have either focused on the development of the cathode host material, such as by employing an activated carbon cloth/sulfur composite,45 or constructing a cathode matrix by dispersing a sulfur/Al[EMI]Cl4 slurry onto free-standing carbon nanofiber (CNF) paper and coating of the single-wall carbon nanotube paper on the separator to slow down the diffusion of polysulfide.46 Or, by using a lithium-ion (Li+-ion)-mediated ionic liquid electrolyte.47 However, these room-temperature Al–S batteries were still observed to either show extremely poor reversibility or suffer from a sluggish electrochemical kinetics due to the lack of a suitable sulfur cathode host. In this regard, very recently, a composite of S on a carbonized Hong Kong University of Science and Technology-1 (HKUST-1) matrix (S@HKUST-1-C) was reported for Al–S batteries,48 which maintained a reversible capacity of 600 mA h g−1 at the 75th cycle and a reversible capacity of 460 mA h g−1 at the 500th cycle with a Coulombic efficiency of ∼95%. The improved efficiency of this battery was credited to the presence of Cu in the electrochemical process that formed an ionic cluster with polysulfide that facilitated the electrochemical reaction and improved the reversibility of S cathode during charging/discharging process in Al–S battery. Additionally, Cu increases the electron conductivity at the HKUST-1-C/S interface. Such Cu-based metal organic frameworks (MOFs) have also been explored for the development of more efficient Li–S batteries, both experimentally and theoretically.50–53 Therefore, for the effective development of Al–S batteries, it is necessary to take inspiration from more developed Li–S batteries as Al–S batteries may also follow a similar kind of working mechanism. Moreover, it is also important to investigate the reason behind the superior anchoring effect of Cu–metal based materials compared to the other experimentally studied carbonaceous host materials in Al–S batteries.
Therefore, in this study, we have investigated a two-dimensional (2D) copper-based MOF (Cu-BHT) monolayer by means of density functional theory (DFT) computation to be used as a potential cathode support material in Al–S batteries,54 which has been recently synthesized by connecting benzenehexathial (BHT) ligands with Cu ions in an extremely dense fashion. To investigate how a MOF could act as a better cathode host than the experimentally reported carbon-based cathode hosts, we have carried out a comparative study of the Cu-BHT monolayer MOF with respect to graphene, a representative of the carbonaceous hosts. We have analysed the corresponding cathode systems on the basis of the detailed binding characteristics of sulfur and Al polysulfides, electronic properties, electrolyte interactions and Al polysulfide deposition behaviour. Our results show that the Cu-BHT MOF could be a promising cathode host for use in Al–S batteries.
2. Computational details
Density functional theory (DFT) as implemented in the Vienna Ab initio Simulation Package (VASP) was used to perform the calculations.55–58 All geometry optimization and electronic property calculations were performed using the Perdew–Burke–Ernzerhof (PBE) functional of the generalized gradient approximation (GGA).59,60 The interactions between ion cores and valence electrons were treated by employing the projector augmented-wave (PAW) method.61,62 The plane wave cut-off energy was fixed to 470 eV and the total energy was converged to 10−5 eV. To optimize the structures, the atomic positions were fully relaxed until the Hellmann–Feynman forces on all atoms were smaller than 0.01 eV Å−1. Moreover, all the structure optimizations were carried out using van der Waals corrected density functional theory (DFT-D3) to overcome the deficiencies of DFT in treating dispersion interactions.63 A 2 × 2 × 1 supercell of Cu-BHT monolayer and 7 × 7 × 1 supercell of graphene having the lattice parameters a = b = 17.51 Å and a = b = 17.27 Å, respectively, were used for the calculations and the Brillouin zone was represented by a Monkhorst-k-point grid of 5 × 5 × 1 for both systems. To avoid periodic image interaction between the two nearest neighbour unit cells, the vacuum was set to 20 Å in the z-direction.3. Results and discussion
3.1. Binding characteristics of sulfur (S8) and Al polysulfides (Al2Sx) on the Cu-BHT MOF and graphene
It has been observed that in Al–S batteries, the charging and discharging processes proceed through the formation of Al polysulfide intermediates, as given by eqn (4) and (5), | (4) |
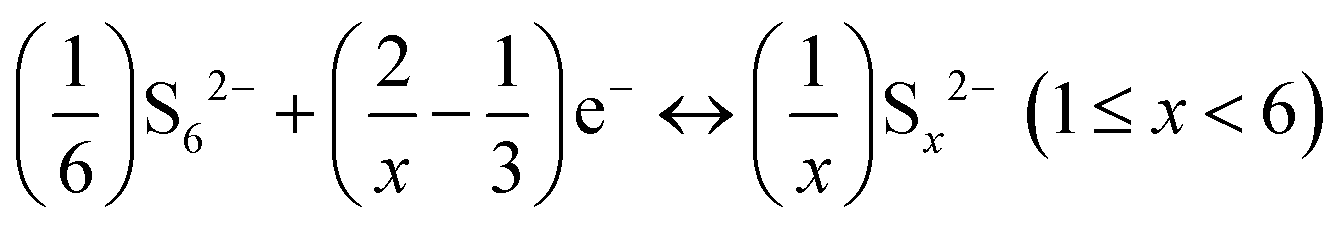 | (5) |
These Al polysulfide molecules can be represented with the formula unit of Al2Sx. Therefore, to study the binding behaviour of these Al polysulfides on the Cu-BHT monolayer and graphene, we have to first find the stable structures of the set of S-containing species, Al2(S6)3 or Al2S18, Al2(S4)3 or Al2S12, Al2(S2)3 or Al2S6, and Al2(S)3 or Al2S3 along with S8. Out of all the possible structures studied, the most stable structures are presented in Fig. 1a.
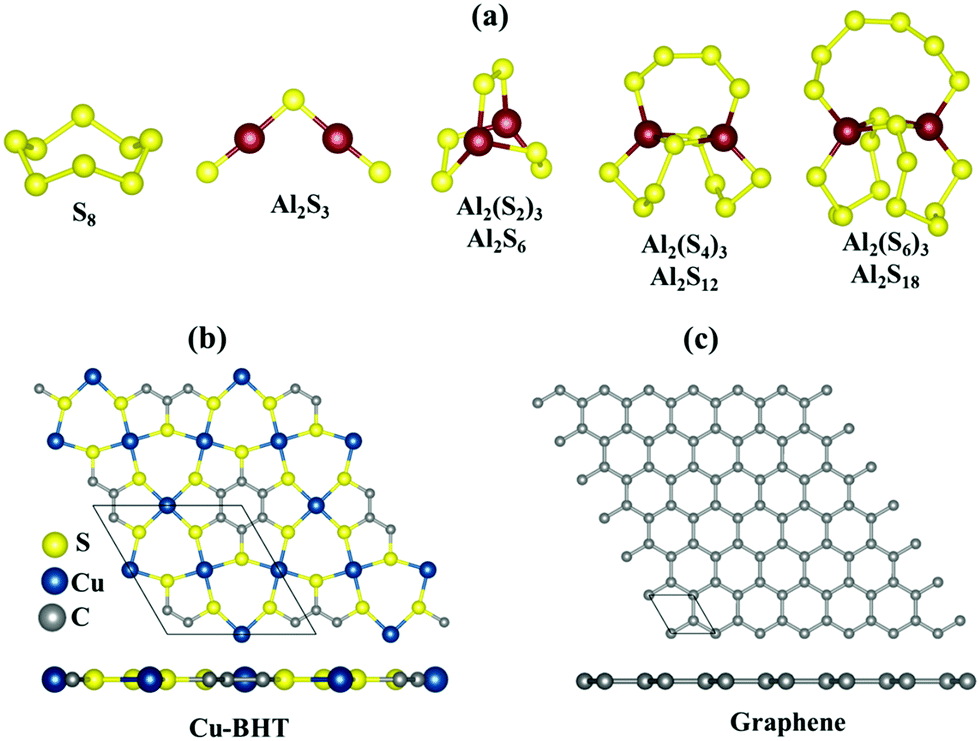 | ||
| Fig. 1 (a) Most stable molecular structures of S8 and Al2Sx (x = 3, 6, 12 and 18). Top and side views of the fully optimized structure of the (b) Cu-BHT and (c) graphene monolayer surfaces. | ||
The S8 molecule stabilizes in its most stable allotrope form at room temperature, which is a 3D crown like orthorhombic α-S8 structure, whereas Al2S3 prefers to form a planar bent structure compared to the linear structure with the S atom taking the centre position with side S–Al–S and centre Al–S–Al bond angles of 179.9° and 100°, respectively, which is in close accordance with a previous study (179.2° and 101.7°, respectively).64,65 For higher order Al2Sx with more S atoms, the 3D cluster shapes are more stable compared to the 2D chains with Al or S atoms at the terminal ends, and the cluster shape can effectively avoid the dangling bonds observed in Al2S3 and contribute to the structure stability. Similar results have also been observed for higher order Li polysulfides in Li–S batteries.66 On closer observation, we found that the S atoms in these Al2Sx molecules are not all equivalent in their binding nature, and the S–S and Al–S bond lengths fall in the ranges of 2.02–2.14 Å and 2.18–2.38 Å, respectively (Fig. S1, ESI†). Also, as the size of Al2Sx molecule increases, the average Al–S bond length increases from Al2S3 (2.09 Å) to Al2S18 (2.27 Å), which could lead to the easy dissociation of the Al–S bond in higher order Al2Sx molecules compared to lower order Al polysulfides. This could be one of the reasons for the irreversibility observed in the charging process in Al–S batteries, which involves the conversion of lower order to higher order Al2Sx.45,46
The Cu-BHT monolayer possesses a hexagonal planar structure, where the Cu atoms and BHT are connected together by Cu–S covalent bonds (2.28 Å), as shown in Fig. 1b. Our optimized lattice constant of 8.76 Å is in good agreement with the previous reported values (8.76 Å and 8.80 Å).54,67 The monolayer of the Cu-BHT MOF has an intrinsic metallic nature mainly contributed by the electronic states of C 2p, S 3p, and Cu 3d orbitals at the Fermi level and it is dynamically stable.68 This metallic characteristic of the Cu-BHT MOF could be quite beneficial in improving the rate performance of Al–S batteries. The graphene monolayer optimized for the comparative study is also shown in Fig. 1c with lattice parameters (a = b = 2.46 Å) in good accordance with the experimental data for graphite.69,70
To investigate the reason behind the poor performance of carbon based cathode hosts for Al–S batteries, we have systematically compared the binding behaviour of S8 and the corresponding Al2Sx molecules on a graphene surface as well as on the Cu-BHT MOF. Various initial structures of S8 and Al2Sx molecules were investigated at possible binding sites on both Cu-BHT and graphene monolayers and the corresponding binding energies were calculated using the following equation:
| EBinding= (EHost+M − EHost − EM) | (6) |
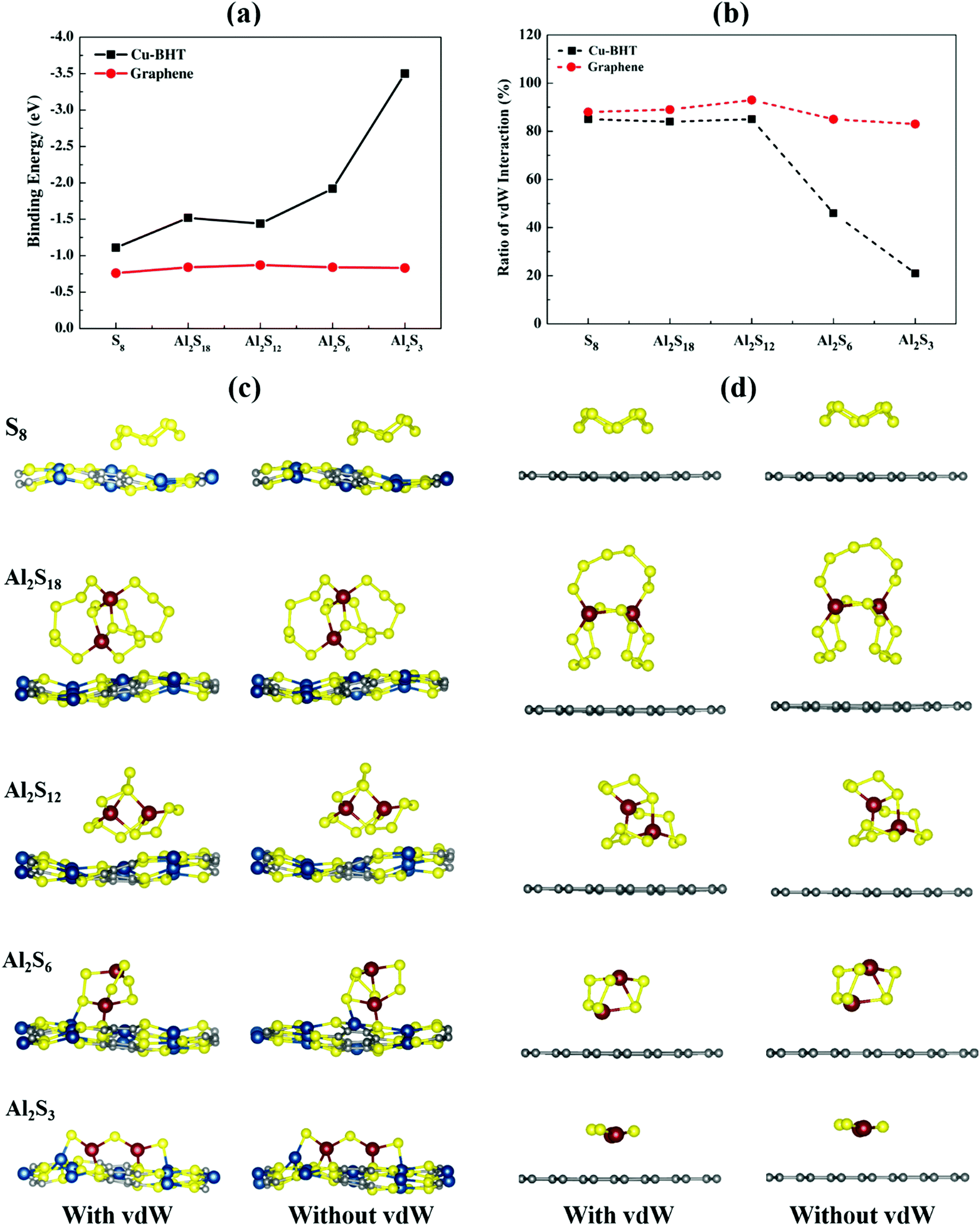 | ||
| Fig. 2 (a) Binding energies for S8 and Al2Sx (x = 3, 6, 12, and 18) on Cu-BHT and graphene monolayers, and (b) the corresponding ratio (r%) for vdW interaction for the Cu-BHT and graphene monolayers. Optimized conformations of S8 and Al2Sx (x = 3, 6, 12, and 18) on the (c) Cu-BHT and (d) graphene monolayers, simulated with and without the vdW functional. | ||
From the binding energy plot shown in Fig. 2a, we observed that graphene shows quite low binding affinity towards S8 (−0.76 eV) and Al2Sx molecules (−0.83 to −0.87 eV) with very little variation. Whereas, for the Cu-BHT MOF, the binding strength remarkably increased, and, in contrast to the graphene, the binding energy was not constant but increased from S8 (−1.11 eV) to the Al2Sx molecules, having a maximum value (−3.56 eV) for Al2S3. Our results reveal that the binding energy of Al2Sx molecules varies in the range of −1.44 to −3.56 eV and this stronger binding strength for lower order Al polysulfides could be because of the closer binding approach of the optimized structures forming Al–SCu-BHT (S of Cu-BHT) and Cu–SM (S of the Al2Sx molecule) bonds, as shown on the “with vdW” side in Fig. 2c. This overall increase in the binding strength on the Cu-BHT monolayer compared to graphene indicates its improved anchoring effect as a cathode host in Al–S batteries.
Further, to get more valuable insights into the anchoring effects of the Cu-BHT MOF and graphene, we evaluated the contribution of the physisorption (given by the vdW interactions) and chemisorption to the net binding strengths of S8 and Al2Sx molecules. The percentage ratio of the vdW interaction in the overall binding strength can be calculated using 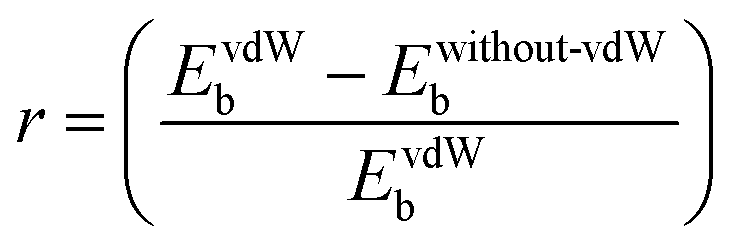 , where EvdWb and Ewithout−vdWb are the binding energies with and without van der Waals corrections. Fig. 2b shows the variation in the van der Waals contributions for the binding of S8 and other Al2Sx molecules on graphene and Cu-BHT monolayers. For graphene, the physisorption dominates the whole process of binding, with the maximum for S8 and the higher order Al2S18 molecule, then Al2S12 (85–87%), and reducing to 78% for the lower order Al2S6 and Al2S3 systems. From this, we can conclude that the physisorption is mainly contributed by the S content and promotes the binding of S8, Al2S18, and Al2S12, whereas for the lower order Al2S6 and Al2S3, chemisorption also starts to contribute. For the case of graphene, the contributions from the vdW functional do not change the nature of interactions of S8 and Al2Sx (Fig. 2d), other than by decreasing the binding distance values from 3.31–4.34 Å to 3.17–3.83 Å. However, the overall binding energies remain quite constant for graphene throughout the charge/discharge process. On the other hand, for the Cu-BHT MOF, physisorption plays a major role for S8 and the higher order Al polysulfides (Al2S12 and Al2S18) with the range of 84–85%, but for Al2S6 and Al2S3, chemisorption dominates, having a vdW ratio of 46 and 21%, respectively. The higher contribution of the chemical interaction for the cases of Al2S3 and Al2S6 can also be clearly understood from Fig. 2c, where Al–SCu-BHT and Cu–SM bond formations are observed both with and without inclusion of the vdW functional.
, where EvdWb and Ewithout−vdWb are the binding energies with and without van der Waals corrections. Fig. 2b shows the variation in the van der Waals contributions for the binding of S8 and other Al2Sx molecules on graphene and Cu-BHT monolayers. For graphene, the physisorption dominates the whole process of binding, with the maximum for S8 and the higher order Al2S18 molecule, then Al2S12 (85–87%), and reducing to 78% for the lower order Al2S6 and Al2S3 systems. From this, we can conclude that the physisorption is mainly contributed by the S content and promotes the binding of S8, Al2S18, and Al2S12, whereas for the lower order Al2S6 and Al2S3, chemisorption also starts to contribute. For the case of graphene, the contributions from the vdW functional do not change the nature of interactions of S8 and Al2Sx (Fig. 2d), other than by decreasing the binding distance values from 3.31–4.34 Å to 3.17–3.83 Å. However, the overall binding energies remain quite constant for graphene throughout the charge/discharge process. On the other hand, for the Cu-BHT MOF, physisorption plays a major role for S8 and the higher order Al polysulfides (Al2S12 and Al2S18) with the range of 84–85%, but for Al2S6 and Al2S3, chemisorption dominates, having a vdW ratio of 46 and 21%, respectively. The higher contribution of the chemical interaction for the cases of Al2S3 and Al2S6 can also be clearly understood from Fig. 2c, where Al–SCu-BHT and Cu–SM bond formations are observed both with and without inclusion of the vdW functional.
To get more insights into the interaction mechanism between the Cu-BHT MOF and Al polysulfides, we have analysed the charge density difference (ρCDD), as expressed by the following equation:
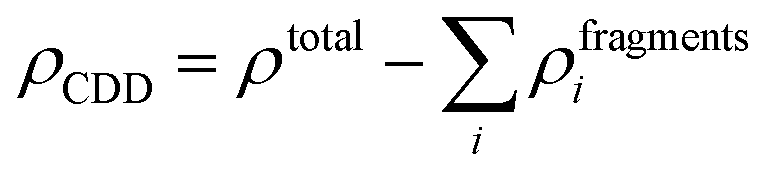 | (7) |
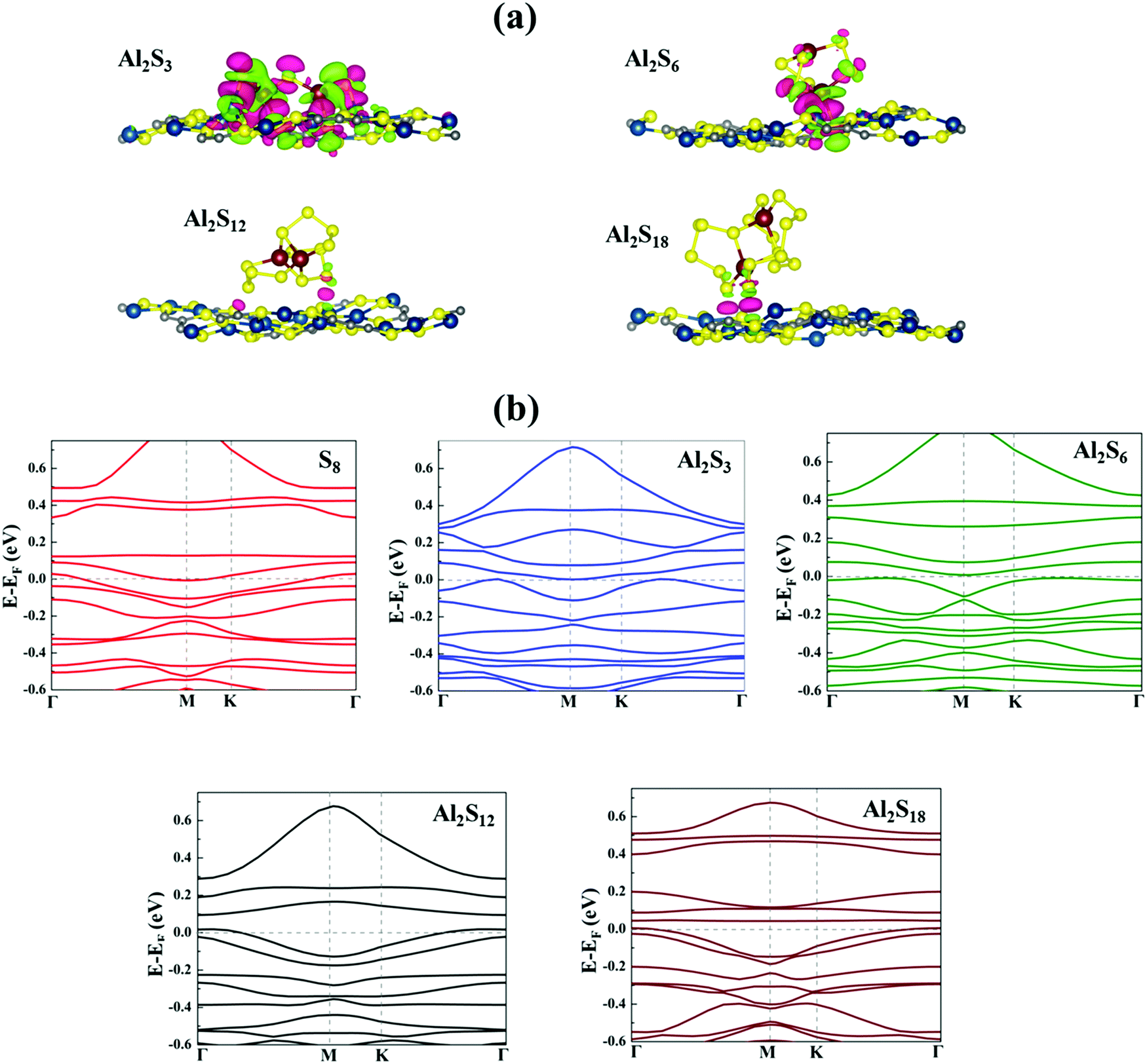 | ||
| Fig. 3 (a) Charge density difference (ρCDD) plots of Al2Sx molecules on the Cu-BHT MOF surface (isosurface value = 0.0015 e Å−3). Here, the pink and green colors represent charge accumulation and depletion, respectively. (b) Electronic band structures of S8 and Al2Sx molecules on the Cu-BHT MOF. The Fermi level is set to zero and presented in grey. | ||
| System | Al2S18 | Al2S12 | Al2S6 | Al2S3 |
|---|---|---|---|---|
| Charge transfer (|e|) | 0.05 | −0.05 | 0.24 | 0.73 |
Further, to evaluate the suitability of the Cu-BHT MOF as a cathode host in Al–S batteries, it is important to check whether it is able to overcome the insulating nature of the S cathode and the corresponding Al polysulfides formed during charging and discharging processes to provide constant conductivity in the system. Cu-BHT is superconducting in nature with Tc ∼ 3 K.75Fig. 3b shows the electronic band structures of the S8 and Al2Sx containing Cu-BHT monolayer systems. All the systems retain their metallic character in the presence of the Cu-BHT monolayer. We further observed that for all S8 and Al2Sx containing Cu-BHT MOF systems except the case of Al2S3, the bands at the Fermi level are quite flat, indicating no significant interaction between S8/Al2Sx and Cu-BHT MOF. Moreover, for the Al2S3 system along the high symmetry k-point path, from Γ point to K point, the nature of the bands at the Fermi level is not flat. And, this further signifies the strong interaction between Al2S3 and the Cu-BHT MOF as observed from the binding energy and charge transfer calculations. This metallic nature may be suitable to provide electron conduction in the whole system, making a path for electrons to participate in the redox reactions of S8 and Al polysulfides on the Cu-BHT surface during charge/discharge reactions. Therefore, the Cu-BHT system may work in a similar manner to the experimentally studied S@HKUST-1-C system,48 where a significant decrease in the kinetic barrier for the conversion of sulfur species during battery operation is observed due to the presence of Cu atoms.
The strong interaction between Al2S3 and Cu-BHT may not be good for the performance of the Al–S battery as it can lead to decomposition of Al2S3 and reduce the reversibility of the charging process. Therefore, to check the possibility of Al2S3 decomposition on the Cu-BHT surface, we calculated the binding energy difference between the intact and decomposed structures. The following decomposition reactions were studied:
| Al2S3 → Al + AlS3; ΔE1 = EAl2S3 − EAl+AlS3 = −0.21 eV | (8) |
| Al2S3 → Al + Al + S3; ΔE2 = EAl2S3 − EAl+Al+S3 = −0.11 eV | (9) |
3.2. Interaction with the electrolyte
One of the primary concerns for sulfur based batteries is the low confinement of elemental sulfur and polysulfides in the cathode host compared to their increased attraction towards the involved electrolyte, which leads to shuttle reactions that cause poor cycle lifetimes, high self-discharge rates and low Coulombic efficiency. In Al–S batteries, the carbonaceous cathode host (S@C) is also observed to show pronounced shuttling effects of Al polysulfides from cathode to anode and fast capacity decay, poor S utilization, and low reversibility.48 We have calculated the binding strengths of higher order Al2Sx (x = 6, 12, and 18) with an ionic liquid electrolyte and compared the results for the graphene and Cu-BHT hosts. To take a reliable approach and to uncover the solvation effect, we have employed the bulk liquid electrolyte model (Fig. 4) to calculate the binding energies of Al2Sx with the ionic liquid electrolyte. The bulk electrolyte model was constructed using a cubic supercell of 22.58 Å length containing 32 EMIM+AlCl4− molecules consistent with the density of about 1.3 g cm−3 EMIM+AlCl4− electrolyte. For simulation of the higher order polysulfides, Al2S6/Al2S12 and Al2S18, the cubic supercell contained 31 EMIM+AlCl4− with 1 Al2S6/Al2S12 and 30 EMIM+AlCl4− with 1 Al2S18, respectively. We allowed the AIMD simulation to run for 2 ps with the NVT ensemble at 300 K and the Nosé thermostat76 was used to control the temperature. The corresponding total energy vs. time plots are shown in Fig. S4, ESI,† and the resultant bulk systems obtained after 2 ps are presented in Fig. 4.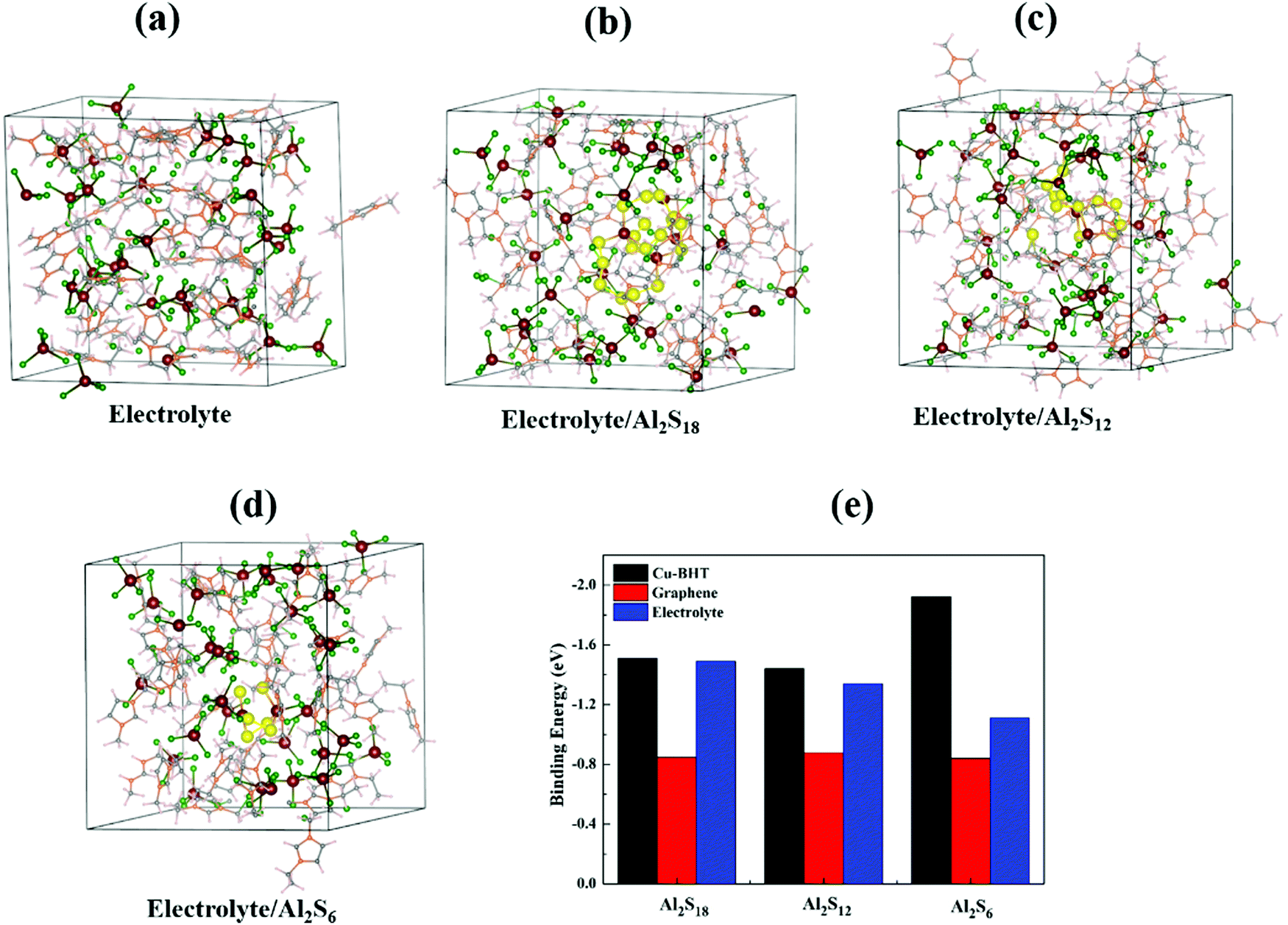 | ||
| Fig. 4 Optimized supercells of (a) EMIM+AlCl4− electrolyte, (b) EIM+AlCl4−/Al2S18, (c) EMIM+AlCl4−/Al2S12, and (d) EMIM+AlCl4−/Al2S6 after 2 ps of AIMD simulations. Al2S18, Al2S12, and Al2S6 immersed in an ionic liquid are highlighted by larger spheres. (d) Comparative binding energies of Al2Sx (x = 6, 12, and 18) molecules with graphene, the Cu-BHT monolayer and EMIM+AlCl4− electrolyte. | ||
The last 1 ps of simulation was used to calculate the average total energy of the simulated bulk systems (Fig. 4a–d). The average binding energy for each system was obtained using the following equation:
| EBinding = (EElecrolyte+M − EElectrolyte − EM) | (10) |
We observed that the EMIM+AlCl4− ionic liquid electrolyte shows a higher binding strength for Al2S18 (−1.49 eV), Al2S12 (−1.34 eV) and Al2S6 (−1.11 eV) compared to the graphene monolayer (−0.84 to −0.87 eV). Moreover, we also observed that the electrolyte itself shows stronger binding to the graphene surface with the binding energy of −0.95 eV, much higher than those of S8 and Al2Sx molecules on graphene. Based on these results, we can conclude that carbonaceous materials do not make suitable anchoring hosts for Al–S batteries, as they not only are able to bind elemental sulfur and Al–polysulfides strongly, but the involved ionic liquid electrolyte also competes to bind with the carbonaceous host. And, all of these reasons lead to shutting effects of intermediate polysulfides, observed in experimental applications of carbon-based cathode hosts.48
However, in the case of the Cu-BHT monolayer, even after consideration of the solvation effect in the bulk electrolyte environment, the binding energies of Al polysulfides (−1.51 to −1.92 eV) on the Cu-BHT host are higher than that of the electrolyte. The stronger binding is a result of soft acid–base interactions between the S atoms (Lewis base) of Al polysulfides and Cu(I) atoms (Lewis acid) of the Cu-BHT MOF compared to the Cl atoms of the EMIM+AlCl4− electrolyte. In addition, the EMIM+AlCl4− electrolyte was also observed to have a lower interaction energy (−0.98 eV) with the Cu-BHT monolayer host in comparison with the Al polysulfides. Therefore, there is a lower probability of S8 and Al2Sx molecules dissolving in the electrolyte. We expect that the Cu-BHT MOF can act as a potential anchoring host with reduced polysulfide shuttling compared to carbonaceous hosts to provide an improved cycle life in Al–S batteries. In a similar way, the vital effect of Cu has also been observed in a recent experimental report on a HKUST-1-C/S cathode for Al–S batteries,48 where, in the electrochemical process, Cu forms an ionic cluster with the polysulfide, facilitates the electrochemical reaction and shows improved reversibility of S cathode during charging/discharging process.
3.3. Deposition of Al2S3 on the Cu-BHT monolayer surface
Several experimental and theoretical reports on Li–S batteries have expressed that a cathode host material able to regulate uniform deposition of the discharge product Li2S is useful to reduce the cell resistance as well as manifest a rapid kinetics in the redox reaction of Li polysulfides.68,77,78 Therefore, to check the suitability of Cu-BHT as an anchoring material in Al–S batteries, we investigated the deposition behaviour of Al2S3 molecules on the Cu-BHT monolayer surface. We compared the binding energies of various possible initial configurations of two and then three Al2S3 molecules on the surface of the Cu-BHT MOF. Fig. 5 shows the most stable configurations of the isolated Al2S3 molecules and the agglomeration networks for both two and three Al2S3 molecule depositions. The relative energies of these configurations show that Al2S3 molecules are more inclined to form an agglomerated network and the network size further increases as more Al2S3 molecules are deposited on the Cu-BHT surface (Fig. 5c). This uniform deposition of Al2S3 on the Cu-BHT MOF is a result of the synergistic dual interactions of Al–SCu-BHT and Cu–SM, which enhance the tendency of Cu-BHT to form an electrical contact with Al2Sx. And, as a result, Cu-BHT can reduce the kinetic barrier of Al polysulfide redox reactions. Moreover, this uniform deposition of Al2S3 increases the surface utilization of the Cu-BHT monolayer, as shown in Fig. 5c.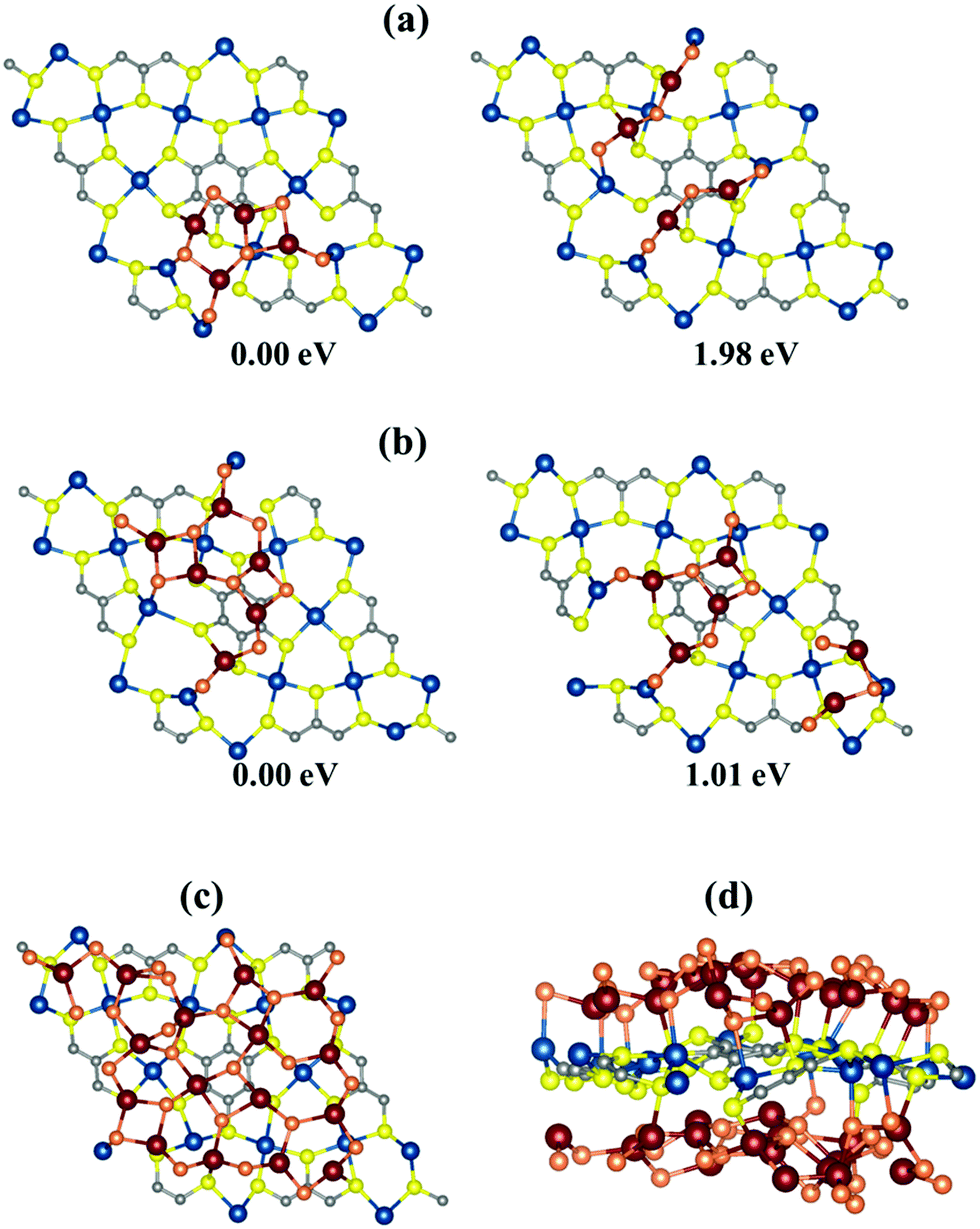 | ||
| Fig. 5 Optimized configurations of (a) two Al2S3 and (b) three Al2S3 molecules on the Cu-BHT monolayer with their relative energies, situated near and far from each other. (c) Uniform Al2S3 layer deposition on the Cu-BHT monolayer surface. (d) Side view of the optimized configuration with the maximum stable loading of Al2S3 molecules on the Cu-BHT monolayer surface. Here, for clear understanding, the sulfur atoms of the Al2S3 molecules and the Cu-BHT monolayer surface are shown in orange and yellow colors, respectively. | ||
Further, to check the capacity retention in an Al–S battery, we calculated the amount of sulfur that can be incorporated into the Cu-BHT MOF host. Al2S3 molecules were allowed to bind on both sides of a 2 × 2 × 1 Cu-BHT monolayer to form an energetically stable system. We observed that the Al2S3 molecules bind in a layered framework on both sides of the Cu-BHT monolayer (Fig. 5c and d) with average binding energies of −5.36 eV and −5.18 eV for one side and both sides of Cu-BHT, respectively. Upon allowing the binding of a third layer of Al2S3 molecules, the system becomes energetically unstable, favouring the binding of 16 Al2S3 molecules in the 2 × 2 × 1 Cu-BHT monolayer host. Therefore, the maximum amount of sulfur that can be incorporated into the Cu-BHT monolayer is 45.81 weight percent (wt%). The capacity of the Cu-BHT MOF is quite a lot higher than that of the experimentally studied S@HKUST-1-C host, which holds 34 wt% sulfur.48 However, the sulfur capacity of the Cu-BHT MOF reduces for multi-layered systems due to a decrease in the number of exposed sites for Al2S3 binding. The 2 × 2 × 2 bilayer and 2 × 2 × 3 trilayer Cu-BHT hosts were observed to hold 38.80 wt% and 36.00 wt% sulfur with the average binding energies of −4.98 eV and −4.65 eV, respectively (Fig. S5, ESI†).
Overall, for both HKUST and Cu-BHT MOFs, the structure along with the presence of Cu plays quite an important role in the Al–S battery by enhancing the stability of sulfur and Al2Sx molecules, and improving reaction reversibility and electronic conductivity. The Cu-BHT monolayer MOF may work as a more suitable cathode host for Al–S batteries, furnishing a higher sulfur capacity.
4. Conclusion
In this work, we have investigated the preferential applicability of a 2D Cu-benzenehexathial (Cu-BHT) metal organic framework for use as a potential anchoring cathode host for Al–S batteries as compared to a carbonaceous host such as graphene. We observed that the favourable electrical conductivity of the Cu-BHT MOF eliminates the insulating nature of S8 and other Al polysulfide (Al2Sx) molecules and could be helpful in supporting the cathode charge/discharge processes. From the binding calculations of S8 and Al2Sx molecules, we observed that graphene shows consistently low binding energies, where physisorption dominates throughout the whole process of binding (78–87%). Whereas, the Cu-BHT MOF exhibits improved binding of Al polysulfides (−1.44 to −3.56 eV), because of the presence of copper and sulfur, which results in increased charge transfer to the Al polysulfides. Moreover, from the comparative study of S8 and Al2Sx molecules binding with graphene, Cu-BHT and EMIM+AlCl4− electrolyte, we concluded that with the relatively stronger binding of Al2Sx molecules, an Al–S battery with a Cu-BHT MOF host cathode could show reduced overall dissolution of Al polysulfides into the electrolyte compared with the case of a carbonaceous host to provide an improved cycle life. The Cu-BHT MOF permits synergic dual interaction based on the Al–S (of the sulfur host) bond and S (of the Al polysulfide)–Cu bond, leading to the uniform deposition of insoluble Al2S3 over the Cu-BHT surface. This improves the utilization of the active cathode host, allowing a better sulfur incorporation of 45.81 wt% sulfur in the Cu-BHT MOF host, which is even higher compared to the HKUST MOF (34 wt%) studied in a very recent experimental report. Overall, all of these results suggest that the Cu-BHT MOF can be used as a promising anchoring cathode host compared to graphene in Al–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IIT Indore for providing the lab and computing facilities. This work is supported by DST-SERB (Project Number CRG/2018/001131) and SPARC (Project Number SPARC/2018-2019/P116/SL), New Delhi. P. B. thanks MHRD for a research fellowship.References
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J.-F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS
.
- Y. Wang, R. Chen, T. Chen, H. Lv, G. Zhu, L. Ma, C. Wang, Z. Jin and J. Liu, Energy Storage Mater., 2016, 4, 103–129 CrossRef
- W. I. A. Sadat and L. A. Archer, Sci. Adv., 2016, 2, e1600968 CrossRef
- L. Bai, J. Electrochem. Soc., 1990, 137, 3737 CrossRef CAS
- P. V. Suneesh, T. G. Satheesh Babu and T. Ramachandran, Int. J. Miner., Metall. Mater., 2013, 20, 909–916 CrossRef CAS
- W. Wang, B. Jiang, W. Xiong, H. Sun, Z. Lin, L. Hu, J. Tu, J. Hou, H. Zhu and S. Jiao, Sci. Rep., 2013, 3, 3383 CrossRef
- M. Chiku, H. Takeda, S. Matsumura, E. Higuchi and H. Inoue, ACS Appl. Mater. Interfaces, 2015, 7, 24385–24389 CrossRef CAS
- H. Wang, Y. Bai, S. Chen, X. Luo, C. Wu, F. Wu, J. Lu and K. Amine, ACS Appl. Mater. Interfaces, 2015, 7, 80–84 CrossRef CAS
- H. Wang, X. Bi, Y. Bai, C. Wu, S. Gu, S. Chen, F. Wu, K. Amine and J. Lu, Adv. Energy Mater., 2017, 7, 1602720 CrossRef
- J. Jiang, H. Li, J. Huang, K. Li, J. Zeng, Y. Yang, J. Li, Y. Wang, J. Wang and J. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 28486–28494 CrossRef CAS
- W. Kaveevivitchai, A. Huq, S. Wang, M. J. Park and A. Manthiram, Small, 2017, 13, 1701296 CrossRef
- L. Mei, J. Xu, Z. Wei, H. Liu, Y. Li, J. Ma and S. Dou, Small, 2017, 13, 1701441 CrossRef
- Z. Li, B. Niu, J. Liu, J. Li and F. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 9451–9459 CrossRef CAS
- L. Geng, J. P. Scheifers, C. Fu, J. Zhang, B. P. T. Fokwa and J. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 21251–21257 CrossRef CAS
- M. A. Vahid, A. Hadjikhani, S. Shahbazmohamadi and M. Beidaghi, ACS Nano, 2017, 11, 11135–11144 CrossRef
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743–9746 RSC
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 324–328 CrossRef CAS
- K. V. Kravchyk, S. Wang, L. Piveteau and M. V. Kovalenko, Chem. Mater., 2017, 29, 4484–4492 CrossRef CAS
- P. Bhauriyal, A. Mahata and B. Pathak, Phys. Chem. Chem. Phys., 2017, 19, 7980–7989 RSC
- S. Wang, K. V. Kravchyk, F. Krumeich and M. V. Kovalenko, ACS Appl. Mater. Interfaces, 2017, 9, 28478–28485 CrossRef CAS
- N. P. Stadie, S. Wang, K. V. Kravchyk and M. V. Kovalenko, ACS Nano, 2017, 11, 1911–1919 CrossRef CAS
- A. S. Childress, P. Parajuli, J. Zhu, R. Podila and A. M. Rao, Nano Energy, 2017, 39, 69–76 CrossRef CAS
- H. Chen, H. Xu, S. Wang, T. Huang, J. Xi, S. Cai, F. Guo, Z. Xu, W. Gao and C. Gao, Sci. Adv., 2017, 3, eaao7233 CrossRef
- H. Chen, F. Guo, Y. Liu, T. Huang, B. Zheng, N. Ananth, Z. Xu, W. Gao and C. Gao, Adv. Mater., 2017, 29, 1605958 CrossRef
- Y. Wu, M. Gong, M.-C. Lin, C. Yuan, M. Angell, L. Huang, D.-Y. Wang, X. Zhang, J. Yang and B.-J. Hwang,
et al.
, Adv. Mater., 2016, 28, 9218–9222 CrossRef CAS
- P. Bhauriyal, A. Mahata and B. Pathak, J. Phys. Chem. C, 2017, 121, 9748–9756 CrossRef CAS
- P. Bhauriyal, A. Mahata and B. Pathak, Chem. – Asian J., 2017, 12, 1944–1951 CrossRef CAS
- P. Bhauriyal, P. Garg, M. Patel and B. Pathak, J. Mater. Chem. A, 2018, 6, 10776–10786 RSC
- P. Bhauriyal, G. Bhattacharyya, K. S. Rawat and B. Pathak, J. Phys. Chem. C, 2019, 123, 3959–3967 CrossRef CAS
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203–5215 CrossRef CAS
- Z. A. Zafar, S. Imtiaz, R. Razaq, S. Ji, T. Huang, Z. Zhang, Y. Huang and J. A. Anderson, J. Mater. Chem. A, 2017, 5, 5646–5660 RSC
- T. Mori, Y. Orikasa, K. Nakanishi, C. Kezheng, M. Hattori, T. Ohta and Y. Uchimoto, J. Power Sources, 2016, 313, 9–14 CrossRef CAS
- S. Wang, Z. Yu, J. Tu, J. Wang, D. Tian, Y. Liu and S. Jiao, Adv. Energy Mater., 2016, 6, 1600137 CrossRef
- Z. Yu, Z. Kang, Z. Hu, J. Lu, Z. Zhou and S. Jiao, Chem. Commun., 2016, 52, 10427–10430 RSC
- S. Wang, S. Jiao, J. Wang, H.-S. Chen, D. Tian, H. Leia and D.-N. Fang, ACS Nano, 2017, 11, 469–477 CrossRef CAS
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zua and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS
- M. J. Klein, G. M. Veith and A. Manthiram, J. Am. Chem. Soc., 2017, 139, 10669–10676 CrossRef CAS
- Q. Pang, X. Liang, C. Y. Kwok and F. N. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS
- S. Xin, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2014, 26, 1261–1265 CrossRef CAS
- S. Wenzel, H. Metelmann, C. Raiß, A. K. Dürr, J. Janek and P. Adelhelm,, J. Power Sources, 2013, 243, 758–765 CrossRef CAS
- T. Gao, M. Noked, A. J. Pearse, E. Gillette, X. Fan, Y. Zhu, C. Luo, L. Suo, M. A. Schroeder and K. Xu,
et al.
, J. Am. Chem. Soc., 2015, 137, 12388–12393 CrossRef CAS
- W. Li, S. Cheng, J. Wang, Y. Qiu, Z. Zheng, H. Lin, S. Nanda, Q. Ma, Y. Xu, F. Ye, M. Liu, L. Zhou and Y. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6406 CrossRef CAS
- G. Cohn, L. Ma and L. A. Archer, J. Power Sources, 2015, 283, 416–422 CrossRef CAS
- T. Gao, X. Li, X. Wang, J. Hu, F. Han, X. Fan, L. Suo, A. J. Pearse, S. B. Lee, G. W. Rubloff, K. J. Gaskell, M. Noked and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 9898–9901 (
Angew. Chem., Int. Ed.
, 128
, 10052–10055
) CrossRef CAS
- X. Yu and A. Manthiram, Adv. Energy Mater., 2017, 7, 1700561 CrossRef
- X. Yu, M. J. Boyer, G. S. Hwang and A. Manthiram, Chem, 2018, 4, 586–598 CAS
- Y. Guo, H. Jin, Z. Qi, Z. Hu, H. Ji and L.-J. Wan, Adv. Funct. Mater., 2019, 29, 1807676 CrossRef
- P. Bhauriyal, S. Das and B. Pathak, J. Phys. Chem. C, 2020, 124, 11317–11324 CrossRef
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Z. Wang, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CrossRef CAS
- G.-P. Hao, C. Tang, E. Zhang, P.-Y. Zhai, J. Yin, W. Zhu, Q. Zhang and S. Kaskel, Adv. Mater., 2017, 29, 1702829 CrossRef
- Y. Mao, G. Li, Y. Guo, Z. Li, C. Liang, X. Peng and Z. Lin, Nat. Commun., 2017, 8, 14628 CrossRef
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Energy Environ. Sci., 2014, 7, 2715–2724 RSC
- X. Huang, P. Sheng, Z. Tu, F. Zhang, J. Wang, H. Geng, Y. Zou, C.-an. Di, Y. Yi, Y. Sun, W. Xu and D. Zhu, Nat. Commun., 2015, 6, 7408 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS
- P. E. BlÖchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 1795–17979 CrossRef
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef
- E. D. Jemmis, K. T. Giju and J. Leszczynski, J. Theor. Chem., 1997, 2, 130–138 CAS
- N. Zhang, Y. Shi, Z. Gao, F. Kong and Q. Zhu, J. Chem. Phys., 1994, 101, 1219 CrossRef CAS
- T. Yu, F. Li, C. Liu, S. Zhang, H. Xu and G. Yang, J. Mater. Chem. A, 2017, 5, 9293–9298 RSC
- X. Zhang, Y. Zhou, B. Cui, M. Zhao and F. Liu, Nano Lett., 2017, 17, 6166–6170 CrossRef CAS
- F. Li, X. Zhang, X. Liu and M. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 15012–15020 CrossRef CAS
- Y. X. Zhao and I. L. Spain, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 993 CrossRef CAS
- A. Bosak, M. Krisch, M. Mohr, J. Maultzsch and C. Thomsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153408 CrossRef
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef
- E. Sanville, S. D. Kenny, R. Smith and G. J. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS
- W. Tang, E. Sanville and G. J. Henkelman, J. Condens. Matter Phys., 2009, 21, 084204 CrossRef CAS
- X. Huang, S. Zhang, L. Liu, L. Yu, G. Chen, W. Xu and D. Zhu, Angew. Chem., Int. Ed., 2018, 57, 146 CrossRef CAS
- S. Nosé, A unified formulation of the constant temperature molecular dynamics methods, J. Chem. Phys., 1984, 81, 511 CrossRef
- L. Kong, X. Chen, B.-Q. Li, H.-J. Peng, J.-Q. Huang, J. Xie and Q. Zhang, Adv. Mater., 2018, 30, 1705219 CrossRef
- J. Zhao, Y. Yang, R. S. Katiyar and Z. Chen, J. Mater. Chem. A, 2016, 4, 6124–6130 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00546k |
| This journal is © The Royal Society of Chemistry 2020 |