
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA twist in the non-slanted H-mers to control π-conjugation in 2-dimensions and optical properties†
Mrinal Kanti
Das‡
,
Fatima
Hameed‡
,
Ryan
Lillis
and
Nagarjuna
Gavvalapalli
 *
*
Department of Chemistry, Institute for Soft Matter Synthesis and Metrology, Georgetown University, 3700 O St NW, Washington, DC 20057, USA. E-mail: ng554@georgetown.edu
First published on 19th October 2020
Abstract
Understanding the structural parameters that determine the extension of π-conjugation in 2-dimensions is key for controlling the optical, photophysical, and electronic properties of 2D-π-conjugated materials. In this article, three non-slanted H-mers including a donor–acceptor H-mer (H-mer-3) with an increase in dihedral angle (twist) between the strands and rungs are synthesized and studied. These non-slanted H-mers represent the repeat units of 2D-π-conjugated materials. H-mer-3, containing donor-strands and an acceptor-rung, is an unexplored donor–acceptor architecture in both slanted and non-slanted H-mers. The H-mers displayed both acid and base dependent optical properties. While the rungs have a little impact on the H-mer absorption spectra they play a key role in the emission and fluorescence lifetime. H-mer-3 (i.e., donor–acceptor H-mer) shows a higher Stokes shift and fluorescence lifetime than the other two H-mers. The twist and the presence of an electron deficient rung in H-mer-3 facilitated an intramolecular charge transfer in the excited state from the strands to the electron deficient rung, and therefore control over the H-mer emission properties. The lack of insulating pendant chains, reduced π–π interactions in thinfilms, and longer fluorescence lifetimes make these H-mers interesting candidates for various electronic and optoelectronic applications.
Introduction
The synthesis and study of structurally well-defined, monodisperse π-conjugated oligomers of diverse shapes that are conjugated in 2-dimensions provide insights into the impact of molecular shape on the extension of π-conjugation in 2-dimensions.1–19 Also, nanosized 2D-π-conjugated oligomers lend an opportunity to realize unique physical and electronic properties that are unattainable with linearly conjugated oligomers. For example, cruciform-shaped (also reported as X-mers)3–5,20 optically active π-conjugated oligomers developed by the Bunz group and others exhibit spatially non-overlapping frontier molecular orbitals, which bestowed them with remarkable sensory behavior. The Zhao group has developed slanted H-mers, which are another class of 2D-π-conjugated oligomers, in which two strands are connected via a rung.1,21–23 In the slanted H-mers, the rungs are directly grafted onto strands and therefore only half of the strands are in conjugation with each other resulting in a conjugation path that resembles the letter Z. The charge and exciton delocalization in H-mers can be further extended by involving the whole strand in the π-conjugation rather than just half of the strand. We envisioned that this can be accomplished by connecting the rung to the strand via a fused heterocycle such as imidazole,24,25 thiazole,26 or triazole ring.27 These fused heterocyclic rings provide π-conjugation paths for the whole strand to be in conjugation with each other. Out of these, the benzimidazole (the fused imidazole) containing H-mers are synthesized in one step via dialdehyde and diamine condensation reaction, which is relatively easy to synthesize unlike the other fused heterocycle based H-mers.24,25,28 The non-slanted H-mers represent the repeat units of 2D-π-conjugated materials.29,30 Therefore, in addition to extended π-conjugation between the strands, these H-mers are also appropriate to grow into polymeric structures due to the presence of mirror-plane symmetry compared to the slanted-H-mers (Scheme 1). Even though the fused imidazole containing H-mers have the potential to generate interesting materials for optical and electronic applications, most of the studies are focused on their self-assembled monolayers on solid substrates23,24,31,32 and use in molecular rotors25,28 as axles. To fill in this knowledge gap, herein we have synthesized three H-mers to understand the impact of twist between the strands and rungs as well as the electronic nature of the rung on the H-mer optical, sensing, and photophysical properties.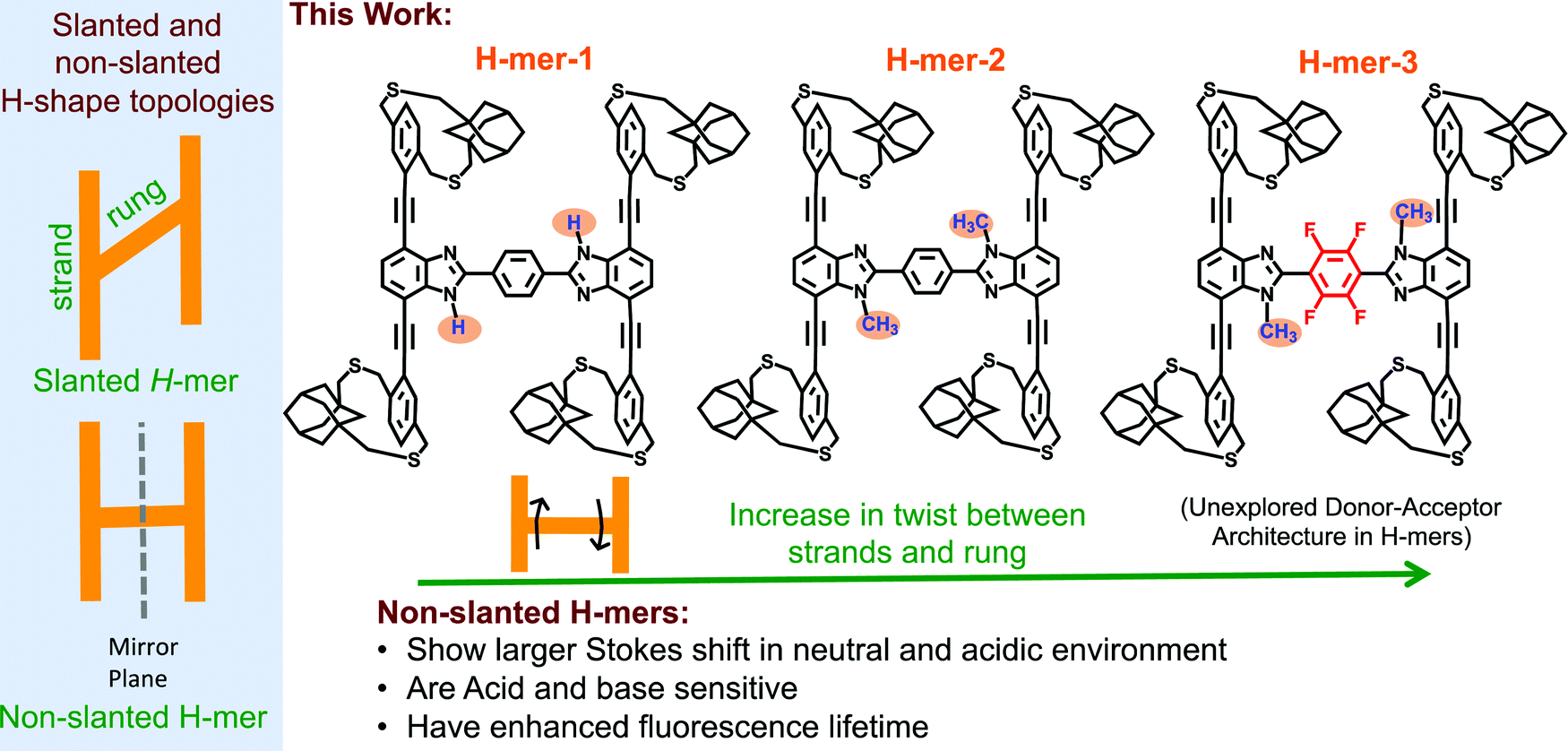 | ||
| Scheme 1 (left) Slanted and non-slanted H-mer topology; (right) three H-mers synthesized in this study with varying twists between strands and rung, as well as an electron deficient rung. | ||
In the H-mers, a rung acts as a bridge between the strands and play a key role in extending the π-conjugation across the strands and therefore determining the optical and electronic properties of H-mers. Thus, the dihedral angle (twist) between the strands and rung determines the extension of π-conjugation between the strands. A twist will disrupt the extension of π-conjugation between the strand and rung, and therefore causes a disruption in π-conjugation between the two strands. To investigate the impact of the twist on the optical properties, a 1H-benzimidazole containing H-mer (H-mer-1), a N-methylated benzimidazole H-mer (H-mer-2), and a N-methylated benzimidazole H-mer with a tetrafluoro substituted phenyl rung (H-mer-3) are synthesized and their optical properties are studied (Scheme 1). H-mer-3 will also help shed light on the impact of an electron deficient rung on the optical properties. Slanted-H-mers containing electron withdrawing and donating substituents on the strands have been synthesized to generate materials with unique optical and optoelectronic properties.1,33 Nonetheless, H-mer-3 with donor-strands and acceptor-rung is an unexplored donor–acceptor architecture in both slanted and non-slanted H-mers and this is the first report of exploring the impact of the electronic nature of rungs on the optical properties of H-mers. Detailed understanding of the factors that dictate the optical and electronic properties of H-mers will help to better design 2D-π-conjugated oligomers and polymers for desired applications.
The benzimidazole H-mers synthesized so far use conventional pendant solubilizing chains to impart solubility.24 To obtain soluble H-mers, herein, we have employed bifacial cyclophanes that were reported by our group recently34 and were shown to impart solubility to polymers without the need for pendant solubilizing chains. All the H-mers are readily soluble without heating in chloroform and dichloromethane. UV-vis absorption and emission spectra of all the three H-mers are recorded in neutral, acidic and basic environments. Studied structural changes and environment had minimal impact on the UV-vis absorption spectra of the H-mers. Interestingly, H-mer-3 with an electron deficient rung and twist displays a larger Stokes shift compared to other H-mers. The red shift in emission upon protonation is significantly higher compared to T-mers that have analogous absorption and emission spectra in a neutral state.35 The larger Stokes shift in the H-mers is due to efficient intramolecular charge transfer in the excited state from the strands to the electron deficient rung. H-mer-1, unlike H-mer-2 and H-mer-3, also shows spectral changes in a basic environment. The photoluminescence of H-mer-3 is quenched by an electron acceptor such as tetracyanoethylene (TCNE). More importantly, the excited state lifetime of H-mer-3 is at least 5 times higher than H-mers-1 and 2, which highlights the importance of the rung electronic nature.
Results and discussion
The H-mers are synthesized as shown in Scheme 2. Rungs for the H-mers are synthesized by condensation of 1,2-dibromo-3,6-diaminobenzene with terephthalaldehyde. In a typical procedure, the diamine and dialdehyde are reacted in the presence of ZrCl4 in chloroform to generate a bisbenzimidazole tetrabromo intermediate (9a). The Sonogashira coupling of 9a with racemic (±) ethynyl adamantanocyclophane (8) resulted in H-mer-1. H-mer-2 is synthesized following similar protocols except that the amines on the tetrabromo-rung were methylated to generate 10a. The methylation of amines helped to improve the solubility of the rung slightly. H-mer-3 was synthesized following similar protocols using tetrafluoro terephthalaldehyde instead of terephthalaldehyde. All the generated H-mers are atactic in nature i.e., there is no control over orientation of the cyclophane units along the strand since a racemic mixture of 8 was used to synthesize the H-mers. Compound 8 was synthesized as shown in Scheme 2. Esterification of adamantane-1,3-dicarboxylic acid followed by reduction yielded adamantane-1,3-dimethanol (3). Thioesterification and reduction of 3 gave adamantane-1,3-dimethanethiol (5). Compound 5 was reacted with 2-bromoxylylene dibromide under high dilution conditions to form (±)-bromo adamantanocyclophane (6). Compound 6 is obtained as a racemic mixture due to planar chirality. Sonogashira coupling of (±)6 with TMS acetylene followed by desilylation resulted in compound (±)8. Tetrabromo intermediates (9a, 9b, 10a and 10b) are insoluble in organic non-polar solvents including chloroform and dichloromethane but are soluble in organic polar solvents including DMSO and DMF. Nonetheless, the final H-mers are readily soluble without heating in low-polarity chlorinated solvents including chloroform and dichloromethane because of the cyclophanes, which impart solubility without the need for pendant solubilizing chains. Absence of pendant solubilizing chains gets rid of the insulating alkyl chain layers between the H-mers in thin films and therefore facilitates enhanced charge and energy transport between the molecules. Also, the ready solubility of H-mers in organic solvents coupled with the absence of pendant solubilizing chains further enables the use of these H-mers as building blocks for 2D-π-conjugated networks.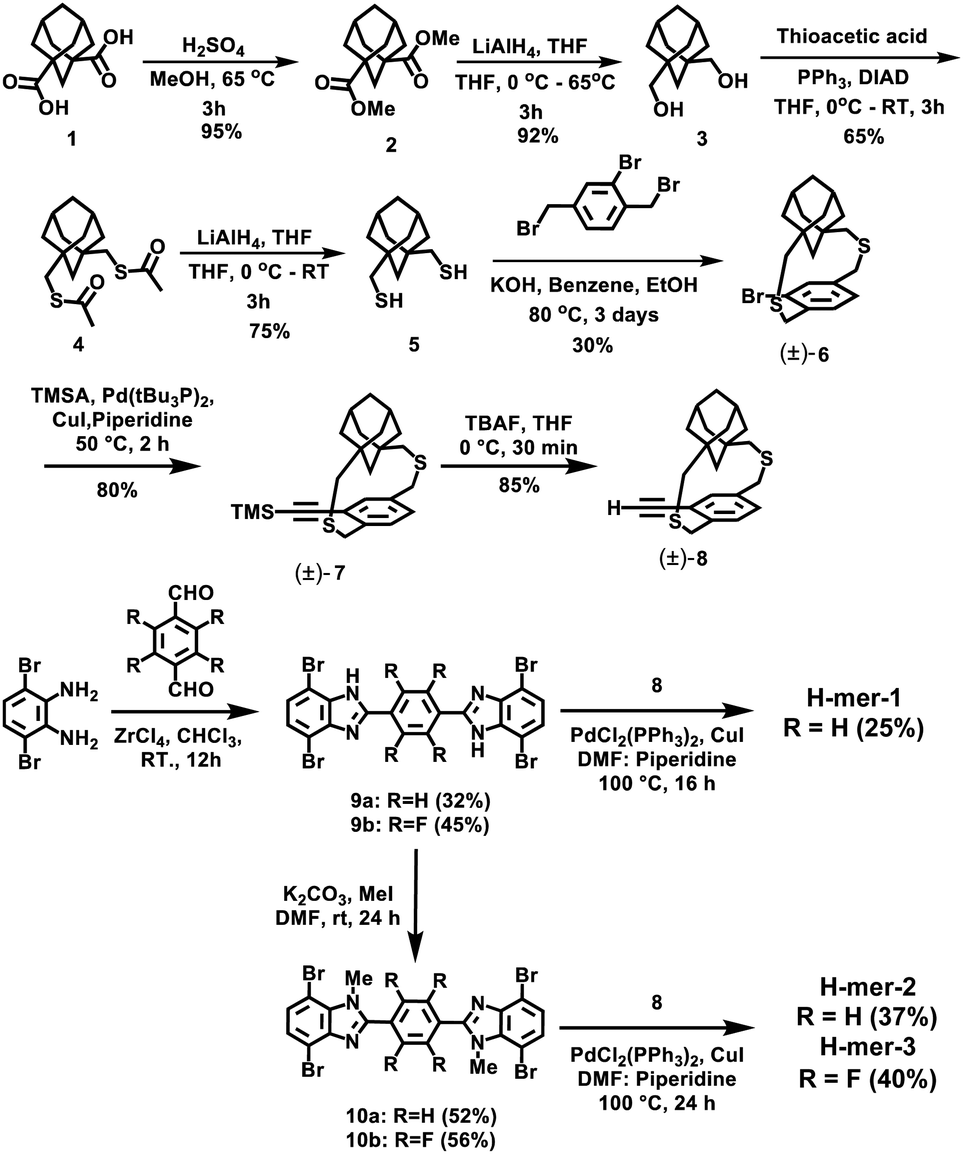 | ||
| Scheme 2 Synthesis of H-mers. | ||
The twist along the rung determines the extension of conjugation between the strands. 1H-NMR spectra of the tetrabromo intermediates provided insight into the twist along the rung and hence the extension of π-conjugation between the benzimidazole and the rung. In the case of 9a, the protons on the phenyl ring are highly deshielded (δ = 8.51 ppm) due to the electron with drawing nature of imidazoles (Fig. 1 and ESI†). Whereas in the case of 10a, the protons are relatively less deshielded (δ = 8.03 ppm) than in 9a indicating that the electron withdrawing effect of imidazoles is slightly impeded after N-methylation (Fig. 1). This is due to the increased twist between the benzimidazole and phenyl ring, which reduces the p-orbital overlap between the imidazoles and phenyl ring. Simulations using B3LYP/6-31G* also showed that the strand is twisted out of plane by 35.3° in the case of the N-methylated H-mer-2 whereas there is no twist in the unmethylated H-mer-1 (0.002°). In the case of H-mer-3, the steric repulsion between the N-methyl group and fluorine further increased the twist to a 54.7° dihedral angle (Fig. 2). Based on the NMR studies and simulations, the twist in the H-mers follow the order: H-mer-3 > H-mer-2 > H-mer-1. The twist between strands and rung helps the H-mers to not pack closely in thin films, which is a desired quality for electroluminescence devices.22
 | ||
| Fig. 1 1H-NMR spectra of 9a and 10a depicting the change in chemical shift of phenyl rung protons. | ||
 | ||
| Fig. 2 Front (top) and side-view (bottom) of H-mers. | ||
In order to determine the impact of twist and electron withdrawing rung on the H-mer optical properties, UV-vis absorption spectra, fluorescence spectra, and fluorescence lifetime of the H-mers were recorded (Fig. 3 and 4). All the H-mers have an absorption maximum at ca. 350–370 nm and a shoulder at ca. 385–390 nm (Fig. 3). The optical band gap of the H-mers, determined from the onset of UV-vis absorption, are within the range of 2.92–3.08 eV indicating that neither twist between the strands and rung nor the presence of electron deficient tetrafluoro phenyl rung has an effect on the band gap (Table 1). The band gap values determined from simulations (B3LYP/6-31G*) are also close for all the three H-mers and concur with the experimental observations (Table 1).
 | ||
| Fig. 3 (a) UV-vis absorption and (b) emission spectra of H-mers in chloroform (c) UV-vis absorption and (d) emission spectra of H-mer-2 and H-mer-3 in hexanes (low polarity solvent) and chloroform (relatively high-polarity solvent). | ||
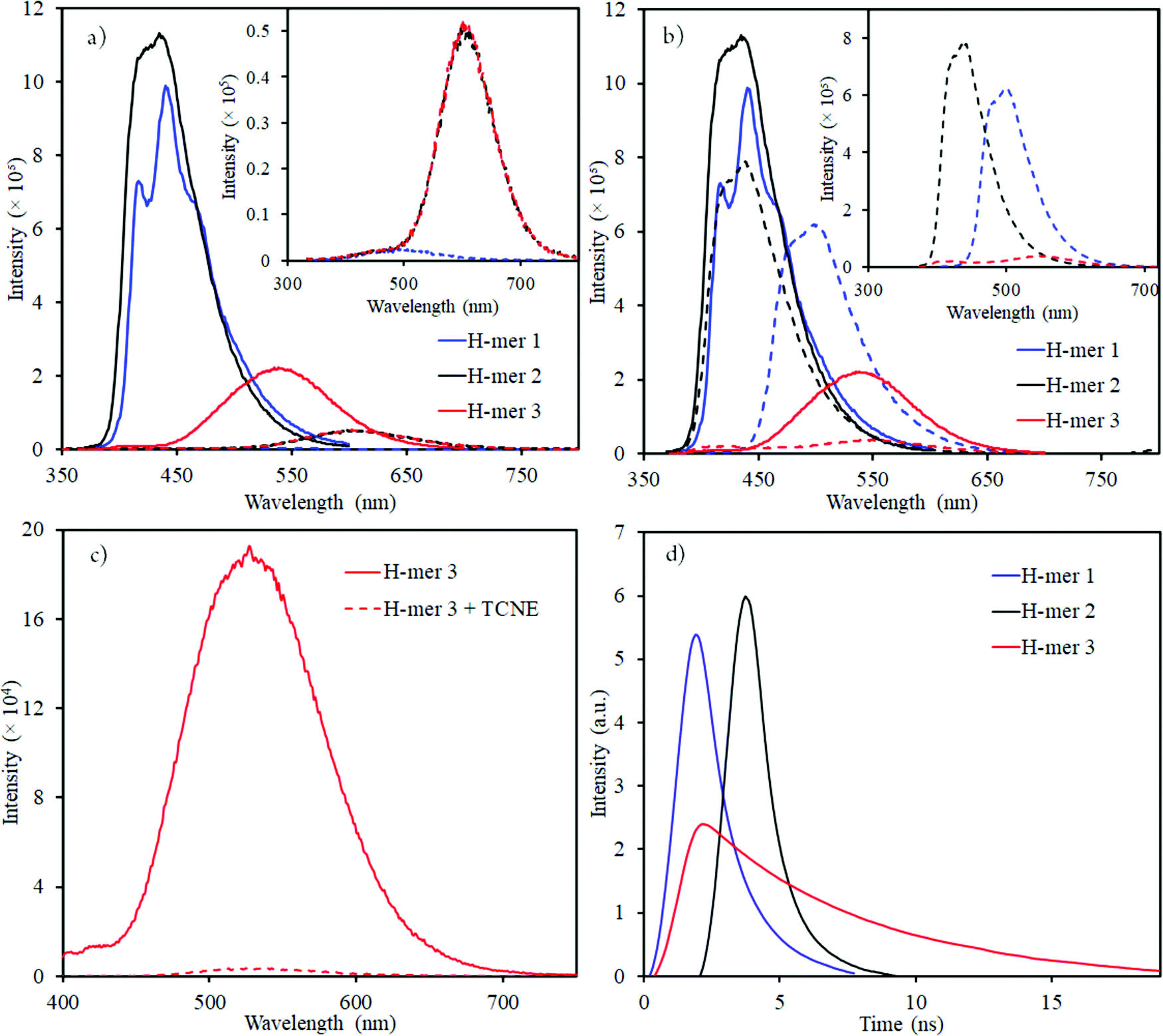 | ||
| Fig. 4 Emission spectra of H-mers in (a) acidic (dashed lines); and (b) basic (dashed lines) environment (inset shows the emission spectra of H-mers in acid or base environment); emission spectra of neutral H-mers are shown in solid lines; (c) H-mer-3 fluorescence quenching by TCNE acceptor; (d) Re-plotted fluorescence lifetime measurements for H-mers (the x-axis was modified with time = 0 ns) where the intensity starts to increase (unmodified fluorescence lifetime measurements data is shown in Fig. S11, ESI†). | ||
| H-mer | λ absmax (CHCl3) (nm) | λ emmax (CHCl3) (nm) | λ emmax (hexane) (nm) | λ absmax (film) (nm) | Bandgap (DFT studies) (eV) | Bandgap (UV-vis absorption) (eV) | Quantum yielda | Fluorescence lifetime | |
|---|---|---|---|---|---|---|---|---|---|
| τ 1 (ns) | τ 2 (ns) | ||||||||
| a Relative quantum yield using 9,10-diphenylanthracene in ethanol as a reference. | |||||||||
| H-mer-1 | 368 | 417, 440 | — | — | 3.023 | 2.90 | 0.21 | 0.78 (80%) | 2.24 (20%) |
| H-mer-2 | 361 | 416, 435 | 411, 437 | 366 | 3.122 | 2.99 | 0.24 | 0.46 (69%) | 1.24 (31%) |
| H-mer-3 | 351, 362 | 535 | 484, 510 | 364 | 3.113 | 3.06 | 0.12 | 3.81 (67%) | NA |
It has been shown that benzimidazoles and 2,2′-bisbenzimidazoles exhibit imine-amine tautomerization and the rate of interconversion is substituent dependent. In general, the conversion is fast above ca. 280 K based on 1H-NMR studies.36,37 H-mer-1 is expected to exhibit imine-amine tautomerization, whereas tautomerization is not possible in H-mer-2 and H-mer-3 due to the N-methylation. Thus, the conjugation path and absorption spectrum of H-mer-1 may vary depending on temperature due to tautomerization whereas temperature should have a negligible effect on the conjugation path in H-mer-2. 1H-NMR spectra of H-mer-1 at both room temperature (298 K) and high temperature (353 K) are recorded and compared (Fig. S1 and S2, ESI†). At high temperature the peaks in the 1H-NMR became slightly sharper. This behavior is also observed in H-mer-2 and H-mer-3 that do not have the possibility to tautomerize (Fig. S3 and S4, ESI†). So, the change in the shape and intensity of the peak at δ: 7.5 ppm in H-mer-1 could be simply due to increase in the solubility of H-mer-1 at elevated temperature and not due to tautomerization. Moreover, due to the presence of different diastereomers in H-mers (see ESI,† page-3) it will be very challenging to clearly assign which protons are responsible for the change in δ: 7.5 ppm peak shape and intensity. 1H-NMR experiments are not helpful here to learn about the possible tautomerization in H-mer-1. It has been shown that some of the substituted benzimidazoles UV-vis peak pattern is useful to identify and quantify the percentage of each tautomers.38–40 Tautomers of H-mer-1 are degenerate, hence their ratio in solution should have minimum effect on the UV-vis peak pattern. Indeed, the UV-vis peak pattern of H-mer-1 did not change at 363 K (Fig. S5, ESI†) indicating that both the tautomers are degenerate. High temperature UV-vis absorption spectrum of H-mer-2 was also recorded to determine if N-methylation has any effect on the extension of π-conjugation within H-mer-2 compared to unmethylated H-mer-1 (Fig. S6, ESI†). No significant difference in the absorption spectra of H-mer-1 and H-mer-2 was observed within the temperature window of 298–363 K, indicating that both secondary (unsubstituted) and tertiary amines (N-methylated) have similar capability in extending the π-conjugation between the strands via the rung.
The absorption maxima of H-mers in thin films (Fig. S7, ESI†) are similar to those in the solution state. Similar absorption spectra in both solution and thin films indicate that π–π aggregation between H-mers is hindered in thin films. Based on our previous studies on cyclophane-based polymers, bifacial cyclophanes are shown to hinder π–π-interactions.34 Even though the cyclophanes in H-mers are located only at the periphery and not on the benzimidazole and the rungs, they are efficient in masking the π-surface of H-mers and hindering π–π interactions in the aggregated state. In addition to this, the twist between the strands and rung in the H-mers also hinders the intermolecular π–π aggregation in thin films and therefore results in no significant changes in the absorption spectra of H-mers in solution and thin films. Thus, the twist in H-mers helps them to not pack closely in thin films, which is a desired quality for electroluminescence devices.22
The emission maxima of H-mers 1 and 2 are 440 and 435 nm, respectively, which correspond to a ca. 72 and 74 nm Stokes shift in chloroform (Fig. 3). On the other hand, the emission maximum of H-mer-3 is 535 nm, which corresponds to ca. 174 nm Stokes shift in chloroform (Fig. 3). The large Stokes shift in the emission maximum of H-mer-3 compared to H-mers-1 and 2 indicates an efficient intramolecular charge transfer in the excited state from the strands to the electron deficient tetrafluorophenyl rung. Emission from the low-energy excited charge transfer state results in the larger Stokes shift. Linear molecules and polymers containing donor–acceptor units have, in general, shown a higher Stokes shift due to intramolecular charge transfer from donor to acceptor moieties in the excited state.41–45 In order to confirm the emission occurring from the charge-transfer state, the polarity of the solvent is reduced. The low polarity solvents are not effective in stabilizing the excited charge-transfer state, and therefore hamper the formation of the charge-transfer excited state and reduce the Stokes shift. The absorption and emission spectra of H-mer-2 and H-mer-3 are recorded in hexane, a low polarity solvent (Fig. 3). Indeed the low polarity hexane has no effect on the absorption and emission spectra of H-mer-2. Similarly, the low polarity hexane has no effect on the absorption spectrum of H-mer-3. The emission spectrum of H-mer-3 in hexane, however, is blue-shifted by 25 nm as expected. Also, the emission spectrum has two maxima at 484 nm and 510 nm. The similar absorption spectrum but different emission spectrum of H-mer-3 compared to H-mers-1 and 2 indicate that rungs have a little impact on the absorption spectra whereas they play a key role in the emission and fluorescecne lifetime (vide infra). Simulations (B3LYP/6-31G*) also show that the HOMO is mostly localized on the strands whereas the LUMO is localized on both the strands and rungs (Fig. 5). This is the first report in which the impact of the electronic nature of the rung is studied in not only the non-slanted-H-mers but the slanted-H-mers, as well. The emission intensities of the H-mers are highly dependent on the structure. The quantum yields of H-mers are recorded relative to the 9,10-diphenylanthracene46,47 and are reported in Table 1 and Fig. S8 (ESI†). The quantum yield of H-mers follow the order H-mer-2 ≈ H-mer-1 > H-mer-3. Amines are known to quench the fluorescence of molecules, which explains the slightly lower quantum yield of H-mer-1 compared to H-mer-2. In general the emission from charge transfer state is weaker and explains the low quantum yield of H-mer-3 compared to other H-mers.
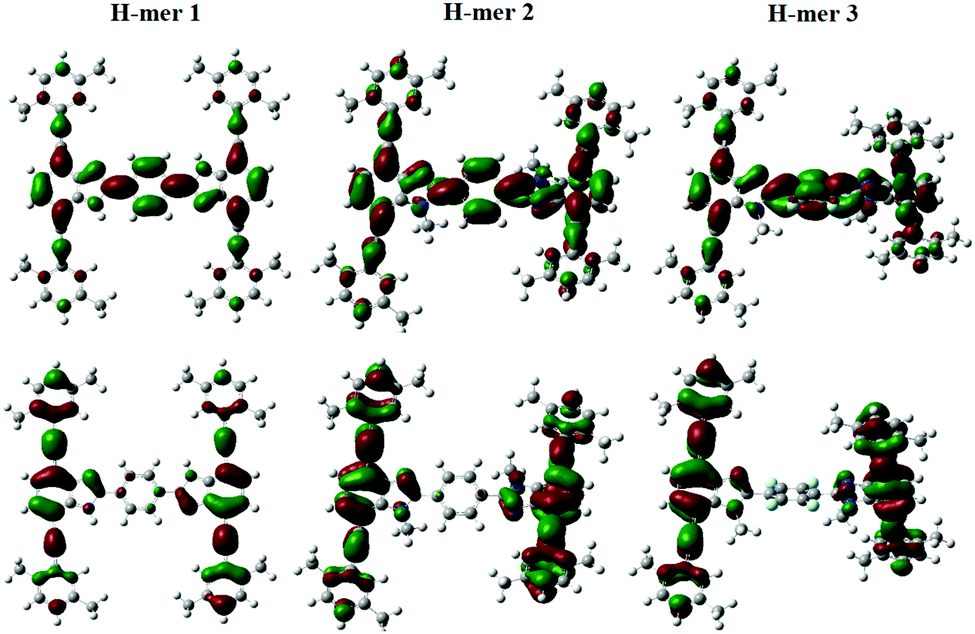 | ||
| Fig. 5 Frontier molecular orbitals (HOMO [bottom]; LUMO [top]) of H-mers 1–3. | ||
The presence of benzimidazoles affects the absorption and emission spectra of H-mers in acidic and basic environment. Toward this, the absorption and emission spectra of H-mers in trifluoroacetic acid (TFA) and tetrabutylammonium hydroxide solutions were recorded. All three H-mers show a slight blue shift in absorption in the acidic environment and the magnitude of the blue shift follows the order: H-mer-1 (31 nm) ≈ H-mer-2 (36 nm) > H-mer-3 (11 nm) (Table 2 and Fig. S9, ESI†). Interestingly, the H-mers displayed a significant red shift in emission and the magnitude of the red shifts follows the order: H-mer-2 (187 nm) > H-mer-1 (104 nm) > H-mer-3 (67 nm) (Table 2 and Fig. 4). In addition to the Stokes shift, the emission intensity of the H-mers also reduced upon protonation. The imine nitrogen, which is the most basic nitrogen in the imidazole, gets protonated in the acidic environment. This enhances the electron withdrawing nature of the rungs and stabilizes the LUMO, which is mostly located on the rungs. The large Stokes shift in the emission maximum of H-mers in acidic environment is due to an efficient intramolecular charge transfer in the excited state from strands (HOMO) to the electron deficient rungs (LUMO) as well as stabilization of the LUMO. Emission from the low-energy excited charge transfer state results in a larger Stokes shift. Within the H-mers, H-mer-2 had a higher shift due to the drastic change in the electronic nature of the rung compared to H-mer-3; also the larger twist in the H-mer-2 isolates the frontier molecular orbitals and promotes intramolecular charge transfer in the excited state leading to higher red shift in H-mer-2 compared to H-mer-1. The benzimidazole containing T-mer's35 and L-mer's48 show absorption and emission trends similar to H-mers’ upon protonation. The key difference is the magnitude of the red shift in the emission spectra upon protonation, which is high in H-mers compared to T-mer's35 and L-mer's.2,48 In the case of a T-mer that has similar absorption and emission spectra to H-mer-2 in the neutral state showed only a ca. 75 nm red shift in emission upon protonation compared to the ca. 187 nm red shift observed in H-mer-2. Comparison of the magnitude of the red shift in upon protonation highlights the advantage of the non-slanted H-mer topology over the T-mer topology.
| H-mer | λ absmax (CHCl3) (nm) | TFA λabsmax (nm) | Δλabsmax (nm) | λ emmax (CHCl3) (nm) | TFA λemmax (nm) | Δλemmax (nm) | TBAOH λabsmax (nm) | Δλabsmax (nm) | TBAOH λemmax (nm) | Δλemmax (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| H-mer-1 | 368 | 337 | −31 | 417, 440 | 521 | 104 | 394 | 26 | 498 | 50 |
| H-mer-2 | 361 | 325 | −36 | 416, 435 | 603 | 187 | 365 | 4 | 438 | 3 |
| H-mer-3 | 351, 362 | 340 | −11 | 535 | 602 | 67 | 365 | 3 | 554 | 19 |
In the basic environment H-mer-1 shows a significant red shift in both the absorption (26 nm) (Fig. S10, ESI†) and emission (50 nm) (Fig. 4) spectra. On the other hand, H-mer-2 and H-mer-3 show minimal changes in the absorption and emission spectra. Benzimidazolate anion generated upon deprotonation of the benzimidazole, destabilizes the HOMO and reduces the band gap resulting in a red shift in both the absorption and emission spectra. Since there is no acidic imide proton in both H-mer-2 and H-mer-3, the base has minimal effect on their absorption and emission spectra. These results show the H-mers as potential candidates for chemical sensors. In addition to acid and base sensing, the H-mers are also capable of transferring electrons to suitable acceptors in the photoexcited state. The photoluminescence of H-mer-3 is quenched by the TCNE acceptor (Fig. 4), indicating that H-mers are suitable for optoelectronic and electronic applications.
The fluorescence lifetimes of all the three H-mers were recorded to determine the impact of the H-mer structure on the excited state lifetime (Fig. 4). The lifetime decays of all the three H-mers are described by a biexponential decay model with two lifetimes as shown in Table 1. The fast and slow decay lifetimes and their corresponding relative amplitudes are 0.78 ns (79.6%) and 2.24 ns (20.4%) for H-mer-1, 0.46 ns (69.1%) and 1.24 ns (30.9%) for H-mer-2, and 3.81 (67.3%) for H-mer-3. The slow decay for H-mer-3 could not be determined due to low emission intensity. More importantly, the excited state lifetime of H-mer-3 is at least 5 times higher than that of H-mers-1 and 2. We are thrilled to see such a long lifetime for H-mer-3 since higher lifetime is desired in solar cells because it enhances the efficiency of photo-excited exciton splitting.49
Conclusions
We show that the twist between the strands and rung is controllable in non-slanted H-mers and the twist increases as the substituents size increases. The twist, along with the electronic nature of the rung, plays a significant role in determining the optical and photophysical properties of the H-mers. Also, this work highlights the importance of the type of donor–acceptor architecture in H-mers. H-mers with an electron deficient rung and twist showed larger Stokes shift in neutral and acidic environments. H-mer-1, unlike the other two H-mers also showed spectral changes in a basic environment due to the presence of acidic proton on the imide nitrogen. The non-slanted H-mers showed a larger red shift in emission spectra and fluorescence lifetime compared to T-mers highlighting the advantages of the non-slanted H-mer design. The larger Stokes shift is due to an efficient intramolecular charge transfer in the excited state from strands to the electron deficient rung. Therefore, rungs have a little impact on the absorption spectra whereas they play a key role in the emission and fluorescence lifetime. Bifacial cyclophanes on the periphery H-mer render solubility without conventional solubilizing chains. The lack of insulating pendant chains, reduced π–π interactions in thinfilms, longer fluorescence lifetime make these H-mers suitable for various electronic and optoelectronic applications including electroluminescence, photovoltaics, and sensors. The developed H-mer design criteria also helps to design 2D-π-conjugated materials with control over the π-conjugation in two dimensions.Funding sources
This work was supported by a National Science Foundation CAREER Grant (NSF-1944184) and Georgetown University start-up funds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. J. D. Tovar and Sayak Panda for help with the time resolved fluorescence measurements on H-mers.References
- N. Zhou, L. Wang, D. W. Thompson and Y. Zhao, OPE/OPV H-mers: synthesis, electronic properties, and spectroscopic responses to binding with transition metal ions, Tetrahedron, 2011, 67, 125–143 CrossRef CAS.
- M. A. Saeed, H. T. Le and O. S. Miljanic, Benzobisoxazole cruciforms as fluorescent sensors, Acc. Chem. Res., 2014, 47, 2074–2083 CrossRef CAS.
- A. J. Zucchero, P. L. McGrier and U. H. F. Bunz, Cross-Conjugated Cruciform Fluorophores, Acc. Chem. Res., 2010, 43, 397–408 CrossRef CAS.
- J. N. Wilson and U. H. F. Bunz, Switching of intramolecular charge transfer in cruciforms: Metal ion sensing, J. Am. Chem. Soc., 2005, 127, 4124–4125 CrossRef CAS.
- A. J. Zucchero, J. N. Wilson and U. H. F. Bunz, Cruciforms as functional fluorophores: Response to protons and selected metal ions, J. Am. Chem. Soc., 2006, 128, 11872–11881 CrossRef CAS.
- S. R. Bheemireddy, P. C. Ubaldo, P. W. Rose, A. D. Finke, J. P. Zhuang, L. C. Wang and K. N. Plunkett, Stabilizing Pentacene By Cyclopentannulation, Angew. Chem., Int. Ed., 2015, 54, 15762–15766 CrossRef CAS.
- A. C. Whalley, K. N. Plunkett, A. A. Gorodetsky, C. L. Schenck, C. Y. Chiu, M. L. Steigerwald and C. Nuckolls, Bending contorted hexabenzocoronene into a bowl, Chem. Sci., 2011, 2, 132–135 RSC.
- S. R. Bheemireddy, M. P. Hautzinger, T. Li, B. Lee and K. N. Plunkett, Conjugated Ladder Polymers by a Cyclopentannulation Polymerization, J. Am. Chem. Soc., 2017, 139, 5801–5807 CrossRef CAS.
- W. L. Yang, A. Lucotti, M. Tommasini and W. A. Chalifoux, Bottom-Up Synthesis of Soluble and Narrow Graphene Nanoribbons Using Alkyne Benzannulations, J. Am. Chem. Soc., 2016, 138, 9137–9144 CrossRef CAS.
- K. M. Magiera, V. Aryal and W. A. Chalifoux, Alkyne benzannulations in the preparation of contorted nanographenes, Org. Biomol. Chem., 2020, 18, 2372–2386 RSC.
- R. Bam, W. L. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Four-Fold Alkyne Benzannulation: Synthesis, Properties, and Structure of Pyreno a pyrene-Based Helicene Hybrids, Org. Lett., 2019, 21, 8652–8656 CrossRef CAS.
- F. Shao, W. Y. Dai, Y. Zhang, W. Zhang, A. D. Schluter and R. Zenobi, Chemical Mapping of Nanodefects within 2D Covalent Monolayers by Tip-Enhanced Raman Spectroscopy, ACS Nano, 2018, 12, 5021–5029 CrossRef CAS.
- H. S. Yang, Y. Du, S. Wan, G. D. Trahan, Y. H. Jin and W. Zhang, Mesoporous 2D covalent organic frameworks based on shape-persistent arylene–ethynylene macrocycles, Chem. Sci., 2015, 6, 4049–4053 RSC.
- S. Thomas, H. Li, C. Zhong, M. Matsumoto, W. R. Dichtel and J. L. Bredas, Electronic Structure of Two-Dimensional pi-Conjugated Covalent Organic Frameworks, Chem. Mater., 2019, 31, 3051–3065 CrossRef CAS.
- S. R. M. Marques, R. C. Selhorst, D. Venkataraman and M. D. Barnes, Probing the Evolution of Molecular Packing Underlying HJ-Aggregate Transition in Organic Semiconductors Using Solvent Vapor Annealing, J. Phys. Chem. C, 2019, 123, 28948–28957 CrossRef.
- C. M. Thompson, G. Occhialini, G. T. McCandless, S. B. Alahakoon, V. Cameron, S. O. Nielsen and R. A. Smaldone, Computational and Experimental Studies on the Effects of Monomer Planarity on Covalent Organic Framework Formation, J. Am. Chem. Soc., 2017, 139, 10506–10513 CrossRef CAS.
- D. J. Rizzo, Q. Q. Dai, C. Bronner, G. Veber, B. J. Smith, M. Matsumoto, S. Thomas, G. D. Nguyen, P. R. Forrester, W. Zhao, J. H. Jorgensen, W. R. Dichtel, F. R. Fischer, H. Li, J. L. Bredas and M. F. Crommie, Revealing the Local Electronic Structure of a Single-Layer Covalent Organic Framework through Electronic Decoupling, Nano Lett., 2020, 20, 963–970 CrossRef CAS.
- Z. Zhang and Y. Qin, Cross-conjugated poly(selenylene vinylene)s, Polym. Chem., 2019, 10, 1018–1025 RSC.
- C. Z. Zhu, Z. H. Guo, A. U. Mu, Y. Liu, S. E. Wheeler and L. Fang, Low Band Gap Coplanar Conjugated Molecules Featuring Dynamic Intramolecular Lewis Acid-Base Coordination, J. Org. Chem., 2016, 81, 4347–4352 CrossRef CAS.
- H. C. Zhang, E. Q. Guo, Y. L. Zhang, P. H. Ren and W. J. Yang, Donor–Acceptor-Substituted Anthracene-Centered Cruciforms: Synthesis, Enhanced Two-Photon Absorptions, and Spatially Separated Frontier Molecular Orbitals, Chem. Mater., 2009, 21, 5125–5135 CrossRef CAS.
- F. P. Jørgensen, J. F. Petersen, C. L. Andersen, A. B. Skov, M. Jevric, O. Hammerich and M. B. Nielsen, Synthesis of Covalently Linked Oligo(phenyleneethynylene) Wires Incorporating Dithiafulvene Units: Redox-Active H-Cruciforms, Eur. J. Org. Chem., 2017, 1253–1261 CrossRef.
- Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, Parallel H-shaped phenylenevinylenes: Quasi-coplanar molecules for highly luminescent blue OLEDs, Org. Electron., 2009, 10, 1082–1090 CrossRef CAS.
- Z. Wang, K. Zhong, Y. Liang, T. Chen, B. Yin, M. Lee and L. Y. Jin, Ordered nanostructures from self-assembly of H-shaped coil-rod-coil molecules, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 85–92 CrossRef CAS.
- M. Hundgen, K. A. Maier, S. Hoger and S. S. Jester, Supramolecular nanopatterns of H-shaped molecules, Chem. Commun., 2018, 54, 10558–10561 RSC.
- K. Zhu, G. Baggi and S. J. Loeb, Ring-through-ring molecular shuttling in a saturated [3]rotaxane, Nat. Chem., 2018, 10, 625–630 CrossRef CAS.
- F. Li, F. Meng, Y. Wang, C. Zhu and Y. Cheng, Polymer-based fluorescence sensor incorporating thiazole moiety for direct and visual detection of Hg2+ and Ag+, Tetrahedron, 2015, 71, 1700–1704 CrossRef CAS.
- I. Torres, J. R. Carrillo, A. Díaz-Ortiz, R. Martín, M. V. Gómez, L. Stegemann, C. A. Strassert, J. Orduna, J. Buendía, E. E. Greciano, J. S. Valera, E. Matesanz, L. Sánchez and P. Prieto, Self-assembly of T-shape 2H-benzo[d][1,2,3]-triazoles. Optical waveguide and photophysical properties, RSC Adv., 2016, 6, 36544–36553 RSC.
- K. Zhu, V. N. Vukotic and S. J. Loeb, Molecular shuttling of a compact and rigid H-shaped [2]rotaxane, Angew. Chem., Int. Ed., 2012, 51, 2168–2172 CrossRef CAS.
- S. L. Cai, K. Zhang, J. B. Tan, S. Wang, S. R. Zheng, J. Fan, Y. Yu, W. G. Zhang and Y. Liu, Rationally Designed 2D Covalent Organic Framework with a Brick-Wall Topology, ACS Macro Lett., 2016, 5, 1348–1352 CrossRef CAS.
- W. Liu, X. Luo, Y. Bao, Y. P. Liu, G. H. Ning, I. Abdelwahab, L. J. Li, C. T. Nai, Z. G. Hu, D. Zhao, B. Liu, S. Y. Quek and K. P. Loh, A two-dimensional conjugated aromatic polymer via C–C coupling reaction, Nat. Chem., 2017, 9, 563–570 CrossRef CAS.
- S. S. Jester, D. Schmitz, F. Eberhagen and S. Hoger, Self-assembled monolayers of clamped oligo(phenylene-ethynylene-butadiynylene)s, Chem. Commun., 2011, 47, 8838–8840 RSC.
- S. S. Jester, N. Shabelina, S. M. Le Blanc and S. Hoger, Oligomers and cyclooligomers of rigid phenylene-ethynylene-butadiynylenes: synthesis and self-assembled monolayers, Angew. Chem., Int. Ed., 2010, 49, 6101–6105 CrossRef CAS.
- N. Zhou, L. Wang, D. W. Thompson and Y. J. O. L. Zhao, H-shaped OPE/OPV oligomers: A new member of 2D-conjugated fluorophore cores, Org. Lett., 2008, 10, 3001–3004 CrossRef CAS.
- S. Chaudhuri, M. Mohanan, A. V. Willems, J. A. Bertke and N. Gavvalapalli, beta-Strand inspired bifacial pi-conjugated polymers, Chem. Sci., 2019, 10, 5976–5982 RSC.
- T. Inouchi, T. Nakashima, M. Toba and T. Kawai, Preparation and Acid-Responsive Photophysical Properties of T-Shaped pi-Conjugated Molecules Containing a Benzimidazole Junction, Chem. – Asian J., 2011, 6, 3020–3027 CrossRef CAS.
- E. Dall'Oglio, M. B. Caro, J. C. Gesser, C. Zucco and M. C. J. Rezende, The influence of substituents on the tautomerism of symmetrically substituted 2,2′-bis-benzimidazoles, J. Braz. Chem. Soc., 2002, 13, 251–259 CrossRef.
- C. Zucco, E. L. Dall'Oglio, G. V. Salmória, H. Gallardo, A. Neves and M. C. Rezende, NMR and semi-empirical study of the tautomerism of 2,2′-bisbenzimidazolyl, J. Phys. Org. Chem., 1998, 11, 411–418 CrossRef CAS.
- C. Diaz, L. Llovera, L. Echevarria and F. E. Hernández, Assessment of the tautomeric population of benzimidazole derivatives in solution: a simple and versatile theoretical–experimental approach, J. Comput.-Aided Mol. Des., 2014, 29, 143–154 CrossRef.
- F. R. Prieto, M. C. R. Rodriguez, M. M. Gonzalez and M. A. R. Fernandez, Ground- and Excited-State Tautomerism in 2-(3′-Hydroxy-2′-pyridyl)benzimidazole, J. Phys. Chem., 1994, 98, 8666–8672 CrossRef.
- F. Zutterman, O. Louant, G. Mercier, T. Leyssens and B. Champagne, Predicting Keto–Enol Equilibrium from Combining UV/Visible Absorption Spectroscopy with Quantum Chemical Calculations of Vibronic Structures for Many Excited States. A Case Study on Salicylideneanilines, J. Phys. Chem. A, 2018, 122, 5370–5374 CrossRef CAS.
- S. Holliday, Y. L. Li and C. K. Luscombe, Recent advances in high performance donor-acceptor polymers for organic photovoltaics, Prog. Polym. Sci., 2017, 70, 34–51 CrossRef CAS.
- E. R. Darzi, E. S. Hirst, C. D. Weber, L. N. Zakharov, M. C. Lonergan and R. Jasti, Synthesis, Properties, and Design Principles of Donor–Acceptor Nanohoops, ACS Cent. Sci., 2015, 1, 335–342 CrossRef CAS.
- N. M. Dickson-Karn, C. M. Olson, W. C. W. Leu and C. S. Hartley, Intramolecular charge transfer in donor-bridge-acceptor compounds with paired linearly conjugated or cross-conjugated pathways, J. Phys. Org. Chem., 2014, 27, 661–669 CrossRef CAS.
- M. Ortiz, S. Cho, J. Niklas, S. Kim, O. G. Poluektov, W. Zhang, G. Rumbles and J. Park, Through-Space Ultrafast Photoinduced Electron Transfer Dynamics of a C-70-Encapsulated Bisporphyrin Covalent Organic Polyhedron in a Low-Dielectric Medium, J. Am. Chem. Soc., 2017, 139, 4286–4289 CrossRef CAS.
- A. M. Sanders, T. J. Magnanelli, A. E. Bragg and J. D. Tovar, Photoinduced Electron Transfer within Supramolecular Donor–Acceptor Peptide Nanostructures under Aqueous Conditions, J. Am. Chem. Soc., 2016, 138, 3362–3370 CrossRef CAS.
- A. M. Brouwer, Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report), Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
- A. T. R. Williams and S. A. Winfield, Relative Fluorescence Quantum Yields Using a Computer-Controlled Luminescence Spectrometer, Analyst, 1983, 108, 1067–1071 RSC.
- R. C. Lirag, H. T. M. Le and O. S. Miljanic, L-shaped benzimidazole fluorophores: synthesis, characterization and optical response to bases, acids and anions, Chem. Commun., 2013, 49, 4304–4306 RSC.
- K. A. Mazzio and C. K. Luscombe, The future of organic photovoltaics, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetis, characterization details, and characterization of molecules and H-mers. See DOI: 10.1039/d0ma00424c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |