
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKeratin–cinnamon essential oil biocomposite fibrous patches for skin burn care†
Despoina
Kossyvaki‡
 a,
Giulia
Suarato‡
ab,
Maria
Summa
b,
Arianna
Gennari
c,
Nora
Francini
c,
Iosifina
Gounaki
d,
Danae
Venieri
d,
Nicola
Tirelli
c,
Rosalia
Bertorelli
b,
Athanassia
Athanassiou
*a and
Evie L.
Papadopoulou
*a
a,
Giulia
Suarato‡
ab,
Maria
Summa
b,
Arianna
Gennari
c,
Nora
Francini
c,
Iosifina
Gounaki
d,
Danae
Venieri
d,
Nicola
Tirelli
c,
Rosalia
Bertorelli
b,
Athanassia
Athanassiou
*a and
Evie L.
Papadopoulou
*a
aSmart Materials Group, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy. E-mail: paraskevi.papadopoulou@iit.it; athanassia.athanassiou@iit.it
bTranslational Pharmacology, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy
cPolymers and Biomaterials, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy
dSchool of Environment Engineering, Technical University of Crete, Akrotiri Campus, 73100 Chania, Greece
First published on 24th July 2020
Abstract
Novel electrospun fibrous biocomposites have been fabricated, based on two naturally derived materials, namely wool keratin and cinnamon essential oil, and their efficacy for the treatment of skin burns caused by UVB exposure is demonstrated. The cinnamon essential oil, successfully encapsulated in uniform, bead-free fibers, enhances the mechanical compliance of the composite, retains its antioxidant properties and is released from the keratin matrix upon its dissolution in an aqueous environment. The low-cytotoxicity material reduces the risk of infection from Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa. Additionally, upon contact with UV-inflammated skin, keratin–cinnamon essential oil fibers are able to down-regulate in vivo IL-6 and IL-1β cytokine production, thus limiting the inflammatory response induced by UVB radiation exposure and confirming the potential of our designed dressing as a biomedical device for burn wound care.
1. Introduction
Skin burns are characterized by severe tissue damage, involving morphological alterations, erythema, apoptosis, and impairment of the physiological epidermal barrier function,1,2 which may result in malfunction of microcirculation, generation of oxidative stress, and release of proinflammatory cytokines at the injured site.3 Moreover, the burnt skin loses its protective function, becoming the perfect environment for bacterial growth.4 In fact, phenomena such as infection and chronic inflammation can undermine a successful healing of the tissue.5,6 In the past decade, the design of smart dressings able to accelerate the skin burn healing process by providing antibacterial, antioxidant or anti-inflammatory features has been intensively pursued.7–11 Generally, an active wound dressing should be biocompatible, self-adhere to both moist and dry skin surfaces, and keep the wound bed clean.12 Ideally, it should also be resorbable, in order to avoid the risk of repeated injury upon removal.11 More importantly, the dressing should be able to release drugs or bioactive compounds, to counteract the onset of bacterial infections and actively support the tissue healing process.13–16In an attempt to respond to these challenges, scientists have been using nature as a source of inspiration. Keratin, a ubiquitous biological material, can be found in the hair, wool, quills and horns of mammals (mostly composed of α-keratins), and the feathers, claws and beaks of birds and reptiles (mostly containing β-keratins).17,18 Its abundance in nature and its ability to enhance cell proliferation and attachment19–22 make keratin an ideal material for a variety of biomedical applications, ranging from scaffolds for cell growth23–25 to drug delivery.26,27 In the field of wound management, keratin-based products have been developed and some of them have been used in acute and chronic wound management, and skin disorders.28 Given the rather poor mechanical properties of keratin and its low molecular weight (ranging from 10 to 60 kDa), the protein is often combined with synthetic polymers, such as PEO,29,30 or PCL and PVA,25,31 which serve as adjuvants to enhance processability.
Natural medicinal extracts can be implemented for several pharmaceutical applications.16,32 For example, Hajiali et al.15 have successfully encapsulated lavender essential oil in sodium alginate nanofibers, which led to a remarkable reduction in pro-inflammatory cytokines in a mouse skin burn model. A plant extract that since the ancient years has been exploited for its biological activities is cinnamon essential oil, an oil derived from the leaves or the bark of different types of cinnamon plants. Amongst them, the essential oil extracted from the bark of the Cinnamomum verum plant exhibits a wide variety of therapeutic effects, including antimicrobial activity33–35 and antioxidant properties36 that may actively contrast skin inflammation.37 Cinnamon essential oil, mainly composed of cinnamaldehyde and eugenol aromatic compounds, is also rich in phenolic molecules and flavonoids,38 responsible for the resulting biological features.37,39,40
Herein, we present the fabrication of electrospun fibers of wool-extracted keratin with polyvinylpyrrolidone (PVP) and different concentrations of cinnamon essential oil and their assessment as dressings for skin burns. Keratin–PVP aqueous emulsions with the cinnamon essential oil were prepared for subsequent water-based, ecofriendly electrospinning, resulting in biocompatible fibrous mats. Thanks to its high surface to volume ratio, the fibrous structure would result in an adequate drug loading and release, a crucial aspect for efficient wound healing. The homogeneous distribution of the cinnamon essential oil in the polymer conveyed strong antioxidant and antimicrobial activity to the fibrous mats. Furthermore, the fibrous mats were able to release the bioactive essential oil and promote the treatment of UVB skin burns. The obtained fibers show anti-bacterial activity against both Gram (+) and Gram (−) representative bacterial species, as well as in vivo anti-inflammatory effects following UVB exposure. The fibrous biocomposites and their ability to contain radiation-induced inflammation qualify our material as a potential wound dressing for skin burns.
2. Experimental
2.1 Materials
Keratin was extracted from Argentinian sheep wool (Olimpias, Italy). Cinnamon essential oil of the Cinnamomum verum species was purchased from Maitreya-Natura (Italy). Poly(N-vinyl pyrrolidone) (PVP) (nominally with ; from GPC analysis in DMF
; from GPC analysis in DMF  and from AF4 analysis in water
and from AF4 analysis in water 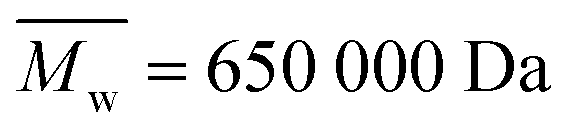 , always with dispersity index Đ ≫ 3; see Section S1 in the ESI,† Table S1 and Fig. S1), phosphate buffered saline (PBS), ethanol, 2,2-diphenyl-1-picrylhydrazyl (DPPH), acetone, methanol, urea, β-mercaptoethanol, sodium dodecyl sulfate (SDS) and sodium azide (NaN3) were purchased from Sigma-Aldrich.
, always with dispersity index Đ ≫ 3; see Section S1 in the ESI,† Table S1 and Fig. S1), phosphate buffered saline (PBS), ethanol, 2,2-diphenyl-1-picrylhydrazyl (DPPH), acetone, methanol, urea, β-mercaptoethanol, sodium dodecyl sulfate (SDS) and sodium azide (NaN3) were purchased from Sigma-Aldrich.
2.2 Keratin extraction protocol
Argentinian sheep wool fibers were washed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of acetone:methanol (6 h under mild agitation), thoroughly washed with MilliQ water and let dry overnight at room temperature. Extraction of keratin from wool was performed following the protocol presented by Suarato et al.41 Briefly, 2 g of washed wool fibers were dissolved in a 7 M urea solution (36 mL) with SDS (0.107 M), using β-mercaptoethanol (1.2 M) to remove the intra-keratin disulfide bonds. The fibers were immersed in the reagent mixtures and heated at 60 °C for 4 h in an oven. Subsequently, the supernatant was collected, and the fibers were pressed onto a metallic net to allow further extraction of the protein. The extracted material was dialyzed against distilled water for 48 h with dialysis tubes (Mw cut-off of 3500 Da, SpectrumLabs) and then centrifuged (three times at 9000 rpm and 4 °C for 20 min), to eliminate residues and fibrous debris. The final keratin concentration was 75–80 mg mL−1, assessed by air evaporation.
:1 solution of acetone:methanol (6 h under mild agitation), thoroughly washed with MilliQ water and let dry overnight at room temperature. Extraction of keratin from wool was performed following the protocol presented by Suarato et al.41 Briefly, 2 g of washed wool fibers were dissolved in a 7 M urea solution (36 mL) with SDS (0.107 M), using β-mercaptoethanol (1.2 M) to remove the intra-keratin disulfide bonds. The fibers were immersed in the reagent mixtures and heated at 60 °C for 4 h in an oven. Subsequently, the supernatant was collected, and the fibers were pressed onto a metallic net to allow further extraction of the protein. The extracted material was dialyzed against distilled water for 48 h with dialysis tubes (Mw cut-off of 3500 Da, SpectrumLabs) and then centrifuged (three times at 9000 rpm and 4 °C for 20 min), to eliminate residues and fibrous debris. The final keratin concentration was 75–80 mg mL−1, assessed by air evaporation.
2.3 Preparation of the composite fibers by electrospinning
PVP/keratin aqueous solutions prepared at different weight ratios (3:1, 2:1, and 1:1, w/w) were stirred overnight at room temperature and were electrospun under ambient conditions. In addition, for the PVP:keratin solution with a 1:1 weight ratio, various volumes of cinnamon essential oil were added (5%, 10% and 15% v/v with respect to the volume of the PVP/keratin solution – designated as KC5, KC10 and KC15, respectively). The resulting systems were stirred with a vortex mixer, in order to emulsify the oil-in-water phase, prior to electrospinning. The emulsions were stable for ca. 1 week. Electrospinning was carried out immediately after the emulsion preparation, as follows. A syringe was filled with the final solution (KC0) or emulsion (KC5, KC10 or KC15) and connected to a syringe pump (NE-1000 New Era Pump Systems, Inc.) working at a flow rate between 350 and 850 μL h−1. The stainless-steel spinneret (18-gauge needle) was clamped to the positive electrode of a high-voltage power supply (EH40R2.5, Glassman High Voltage, Inc.) applying a voltage of 24 kV and the ground electrode was connected to an aluminum collector placed at 25 cm from the spinneret tip.
2.4 Asymmetric flow field-flow fractionation (AF4)
The composition of electrospun KC0 fibers, containing different PVP:keratin ratios, was analysed after dissolution of the fibers in deionized water (10 mg mL−1) for 2 h at room temperature. The AF4 system AF2000 TM (Postnova Analytics, Landsberg, Germany) was coupled online to a UV-VIS spectrophotometer at 280 nm (S3210, Laserchrom, Rochester, UK), a PN3609 multi-angle light scattering (MALS) system (Postnova Analytics, Landsberg, Germany), and a PN3150 refractive index (RI, Postnova Analytics, Landsberg, Germany) detector in the given order. The AF4 channel was equipped with a 350 μm spacer and a 10 kDa MWCO membrane of regenerated cellulose as an accumulation wall. A 7 mM SDS solution supplemented with 0.02% (w/v) sodium azide (NaN3) was filtered through a 0.1 μm filter and used as the eluent. For the calibration curve, the single compounds (PVP and keratin) were individually dissolved in water with concentrations ranging from 20 to 1 mg mL−1. In a typical experiment, the detector flow rate was set at 1 mL min−1 and 20 μL of sample was injected over 4 minutes, setting 0.2 mL min−1 as the injection flow rate. The initial cross flow and focusing flow were set at 4.5 and 5.3 mL min−1, respectively (focusing step). For the elution step the cross flow was maintained constant at 4.5 mL min−1 for 3 min and then exponentially (exponent = 0.30) decreased to 0 mL min−1 over 20 min. Subsequently, it was kept at 0 mL min−1 for 8 additional min. Lastly, a rinse step was performed for 0.5 min. The polymer concentration was obtained by integrating the UV/Vis signal between 7 and 35 minutes for keratin and the refractive index signal between 25 and 30 min for PVP.
2.5 Sample characterisation
2.6 Drug release
The fiber mats were studied for the release of the essential oil. Samples of 1 mg were placed in quartz cuvettes with 3 mL of PBS:ethanol solution, at a volume ratio of 60:40, according to Hosseini et al.,42 in which the electrospun fibers were not dissolved, and the absorption was measured using a UV-visible spectrophotometer (CARY200 Scan, Varian), over the course of 24 h. Before each measurement, the cuvettes were gently shaken, in order to achieve a homogeneous dispersion of the released active compound in the solution. The release was studied following the increase of the absorption peak of cinnamaldehyde (CA), at 291 nm.42 To ensure that infinite sink conditions are simulated and preserved during the course of the experiments we calculated the solubility of CA to be approximately 0.1 mg mL−1, whereas the samples KC5, KC10 and KC15 reached a release of 0.004 mg mL−1, 0.008 mg mL−1 and 0.010 mg mL−1, respectively. The experiment was carried out in triplicate and the data are expressed as cumulative percentage.
2.7 Antioxidant activity
The antioxidant activity of the fibers was investigated by the standard DPPH˙ free radical scavenging assay (RSA). Samples of 5 mg of KC0, KC5, KC10 and KC15 fiber mats were separately placed in polystyrene cuvettes containing 2 mL of 0.2 mM solution of DPPH˙ radical in ethanol. The decrease in the radical signal absorption was measured at 528 nm using a Cary 300 Scan UV-visible spectrophotometer, in the dark, at room temperature for 24 h. All measurements were performed in triplicates. A sample with 2 mL of DPPH˙ solution was used as a control, to ensure the reagent stability. RSA was expressed as the inhibition percentage of the free radical of the samples and was calculated using eqn (1): | (1) |
2.8 Antibacterial activity
The bacterial strains used for this assay were: (a) Escherichia coli reference strain DSM498 (Gram negative), (b) Pseudomonas aeruginosa (Gram negative), which was isolated from a surface water sample and was biochemically identified using the API® 20NE kit (Biomérieux) and (c) Staphylococcus aureus (Gram positive), which was isolated from a food sample (poultry) and was identified biochemically using the API® Staph test (Biomérieux). The specific bacterial species used were chosen based on their importance for public health and their frequent presence in skin wounds. All isolated strains were stored at −80 °C in glucose solution. Bacterial inactivation for each species was performed in nutrient broth (LABM) with a bacterial concentration of 102 CFU mL−1. The experiment was performed for the fibrous samples (either loaded or unloaded) and the pure cinnamon essential oil as a control, following Zahid et al.43 Briefly, electrospun fibers of KC0 and KC10 were cut into small round pieces with a diameter of 1 cm, weighing 2.8 ± 0.5 mg. Liquid cultures were prepared for each bacterial strain in nutrient broth (LABM) with a concentration of 102 CFU mL−1. Sample disks were added in each liquid culture and bacterial inocula were incubated at 37 °C for 24 h. After the incubation period, the optical density of each liquid culture was measured at 600 nm and viable counts were performed using nutrient agar (LABM) plates and subsequent incubation at 37 °C for 24 h. The reduction of the bacterial population in each case was estimated based on optical density values and colony counting. Furthermore, in another set of experiments, pure cinnamon essential oil was added in the bacterial inoculum, and bacterial inactivation was recorded after a 24 h incubation, following the aforementioned procedure. The quantity of cinnamon oil added in each case was calculated according to the equivalent amount present in the electrospun fibers. Specifically, 1.5, 1.9 and 2.9 μL of oil were added as equivalent of KC5, KC10 and KC15 samples, respectively.2.9 In vitro biocompatibility assay
Primary human dermal fibroblasts (HDFa, Thermo Fisher Scientific) were cultured in T75 flasks in the presence of Medium 106 supplemented with LSGS Kit (Thermo Fisher Scientific) until confluency, seeded onto 24-well plates at a density of 7000 cells per cm2, and let attach overnight in an incubator at 37 °C and with 5% CO2. Extraction media from the various keratin-based fibrous matrices were prepared following the ISO10993-5 standard test. Briefly, fibrous samples were cut into small pieces (∼10 mg of weight) and UV-light sterilized for 20 minutes (10 minutes per side). The fibers were immersed in 1 mL of cell culture medium for 24 h at 37 °C to obtain a stock solution which was used for the preparation of the various dilutions tested (0.01, 0.05, 0.10, and 0.50 mg mL−1). The attached fibroblasts were then incubated with the different extraction media for 24 h, while untreated cells were considered as control samples. MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega) was used to determine the cell viability following Papadopoulou et al.44 In order to better analyze the effect of the active principle, pure cinnamon essential oil was directly applied in the culture medium, after having estimated the amount present in the corresponding loaded fibers. Prior to cell treatment, the sterile LSGS supplemented Medium 106 and essential oil emulsions were vortexed to favor oil droplet dispersion. After 24 h, the samples were washed, and the wells were replenished with fresh culture medium, to remove the residual essential oil, and an MTS assay was performed.Confocal imaging was carried out to observe the morphology of the cells exposed to the KC fiber extracts. Briefly, fibroblasts were plated onto fibronectin coated-glass coverslips at a density of 7000 cells per cm2 and treated as above-mentioned. After 24 h, the samples were fixed with 3.7% paraformaldehyde for 20 min, and the nuclei were stained with DAPI solution (2.5 μg mL−1) for 15 min in the dark. Afterwards, the samples were permeabilized with 0.3% Triton X-100 for 8 min and washed twice with PBS, prior to incubation in Alexa Fluor 564 Phalloidin (Thermo Fisher Scientific, 1:100 dilutions in PBS) for 20 min in the dark. Stained coverslips were then mounted with Fluoromont-G onto glass slides and imaged with a Nikon A1 confocal microscope.
2.10 In vivo test
Male C57BL/6J mice, 8 weeks old (Charles River, Calco, Italy), were used for the animal experiment.3. Results and discussion
3.1 Electrospinning of PVP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :keratin solutions at different mass ratios
:keratin solutions at different mass ratios
In order to ease the spinnability via a green, all-water electrospinning process, keratin has been combined with the biocompatible PVP, initially at a mass ratio of 3:1 (PVP:keratin), following a protocol previously established in our group.41 The use of the adjuvant polymer allowed the formation of stable and continuous jets under the application of a high voltage, leading to the fabrication of homogenous, bead-free fibers with average diameters of 424 ± 137 nm (Fig. 1a). Increasing the concentration of the extracted keratin enabled the reduction of the synthetic and water soluble PVP in the composite solution, preserving the viscosity of the composite solution that allowed electrospinning. By increasing the keratin concentration from 40 mg mL−1 to 80 mg mL−1, electrospinning with different mass ratios (3:1, 2:1 and 1:1) of PVP:keratin was performed. The resulting fibers presented a uniform morphology, and were rather similar in size (466 ± 199 nm and 315 ± 89 nm for the 2:1 and 1:1 PVP:keratin fibers, respectively) (Fig. 1b and c).
 | ||
| Fig. 1 SEM images of PVP:keratin fibers electrospun from solutions at different mass ratios, (a) 3:1, (b) 2:1 and (c) 1:1; (d) representative AF4 analyses for the 1:1 (above) and 3:1 (below) PVP:keratin fibers; the refractive index detector traces (red curves, right axis) provides peaks corresponding to the elution of both materials, but its comparison with UV absorption (black curves, left axis) allows the clearer identification of keratin. (e) The experimental values of the mass PVP/keratin mass ratio were obtained through appropriate calibration of the regions in the AF4 elugrams; the comparison between the composition of the solution used for electrospinning and the electrospun fibers (first two columns from the left) shows no significant deviation of the material composition from that of its feed. Furthermore, the mass ratio between the two components of the fibers was always close to its theoretical value. | ||
The composition of the electrospun materials was studied using a field flow fraction method; specifically, AF4 was employed with different detectors for PVP and keratin, thereby verifying the presence of the two components and simultaneously determining their actual ratio (see the ESI,† Section S2, Fig. S2, S3 and Table S2; this analysis also shows no significant loss/degradation of the materials during the process). First, for each compound a calibration curve relating the sample concentration with a detector signal was obtained. PVP and keratin exhibit absorption peaks at different wavelengths, allowing for the selective detection of the protein. An initial characterization conducted in parallel onto the polymer:protein (1:1, w/w) solution prepared for the electrospinning process and the actual electrospun material revealed that the corresponding mass ratio was maintained (Fig. 1e). Subsequently, different electrospun samples, obtained from the various PVP:keratin mass ratios under study, were analysed. In Fig. 1e the PVP:keratin ratios are plotted against the nominal values. The results showed that the calculated concentration ratios finely corresponded to the theoretical ones (1.09 ± 0.04, 2.24 ± 0.07, and 3.36 ± 0.11, for the 1:1, 2:1, 3:1 PVP:keratin mass ratios, respectively). Nonetheless, the presence of some keratin aggregates was detectable by the UV signal after 28 min of elution. These aggregates would also lead to a slight overestimation of the PVP concentration. Indeed, the PVP recovery appeared to be about 10% higher than the keratin one. The addition of more protein in the working solution resulted in electrospun fibers with varying mass ratios of the two components, indicating a good control of the composite formulation. Moreover, the AF4 technique, employed herein for the first time onto keratin electrospun matrices, allows a precise and straightforward characterization of the material.
More importantly, by modifying the amount of the soluble, synthetic polymer within the composite matrix, the resulting fibers were less prone to an immediate dissolution in water (data not shown). By simply tuning the fiber composition, their handling was facilitated and their application in aqueous milieu was resolved, without using any chemical or thermal crosslinking. Hence, the following characterization as well as the final in vivo assessment were carried out employing matrices obtained from PVP:keratin 1:1 mass ratios.
3.2 Electrospinning of PVP:keratin 1:1 with cinnamon essential oil
In order to functionalize the PVP:keratin fibers, cinnamon essential oil was introduced at different quantities (5, 10, and 15% v/v with respect to the PVP:keratin solution). The small amount of SDS, residual from the protein extraction, acted as a surfactant and ameliorated the emulsification of the cinnamon essential oil in the aqueous phase, improving the stability of the emulsions, up to 1 week. All of the above led to well-defined, beadless fibers, as seen in the SEM micrographs in Fig. 2a, c and e. The average diameters were measured for KC5, KC10 and KC15, with the values of 415 ± 98 nm, 512 ± 194 nm, and 531 ± 112 nm, respectively (Fig. 2b, d and f). This suggests a direct correlation between the amount of oil added in the emulsion systems and the size of the fabricated fibers.
 | ||
| Fig. 2 SEM images and corresponding diameter distribution for (a and b) KC5, (c and d) KC10 and (e and f) KC15 electrospun fibers. | ||
Furthermore, in the confocal microscope, the autofluorescence signal of the cinnamon essential oil (in the green channel) was visibly colocalized with the bright field signal of the electrospun fibers (Fig. S4, ESI†), indicating the presence of the essential oil and its homogeneous distribution within the protein-based fibers. In addition, the prepared fibrous mats presented a light-yellow color and were characterized by a rather persisting cinnamon scent, further validating the encapsulation.
3.3 Physico-chemical analysis of the composite fibers
The FTIR spectra of the different composite samples are shown in Fig. 3, where pristine PVP is also presented for comparison. The pristine PVP shows the main vibrational peaks at 3399 cm−1 due to the O–H stretching, the asymmetric and symmetric CH2 stretching modes at 2950 cm−1 and 2920 cm−1, respectively, and the amide I peak at 1643 cm−1. In addition, the bands at 1493 cm−1, 1316 cm−1 and 1286 cm−1 have been assigned to the PVP backbone structure.13 Comparison of the PVP spectrum with that of KC0 reveals the presence of an overlapping of the absorption peaks between the two components. The amide I (1643 cm−1) band in KC0 is likely still overwhelmingly due to PVP; however, the fibers also show a clear amide II peak at 1543 cm−1, which is typical of primary amides and therefore can only be assigned to keratin. This further confirms the presence of the latter within the fibrous mats.46 | ||
| Fig. 3 (a) Comparison between the FTIR spectra of pristine Keratin, pristine PVP, and the KC0 fibers; (b) FTIR spectra of the cinnamon oil-loaded composite KC mats; (c) Raman spectra of the KC0 mats and the composite KC mats. | ||
In Fig. 3b the vibrational peaks of KC5, KC10 and KC15 are depicted and compared to KC0. All samples exhibit vibrational peaks due to the cinnamon essential oil at 1124 cm−1 (C–O stretching), 973 cm−1 (C–H bending vibration), 749 cm−1 (aromatic out of plane C–H bending) and 690 cm−1 (vibration absorption of olefins).47 In the carbonyl region, cinnamon oils present a weak peak at 1727 cm−1, a strong one at 1679 cm−1 and a weak one 1626 cm−1 and the first and the third become increasingly visible at cinnamon concentrations higher than 10% (v/v), whereas the second is overshadowed by the PVP/keratin amide I.
The μRaman spectra of PVP, KC0, KC5, KC10 and KC15 fibrous mats are presented in Fig. 3c. The complementary Raman technique has been used in this study to investigate the –SS– groups of keratins. Similar to the FTIR results, comparing the spectra of KC0 and PVP the overlapping of the bands is evident. The Raman resonances associated with disulfides in pristine keratin localize at 510–520 cm−1 (S–S stretching) and 650–670 cm−1 (C–S stretching);48 both of them have relatively low-intensity peaks, in particular the latter. Indeed, in most keratin-containing samples it is easy to recognize a broad peak at 500–530 cm−1 that can be assigned to keratin disulfides (either reformed during processing or –less likely– mixed with disulfides of residual mercaptoethanol). The second peak is not seen due to low intensity.49
In addition, the peaks from cinnamon essential oil are quite prominent in the spectra of KC5, KC10 and KC15. The most intense bands are present at 1003 cm−1 (2,4,6 radial in-phase stretching mode), 1599 cm−1 (ring vibration), 1631 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching), 1675 cm−1 (CO stretching). For cinnamon content higher than 10% v/v, more peaks appear at 1163 cm−1, 1182 cm−1, 1254 cm−1, 1580 cm−1, that are assigned to cinnamaldehyde.50
C stretching), 1675 cm−1 (CO stretching). For cinnamon content higher than 10% v/v, more peaks appear at 1163 cm−1, 1182 cm−1, 1254 cm−1, 1580 cm−1, that are assigned to cinnamaldehyde.50
The mechanical characterization of the fibrous mats revealed that the encapsulation of the essential oils improves the ductility of the matrices.51,52 In fact, the Young's modulus decreased from 200 ± 62 MPa for KC0 to 48 ± 29 MPa for KC15, while the elongation at break remained between 2–6% for all fibrous mats, thereby showing that these systems were able to withstand handling while being conformable to a surface (i.e. a skin wound). Table S3 (ESI†) reports the detailed results of the stress–strain analysis of the materials.
3.4 Bioactive properties of the composite fiber mats
The release of cinnamaldehyde (CA) from KC5, KC10 and KC15 fibers was investigated by UV-vis spectroscopy at different time points. The release profiles (Fig. 4a) showed that CA presented a burst release in the first 30 min, followed by a very slow but sustained release profile for the next 24 h. This behavior is of great advantage when an immediate response of the dressing is needed to deliver active principles at the wound site.6,9,53,54 After 24 h, the KC5, KC10 and KC15 fibrous mats were able to release up to 19.6 ± 1.9%, 21.1 ± 3%, and 34.6 ± 0.5% of their total initial amount of CA, respectively (Fig. 4a). Hence, by tuning the amount of embedded active molecule, one can tune its release. | ||
| Fig. 4 (a) Release profile of cinnamaldehyde from KC5, KC10 and KC15 fibers up to 24 h; (b) DPPH˙ radical scavenging activity of KC0, KC5, KC10 and KC15 fibers; (c) images of a cuvette before the DPPH˙ scavenging assay; (d and e) images of cuvettes after the DPPH˙ scavenging assay using fibers without and with cinnamon essential oil, respectively. The color variation is indicative of the radical inactivation. (f) Bacterial reduction of the tested bacterial strains upon incubation with the fibrous patches (KC0 and KC10), and the pure essential oil of the corresponding concentration to KC10. | ||
The DPPH˙ scavenging activity of the electrospun mats is reported in Fig. 4b. Surprisingly, the KC0 matrices presented an RSA of 77.3 ± 2.5% after 24 h. The antioxidant activity of keratin has been scarcely reported in the literature, and only specifically for keratin extracted from chicken feathers.18,55 Moreover, the PVP component does not present any radical scavenging ability.56 As expected, the presence of the cinnamon essential oil within the composite fibers enhanced their antioxidant activity by drastically decreasing the concentration of the DPPH˙ by 94.7 ± 1.5%, 96.8 ± 0.7% and 96.3 ± 0.1% for the KC5, KC10 and KC15 samples, respectively, after 24 h. Therefore, the addition of the bioactive compound (the DPPH˙ assay for the free cinnamon oil is reported in Fig. S5, ESI,† as reference) further enhanced the scavenging activity of the fibrous matrices by 19%. For both KC10 and KC15, RSA reaches values higher than 90% within 3 h, much sooner than for KC0 or KC5. Furthermore, complete free radical inactivation takes place with 10% v/v of encapsulated cinnamon essential oil, so further increase of the amount of the active molecule does not lead to higher RSA. This antioxidant boost is due to the presence of several phenolic compounds, such as cinnamaldehyde, eugenol and flavonoids.38,40,57,58
 | ||
| Fig. 5 Composite fiber biocompatibility study onto primary human dermal fibroblasts adult (HDFa). (a) Effect of the unloaded and loaded fiber extracts on the cell viability determined via MTS, p < 0.01 (**) according to a Student's t-test with unequal variances. Confocal imaging analysis depicting the fibroblast morphology in the (b) control samples, and in the samples treated either with 0.05 mg mL−1 of the (c) unloaded fiber extract, or with 0.05 mg mL−1 of the extract of fibers containing cinnamon essential oil fibers at a concentration of (d) 5%, (e) 10%, and (f) 15%. Actin fibers highlighted in red (Alexa Fluor Phalloidin 546 staining) and cell nuclei in blue (DAPI staining). Scale bar 50 μm. | ||
In order to assess the antibacterial features of the fabricated matrices, bacterial reduction tests were performed, with E. coli, P. aeruginosa (Gram (−)), and S. aureus (Gram (+)) bacterial strains, as shown in Fig. 4f. Considering first the effect of the plain PVP:keratin fibrous matrices (KC0), no reduction in the initial bacterial populations was observed for E. coli and P. aeruginosa. However, S. aureus exhibited a different behavior, as 24 h-incubation with the control sample (KC0) resulted in the complete inactivation of this bacterial species (Fig. 4f, black bar). In the updated literature, studies on the antibacterial properties of keratinous materials (either extracted from wool, chicken feathers or human hair) had not led to conclusive results. For example, Caven et al.59 investigated the antibacterial nature of wool against S. aureus, which was observed only for the reduced components and the cuticle scales, while the cortical cells of the wool fibers appeared to be a suitable substrate for bacterial growth. In contrast, in other studies55,60 the analysis of keratin-based composite films and nanoparticles did not reveal a strong bactericidal activity. As a consequence, various interpretations of the antibacterial activity of the keratin macromolecules have been recently proposed, either based on the presence of the disulfide bonds and the content of coiled-coil random domains60 or depending on the amount of reduced protein molecules and the extraction methods employed.59 Moreover, PVP-based membranes may inhibit the biofilm formation of Staphylococcus epidermidis, affecting the transfer of nutrients and oxygen into the bacterial cells.61 In our case, the PVP–keratin fibers KC0, obtained with reduced wool keratin (mostly presenting α-helix fractions) appeared to induce a bactericidal effect only towards S. aureus, in accordance with Caven et al.59
Next, the effect of the cinnamon essential oil alone on the strains was analyzed, resulting in bacterial reduction for all species, as expected (Fig. 4f, yellow bar). In general, the effectiveness of the essential oils may differ depending on their various chemical constituents and the structure and composition of the cell membrane pertaining to the target bacteria (Gram (+) or Gram (−)). Thanks to their lipophilic nature, the essential oils are able to penetrate within the bacterial membrane, destabilizing the cellular architecture, and impairing cellular activities such as energy production and respiration.62 After 24 h of incubation with cinnamon essential oil alone, 100% of bacterial reduction was achieved towards S. aureus, while the population reduction of E. coli and P. aeruginosa was 37.9 and 47.2%, respectively. This difference is likely due to the hydrophobic nature of the essential oil, which can mainly and more easily penetrate the cell wall of Gram (+) bacteria (i.e. S. aureus), making the Gram (−) ones more resistant to it.63
Lastly, the effect of the cinnamon essential oil-loaded fibrous matrices (KC10, Fig. 4f, red bars) led to a population reduction of 10.1 ± 1.5% and 26.9 ± 7.5% for E. coli and P. aeruginosa, respectively, while the Gram (+) strain S. aureus inactivation was reduced by 18.8%. The peculiar behavior of the KC10 fibers, which show reduced S. aureus inactivation compared to pure essential oil and KC0 (which both show 100% inactivation), could be explained by the efficient radical scavenging of the loaded fibers. It has been previously reported64–66 that a strong antibacterial activity can be related to the presence of free radicals, able to exert a killing oxidative stress onto the bacterial cells.65 Bacteria, like S. aureus, are very prone to Reactive Oxygen Species (ROS)-induced bactericidal mechanisms.66 In our case, the high antioxidant activity of KC10 (Fig. 4b, red line) results in ROS inactivation within the bacterial broth, thereby reducing the oxidative stress experienced by the bacterial cells. However, it should be pointed out that the biocidal activity of ROS is highly affected by the special features and structures of the target microorganism employed each time. For instance, Gram (+) and Gram (−) bacteria may exhibit different behavior, according to the operating conditions of the process and the materials that are used. In this sense, the thick cell wall, which is present in Gram (+) bacteria, may act as a very successful defense mechanism against the penetration of ROS, while Gram (−) species prove to be more sensitive. On the other hand, this precedence order could be reversed, due to the extra outer membrane of Gram (−) bacteria.67,68 This complex behavior is reflected in our results, as bacterial populations were reduced at various levels, whether the bacterium was Gram (+) or Gram (−) (Fig. 4f). Moreover, it is highly plausible that since ROS are no longer present in the microenvironment to carry out their bactericidal effect on the more susceptible Gram (+) S. aureus, the bacterial population experiences a smaller reduction (18.8%).
Taken together, our data suggest that the effective antibacterial activity of the cinnamon essential oil-loaded PVP:keratin matrices might be the result of an interplay between the antioxidant features (via the ROS-mediated mechanism) and the presence of lipophilic chemical compounds (via the membrane-destabilization mechanism). Cinnamon essential oil clearly exhibits antibacterial properties towards several bacterial species, as previously reported in the literature.16,69–73 However, direct use on burnt skin might cause sudden irritation and fast evaporation. In the herein reported strategy, the encapsulation within a fibrous matrix with high surface area would prevent the above-mentioned side effects and prolong the beneficial activity.
3.5 In vitro biocompatibility assay
In order to assess the cytotoxicity of the prepared matrices and their suitability for cell culture applications, primary human dermal fibroblasts (HDFa cells) were used as an in vitro model. The fibroblasts exposed for 24 h to different concentrations of matrix extraction media (obtained following the ISO10993-5 standard test) were completely viable, if compared to the control, untreated cells (Fig. 5a). More specifically, via MTS assay cell viabilities of (105.8 ± 1.6)%, (128.1 ± 1.0)%, (110.4 ± 0.7)% and (116.7 ± 0.9)% were registered for treatments with 0.1 mg mL−1 of KC0, KC5, KC10 and KC15 fiber extracts, respectively. At the highest concentration tested (0.5 mg mL−1), the KC0 and KC5 matrices did not show any cytotoxic effect, while KC10 and KC15 fibers decreased the cell survival, leading to values of (65.9 ± 1.5)% and (76.4 ± 1.7)%. The effects of direct application of free cinnamon essential oil were also considered (Fig. S6, ESI†). The experiments were performed by calculating the equivalent concentrations of essential oil remaining in the various fibrous sample extracts. The results revealed a more pronounced cytotoxicity, as for higher concentrations (0.5 mg mL−1, equivalent) the cell viability dropped down to (28.5 ± 0.4)%, (38.9 ± 0.5)%, and (8.15 ± 0.2)%, for the corresponding KC5, KC10 and KC15 samples, respectively. Therefore, the encapsulation within the protein-based matrix mitigates the adverse biological effects in vitro of the free compounds. A confocal imaging investigation was conducted in parallel to observe the impact of the biocomposite fiber extracts (0.05 mg mL−1) onto the cellular morphology (Fig. 5b–f). For every treatment, the HDFa cells uniformly coated the glass substrates in a spread-out fashion, and presented an elongated morphology, with stretched actin fibers. No differences were noticeable between the control, untreated samples and the cells grown in the presence of either the plain or cinnamon-containing fibers, thus confirming their suitable implementation as wound dressing materials.3.6 In vivo efficacy of the fibrous dressings
Amongst the typical cutaneous manifestations after UVB-induced inflammation, there are erythema and redness, which result from vasodilation of the cutaneous blood vessels. These particular states are characterized by an upregulated production of inflammatory mediators, alteration of vascular responses, and inflammatory cell infiltration.74,75 Therefore, the use of natural products with anti-inflammatory activity, such as cinnamon essential oil,76,77 to alleviate the negative effects of UVB radiation could be of high interest.The satisfying physico-chemical characterization presented above, the most promising antimicrobial activity, and the good biocompatibility results, led to the identification of the KC10 fibrous matrix as the most suitable for in vivo assessment. A preliminary study was conducted to determine the therapeutic properties of the fabricated KC0 (as reference) and KC10 fibrous patches in an irradiated mouse skin model, by examining the cutaneous levels of specific inflammatory cytokines, such as IL-6 and IL-1β, at their peak production effect after 48 h from the irradiation (acute phase78). During the in vivo experiment, the exposure of the animal skin to UVB irradiation produced evident erythema with signs of mild burn, belonging to a first degree of burn that led to visible skin injury and peak expression of cytokines within 48 h, as previously described.15 When the burns were treated with one sole application of dressings, fibers were still present on the skin 48 h later, while burn marks were less visible and the erythema disappeared from the skin of the biocomposite matrices treated animals, compared to SHAM.
In Fig. 6, a significant increment of the produced cytokines can be observed for mice exposed to the UVB light without subsequent treatment (SHAM) in comparison with the ones not UVB-exposed (naïve), indicating the presence of an inflammatory state caused by the burn wound. When the biocomposite patches were applied, a significant reduction in the production of inflammatory factors occurred in the mice treated with KC10 fibers compared to the untreated group. More specifically, the expression of IL-1β (Fig. 6a) and IL-6 (Fig. 6b) was up to 7 and 5 times lower than the UVB-irradiated group, respectively. Similar results were obtained for the group treated with KC0 fibers (6 and 4 times lower than the UVB-irradiated mice, respectively). Hence, the reduction of redness and injury correlated very well with the anti-inflammatory activities of the fibers, and the healing processes, as already demonstrated also for other biomaterials.15
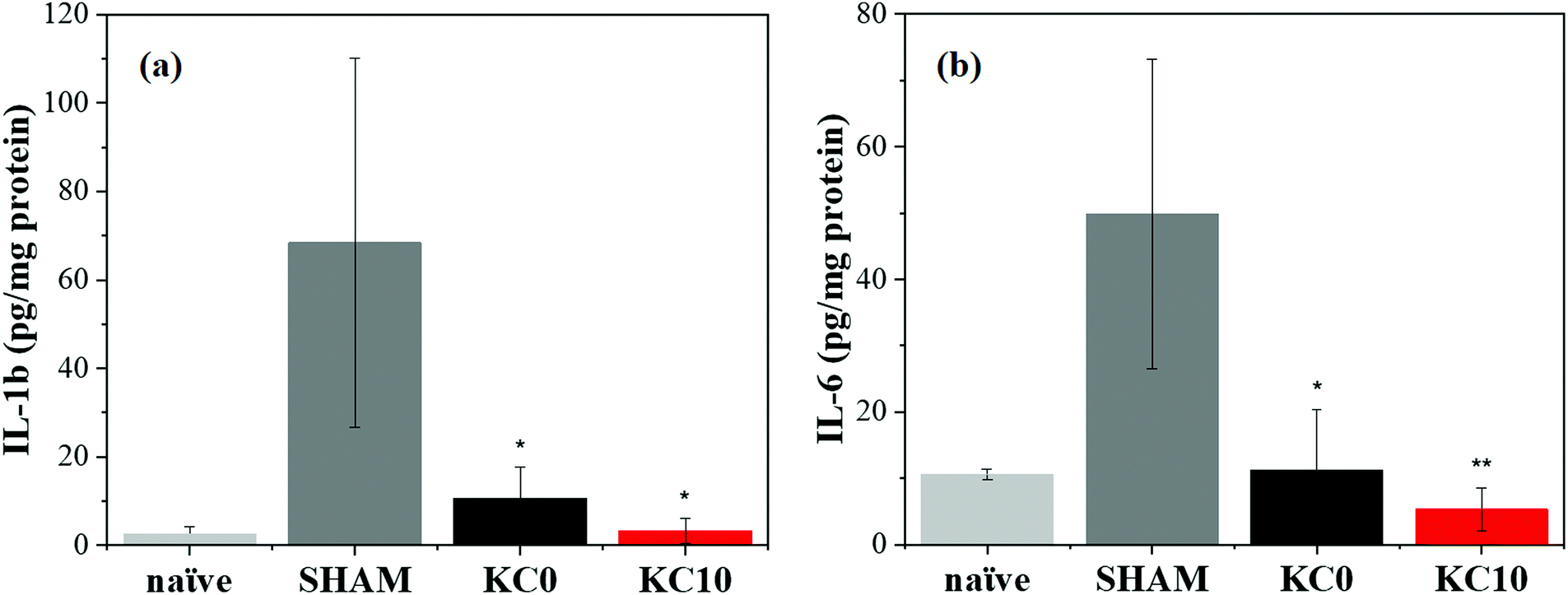 | ||
| Fig. 6 In vivo wound healing results from a UV-B skin damage mouse model. Expression of the inflammatory cytokines (a) IL-1b and (b) IL-6 for non-UVB-exposed, untreated animals (naïve), UV-B exposed, untreated animals (SHAM), UV-B exposed animals treated with KC0 and UV-B exposed animals treated with KC10 48 h after the irradiation. Results are presented as the average ± S.E.M. (n = 5 for each group). *p < 0.05 vs. SHAM; **p < 0.01 vs. SHAM. | ||
The observed reduction in the expression of both the inflammatory factors considered in the study, following a single application of the composite fibers, showed that the biological reaction was contained and even prevented by our keratin-based fibers, suggesting a potential use without changing too frequently the patch. This highlights the promising and central role of the designed cinnamon–keratin-based dressings, as a platform for burn management, although further studies are needed to better understand their involvement in the management of deep burn wounds.
4. Conclusions
In this work, we designed and implemented a biocompatible cinnamon oil/wool keratin-based composite dressing for skin burn management. The successful dispersion of cinnamon essential oil within the polymeric matrix was confirmed with FTIR, μRaman and confocal microscopy. When in contact with a water-like environment, the encapsulated active compounds were released from the matrices, as measured via UV-vis absorption. Our biocomposite constructs presented a notable antibacterial activity against S. aureus, E. coli and P. aeruginosa, and the antioxidant features of the fibers were enhanced with the addition of cinnamon essential oil. Furthermore, keratin-based patches with and without cinnamon essential oil were able to reduce in vivo the expression of pro-inflammatory factors by 5–7 fold. Therefore, this innovative natural-based material holds high potential for the healing of skin burns.Funding sources
D. K. was supported by the Greek State Scholarships Foundation (IKY), via the Erasmus+ Program.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Olimpias s.r.l. (Ponzano Veneto, TV, Italy) is acknowledged for kindly donating the Argentinian sheep wool. Doriana Debellis is acknowledged for the SEM images. Marco Scotto is acknowledged for his help with the confocal microscopy. D. K. acknowledges Thi Nga Tran and Marco Contardi for the fruitful discussions and the Greek State Scholarship Foundation (IKY) for financial support.References
- H. Bak, S. Phil Hong, S. K. Jeong, E. H. Choi, S. E. Lee, S. H. Lee and S. K. Ahn, Int. J. Dermatol., 2011, 50, 832–837 CrossRef PubMed.
- W. M. Holleran, Y. Uchida, L. Halkier-Sorensen, A. Haratake, M. Hara, J. H. Epstein and P. M. Elias, Photodermatol., Photoimmunol. Photomed., 1997, 13, 117–128 CrossRef CAS PubMed.
- B. Son, S. Lee, H. Kim, H. Kang, J. Kim, H. Youn, S. Young and B. Youn, J. Dermatol. Sci., 2019, 96, 81–89 CrossRef CAS PubMed.
- S. Kargozar, M. Mozafari, S. Hamzehlou and F. Baino, Front. Bioeng. Biotechnol., 2019, 7, 62 CrossRef PubMed.
- H. Derakhshandeh, S. S. Kashaf, F. Aghabaglou, I. O. Ghanavati and A. Tamayol, Trends Biotechnol., 2018, 36, 1259–1274 CrossRef CAS PubMed.
- G. Broughton, J. E. Janis and C. E. Attinger, Plast. Reconstr. Surg., 2006, 117, 1–32 CrossRef PubMed.
- K. A. Rieger, N. P. Birch and J. D. Schiffman, J. Mater. Chem. B, 2013, 1, 4531–4541 RSC.
- M. Norouzi, S. M. Boroujeni, N. Omidvarkordshouli and M. Soleimani, Adv. Healthcare Mater., 2015, 4, 1114–1133 CrossRef CAS PubMed.
- M. B. Dreifke, A. A. Jayasuriya and A. C. Jayasuriya, Mater. Sci. Eng., C, 2015, 48, 651–662 CrossRef CAS PubMed.
- E. G. Loboa, 23 – Nanofibrous smart bandages for wound care, Elsevier Ltd, 2016, vol. 2 Search PubMed.
- Y. Tang, X. Lan, C. Liang, Z. Zhong, R. Xie, Y. Zhou, X. Miao, H. Wang and W. Wang, Carbohydr. Polym., 2019, 219, 113–120 CrossRef CAS PubMed.
- R. S. Ambekar and B. Kandasubramanian, Eur. Polym. J., 2019, 117, 304–336 CrossRef CAS.
- E. L. Papadopoulou, P. Valentini, F. Mussino and P. P. Pompa, Chem. Eng. J., 2018, 347, 19–26 CrossRef CAS.
- M. Contardi, D. Russo, G. Suarato, J. A. Heredia-Guerrero, L. Ceseracciu, I. Penna, N. Margaroli, M. Summa, R. Spanò, G. Tassistro, L. Vezzulli, T. Bandiera, R. Bertorelli, A. Athanassiou and I. S. Bayer, Chem. Eng. J., 2019, 358, 912–923 CrossRef CAS.
- H. Hajiali, M. Summa, D. Russo, A. Armirotti, V. Brunetti, R. Bertorelli, A. Athanassiou and E. Mele, J. Mater. Chem. B, 2016, 4, 1686–1695 RSC.
- I. Liakos, L. Rizzello, D. J. Scurr, P. P. Pompa, I. S. Bayer and A. Athanassiou, Int. J. Pharm., 2014, 463, 137–145 CrossRef CAS PubMed.
- B. Wang, W. Yang, J. McKittrick and M. A. Meyers, Prog. Mater. Sci., 2016, 76, 229–318 CrossRef CAS.
- S. Sharma, A. Gupta, S. M. S. T. Chik, C. G. Kee, B. M. Mistry, D. H. Kim and G. Sharma, Int. J. Biol. Macromol., 2017, 104, 189–196 CrossRef CAS PubMed.
- Z. S. Thompson, N. P. Rijal, D. Jarvis, A. Edwards and N. Bhattarai, J. Visualized Exp., 2016, 108, 1–8 Search PubMed.
- F. Costa, R. Silva and A. R. Boccaccini, Fibrous protein-based biomaterials (silk, keratin, elastin, and resilin proteins) for tissue regeneration and repair, Elsevier Ltd, 2017 Search PubMed.
- A. Tachibana, Y. Furuta, H. Takeshima, T. Tanabe and K. Yamauchi, J. Biotechnol., 2002, 93, 165–170 CrossRef CAS PubMed.
- P. Sierpinski, J. Garrett, J. Ma, P. Apel, D. Klorig, T. Smith, L. A. Koman, A. Atala and M. Van Dyke, Biomaterials, 2008, 29, 118–128 CrossRef CAS PubMed.
- J. R. Richter, R. C. De Guzman, O. K. Greengauz-Roberts and M. Van Dyke, Acta Biomater., 2012, 8, 274–281 CrossRef CAS PubMed.
- V. Verma, P. Verma, P. Ray and A. R. Ray, Biomed. Mater., 2008, 3, 025007 CrossRef PubMed.
- G. Suarato, R. Bertorelli and A. Athanassiou, Front. Bioeng. Biotechnol., 2018, 6, 1–11 CrossRef PubMed.
- G. Perotto, G. Sandri, C. Pignatelli, G. Milanesi and A. Athanassiou, J. Mater. Chem. B, 2019, 7, 4385–4392 RSC.
- T. R. Ham, R. T. Lee, S. Han, S. Haque, Y. Vodovotz, J. Gu, L. R. Burnett, S. Tomblyn and J. M. Saul, Biomacromolecules, 2016, 17, 225–236 CrossRef CAS PubMed.
- R. Kelly, Keratins in wound healing, Elsevier Ltd, 2016, vol. 2 Search PubMed.
- A. Aluigi, C. Vineis, A. Varesano, G. Mazzuchetti, F. Ferrero and C. Tonin, Eur. Polym. J., 2008, 44, 2465–2475 CrossRef CAS.
- J. Fan, T. Lei, J. Li, P. Zhai, Y. Wang, F. Cao and Y. Liu, Mater. Des., 2016, 104, 60–67 CrossRef CAS.
- C. Wang, J. Wang, L. Zeng, Z. Qiao, X. Liu, H. Liu, J. Zhang and J. Ding, Molecules, 2019, 24, 834 CrossRef PubMed.
- M. Contardi, J. A. Heredia-Guerrero, S. Guzman-Puyol, M. Summa, J. J. Benitez, L. Goldoni, G. Caputo, G. Cusimano, P. Picone, M. Di Carlo, R. Bertorelli, A. Athanassiou and I. Bayer, J. Mater. Chem. B, 2019, 7, 1384–1396 RSC.
- D. Kalemba and A. Kunicka, Curr. Med. Chem., 2003, 10, 813–829 CrossRef CAS PubMed.
- N. Goel, H. Rohilla, G. Sing and P. Punia, J. Clin. Diagn. Res., 2016, 10, DC09–DC11 CAS.
- A. Brochot, A. Guilbot, L. Haddioui and C. Roques, MicrobiologyOpen, 2017, 6, 1–6 CrossRef PubMed.
- E. Schmidt, K. K. Co, H. A. Schloss and D. Wallerstein, J. Essent. Oil-Bear. Plants, 2006, 9, 170–182 CrossRef CAS.
- I. Husain, R. Ahmad, A. Chandra, S. T. Raza, Y. Shukla and F. Mahdi, J. Ethnopharmacol., 2018, 219, 110–116 CrossRef CAS PubMed.
- R. Ribeiro-Santos, M. Andrade, D. Madella, A. P. Martinazzo, L. de Aquino Garcia Moura, N. R. de Melo and A. Sanches-Silva, Trends Food Sci. Technol., 2017, 62, 154–169 CrossRef CAS.
- H. Chen, X. Hu, E. Chen, S. Wu, D. Julian, S. Liu, B. Li and Y. Li, Food Hydrocolloids, 2016, 61, 662–671 CrossRef CAS.
- H. Fasihi, N. Noshirvani, M. Hashemi, M. Fazilati and H. Salavati, Food Packag. Shelf Life, 2019, 19, 147–154 CrossRef.
- G. Suarato, M. Contardi, G. Perotto, J. A. Heredia-Guerrero, F. Fiorentini, L. Ceseracciu, C. Pignatelli, D. Debellis, R. Bertorelli and A. Athanassiou, Mater. Sci. Eng., C, 2020, 116, 111151 CrossRef CAS.
- S. F. Hosseini, M. Zandi, M. Rezaei and F. Farahmandghavi, Carbohydr. Polym., 2013, 95, 50–56 CrossRef CAS PubMed.
- M. Zahid, E. L. Papadopoulou, G. Suarato, V. D. Binas, G. Kiriakidis, O. Moira, D. Venieri and I. S. Bayer, ACS Appl. Bio Mater., 2018, 1, 1154–1164 CrossRef CAS.
- E. L. Papadopoulou, U. C. Paul, T. N. Tran, G. Suarato, L. Ceseracciu, S. Marras, R. Arcy and A. Athanassiou, ACS Appl. Mater. Interfaces, 2019, 11, 31317–31327 CrossRef CAS PubMed.
- I. Romano, M. Summa, J. A. Heredia-Guerrero, R. Spanó, L. Ceseracciu, C. Pignatelli, R. Bertorelli, E. Mele and A. Athanassiou, Biomed. Mater., 2016, 11, 041001 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- Y. Q. Li, D. X. Kong and H. Wu, Ind. Crops Prod., 2013, 41, 269–278 CrossRef CAS.
- A. Kuzuhara and T. Hori, J. Mol. Struct., 2013, 1037, 85–92 CrossRef CAS.
- A. Shavandi, T. H. Silva, A. A. Bekhit and A. E. D. A. Bekhit, Biomater. Sci., 2017, 5, 1699–1735 RSC.
- G. N. Andreev, B. Schrader, H. Schulz, R. Fuchs, S. Popov and N. Handjieva, Anal. Bioanal. Chem., 2001, 371, 1009–1017 CAS.
- H. Atri, E. Bidram and D. E. Dunstan, Materials, 2015, 8, 7472–7485 Search PubMed.
- M. Yahyaoui, O. Gordobil, R. Herrera, M. Abderrabba and J. Labidi, React. Funct. Polym., 2016, 109, 1–8 CrossRef CAS.
- J. Wang, V. Planz, B. Vukosavljevic and M. Windbergs, Eur. J. Pharm. Biopharm., 2018, 129, 175–183 CrossRef CAS PubMed.
- S. Guo and L. A. DiPietro, J. Dent. Res., 2010, 89, 219–229 CrossRef CAS PubMed.
- M. Sundaram, R. Legadevi, N. Afrin Banu and V. Gayathri, Eur. J. Biotechnol. Biosci., 2015, 3, 1–5 Search PubMed.
- M. Ignatova, N. Manolova, I. Rashkov and N. Markova, Int. J. Pharm., 2018, 545, 342–356 CrossRef CAS PubMed.
- P. V. Rao and S. H. Gan, Evidence-Based Complementary Altern. Med., 2014, 2014, 1–2 CrossRef PubMed.
- K. N. Prasad, B. Yang, X. Dong, G. Jiang, H. Zhang, H. Xie and Y. Jiang, Innov. Food Sci. Emerg. Technol., 2009, 10, 627–632 CrossRef CAS.
- B. Caven, B. Redl and T. Bechtold, Text. Res. J., 2019, 89, 510–516 CrossRef CAS.
- C. D. Tran, F. Prosencyes, M. Franko and G. Benzi, Carbohydr. Polym., 2016, 151, 1269–1276 CrossRef CAS PubMed.
- N. Bou Haidar, S. Marais, E. Dé, A. Schaumann, M. Barreau, M. G. J. Feuilloley and A. C. Duncan, Mater. Sci. Eng., C, 2020, 106, 110130 CrossRef CAS PubMed.
- M. K. Swamy, M. S. Akhtar and U. R. Sinniah, Evidence-Based Complementary Altern. Med., 2016, 2016, 3012462 Search PubMed.
- A. Man, L. Santacroce, R. Jacob, A. Mare and L. Man, Pathogens, 2019, 8, 1–11 Search PubMed.
- T. Paul, A. Mandal, S. M. Mandal, K. Ghosh, A. K. Mandal, S. K. Halder, A. Das, S. K. Maji, A. Kati, P. K. D. Mohapatra, B. R. Pati and K. C. Mondal, Appl. Biochem. Biotechnol., 2015, 175, 3371–3386 CrossRef CAS PubMed.
- M. Y. Memar, R. Ghotaslou, M. Samiei and K. Adibkia, Infect. Drug Resist., 2018, 11, 567–576 CrossRef CAS PubMed.
- D. Díaz-García, P. R. Ardiles, M. Díaz-Sánchez, I. Mena-Palomo, I. del Hierro, S. Prashar, A. Rodríguez-Diéguez, P. L. Páez and S. Gómez-Ruiz, J. Inorg. Biochem., 2020, 203, 110912 CrossRef PubMed.
- T. Makropoulou, P. Panagiotopoulou and D. Venieri, J. Chem. Technol. Biotechnol., 2018, 93, 2518–2526 CrossRef CAS.
- I. Levchuk, T. Homola, J. Moreno-Andres, J. J. Rueda-Marquez, P. Dzik, M. A. Morinigo, M. Sillanpaa, M. A. Manzano and R. Vahala, Sol. Energy, 2019, 191, 518–529 CrossRef CAS.
- S. Chouhan, K. Sharma and S. Guleria, Medicines, 2017, 4, 58 CrossRef PubMed.
- Z. I. Yildiz, M. E. Kilic, E. Durgun and T. Uyar, J. Agric. Food Chem., 2019, 67, 11066–11076 CrossRef CAS PubMed.
- M. Laranjo, A. M. Fernández, L. Ana, C. A. Santos, M. E. Potes and M. Elias, J. Food Process. Preserv., 2019, e14278 Search PubMed.
- D. Kaic and A. Rosario, Int. Food Res. J., 2019, 26, 1103–1110 Search PubMed.
- Y. El Atki, I. Aouam, K. Nayme and B. Lyoussi, J. Adv. Pharm. Technol. Res., 2019, 10, 63–67 CrossRef CAS PubMed.
- L. L. Hruza and A. P. Pentland, J. Invest. Dermatol., 1993, 100, S35–S41 CrossRef.
- C. Kuanpradit, Y. Jaisin, S. Jungudomjaroen and S. A. Mitu, Int. J. Mol. Med., 2017, 39, 1083–1090 CrossRef CAS PubMed.
- M. R. Farahpour, A. Amniattalab and H. Hajizadeh, Afr. J. Biotechnol., 2012, 11, 15068–15071 Search PubMed.
- Z. Hussain, J. A. Khan, A. Arshad, P. Asif, H. Rashid and M. I. Arshad, Biomed. Pharmacother., 2019, 109, 2285–2292 CrossRef CAS PubMed.
- I. W. Nasr, C. A. Massaad, B. Sa, N. E. Saade, S. J. Jabbur and S. A. Kanaan, Br. J. Pharmacol., 2000, 131, 1317–1324 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00416b |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |