
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly pattern directed sustained release from porous microspheres of discotic tripeptides†
Santosh
Kumar
,
Santu
Bera
 ,
Sujay Kumar
Nandi
and
Debasish
Haldar
*
,
Sujay Kumar
Nandi
and
Debasish
Haldar
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, West Bengal 741246, India. E-mail: deba_h76@yahoo.com; deba_h76@iiserkol.ac.in; Fax: +91 3325873020; Tel: +91 3325873119
First published on 14th November 2020
Abstract
Porous microspheres from discotic tripeptides have been investigated as potential candidates for drug delivery vehicles. The C3 symmetric discotic tripeptide adopts a supramolecular helical column structure by three fold intermolecular hydrogen bonding interactions as well as face to face π–π stacking interactions. But the C2 symmetric discotic tripeptide adopts a supramolecular dimer like structure by six-fold intermolecular hydrogen bonding interactions and face to face π–π stacking interactions. Field emission scanning electron microscopy (FE-SEM) revealed that the C3 symmetric discotic tripeptide exhibits bird nest-like porous microsphere morphology formed by the assembly of the individual columns. However the C2 symmetric discotic tripeptide forms round clay pitcher like porous microspheres. These porous microspheres have been used as potential carriers for the sustained release of a bacteriostatic antibiotic sulfamethoxazole. The spectroscopic studies as well as the growth inhibition of E. coli reveal that the round clay pitcher-like porous microspheres are more efficient than the bird nest-like porous microspheres for the sustained release of drugs. The report highlights the importance of the self-assembly pattern for the fabrication of advanced functional materials.
Introduction
Supramolecular microstructures developed via self-assembly of building blocks have received considerable attention because these structures can lead to advanced functional materials.1–5 The self-assembly process is governed by various non covalent interactions, including hydrogen-bonding interactions, ion-pair interactions, and π–π stacking and hydrophobic interactions.6–8 Normally, this self-assembly process is fully reversible which leads to numerous possible applications of the supramolecular material in the fields of environmental sciences, material sciences, biomedical sciences and chemistry.9–12 The reversibility of the supramolecular systems has led to a change in their topologies and properties upon exposure to external stimuli such as temperature, light, pH, and redox potential.13–15 Among these diverse self-assembly systems, the amphiphilic microspheres offer potential applications for drug encapsulation, delivery, and sustained release.16Unwanted side effects and systemic toxicity are common hazards of using naked drugs.17 So, rapping and pharmaceutical formulation is important.18 But this should work in synergy and will not generate toxic chemicals by further reaction. An ideal delivery vehicle should deliver the required amount of drug to the target site and confirm the sustained release of the encapsulated drug under physiological conditions.19 To date, diverse compounds such as polymers and polymer-based micelles,20 metal–organic frameworks (MOFs),21 covalent organic frameworks (COFs),22 carbon nanotubes,23 metal nanoparticles,24 silica,25 iron oxide nanoparticles,26 or dendrimer nanoparticles have been used for drug formulation and delivery.27 But, due to surface modification most of these reported delivery systems have side effects including intrinsic toxicity or immunogenicity.28 In this regard, peptide-based self-assembled microspheres29 are promising due to their biocompatibility30 and recognition properties.31 Also, due to their hydrophilic–hydrophobic balance for solubilising and targeted delivery of highly hydrophobic anti-tumour32 and antidepressant drugs,33 and statins,34 peptide-based delivery vehicles are very popular. Moreover, the hybrid peptides have stability towards enzymatic degradation.35 Hence, the design and synthesis of new peptide building blocks and delivery vehicles are of great interest. P. K. Das and co-workers have reported vesicles from discotic amphiphiles that can encapsulate and deliver doxorubicin inside mammalian cells.36
We are developing microspheres and microvesicles as delivery vehicles, from peptidomimetic compounds.37 Herein, we have designed and synthesized two discotic tripeptides with different symmetries, from commercially available sources. The discotic tripeptide having C3 symmetry adopts a supramolecular column structure stabilized by three-fold intermolecular hydrogen bonds. However, the discotic tripeptide having C2 symmetry forms a supramolecular dimer stabilized by six-fold intermolecular hydrogen bonding interactions. The FE-SEM and the Atomic Force Microscopy (AFM) studies revealed that the C3 symmetric discotic tripeptide shows bird nest like porous microsphere morphology but the C2 symmetric discotic tripeptide exhibits round clay pitcher like porous microsphere morphology. The microspheres were loaded with the bacteriostatic antibiotic sulfamethoxazole. The round clay pitcher-like porous microspheres of the C2 symmetric discotic tripeptide are more efficient than the bird nest-like porous microspheres of the C3 symmetric discotic tripeptide, for the sustained release of encapsulated drugs.
Results and discussion
Synthesis and characterization
We have designed and synthesized two discotic tripeptides having C3 symmetry and C2 symmetry (Scheme 1). The assumption was that compound 1 with C3 symmetry would form a supramolecular columnar structure by three-fold intermolecular hydrogen bonds between amide functionalities, like that reported in earlier reports.36 However, compound 2 with C2 symmetry may fail to form such a columnar structure. Three L-Phe residues should impart hydrophobicity in the compounds and direct donor–acceptor type π–π stacking and further modulate the self-assembly process. The discotic tripeptide 1 was synthesized from benzene-1,3,5-tricarboxylic acid and L-phenylalanine methyl ester using DCC as a coupling reagent in 72% yields. The discotic tripeptide 2 was synthesized by coupling methyl ester of 5-aminoisophthalic acid and Boc-Phe-OH followed by hydrolysis and further coupling with L-phenylalanine methyl ester using DCC as a coupling reagent in 44.5% yield (Scheme S1, ESI†). All the synthesized discotic tripeptides and intermediates were purified by column chromatography and fully characterized by 1H-NMR, 13C-NMR and FTIR spectroscopy and mass spectrometry.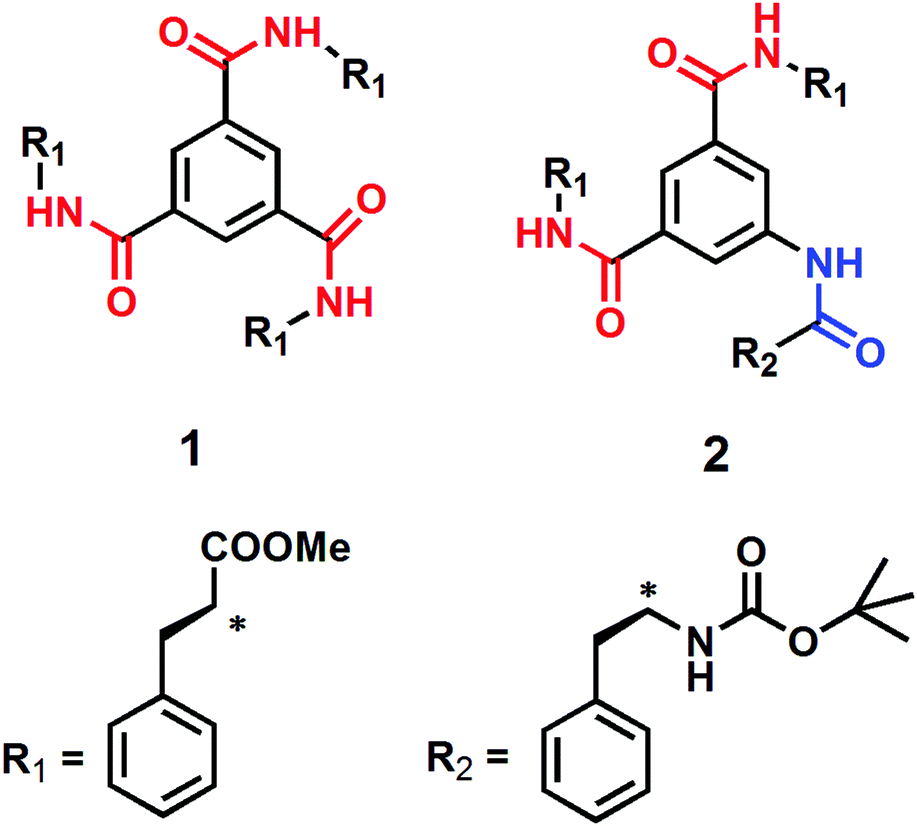 | ||
| Scheme 1 The schematic presentation of discotic tripeptides 1 and 2. | ||
Morphology
The self-assembly propensity of the discotic tripeptides 1 and 2 was examined by field emission scanning electron microscopy (FE-SEM) and atomic force microscopy (AFM). For FE-SEM studies, a small amount of solution (0.02 M) of the discotic tripeptides 1 and 2 in methanol was placed on a clean glass slide and then dried by slow evaporation at room temperature. The samples were then allowed to dry under vacuum at 30 °C for 24 h. The dried samples were coated with gold, and the micrographs were taken in a Zeiss DSM 950 scanning electron microscope. The FE-SEM images of the discotic tripeptide 1 exhibit the formation of bird nest-like polydisperse porous microsphere morphology (Fig. 1a). The average diameter of the microspheres is about 1.5 μm. A careful observation revealed that the bird nest-like porous microspheres are formed by assembly of nanorods (Fig. 1b). The average diameter of the nanorods is ca. 130 nm. The size of the pores on the surface of the microspheres is ca. 50 nm. We were also able to capture the image of bird nest-like porous microspheres formed from the nanorods (Fig. S1, ESI†), which shows that the shape and size of the nanorods are very regular. However, the FE-SEM images of the discotic tripeptide 2 show the formation of round clay pitcher like polydisperse porous microsphere morphology (Fig. 1c). The average diameter of the microspheres is ca. 1.2 μm. The internal diameter of the hollow inside the microspheres is ca. 800 nm. The thickness of the microsphere wall is ca. 150 nm. The inset of Fig. 1d showing an enlarged image of a microsphere like round clay pitcher clearly indicates the presence of cavities inside the microspheres and pores on the surface. The size of the pore is ca. 50 nm. To know about the topography of the discotic tripeptides 1 and 2, atomic force microscopic studies have been performed. For AFM studies, the solutions (0.02 M) of tripeptides 1 and 2 in methanol were placed on a microscopic glass slide and allowed to dry under vacuum at 30 °C for two days and studied using a NT-MDT atomic force microscope. The AFM micrograph of discotic tripeptide 1 shows the polydisperse microsphere like morphology where the surface of the microspheres is very rough (Fig. S2a, ESI†). Fig. S2b (ESI†) shows the 3D AFM image of the microsphere morphology. However, the AFM image of discotic tripeptide 2 shows the polydisperse microsphere like morphology where the surface of the microspheres is quite smooth (Fig. S2c, ESI†). Fig. S2d (ESI†) shows the 3D AFM image of the microsphere morphology with a smooth surface. | ||
| Fig. 1 (a) The FE-SEM images showing bird nest-like porous microsphere morphology of discotic tripeptide 1. (b) The FE-SEM images showing the assembly of nanorods to form the bird nest-like porous microspheres of discotic tripeptide 1. (c) and (d) The FE-SEM images showing round clay pitcher like porous microsphere morphology of discotic tripeptide 2. (d) The inset showing the pores of the microspheres of discotic peptide 2. | ||
Structure analysis
For discotic tripeptide 2, mass spectrometry points to the existence of both the monomer and dimer. Distinct signals for the monomer and dimer were detected using a Q-Tof Micro YA263 high-resolution mass spectrometer (Fig. 2).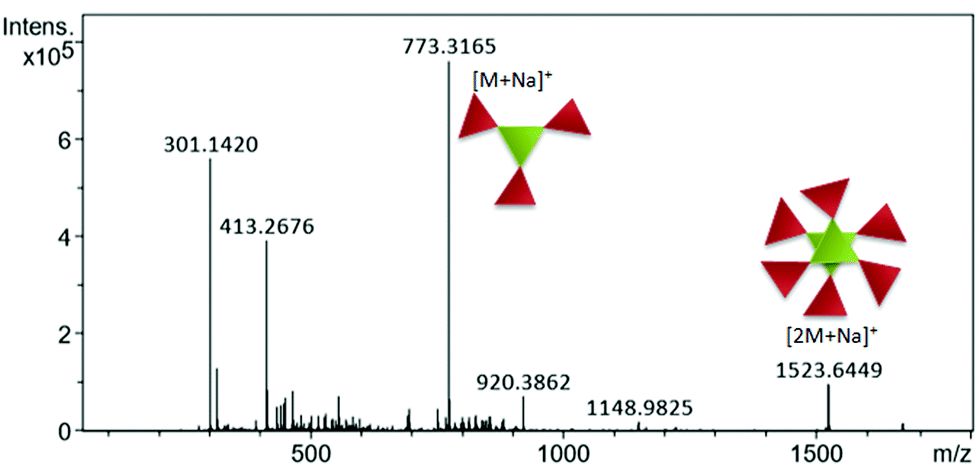 | ||
| Fig. 2 Mass spectrometry data showing the coexistence of both the monomer and dimer of discotic tripeptide 2. | ||
Circular dichorism (CD) spectroscopy in the solution state was performed to study the aggregation propensity of the discotic tripeptides 1 and 2. The shape and intensity of the CD spectra differ significantly for discotic tripeptides 1 and 2 in methanol. Discotic tripeptide 1 shows a positive cotton effect at 198 nm and negative cotton effects at 208 nm and 228 nm (Fig. 3a), reflecting the formation of helical supramolecular stacks.38 We conclude that such a strong CD signal originates from a rigid, elongated columnar self-assembly of the compound. The discotic tripeptide 2 has a positive band at 200 nm and negative bands at 211 and 226 nm with much lower intensity responsible for a dimer structure of shorter stacking (Fig. 3a).
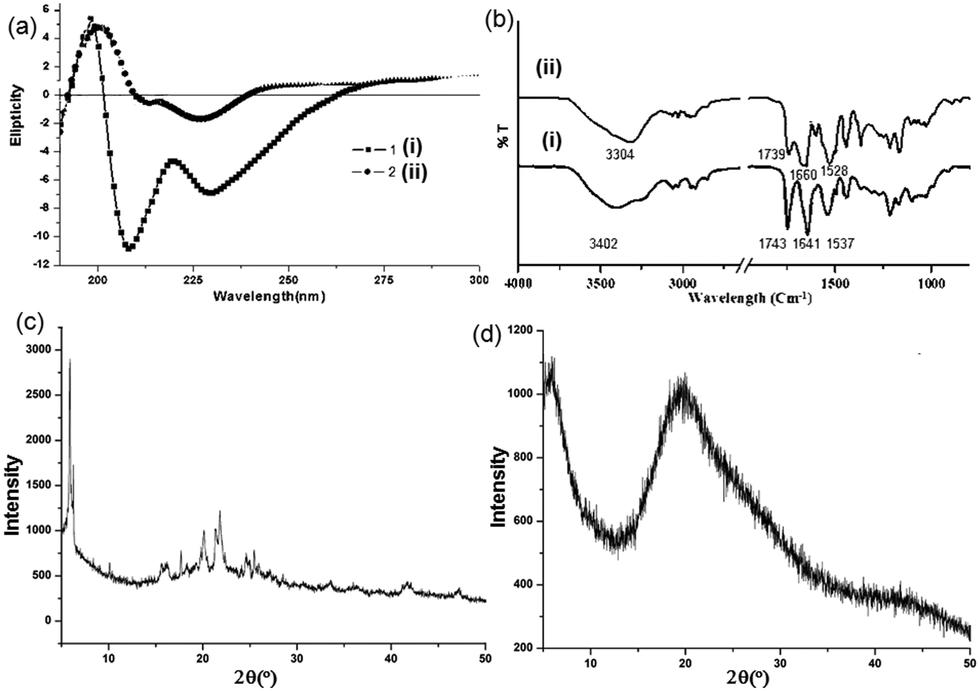 | ||
| Fig. 3 (a) CD spectra of the discotic tripeptides 1 and 2 in methanol. (b) Solid-state FT-IR spectra of the microspheres obtained from discotic tripeptides 1 and 2. (c) and (d) PXRD patterns of discotic tripeptides 1 and 2, respectively. | ||
Solid-state FT-IR spectroscopy was performed to study the structure of the discotic tripeptides 1 and 2 in porous microspheres. For discotic tripeptide 1, an intense band at 1743 cm−1 indicates the presence of three ester groups in the compound (Fig. 3b).39 The peaks at 1641 cm−1 and 1537 cm−1 arise due to the formation of H-bonded stacks of the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (Fig. 3b). For discotic tripeptide 2, the band at 1739 cm−1 indicates the presence of two ester groups (Fig. 3b). The peaks at 1660 cm−1 and 1528 cm−1 arise due to the formation of H-bonded amide I and amide II (Fig. 3b). Another informative frequency range is 3500–3200 cm−1, corresponding to the N–H stretching vibration. The discotic tripeptides 1 and 2 show peaks at 3304 cm−1 for hydrogen-bonded NH (Fig. 3b).
O groups (Fig. 3b). For discotic tripeptide 2, the band at 1739 cm−1 indicates the presence of two ester groups (Fig. 3b). The peaks at 1660 cm−1 and 1528 cm−1 arise due to the formation of H-bonded amide I and amide II (Fig. 3b). Another informative frequency range is 3500–3200 cm−1, corresponding to the N–H stretching vibration. The discotic tripeptides 1 and 2 show peaks at 3304 cm−1 for hydrogen-bonded NH (Fig. 3b).
The structures of the discotic tripeptides 1 and 2 were also studied by powder X-ray diffraction (PXRD). The PXRD pattern of the microspheres of discotic tripeptide 1 shows that the material is crystalline in nature. Sharp reflections were observed in the 5–50° 2θ range (Fig. 3c). Powder X-ray diffraction (PXRD) had reflection at d-spacings of 4.07 Å and 15.98 Å corresponding to the distances between neighbouring aromatic moieties and the laminal spacing, respectively. However, the PXRD pattern of the microspheres of discotic tripeptide 2 shows that the material is non-crystalline in nature (Fig. 3d).
From the above experimental results, we propose that the discotic tripeptide 1 self-assembles by three-fold intermolecular hydrogen bonding interactions to form supramolecular helical columnar stacks which further bundled to develop a nano rod-like structure (Fig. 4a). In higher-order, these nanorods are assembled to form bird nest-like porous microspheres (Fig. 4a). However, the discotic tripeptide 2 self-assembles by six-fold intermolecular hydrogen bonding interactions to form a dimer which further assembles to develop a 2D sheet-like structure that folds and forms the round clay pitcher like porous microspheres (Fig. 4b).
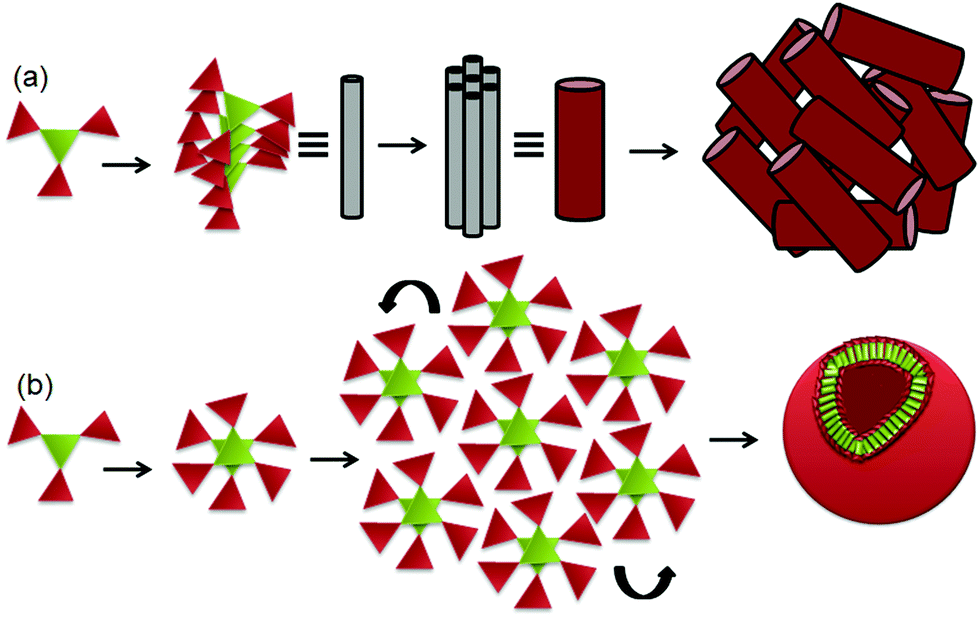 | ||
| Fig. 4 (a) The schematic presentation, showing self-assembly of discotic tripeptide 1 to columnar stacks to nanorods to porous microspheres. (b) The schematic presentation showing self-assembly of discotic tripeptide 2 to dimers to 2D sheets to porous microspheres. | ||
The microspheres from discotic tripeptides are stable and insoluble in water. We have encapsulated a bacteriostatic antibiotic sulfamethoxazole in discotic tripeptide 1 and 2 microspheres. To 3 mL of 0.50 × 10−3 M drug solution in methanol, 5 mg of discotic tripeptides 1 or 2 was added and stirred for 6 h, the solvent was evaporated under vacuum and the drug-loaded microspheres were washed several times with 50 mL water and centrifuged until the drained water after centrifugation exhibited negligible drug fluorescence. The drug-binding property of the microspheres obtained from methanol solution of discotic tripeptides 1 or 2 was studied by absorption and emission spectroscopy as these are quite sensitive techniques to understand any changes in microenvironments. From the absorption spectrum of discotic tripeptide 2 (Fig. 5a), it is clear that with increasing sulfamethoxazole concentration the absorbance at 262 nm gradually increases and the spectrum is red shifted (ca. 6 nm). This implies that the microspheres have some interactions with the drug molecules. The result was further supported by emission spectroscopy. From Fig. 5b, it is evident that when we increase the concentration of the drug the fluorescence intensity of discotic tripeptide 2 at 347 nm gradually decreases and this spectrum is also shifted by 10 nm. Discotic tripeptide 1 also shows similar results with increasing sulfamethoxazole concentration studied by absorption spectroscopy (Fig. S3, ESI†).
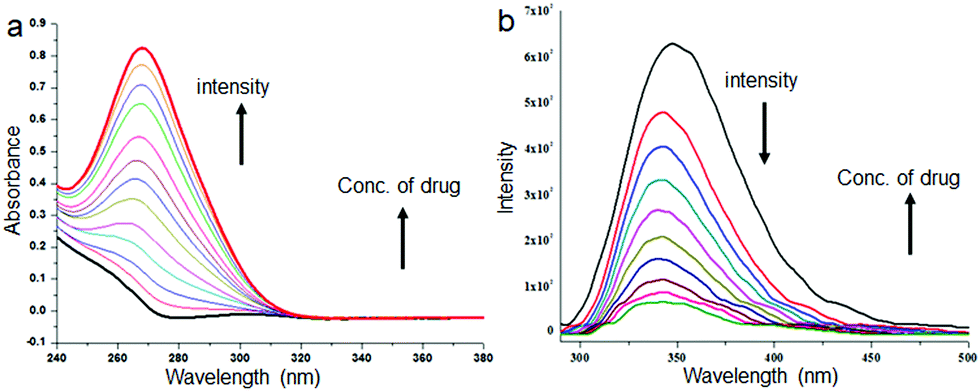 | ||
| Fig. 5 (a) Absorption spectra of discotic tripeptide 2 with increasing concentration of sulfamethoxazole in methanol. Discotic tripeptide 2 concentration = 1 × 10−5 M. (b) Emission spectra of discotic tripeptide 2 with increasing concentration of sulfamethoxazole in methanol. Excitation wavelength is 270 nm. Discotic tripeptide 2 concentration = 1 × 10−5 M. | ||
The encapsulation efficiency (%) was quantified by absorption spectroscopy and calculated as [(amount of drug added − amount of free drug)/amount of drug added] × 100%. The encapsulation efficiency of the drug loaded discotic tripeptide 1 microspheres was found to be 60% and that of discotic tripeptide 2 microspheres was 70%. The drug loading content for the formulation was calculated as [weight of the encapsulated drug in the microspheres/weight of the microspheres used] × 100% and was found to be 4.33% for discotic tripeptide 1 and 5.33% for discotic tripeptide 2.
A further FE-SEM experiment was performed to know whether there is any change in the size or shape of the microspheres after drug loading. The FE-SEM image of the drug-loaded discotic tripeptide 1 clearly indicates that the bird nest-like microsphere morphology is retained and there is no average change in size and shape after drug encapsulation (Fig. 6a). Only the pores are absent in the drug-loaded microspheres. Fig. 6b shows the FE-SEM image of the drug encapsulated discotic tripeptide 2 microspheres, which clearly indicates that the morphology is retained, only the pores of the microspheres are filled up. For reference, an FE-SEM study of only the sulfamethoxazole drug under the same conditions was done. Fig. 6c shows the fiber-like morphology of sulfamethoxazole. The fibers have a diameter of ca. 100 nm and are several micrometers in length.
 | ||
| Fig. 6 (a) FE-SEM image of discotic tripeptide 1 polydisperse bird nest like microspheres after sulfamethoxazole loading. (b) FE-SEM image of discotic tripeptide 2 polydisperse microspheres after sulfamethoxazole encapsulation. (c) FE-SEM image exhibiting fiber like morphology of sulfamethoxazole. | ||
We have also studied the release of the encapsulated sulfamethoxazole from the nanovesicles. For this purpose, sulfamethoxazole loaded nanovesicles were immersed into 5 mL Tris buffer (at pH 7.4) in a 15 mL centrifuge tube, centrifuged at 2800 rpm for 15 minutes, and examined by absorption spectroscopy at different time intervals. Fig. 7a shows the drug release profile which clearly indicates that the microspheres of the discotic tripeptide 2 slowly release the encapsulated drug compared to the microspheres of the discotic tripeptide 1, with a complete release by 30 h (Fig. S4, ESI†).
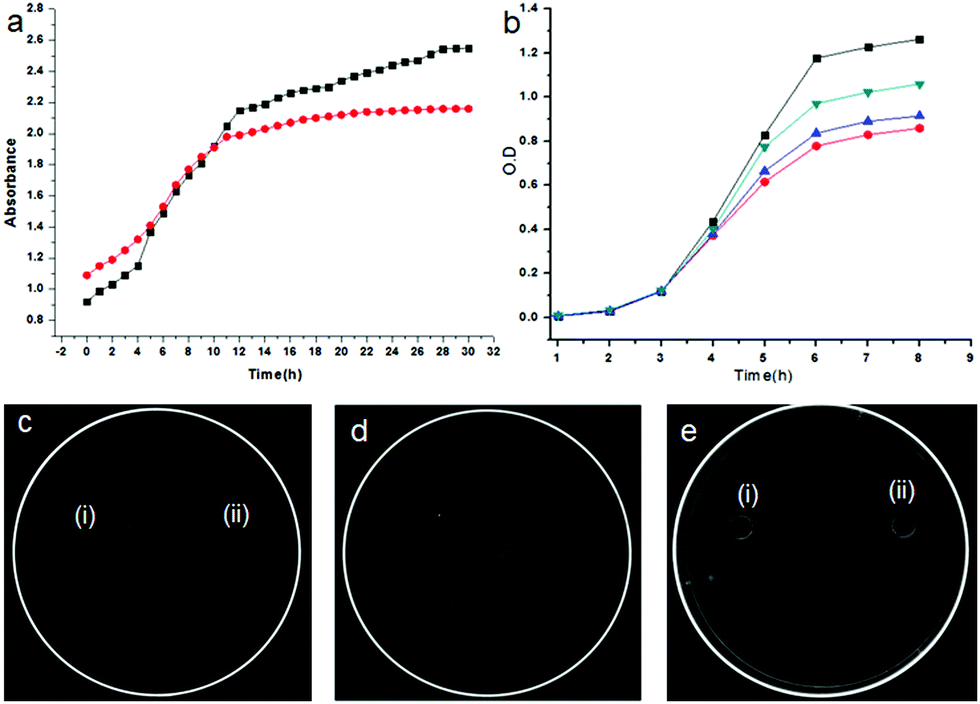 | ||
| Fig. 7 (a) Drug release profile of sulfamethoxazole loaded microspheres of discotic tripeptide 1 (red) and sulfamethoxazole loaded microspheres of discotic tripeptide 2 (black) in tris buffer (pH 7.4) obtained from UV-Vis spectroscopy. (b) The growth inhibition plot of E. coli with only drug (red), discotic tripeptide 1-drug (blue), discotic tripeptide 2-drug (dark cyan), and without any drug (black). The growth inhibition zones of E. coli bacteria against the (c) (i) encapsulated discotic tripeptide 1-drug, (c) (ii) encapsulated discotic tripeptide 2-drug, (d) sulfamethoxazole (drug), (e) (i) discotic tripeptide 1 and (e) (ii) discotic tripeptide 2 in 10% DMSO–water. | ||
The sustainable release of encapsulated sulfamethoxazole and in vitro antibacterial activity in water and DMSO (10%) on E. coli was examined by OD measurement at 600 nm.40Fig. 7b shows the growth inhibition profile of E. coli against the sulfamethoxazole solution in 10% DMSO–water. E. coli is more susceptible in sulfamethoxazole encapsulated microsphere solution than the naked sulfamethoxazole solution (control) (Fig. 7b). It is clear from the diagram that E. coli is more susceptible to the drug-loaded microspheres of discotic tripeptide 2 than the drug-loaded microspheres of discotic tripeptide 1 (Fig. 7b). Fig. 7c(i) and (ii) show the growth inhibition zones of E. coli against the drug-loaded microspheres of discotic tripeptides 1 and 2, respectively, and Fig. 7d shows the same for naked sulfamethoxazole in 10% DMSO–water. Furthermore, Fig. 7e indicates that the discotic tripeptides 1 and 2 do not have any antibacterial activity on the E. coli bacteria.
Conclusions
In conclusion, this study demonstrates the synthesis and self-assembly of C2 and C3 symmetric discotic tripeptides. The C3 symmetric discotic tripeptide forms a supramolecular columnar structure by three fold intermolecular hydrogen bonds. But the C2 symmetric discotic tripeptide adopts a supramolecular dimer by six fold intermolecular hydrogen bonds. FE-SEM images show that the C3 symmetric discotic tripeptide exhibits bird nest-like porous microsphere morphology formed by the assembly of the columns. However the C2 symmetric discotic tripeptide forms round clay pitcher like porous microspheres. Furthermore, these microspheres were loaded with the antibiotic sulfamethoxazole. However, the discotic tripeptides are not growth inhibitors of E. coli. The spectroscopic studies as well as the growth inhibition of E. coli reveal that the round clay pitcher-like porous microspheres are more efficient than the bird nest-like porous microspheres for the sustained release of drugs. The results presented here show the importance of the self-assembly pattern for the fabrication of delivery vehicles.Experimental
General
All chemicals were purchased from Sigma chemicals. The discotic peptides were synthesized by conventional solution phase methodology (ESI†). All the products were purified by column chromatography using silica (100–200 mesh size) gel as a stationary phase and an n-hexane-ethyl acetate mixture as an eluent. The intermediates and final compounds were fully characterized by 400 MHz 1H NMR spectroscopy, 100 MHz 13C NMR spectroscopy and FTIR spectroscopy and mass spectrometry.NMR experiments
All NMR studies were carried out on a JEOL 400 MHz spectrometer at 25 °C. Compound concentrations were in the range of 1–10 mM in CDCl3 and (CD3)2SO.FTIR spectroscopy
The solid-state FTIR spectrum was obtained with a PerkinElmer Spectrum RX1 spectrophotometer with the KBr disk technique.UV/Vis spectroscopy
UV/Vis absorption spectra were recorded on a UV/Vis spectrophotometer (Hitachi).Fluorescence spectroscopy
Fluorescence spectra were recorded on a Cary Eclipse Fluorescence Spectrophotometer. In a typical experiment, compounds were dissolved in methanol. The spectra were recorded by exciting the system at 270 nm, and 274 nm.Atomic force microscopy
For AFM, images were taken with an NT-MDT instrument (model no. AP-0100), in the semi-contact mode.Field emission scanning electron microscopy
Morphologies of the reported compounds were investigated using field emission-scanning electron microscopy (FE-SEM). A small amount of solution (0.02 M) of the compound was placed on a clean glass slide and then dried by slow evaporation. The sample was then allowed to dry under vacuum at 30 °C for 24 h. The materials were gold-coated, and the micrographs were taken in an FE-SEM apparatus (Zeiss DSM 950 scanning electron microscope).Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Kumar and S. K. Nandi acknowledge the CSIR, India, for a research fellowship. S. Bera thanks UGC, India, for a fellowship. We acknowledge the IISER Kolkata, India, for financial assistance.Notes and references
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G. H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS.
- D. Ni, L. Wang, Y. Sun, Z. Guan, S. Yang and K. Zhou, Angew. Chem., Int. Ed., 2010, 49, 4223–4227 CrossRef CAS.
- W. Kim, J. Thvenot, E. Ibarboure, S. Lecommandoux and E. L. Chaikof, Angew. Chem., Int. Ed., 2010, 49, 4257–4260 CrossRef CAS.
- H. Kim, T. Kim and M. Lee, Acc. Chem. Res., 2011, 44, 72–82 CrossRef CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2275–2825 CrossRef.
- K. M. Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–168 CrossRef.
- (a) T. Xiao, L. Xu, J. Wang, Z. Y. Li, X. Q. Sun and L. Wang, Org. Chem. Front., 2019, 6, 936–941 RSC; (b) T. Xiao, L. Xu, J. Götz, M. Cheng, F. Würthner, J. Gu, X. Feng, Z. Y. Li, X. Q. Sun and L. Wang, Mater. Chem. Front., 2019, 3, 2738–2745 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS.
- C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- S.-I. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592–8593 CrossRef CAS.
- (a) B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS; (b) T. Xiao, W. Zhong, L. Xu, X. Q. Sun, X. Y. Hu and L. Wang, Org. Biomol. Chem., 2019, 17, 1336–1350 RSC; (c) T. Xiao, L. Qi, W. Zhong, C. Lin, R. Wang and L. Wang, Mater. Chem. Front., 2019, 3, 1973–1993 RSC.
- (a) P. Vitovič, J. M. Alakoskela and P. K. J. Kinnunen, J. Med. Chem., 2008, 51, 1842–1848 CrossRef; (b) P. George, J. Appl. Pharm. Sci., 2011, 1, 40–44 Search PubMed; (c) W. G. H. Steve and E. Unger, Chem. Res. Toxicol., 2006, 19, 1564–1569 Search PubMed; (d) T. Liu and R. B. Altman, J. Chem. Inf. Model., 2015, 55, 1483–1494 CrossRef CAS.
- (a) M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahramia and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC; (b) S. Kalepu and V. Nekkanti, Acta Pharm. Sin. B, 2015, 5, 442–453 CrossRef; (c) R. Xing, T. Jiao, L. Yan, G. Ma, L. Liu, L. Dai, J. Li, H. Möhwald and X. Yan, ACS Appl. Mater. Interfaces, 2015, 7, 24733–24740 CrossRef CAS; (d) Q. Zou, L. Zhang, X. Yan, A. Wang, G. Ma, J. Li, H. Möhwald and S. Mann, Angew. Chem., Int. Ed., 2014, 53, 2366–2370 CrossRef CAS.
- (a) E. K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS; (b) M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC; (c) H. Zhang, J. Fei, X. Yan, A. Wang and J. Li, Adv. Funct. Mater., 2015, 25, 1193–1204 CrossRef CAS.
- (a) A. O. Eniola and D. A. Hammer, J. Controlled Release, 2003, 87, 15–22 CrossRef CAS; (b) S. S. Chandran, A. Nan, D. M. Rosen, H. Ghandehari and S. R. Denmeade, Mol. Cancer Ther., 2007, 6, 2928–2937 CrossRef CAS; (c) F. Gu, L. Zhang, B. A. Teply, N. Mann, A. Wang, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2586–2591 CrossRef CAS.
- (a) S. Sengupta, A. Goswami and R. Mondal, New J. Chem., 2014, 38, 2470–2479 RSC; (b) J. Zhuang, C.-H. Kuo, L. Y. Chou, D. Y. Liu, E. Weerapana and C. K. Tsung, ACS Nano, 2014, 8, 2812–2819 CrossRef CAS; (c) K. T. Holman, Angew. Chem., Int. Ed., 2011, 50, 1228–1230 CrossRef CAS.
- (a) J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC; (b) M. Dogru, A. Sonnauer, S. Zimdars, M. Döblinger, P. Knochel and T. Bein, CrystEngComm, 2013, 15, 1500–1502 RSC.
- (a) Z. Liu, K. Chen, C. Davis, S. Sherlock, Q. Cao, X. Chen and H. Dai, Cancer Res., 2008, 68, 6652–6660 CrossRef CAS; (b) W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp, J. P. Briand, R. Gennaro, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2005, 44, 6358–6362 CrossRef CAS; (c) S. Kumar, R. Rani, N. Dilbaghi, K. Tankeshwar and K.-H. Kim, Chem. Soc. Rev., 2017, 46, 158–196 RSC.
- C. R. Patra, R. Bhattacharya, D. Mukhopadhyay and P. Mukherjee, Adv. Drug Delivery Rev., 2010, 62, 346–361 CrossRef CAS.
- M. R. Mohammadi, A. Nojoomi, M. Mozafari, A. Dubnika, M. Inayathullah and J. Rajadas, J. Mater. Chem. B, 2017, 5, 3995–4018 RSC.
- (a) S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS; (b) A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- (a) L. M. Kaminskas, B. D. Kelly, V. M. McLeod, G. Sberna, D. J. Owen, B. J. Boyd and C. J. Porter, J. Controlled Release, 2011, 152, 241–248 CrossRef CAS; (b) S. C. Zimmerman, J. R. Quinn, E. Burakowska and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 8164–8167 CrossRef CAS; (c) H. T. Chen, M. F. Neerman, A. R. Parrish and E. E. Simanek, J. Am. Chem. Soc., 2004, 126, 10044–10048 CrossRef CAS.
- (a) B. S. Zolnik, A. G. Fernandez, N. Sadrieh and M. A. Dobrovolskaia, Endocrinology, 2010, 151, 458–465 CrossRef CAS; (b) L. A. Mitchell, F. T. Lauer, S. W. Burchiel and J. D. McDonald, Nat. Nanotechnol., 2009, 4, 451–456 CrossRef CAS.
- (a) S. Cao, Z. Pei, Y. Xu and Y. Pei, Chem. Mater., 2016, 28, 4501–4506 CrossRef CAS; (b) D. G. Fatouros, D. A. Lamprou, A. J. Urquhart, S. N. Yannopoulos, I. S. Vizirianakis, S. Zhang and S. Koutsopoulos, ACS Appl. Mater. Interfaces, 2014, 6, 8184–8189 CrossRef CAS; (c) C. Cai, J. Lin, Y. Lu, Q. Zhang and L. Wang, Chem. Soc. Rev., 2016, 45, 5985–6012 RSC.
- (a) A. J. V. Hell, M. M. Fretz, D. J. A. Crommelin, W. E. Hennink and E. Mastrobattista, J. Controlled Release, 2010, 141, 347–353 CrossRef; (b) C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC; (c) M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS; (d) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS; (e) A. J. V. Hell, C. I. C. A. Costa, F. M. Flesch, M. Sutter, W. Jiskoot, D. J. A. Crommelin, W. E. Hennink and E. Mastrobattista, Biomacromolecules, 2007, 8, 2753–2761 CrossRef.
- C. H. Gorbitz, M. Nilsen, K. Szeto and L. W. Tangen, Chem. Commun., 2005, 4288–4290 RSC.
- Y. Ma, D. Liu, D. Wang, Y. Wang, Q. Fu, J. K. Fallon, X. Yang, Z. He and F. Liu, Mol. Pharmaceutics, 2014, 11, 2623–2630 CrossRef CAS.
- C. Sung, J. E. Raeder and E. W. Merrill, J. Pharm. Sci., 1990, 79, 829–834 CrossRef CAS.
- A. Hughes, M. J. Rogers, A. I. Idris and J. C. Crockett, Calcif. Tissue Int., 2007, 81, 403–413 CrossRef CAS.
- Y. Chen, T. Li, J. Li, S. Cheng, J. Wang, C. Verma, Y. Zhao and C. Wu, Org. Biomol. Chem., 2017, 15, 1921–1929 RSC.
- S. Dinda, M. Ghosh and P. K. Das, Langmuir, 2016, 32, 6701–6712 CrossRef CAS.
- (a) M. Debnath, S. Sasmal and D. Haldar, J. Mater. Chem. B, 2017, 5, 5450–5457 RSC; (b) S. K. Maity, S. Bera, A. Paikar, A. Pramanik and D. Haldar, CrystEngComm, 2014, 16, 2527–2534 RSC; (c) S. K. Maity, S. Bera, A. Paikar, A. Pramanik and D. Haldar, CrystEngComm, 2013, 15, 5860–5866 RSC; (d) S. Maity, P. Jana, S. K. Maity and D. Haldar, Langmuir, 2011, 27, 3835–3841 CrossRef CAS; (e) S. Maity, P. Jana, S. K. Maity and D. Haldar, Soft Matter, 2011, 7, 10174–10181 RSC; (f) A. Pramanik, A. Paikar and D. Haldar, RSC Adv., 2015, 5, 53886–53892 RSC.
- J. J. van Grop, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef.
- (a) C. Toniolo and M. Palumbo, Biopolymers, 1977, 16, 219–224 CrossRef CAS; (b) V. Moretto, M. Crisma, G. M. Bonora, C. Toniolo, H. Balaram and P. Balaram, Macromolecules, 1989, 22, 2939–2944 CrossRef CAS.
- M. Sangra, S. Jaiswal and G. D. Gupta, Indo Am. J. Pharm. Res., 2014, 4, 2943–2950 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of nitropeptides, 1H NMR, 13C NMR, Fig. S1–S4 and S5–S10 or other electronic format. See DOI: 10.1039/d0ma00691b |
| This journal is © The Royal Society of Chemistry 2020 |