
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(N-isopropyl acrylamide)–poly(ethylene glycol)–poly(N-isopropyl acrylamide) as a thermoreversible gelator for topical administration†
P.
Haddow
,
W. J.
McAuley
,
S. B.
Kirton
 and
M. T.
Cook
*
and
M. T.
Cook
*
Centre for Research in Topical Drug Delivery and Toxicology, Department of Clinical and Pharmaceutical Sciences, School of Life and Medical Sciences, University of Hertfordshire, UK. E-mail: m.cook5@herts.ac.uk
First published on 6th May 2020
Abstract
Poly(N-isopropyl acrylamide)-block-poly(ethylene glycol)-block-poly(N-isopropyl acrylamide) is known to exhibit a thermally-induced solution-to-gel transition in water, which may be exploited for biomedical applications. This “thermoreversible gelator” has great potential for application in topical administration to the surfaces of the body such as the skin, eye, and vagina, but this has not yet been evaluated. This study evaluates PNIPAM98–PEG122–PNIPAM98 as a thermoreversible gelator for vaginal administration, for the first time evaluating the effect of polymer concentration on gelation, studying rheological parameters relevant to topicals, measuring dissolution rates, stability and the phenomenon of mucoadhesion. Two drugs relevant to vaginal administration, progesterone and tenofovir disoproxil fumarate are investigated for use in the thermoreversible gelators, studying both hydrophobic and hydrophilic drug solubilisation and release. Throughout the study, comparison is made with poloxamer 407, the most commonly studied thermoreversible gelator. PNIPAM98–PEG122–PNIPAM98 exhibits several advantages for topical administration, having low viscosity at room temperature to allow easy application, then exhibiting a gelation just below body temperature to form a viscous gel which is resistant to dissolution and relatively mucoadhesive. Drug release is highly dependent on temperature, with elevation to body temperature resulting in a dramatic retardation of progesterone release, which may be used in future medicines to provide sustained delivery of hydrophobic xenobiotics.
Introduction
“Smart” materials respond to external stimuli by alteration of one or more of their physical properties. One class of smart material are thermoreversible gelators. These materials undergo a solution-to-gel transition when warmed above a critical temperature (Tgel), exhibiting the reverse gel-to-solution phase change upon cooling.1 Thermoreversible gelators with a Tgel occurring between room temperature (ca. 25 °C) and body temperature (37 °C internally) may pass through an applicator or syringe, then form a viscous gel on contact with the body.2 This in situ gelation may be used for injectables, where the gel may act as a drug depot or scaffold for therapeutic cells,3 and for topical administration to either skin or mucosal membranes.4 Thermoreversible gelators are particularly attractive for vaginal application, as the material may pass through an applicator before forming a viscous gel upon contact with the body which enhances retention of bioactives to prolong local effect or absorption5 at a site where retention is often poor.The most widely studied polymer which exhibits thermoreversible gelation is poloxamer 407 (also called Pluronic F127). Poloxamer 407 is an ABA triblock copolymer with the structure poly(ethylene glycol)101-block-poly(propylene glycol)65-block-poly(ethylene glycol)101.6 Heating concentrated (>15% w/v) poloxamer 407 solutions results in a hierarchical process of desolvation of the poly(propylene glycol) block, which drives micellization and in turn the formation of a face-centred cubic mesophase gel.7 This property makes poloxamer 407 an attractive excipient for topical use, where the body's heat may trigger gelation.8 However, poloxamer 407 has several drawbacks including: a low Tgel of ca. 25 °C and below,9 rapid dissolution, and low mucoadhesion.6 Thus, there is a need for novel thermoreversible gelators with modulated properties.
Poly(N-isopropyl acrylamide)-block-poly(ethylene glycol)-block-poly(N-isopropyl acrylamide) (PNIPAM–PEG–PNIPAM) exhibits thermoreversible gelation in aqueous solution.10 PNIPAM exhibits a lower critical solution temperature (LCST), resulting in a coil-to-globule transition above a critical temperature, typically 32 °C.11 In the PNIPAM–PEG–PNIPAM copolymers, this transition leads to the formation of micelles, believed to have flower-like or fractal-like structures depending on molecular weight.12,13 At high concentrations (above ca. 20% w/v), PNIPAM–PEG–PNIPAM forms physical gels, hypothesized to be a result of entanglement of PNIPAM blocks,14 though this phenomenon has been studied in only a limited number of publications. It has previously been demonstrated that 20% (w/v) solutions of a PNIPAM (2.3 kDa)–PEG (4.6 kDa)–PNIPAM (2.3 kDa) copolymer undergo transition to a gel state when warmed above ca. 26 °C.14 Teodorescu et al.10 studied molecular weight effects on PNIPAM–PEG–PNIPAM gelation at concentrations of 20% (w/v) and below, varying PEG molecular weight between 1 and 6 kDa and PNIPAM between 5 and 30 kDa. It was found that only 4 and 6 kDa PEG with 20 or 30 kDa PNIPAM blocks resulted in gel formation, as determined by rheometry, with Tgels of 38–43 °C. De Graaf et al. have demonstrated PNIPAM (16 kDa)–PEG (6 kDa)–PNIPAM (16 kDa) and PNIPAM (32 kDa)–PEG (6 kDa)–PNIPAM (32 kDa) exhibited gelation temperatures of 35 and 33 °C in phosphate buffered saline, allowing them to form a gel when injected into mice.15 This gel released paclitaxel in a controlled fashion and did not cause acute systemic toxicity. PNIPAM–PEG–PNIPAM is a potential thermoreversible gelator for topical drug delivery. However, understanding of its behaviour in drug delivery is limited and there are no studies evaluating its performance for topical application. Additionally, the solubilisation of drugs in concentrated PNIPAM–PEG–PNIPAM is limited,15 and there are no studies of saturation solubilisation and release in these systems. Furthermore, a critical evaluation of this novel material against poloxamer 407 has not been conducted. In order for novel excipients to translate into biomedical applications, clear advantages of these materials over the current approaches must be demonstrated. While gels are a traditional dosage form for topical application, there is a drive a generate novel materials with added functionality,16 and translation must be considered whilst driving this innovation.
In this research we study thermoreversible gelation in PNIPAM–PEG–PNIPAM with a view to topical drug delivery. The effect of concentration on Tgel and gel strength is studied, along with a critical evaluation of rheological performance against poloxamer 407. The stability of these materials is also reported for the first time. The materials are then compared side-by-side for the preparation of vaginal formulations, as an exemplar mucosal route, revealing a fundamental understanding of the behaviour of hydrophilic and hydrophobic drugs in these thermoreversible gelators.
Experimental
Materials
N-Isopropyl acrylamide (NIPAM) (97%), copper(I) bromide (CuBr) (98%), tetrahydrofuran (THF) (99%) and triethylamine (99.5%) were purchased from Sigma-Aldrich (UK). Tris[2-(dimethylamino)ethyl]amine (Me6TREN) (99%) and 2-bromoisobutyryl bromide (BiBB) (97%) were purchased from Alfa Aesar (UK). Isopropyl alcohol (99%) and dichloromethane (99%) were purchased from Fisher Scientific (UK). Bipyridine (99%), dimethylaminopyridine (DMAP) (97%) and PEG 10 kDa were purchased from Aldrich (UK). Aluminium oxide, neutral, Brockmann I was purchased from Acros Organics (UK). Dialysis tubing with a molecular weight cut off (MWCO) of 3500 Da was purchased from Medicell Membranes Ltd (UK) and soaked in deionised H2O before use. GPC EasiVial poly(methyl methacrylate) mixed standards and a poly(methyl methacrylate) single standard (72 kDa) were purchased from Agilent (UK). Deionised H2O was used in all experiments. All reagents were used as supplied, unless otherwise indicated.Vaginal fluid simulant (VFS) was prepared by dissolution of NaCl (3.510 g, 0.060 mol), KOH (1.400 g, 0.025 mol), Ca(OH)2 (0.222 g, 0.003 mol), bovine serum albumin (0.018 g), lactic acid (2.000 g, 0.022 mol), acetic acid (1.000 g, 0.011 mol), glycerol (0.160 g, 0.002 mol), urea (0.400 g, 0.007 mol) and glucose (5.000 g, 0.028 mol) in 1 L of deionised water. The solution was then adjusted to pH 4 with 1 M hydrochloric acid. This formula follows that recommended by Owen and Katz.17
Porcine vaginal tissue was purchased from WetLab (UK) and supplied frozen. The porcine vaginal mucosa was removed by incision with a scalpel. Once the tissue was removed it was cut into 1 cm2 squares which were attached to the texture analyse probe using adhesive pads.
Synthesis of PEG macroinitiator
A 10 kDa PEG macroinitiator was synthesised using reported methods.18 DMAP (1.17 g, 9.6 mmol) in anhydrous dichloromethane (8 mL) was mixed with triethylamine (0.89 mL, 6.4 mmol) and cooled to 0 °C. BiBB (1.97 mL, 16.0 mmol) in anhydrous dichloromethane (8 mL) was then added to the DMAP and triethylamine solution. A solution of PEG (10 kDa, 16.0 g, 1.6 mmol) in dichloromethane (160 mL) was subsequently added dropwise over 1 h. When the addition of PEG was complete, the reaction was allowed to rise to room temperature and the reaction allowed to proceed with stirring for 18 h. The solution was filtered, and approximately half of the solvent in the filtrate removed in vacuo. The PEG initiator was then precipitated in cold diethyl ether (480 mL) and filtered. The crude PEG macroinitiator in the retentate was then recrystalised from absolute ethanol (300 mL) overnight. The resulting solid was filtered and washed with cold diethyl ether and dried in vacuo (yield = 86.5%).Synthesis of PNIPAM98–PEG122–PNIPAM98 by atom-transfer radical polymerisation (ATRP)
NIPAM was polymerised from the PEG macroinitiator by ATRP following established procedures.12 Firstly, the CuBr catalyst (28.7 mg, 200 μmol) was sealed in a round bottom flask and degassed for 30 min by nitrogen purging. Separately, the Me6TREN ligand (53.5 μL, 200 μmol), NIPAM monomer (2.0 g, 17.7 mmol) and PEG 10 kDa macroinitiator (1.0 g, 100 μmol) were dissolved in deionised water (10 mL), within a sealed round bottomed flask. The solution was then degassed by nitrogen bubbling for 30 min. The solution containing ligand, initiator and monomer was then transferred to the flask containing the catalyst via a degassed syringe. The reaction was allowed to proceed with stirring at room temperature. Samples were taken at regular intervals using a degassed syringe and analysed using GPC to monitor the polymerisation. Once polymerisation ceased (24 h), the reaction mixture was dried in vacuo and dissolved in THF (10 mL). This solution was passed through a short length of neutral aluminium oxide (Brockmann I) to remove the catalyst–ligand complex. The product was then dissolved in H2O (10 mL) and dialysed against water using cellulose membrane (MWCO ∼ 3500 Da) for 48 h. The resulting solution was then freeze dried yielding a white solid, which was analysed using IR, NMR and GPC (yield = 74.3%).Characterisation of polymer structure and molecular weight
1H NMR was performed on an Oxford Instruments ECA600 600 MHz NMR spectrometer with Delta 4.3.6 software. All samples were measured in CDCl3. All spectra were analysed using MNOVA by Mestrelab (Spain). IR spectroscopy was performed on a PerkinElmer Fourier Transform Infra-red (FTIR) Spectrometer Frontier with a PerkinElmer Universal ATR Sample Accessory. A wavelength range of 650–4000 cm−1 was used with a resolution of 4 cm−1. The instrument was cleaned with isopropyl alcohol before and after use.Molecular weights were determined using an Agilent 12600 Infinity II GPC equipped with a refractive index (RI) detector an a Varian PLGel 5 μm mixed D column which ran DMF with 0.1% LiBr as an eluent, at a flow rate of 0.4 mL min−1 with the column and detector held at 30 °C.
Rheological evaluation of polymer solutions
All samples for rheometry were prepared to the required concentration in deionised water or VFS and refrigerated overnight before measurements were taken. Rheology was performed on an AR 1500ex rheometer by TA instruments (USA) with a Julabo AWC100 cooling unit and a 40 mm parallel plate geometry with a gap of 650 μm. Rheological measurements were taken in triplicate.Oscillatory stress sweeps were performed at 1 Hz between 1 and 1000 Pa at 37 °C. Frequency sweeps were measured at 37 °C between 0.1 and 10 Hz at a shear stress of 1 Pa. Temperature ramps were performed at a frequency of 1 Hz and a shear stress of 1 Pa. The temperature was increased at a rate of 2 °C min−1, from 15 to 50 °C.
Reversibility of the phase transition was assessed by rheological measurement at 25 °C for 1 min, followed by 37 °C for 1 min, which was repeated once more. A 2 min equilibration period was included between temperature changes and the measurements were recorded at 1 Pa and 1 Hz.
To determine the gelation time, the temperature of the Peltier plate was held at 25 °C for 1 min, then increased to 37 °C and held for a further 4 min. All measurements were made at a constant shear stress of 1 Pa and a frequency of 1 Hz. The time taken for G′ to exceed G′′ at 37 °C was taken as the gelation time.
Dissolution of polymer gels in VFS
The thermoreversible gelator (20% w/v poloxamer 407 or 50% w/v PNIPAM98–PEG122–PNIPAM98) dissolution rate was identified using a Copley USP II dissolution apparatus held at 37 °C with 400 mL VFS using an immersion cell developed in house consisting of a plastic cylinder with a closed weighted base, where the cylinder had a depth of 8.5 mm and a surface area of 306 mm2. The immersion cell was weighed and 2 mL sample was placed within. The cell was then placed in an oven for 5 minutes at 37 °C prior to starting the experiment to induce gel formation. The immersion cell was then weighed and placed into the AVF. The paddle was set to 50 rpm and the weight of the immersion cell was measured every 5 minutes to identify how much gel remained, until all the gel was dissolved. The weight percent of remaining gel was by expressed relative to the starting mass.Investigation of the influence of temperature-responsive polymers on the solubility of progesterone and tenofovir disoproxil fumarate
The effect of polymer on drug solubility was investigated using polymer concentrations ranging from 0.005 to 10 mg mL−1 at both 25 and 37 °C. Drug (ca. 5 mg) was added to the polymer solutions (1 mL) which were then placed in a water bath at either 25 or 37 °C and were stirred for 24 h. If the polymer solution was clear after 24 h, more drug (ca. 5 mg) was added and the vials were again left for 24 h under constant stirring, this process was repeated until the solutions remained turbid for 24 h. The turbid solutions were then centrifuged, and the clear supernatant was then analysed by HPLC using a validated HPLC procedure (ESI†). If a linear relationship between drug solubility and polymer concentration was observed, the critical micelle concentration (CMC) was calculated (eqn (S4), ESI†). The CMC was then used alongside experimental data to calculate the solubilising power of the polymer (eqn (S5), ESI†). Following the calculation of the polymer solubilising power, the micelle/water partition coefficient was calculated (eqn (S6), ESI†). Each polymer concentration was investigated in triplicate. The effect of progesterone saturation on micellisation was investigated at 10 mg mL−1 using dynamic light scattering on a Zetasizer Nano-NS (Malvern, UK). Undissolved progesterone was removed prior to analysis by passage through a 0.4 μm microfilter.The saturated solubility of progesterone and tenofovir disoproxil fumarate in water and the concentrated thermoreversible gelator solutions was then investigated. The thermoreversible gelators (20% w/v poloxamer 407 or 50% w/v PNIPAM98–PEG122–PNIPAM98) were prepared and stored in the fridge overnight. On the following day, 1 mL of thermoreversible gelator or water was transferred to a sample vial and progesterone or tenofovir disoproxil fumarate (ca. 5 mg) added. The solutions were then stored in a water bath at either 25 or 37 °C with constant stirring. If after 24 h the solution was clear, more drug was added and again left for 24 h in the water bath with constant stirring. This process was repeated until the solution remained turbid for 24 h. Excess drug was then removed by centrifugation (10 min at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm) and the clear supernatant was analysed by HPLC. Each experiment was performed in triplicate.
500 rpm) and the clear supernatant was analysed by HPLC. Each experiment was performed in triplicate.
Release of progesterone and tenofovir disoproxil fumarate from thermoreversible gelators
The release of progesterone and tenofovir disoproxil fumarate from the thermoreversible gelators (20% w/v poloxamer 407 or 50% w/v PNIPAM98–PEG122–PNIPAM98) was investigated using Franz diffusion cells equipped with a cellulose membrane (MWCO 3.5 kDa) at both 25 and 37 °C. The experiments used Franz cells with an average phosphate buffer saline (PBS) receiver fluid volume of 12.3 mL and an average bore area of 174 mm2. The release of 50 μg mL−1 progesterone or 200 μg mL−1 tenofovir disoproxil fumarate was investigated, where this concentration did not violate sink conditions. A 20% w/v ethanol solution in PBS containing 50 μg mL−1 progesterone and a 200 μg mL−1 tenofovir disoproxil fumarate solution in PBS were used as controls. The Franz cells were then placed into a water bath for 30 min to allow the receiver fluid to reach the temperature of the surrounding water (25 or 37 °C). All Franz cells were dosed with 200 μL of sample and the release of drug measured at regular intervals by sampling receiver fluid (500 or 200 μL for the progesterone and tenofovir disoproxil fumarate respectively). Receiver fluid was replaced with an equal volume of pre-warmed PBS. The samples were analysed by HPLC (method and calibration (Fig. S1) in ESI†). The experiment was repeated 4 times.Assessing the adhesion and mucoadhesion of thermoreversible gelators
The adhesion and mucoadhesion of the thermoreversible gelators (20% w/v poloxamer 407 or 50% w/v PNIPAM98–PEG122–PNIPAM98) was assessed using a TA.XT Plus Texture Analyser (Stable Micro Systems, UK) with a poly(propylene) probe with a surface area of 1.25 cm2. This probe was used to determine adhesion values, whereas mucoadhesion was evaluated using porcine vaginal mucosa tissue (WetLab, UK). The thermoreversible gelator was placed into a water bath at 25 °C, the probe was lowered until contact between the probe or vaginal mucosa tissue was made. The temperature of the water bath was then increased to 37 °C and held at this temperature for 2 min to ensure gel formation, and mimic in situ gelation. Once a gel had formed, the probe was withdrawn at a rate of 10.0 mm s−1 until complete detachment was observed, as recommended in prior studies.19 The maximum force of detachment and the area under the force–displacement curve were determined using Texture Exponent 32 software (Stable Micro Systems, UK) and designated the “force of adhesion” and “work of adhesion”, respectively. All adhesion testing was performed 6 times. The mucoadhesion testing was performed using three different porcine vaginal tissues where three samples of vaginal tissue were taken from each vagina. Data was expressed as a mean of triplicate experiments (N = 3), where N was the mean value of the three measurements taken from a single vagina.Stability of thermoreversible gelators
The stability of thermoreversible gelators (20% w/v poloxamer 407 or 50% w/v PNIPAM98–PEG122–PNIPAM98) was assessed over 12 weeks. The polymer solutions were prepared in HPLC vials sealed with parafilm and placed in the refrigerator (4 °C) or in ovens set at 25 and 40 °C representing room temperature and accelerated storage conditions, respectively. At weekly intervals, the samples were lyophilised and the molecular weight determined by GPC, as described previously. Sufficient samples were prepared so that each weekly measurement was taken on a separate sample. The experiment was performed in triplicate.Statistical analysis
All statistical analysis was conducted on Prism5 (GraphPad, USA). Data was analysed by one-way analysis of variance (ANOVA) with Tukey post hoc analysis unless otherwise stated. Data presented as means ± standard deviation.Results and discussion
PNIPAM98–PEG122–PNIPAM98 was synthesised in a two-step process by covalent attachment of an ATRP initiator to PEG122 using a simple SN2 reaction with DMAP-activated BiBB followed by ATRP polymerisation from the macroinitiator using a Cu(I)Br/ME6TREN catalyst/ligand complex (Fig. 1(i)).12,18 GPC confirmed that the resultant copolymer had a monomodal molecular weight distribution with a polydispersity index of 1.88 (Fig. 1(iii)), in line with previous publications.12 The degree of polymerisation was calculated by 1H NMR (Fig. 1(iv)), finding that the polymer had the structure PNIPAM98–PEG122–PNIPAM98. DOSY NMR (Fig. S2, ESI†) unambiguously confirmed attachment of PNIPAM to PEG by demonstrating that the two blocks have the same diffusion coefficient.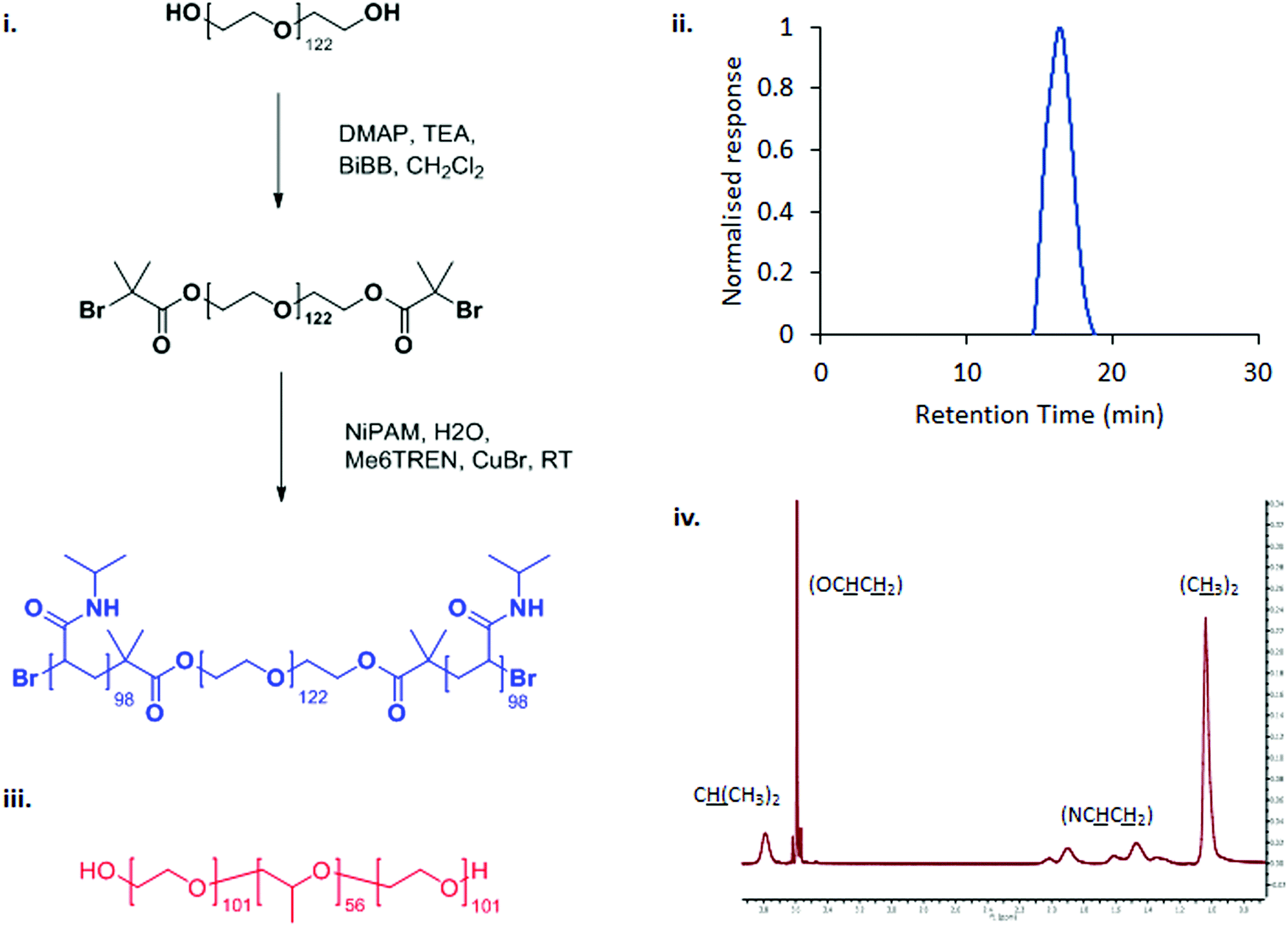 | ||
| Fig. 1 The synthetic route to PNIPAM98–PEG122–PNIPAM98 (i) and chemical structure of poloxamer 407 (iii). PNIPAM98–PEG122–PNIPAM98 was characterised by GPC (ii) and 1H NMR (iv). | ||
All blocks of PNIPAM98–PEG122–PNIPAM98 are water-soluble at room temperature, but the copolymer is expected to transition to an amphiphilic state when warmed due to the desolvation of PNIPAM. The comparator pharmaceutical excipient, poloxamer 407, is shown in Fig. 1(ii). This ABA copolymer exhibits water solubility at temperatures below ca. 20 °C but transitions to an amphiphilic state when warmed due to the desolvation of the central poly(propylene oxide) block. Desolvation of the central block leads to a hierarchical process of micelle formation (hydrodynamic radius ca. 10 nm) and subsequent packing into a face-centred cubic lattice to form a transparent gel mesophase.7,20
The thermoresponsive gelation process exhibited by PNIPAM98–PEG122–PNIPAM98 was studied by rheometry with variation of polymer concentration between 20–50% (w/v) (Fig. 2(i)). The copolymer exhibited dramatic increases in G′ and G′′ above a critical temperature (Tthick), at ca. 30–35 °C believed to be associated with the desolvation of PNIPAM.21 All rheograms exhibited a gelation temperature (Tgel) above ca. 35 °C, at which point the absolute value of G′ surpassed that of G′′, indicative of a transition from a viscous fluid to an elastic gel state. These value of Tthick and Tgel are attractive for biomedical applications where the polymer solutions will have low viscosity at room temperatures, but thicken when warmed by the body. Poloxamer 407 exhibited a markedly different thermoresponsive gelation to PNIPAM98–PEG122–PNIPAM98. Low viscosity gels (G′ ca. 100 Pa) were formed at 15% (w/v), with Tgel at 45 °C. Viscous gels (G′ ca. 10 kPa) were formed at 20 and 25% (w/v) with Tgel < 25 °C. At 30% (w/v) the materials were gels over the whole temperature range studied.
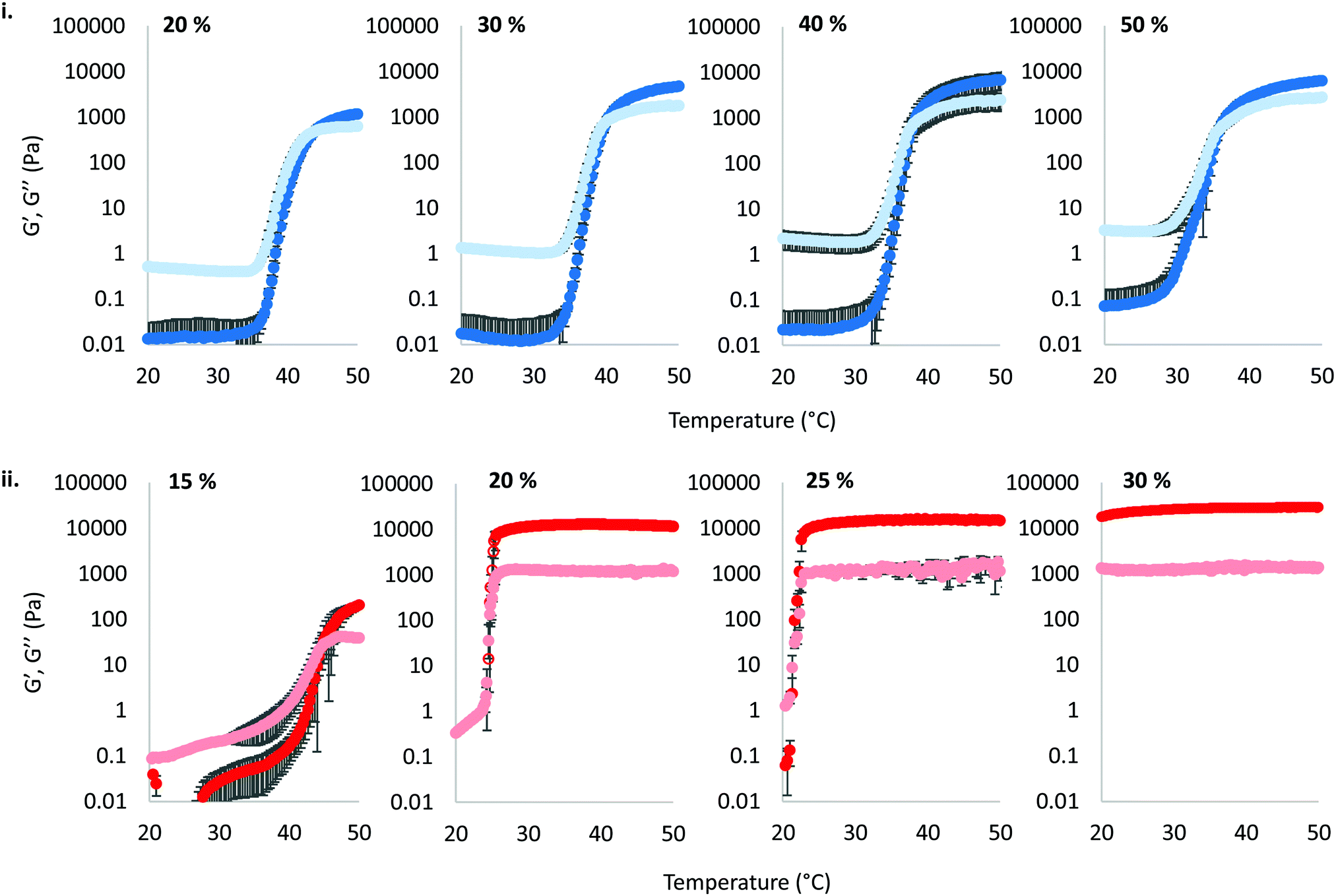 | ||
| Fig. 2 Temperature ramp rheograms of PNIPAM98–PEG122–PNIPAM98 (i) and poloxamer 407 (ii) with variation of concentration (% w/v) at a fixed shear stress (1 Pa) and frequency (1 Hz). G′ is presented as the dark colour, whilst the light colour corresponds to G′′. Data presented as mean ± standard deviation, n = 3. | ||
T
gel, Tthick and the maximum absolute value of G′ reached (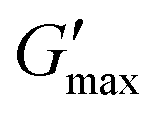 ) were extracted from the rheograms shown in Fig. 2 and Fig. S3 (ESI†) and are presented in Fig. 3. PNIPAM98–PEG122–PNIPAM98 (Fig. 3(i)) exhibited a monotonic, near-linear, decrease in Tgel from 44 to 36 °C across the concentration range studied. A linear fit (R2 = 0.96) indicates that the rate of Tgel depression is −0.25 °C g−1 dL, allowing tight control of Tgel over this range. Tthick decreased from 36 to 29 °C between 20 and 50% (w/v) concentration, allowing thickening to occur upon warming by the body. An inverse proportionality between Tgel and concentration has previously been observed in PNIPAM–poly(N,N-dimethylacrylamide)–PNIPAM solutions, and was attributed to the depressed LCST observed in PNIPAM solutions at high concentrations.22
) were extracted from the rheograms shown in Fig. 2 and Fig. S3 (ESI†) and are presented in Fig. 3. PNIPAM98–PEG122–PNIPAM98 (Fig. 3(i)) exhibited a monotonic, near-linear, decrease in Tgel from 44 to 36 °C across the concentration range studied. A linear fit (R2 = 0.96) indicates that the rate of Tgel depression is −0.25 °C g−1 dL, allowing tight control of Tgel over this range. Tthick decreased from 36 to 29 °C between 20 and 50% (w/v) concentration, allowing thickening to occur upon warming by the body. An inverse proportionality between Tgel and concentration has previously been observed in PNIPAM–poly(N,N-dimethylacrylamide)–PNIPAM solutions, and was attributed to the depressed LCST observed in PNIPAM solutions at high concentrations.22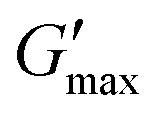 increased with concentration from 1.5 kPa at 20% (w/v) to a maximum of 12.7 kPa at 45% (w/v). An increase in gel strength with concentration was also observed by Kirkland et al.22 when studying PNIPAM–poly(N,N-dimethylacrylamide)–PNIPAM. The authors rationalise this phenomenon using the theory of Semenov et al.,23 that telechelic polymers with associating end-groups form flower-like micelles bridged by polymer chains resulting in elasticity. Kirkland et al. suggest that a greater number of polymer chains results in additional bridges formed between micellar domains, increasing viscosity.22 It is also conceivable that a greater number of micelles increases the degree of overlap between these aggregates, in a manner analogous to that observed in poloxamer 407.24 Values of Tgel for PNIPAM98–PEG122–PNIPAM98 were typically ca. 5 °C greater than Tthick, whereas poloxamer 407 gave a sharper transition at 20 and 25% (w/v) with Tgel within 1 °C of Tthick.
increased with concentration from 1.5 kPa at 20% (w/v) to a maximum of 12.7 kPa at 45% (w/v). An increase in gel strength with concentration was also observed by Kirkland et al.22 when studying PNIPAM–poly(N,N-dimethylacrylamide)–PNIPAM. The authors rationalise this phenomenon using the theory of Semenov et al.,23 that telechelic polymers with associating end-groups form flower-like micelles bridged by polymer chains resulting in elasticity. Kirkland et al. suggest that a greater number of polymer chains results in additional bridges formed between micellar domains, increasing viscosity.22 It is also conceivable that a greater number of micelles increases the degree of overlap between these aggregates, in a manner analogous to that observed in poloxamer 407.24 Values of Tgel for PNIPAM98–PEG122–PNIPAM98 were typically ca. 5 °C greater than Tthick, whereas poloxamer 407 gave a sharper transition at 20 and 25% (w/v) with Tgel within 1 °C of Tthick.
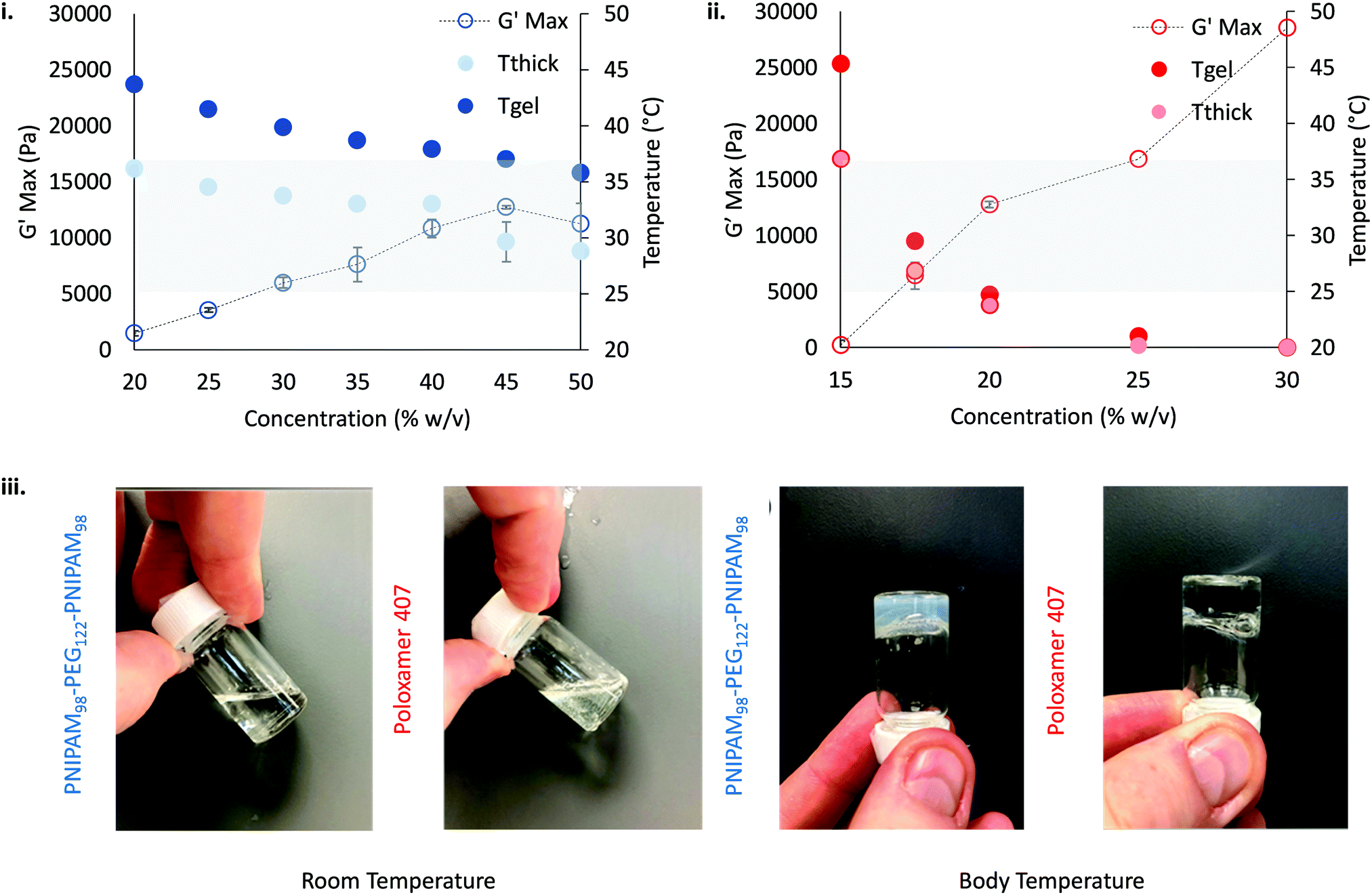 | ||
Fig. 3
T
thick (light circle), Tgel (dark circle), and 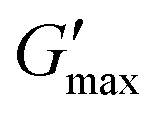 (hollow circle with dashes) as a function of concentration for PNIPAM98–PEG122–PNIPAM98 (i, blue) and poloxamer 407 (ii, red). The temperature range which would allow for in situ thickening of polymer solutions is overlaid in grey. Please note that Tthick and Tgel are omitted for 30% (w/v) poloxamer 407 as they were not present in the rheogram. Data presented as mean ± standard deviation, n = 3. Images of the thermoversible gelators are included (iii) at room temperature and body temperature, 37 °C. (hollow circle with dashes) as a function of concentration for PNIPAM98–PEG122–PNIPAM98 (i, blue) and poloxamer 407 (ii, red). The temperature range which would allow for in situ thickening of polymer solutions is overlaid in grey. Please note that Tthick and Tgel are omitted for 30% (w/v) poloxamer 407 as they were not present in the rheogram. Data presented as mean ± standard deviation, n = 3. Images of the thermoversible gelators are included (iii) at room temperature and body temperature, 37 °C. | ||
Poloxamer 407 exhibited a far greater dependence of Tgel on concentration than PNIPAM98–PEG122–PNIPAM98 (Fig. 3(ii)), with Tgel decreasing from 45 to 21 °C as the concentration was increased from 15 to 25% (w/v). At 30% (w/v) no Tgel was determined, with the rheograms exhibiting G′ > G′′ at all temperatures. These findings are in line with established phase behaviour of pluronics.20 Of the concentrations studied, only 17.5 and 20% (w/v) poloxamer 407 samples (Tgel of 30 and 5 °C, respectively) exhibited Tthick/Tgel at a temperature suitable for in situ thickening upon contact with the body (i.e. 25 °C < Tthick < 37 °C), albeit with a Tgel which may be reached at real room temperatures in warmer climates, e.g. WHO climatic zones III and IV (30 °C).25 The Tthick of 15% poloxamer 407 was at body temperature (37 °C). Poloxamer 407's 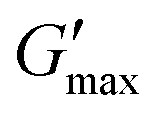 was proportional to concentration, and had a value of 6.5 kPa at 17.5% (w/v) poloxamer 407 and 12.7 kPa at 20% (w/v). Due to the larger gel strength of the 20% (w/v) formulation and its prevalence in the literature,6 this concentration was selected for comparison with PNIPAM98–PEG122–PNIPAM98 in future experiments. It is known that poloxamer 407 forms a gel via the packing of micelles into a cubic structure and increasing concentration leads to an increased volume fraction of micelles, and thus a greater degree of overlap between the micelles.24 The increase in
was proportional to concentration, and had a value of 6.5 kPa at 17.5% (w/v) poloxamer 407 and 12.7 kPa at 20% (w/v). Due to the larger gel strength of the 20% (w/v) formulation and its prevalence in the literature,6 this concentration was selected for comparison with PNIPAM98–PEG122–PNIPAM98 in future experiments. It is known that poloxamer 407 forms a gel via the packing of micelles into a cubic structure and increasing concentration leads to an increased volume fraction of micelles, and thus a greater degree of overlap between the micelles.24 The increase in 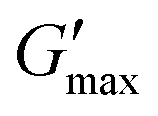 with concentration observed for the poloxamer samples is attributed to this greater degree of overlap and thus internal friction. The gels formed by 50% (w/v) PNIPAM98–PEG122–PNIPAM98 were translucent, whilst the 20% (w/v) poloxamer 407 gels were clear (Fig. 3(iii)). This indicates that the poloxamer micelles are sufficiently small to avoid scattering of visible light, but that the PNIPAM98–PEG122–PNIPAM98 aggregates are larger and the Tyndall effect is observed.
with concentration observed for the poloxamer samples is attributed to this greater degree of overlap and thus internal friction. The gels formed by 50% (w/v) PNIPAM98–PEG122–PNIPAM98 were translucent, whilst the 20% (w/v) poloxamer 407 gels were clear (Fig. 3(iii)). This indicates that the poloxamer micelles are sufficiently small to avoid scattering of visible light, but that the PNIPAM98–PEG122–PNIPAM98 aggregates are larger and the Tyndall effect is observed.
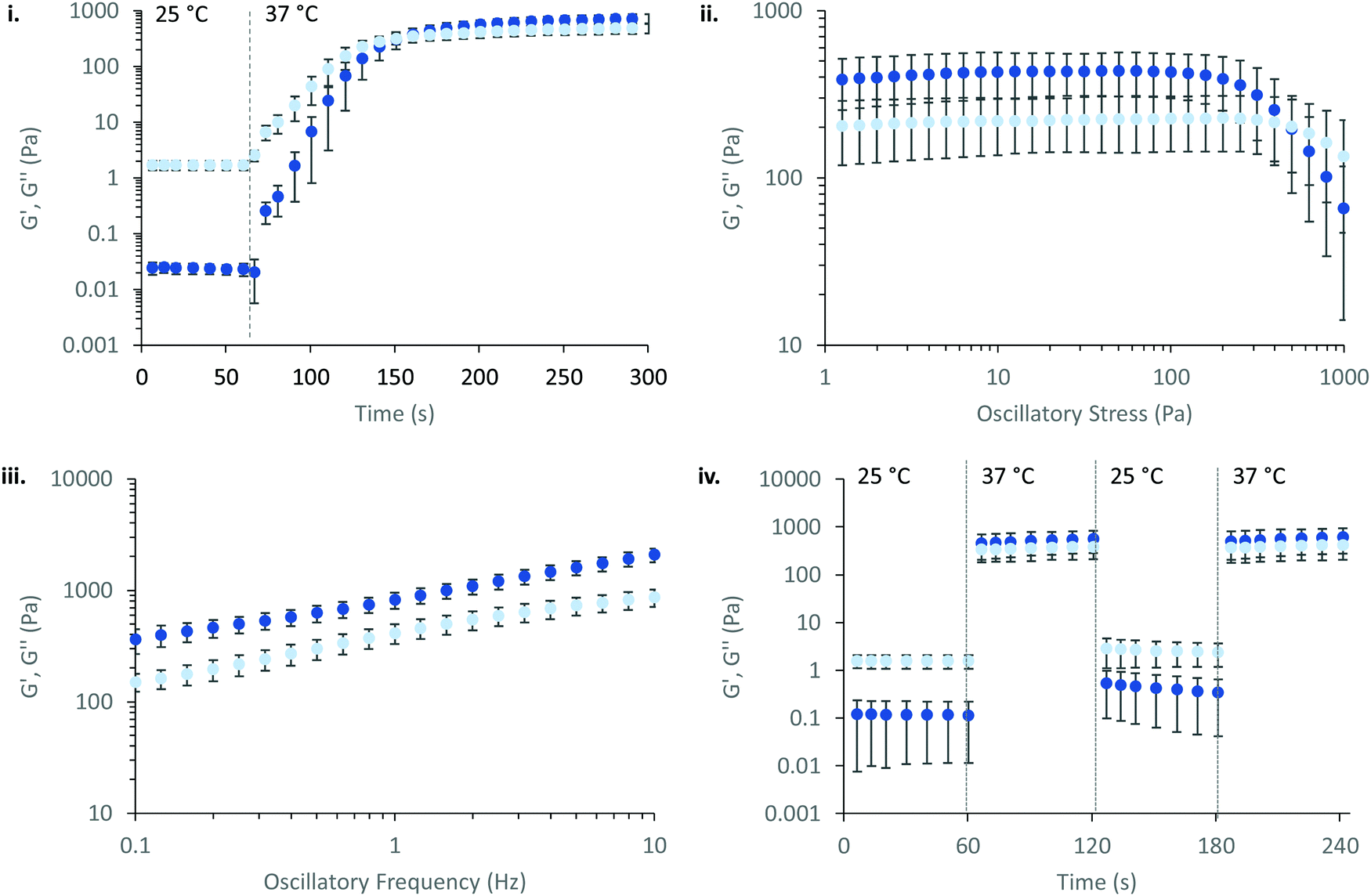
Overall, the rheological behaviour of PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 with temperature are distinct. Where 20–50% (w/v) PNIPAM98–PEG122–PNIPAM98 solutions would increase in viscosity when warmed from room to body temperature, this was observed only in 17.5 and 20% (w/v) poloxamer 407 solutions. The lower concentration dependence of Tgel seen for PNIPAM98–PEG122–PNIPAM98 is attractive where dilution effects have been observed to affect poloxamer 407's gelation in vivo.26 Achieving in situ thickening at high polymer concentrations also allows for viscous gels to be formed with >40% w/v PNIPAM98–PEG122–PNIPAM98, achieving 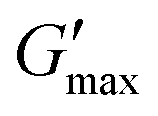 values of 11–13 kPa, whilst retaining a Tthick/Tgel close to body temperature.
values of 11–13 kPa, whilst retaining a Tthick/Tgel close to body temperature.
PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) was further explored as a smart material for drug delivery. Its Tgel of 36 °C is highly attractive for administration onto or into the body at sites where the temperature is 37 °C. In particular, the material may have application in vaginal drug delivery where the local temperature is expected to be 37 °C. Rheology was used to simulate the topical application process and determine the time required for the gel phase to form (Fig. 4(i)). PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) was oscillated at 1 Pa shear stress and a frequency of 1 Hz while temperature was varied. The sample was set constant to room temperature (25 °C) for 60 s before holding at body temperature (37 °C) for 240 s. The transition from 25 to 37 °C lead to immediate thickening of the sample, which formed a gel after 87 ± 5 s and plateaued at a G′ of ca. 700 Pa. This relatively long gelation time may have clinical implications, where a patient may be asked to remain still until gel formation has occurred. The same experimental procedure for poloxamer 407 (20% (w/v)) (Fig. S4, ESI†) gave a time to gel of 27 ± 5 s, with a plateau at ca. 5.5 kPa indicating a more rapid and rigid gel formation for this sample. The PNIPAM98–PEG122–PNIPAM98 gel formed at 37 °C was subjected to an oscillatory stress sweep which demonstrated thinning above ca. 200 Pa with a gel yield observed at 247 ± 72 Pa, which was not significantly different (p > 0.05 by t-test) than poloxamer 407 (20% (w/v)) which exhibited a yield at 256 ± 58 Pa (Fig. S5, ESI†). The yield strength is an indicator of the force required to make the sample undergo viscous flow, and may be taken as the point where G′ becomes lower than G′′ during an oscillatory stress sweep in the gel phase.27 An oscillatory frequency sweep of PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) at 37 °C confirms that the structure is a rheological gel, with G′ > G′′ at all frequencies measured (Fig. 4(iii)). Finally, the PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) was cycled between 25 and 37 °C at 1 Pa and 1 Hz (Fig. 4(iv)), which indicated that the thermoresponsive gelation process was reversible and repeatable.
 | ||
| Fig. 4 (i) Determination of gelation time for PNIPAM98–PEG122–PNIPAM98 (50% (w/v)). (ii) Oscillatory stress sweep of PNIPAM98–PEG122–PNIPAM98 (50% (w/v)), and (iii) oscillatory frequency sweep. (iv) Thermal cycling of PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) to demonstrate reversibility of the gelation process. G′ and G′′ are shown as dark and light blue markers, respectively. Data presented as mean ± standard deviation, n = 3. | ||
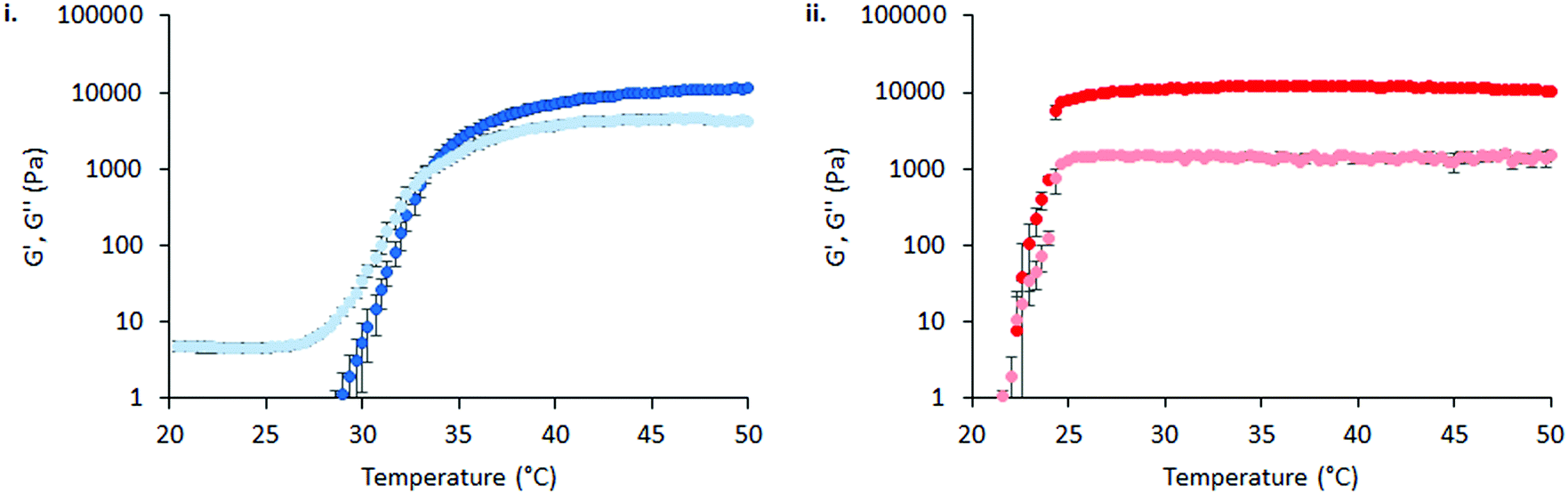
The effect of physiological fluids on the phase transition of PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 was then evaluated by preparing polymer solutions in VFS (Fig. 5).17 The rheological temperature ramp for PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) was altered considerably in VFS relative to deionised water. Tgel shifted to 32.9 ± 0.2 °C in VFS relative to 35.8 ± 0.2 °C in deionised water. This depression of Tgel was also seen in poloxamer 407 which formed a gel at 22.3 ± 0.4 °C in VFS compared to 24.7 ± 0.2 °C in deionised water. The depression of Tgel seen in PNIPAM98–PEG122–PNIPAM98 is attributed to salting-out of the PNIPAM block. Chaotropes have been shown to reduce LCSTs through an increased surface tension between PNIPAM hydrophobic domains (i.e. isopropyl groups and the carbon backbone) and their hydration layer.28Tgel depression in poloxamer 407 is also attributed to salting-out, where salts are know to entropically promote the micellization process.29 VFS did not affect 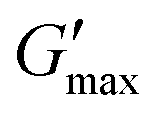 of PNIPAM98–PEG122–PNIPAM98 (50% (w/v)), which was 11.7 ± 1.5 and 11.2 ± 1.8 kPa in VFS and deionised water, respectively (p < 0.05 by t-test). However, the depression in Tgel did result in a greater value of G′ at 37 °C for PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) in VFS compared in deionised water, giving values of 4.8 ± 2.8 kPa and 1.1 ± 0.8 kPa, respectively. G′ of poloxamer 407 (20% (w/v)) at 37 °C was unaffected by the presence of VFS, with both VFS and deionised water leading to values of G′ of ca. 13 kPa at this temperature.
of PNIPAM98–PEG122–PNIPAM98 (50% (w/v)), which was 11.7 ± 1.5 and 11.2 ± 1.8 kPa in VFS and deionised water, respectively (p < 0.05 by t-test). However, the depression in Tgel did result in a greater value of G′ at 37 °C for PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) in VFS compared in deionised water, giving values of 4.8 ± 2.8 kPa and 1.1 ± 0.8 kPa, respectively. G′ of poloxamer 407 (20% (w/v)) at 37 °C was unaffected by the presence of VFS, with both VFS and deionised water leading to values of G′ of ca. 13 kPa at this temperature.
 | ||
| Fig. 5 Temperature ramp rheograms of 50% (w/v) PNIPAM98–PEG122–PNIPAM98 (i) and 20% (w/v) poloxamer 407 (ii) in VFS at a fixed shear stress (1 Pa) and frequency (1 Hz). G′ and G′′ are shown as dark and light markers, respectively. Data presented as mean ± standard deviation, n = 3. | ||
A significant limitation of poloxamer 407 as a thermoresponsive gelator is its rapid dissolution in physiological fluids.6 20% (w/v) solutions of poloxamer 407 dissolved in VFS within 60 min (Fig. 6). PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) had a significantly (p < 0.05) greater resistance to dissolution, requiring 230 min for dissolution to occur. A control of 50% (w/v) poloxamer 407 dissolved after 140 min, which indicated that this difference is not solely explained by concentration. It is hypothesised that where poloxamer 407 gels are composed of non-interacting polymeric micelles, the liberation of micelles into the dissolution medium occurs rapidly. PNIPAM98–PEG122–PNIPAM98 micelles are thought to be bridged by polymeric unimers which reduce the favourability of micelle liberation into the dissolution medium.14,23 These experiments were conducted in a large excess of dissolution media, and the dissolution process in the real, smaller, volumes of physiological fluid present in vivo is likely to occur over a longer period of time.
 | ||
| Fig. 6 Dissolution of PNIPAM98–PEG122–PNIPAM98 50% (w/v) (blue) and 20 (red), 30 (green) and 50% (w/v) (black) poloxamer 407 in VFS. Data presented as mean ± standard deviation (n = 3). | ||
The mucoadhesion of poloxamer 407 is weak,30 which limits its residence time on mucosal membranes. Mucoadhesion is mediated by several different factors, which are not mutually exclusive. Briefly, entanglements may occur between macromolecules in the dosage form and mucin glycoproteins coating the mucosa, which are supported by non-covalent interactions, enhancing adhesion.31 The viscosity of a gel will enhance retention, whilst the movement of moisture from the mucosa to the dosage form will either improve or decrease mucoadhesion depending on levels of hydration.32 Several other theories exist.33 The most common method to determine mucoadhesion is by measuring the force–displacement curve during removal of the dosage form from a membrane, where the peak force is termed the “force of adhesion” and the total area under the curve is the “work of adhesion”.34 These adhesion processes were studied for 50% (w/v) solutions of PNIPAM98–PEG122–PNIPAM98 as well as 20, 30, and 50% (w/v) poloxamer 407. Only 20% (w/v) solutions of poloxamer 407 exhibit Tgel at a relevant temperature (25 °C), but 30 and 50% (w/v) solutions were explored to account for differences in concentration between the two thermoreversible gelators and understand whether adhesion processes are affected by chemical structure or concentration. Firstly, the adhesion of PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 to a poly(propylene) probe was assessed (Fig. 7). In this control experiment, van der Waals forces are believed to be the major contributor to adhesion. The data demonstrates that the adhesion of poloxamer 407 increases with concentration, and that 50% (w/v) solutions of PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 had equivalent (p > 0.05) adhesion to the probe. The mucoadhesion of 20% (w/v) poloxamer 407 and 50% (w/v) PNIPAM98–PEG122–PNIPAM98 is shown in Fig. 7. PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) had greater force and work of adhesion than 20% (w/v) poloxamer 407 (p < 0.01) which is desirable for mucosal drug delivery. A control of 50% (w/v) poloxamer 407 was equivalent to the PNIPAM98–PEG122–PNIPAM98 (50% (w/v)) which demonstrates that this enhanced adhesion is likely to be related to concentration, rather than enhanced specific intermolecular interactions between polymer and mucosa. However, concentrations of poloxamer 407 above 20% (w/v) did not exhibit a Tgel in the range required and thus are not appropriate for in situ gelation with topical administration. Overall, PNIPAM98–PEG122–PNIPAM98 allows the formation of in situ gelators (25 < Tgel < 37 °C) at 50% (w/v), which imparts a greater level of mucoadhesion than seen for 20% (w/v) poloxamer 407. 50% (w/v) poloxamer 407 had equivalent mucoadhesion to 50% (w/v) PNIPAM98–PEG122–PNIPAM98 but it is not a thermoreversible gelator, existing in a gel phase at all temperatures studied.
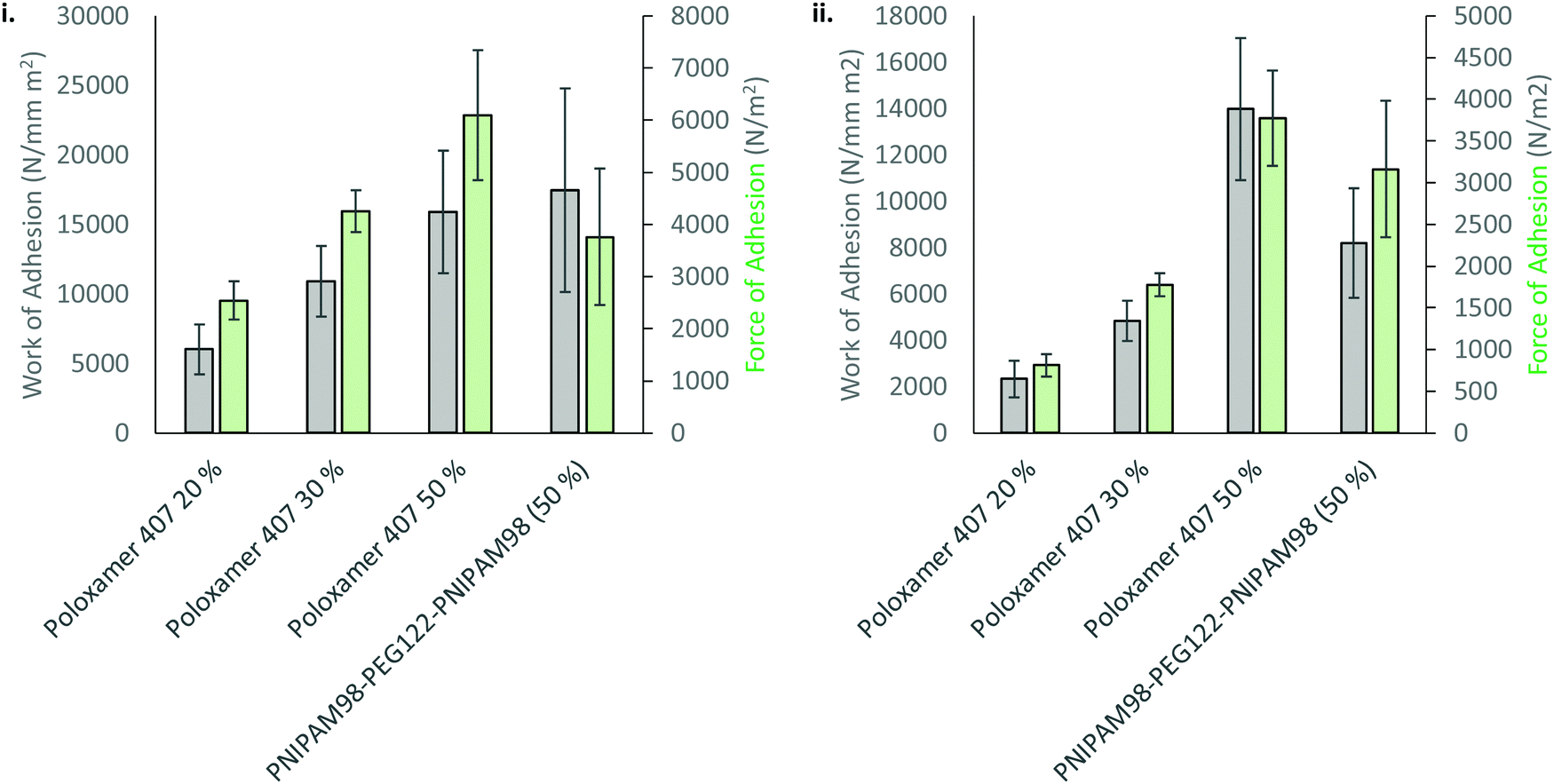 | ||
| Fig. 7 Adhesion (i) and mucoadhesion (ii) of 50% (w/v) PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 onto a poly(propylene) probe or porcine vaginal tissue, respectively. | ||
The thermoresponsive polymers were then evaluated for their ability to solubilise two drugs relevant to intravaginal drug delivery: progesterone and tenofovir disoproxil fumarate. Progesterone is administered intravaginally to treat low fertility, premenstrual syndrome, and puerperal depression, as well as support in vivo fertilisation and reduce the risk of pre-term delivery in pregnancy.35–38 Intravaginal tenofovir disoproxil fumarate has been shown to reduce transmission of HIV in a macaque model.39 In addition to their relevance to vaginal drug delivery, progesterone and tenofovir disoproxil fumarate were selected as they have large differences in their water solubilities. Progesterone was determined to have a water solubility of 9.8 ± 0.1 μg mL−1 at 25 °C, whilst tenofovir disoproxil fumarate's solubility was approximately 1000-fold greater at 9.5 ± 1.0 mg mL−1. The solubility of the two drugs in the presence of the polymers at a range of concentrations between 5 μg mL−1 and 10 mg mL−1 was determined at 25 and 37 °C (Fig. 8). It was found that for both polymers the solubility of progesterone (Fig. 8(i)) was greatly increased above a critical concentration, believed to be the critical micelle concentration (CMC). The CMC could then be extracted from the data, giving values of 0.26 and 1.88 mg mL−1 at 25 °C for PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 respectively, which rose to 0.68 and 1.95 mg mL−1 at 37 °C. The CMC of poloxamer 407 is on the same order of magnitude as that reported previously (ca. 3 mg mL−1) for the pure polymer,40 and the deviation is attributed to the presence of progesterone, as reported for other drugs.41 Tenofovir disoproxil fumarate (Fig. 8(ii)) solubility was not enhanced by increasing concentration of either polymer, even above the CMCs. The data indicates that the formation of micelles occurs for both polymers above a CMC at 25 and 37 °C, at which point the micelles provide a locus for solubilisation for the relatively hydrophobic progesterone, but tenofovir disoproxil fumarate is excluded from the micelle. The solubilities of both drugs are increased at 37 °C, above and below the critical micelle concentration, with a more pronounced effect seen in tenofovir disoproxil fumarate which is attributed to its enhanced water solubility at this temperature.
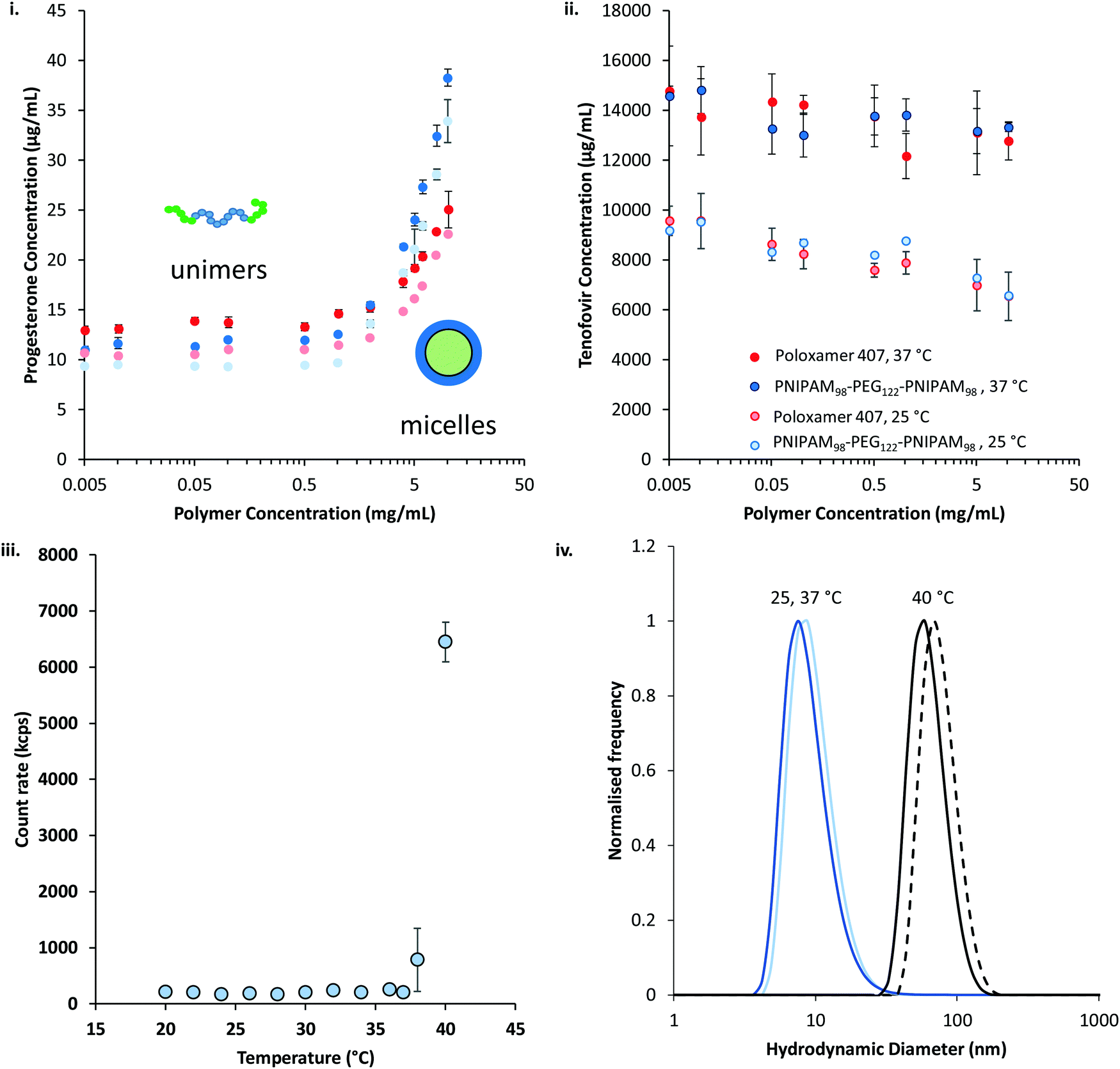 | ||
| Fig. 8 Solubilisation of progesterone (i) and tenofovir disoproxil fumarate (ii) by PNIPAM98–PEG122–PNIPAM98 (blue) and poloxamer 407 (red) at 25 and 37 °C (light and dark colours, respectively). Inset: Schematic representation of the micellization process. (iii) The scattering counts of a 10 mg mL−1 PNIPAM98–PEG122–PNIPAM98 solution saturated with progesterone as a function of temperature. (iv) Hydrodynamic diameter of PNIPAM98–PEG122–PNIPAM98 nanoparticles saturated with progesterone at 25 (light blue), 37 (dark blue), and 40 (black) °C, and PNIPAM98–PEG122–PNIPAM98 without progesterone at 40 °C (black dashed). | ||
Dynamic light scattering revealed a pronounced influence of progesterone on the micellization of PNIPAM98–PEG122–PNIPAM98. 10 mg mL−1 solutions of PNIPAM98–PEG122–PNIPAM98 exhibited no detectable particulates at 25 °C but when warmed to 37 °C transitioned to a 138 nm nanoparticle, which shrunk and remained at a constant hydrodynamic diameter of ca. 90 nm at temperatures at 40 °C and above (Fig. S6, ESI†). In the presence of progesterone, nanoparticle aggregates were detected at 25 °C. Furthermore, at 38 °C and above, a pronounced transition was observed (Fig. 8(iii)). The hydrodynamic diameter of the nanoparticles in the presence of progesterone at 25, 37, and 40 °C were 15 ± 1, 15 ± 1, and 73 ± 0 nm, respectively (Fig. 8(iv)), demonstrating the change in nanostructure associated with the transition at 38 °C. It is hypothesised that the presence of progesterone drives the formation of micellar nanoparticles below the LCST-type transition of PNIPAM, which, when triggered by a rise in temperature, leads to the aggregation or rearrangement of these micelles. The size of these particles is close to those found without progesterone at 40 °C (88 ± 1 nm) (Fig. 8(iv)). Prior study of PNIPAM–PEG–PNIPAM reports PNIPAM (16 kDa)–PEG (6 kDa)–PNIPAM (16 kDa) formed flower-like micelles with a hydrodynamic diameter of 62 nm,13 whereas lower molecular weight PNIPAM (4 kDa)–PEG (6 kDa)–PNIPAM (4 kDa) formed fractal-like structures with hydrodynamic diameters of up to 400 nm.12 Due to the closer molecular weight of the constituent unimers, and similar hydrodynamic diameters, it is believed that PNIPAM98–PEG122–PNIPAM98 forms flower-like micelles in a similar manner to PNIPAM (16 kDa)–PEG (6 kDa)–PNIPAM (16 kDa).
The solubilising power and micelle:water partition coefficients of the polymer/drug mixtures were calculated from the progesterone solubilisation data in Fig. 8(i) and are included in Table 1.42 Briefly, the solubilising power reflects the unit mass increase in progesterone solubilised per unit mass of polymer, whereas the micelle:water partition coefficient reflects the concentration of drug within the micelle relative to that free in solution. PNIPAM98–PEG122–PNIPAM98 had a solubilising power approximately 1.5-fold greater than poloxamer 407 at both temperatures, and a micelle:water partition coefficient around twice as large. These differences were demonstrated to be statistically significant by two-way ANOVA with Bonferroni post hoc testing (p < 0.0001). It is known that the greater the molecular weight of the relatively hydrophobic poly(propylene oxide) domains, the greater the solubilising power of poloxamers.43 The microphase separated NIPAM domains (DP: 196) in PNIPAM98–PEG122–PNIPAM98 have a greater degree of polymerisation (and molecular weight) than the poly(propylene oxide) in poloxamer 407 (DP: 65) which may lead to a greater solubilising power. These considerations are balanced against the free energy of the solubilisation process, which will differ between the two polymers.43
| Polymer | Solubilising power (× 10−3) | Micelle:water partition coefficient | ||
|---|---|---|---|---|
| 25 °C | 37 °C | 25 °C | 37 °C | |
| Poloxamer 407 | 1.53 ± 0.07 | 1.65 ± 0.22 | 1.30 ± 0.06 | 1.21 ± 0.16 |
| PNIPAM98–PEG122–PNIPAM98 | 2.45 ± 0.22 | 2.86 ± 0.09 | 2.37 ± 0.08 | 2.54 ± 0.08 |
Temperature increased the mean value of solubilising power for both polymers, however this increase was only statistically significant (p < 0.05) for PNIPAM98–PEG122–PNIPAM98. As no structural transition was observed between 25 and 37 °C by dynamic light scattering, it is hypothesised that this small increase is the result of entropic effects favouring polymer–progesterone interactions at the elevated temperature. Poloxamer exhibited a structural transition at 26 °C (Fig. S7, ESI†), increasing in hydrodynamic diameter from ca. 9 to 22 nm, however this did not affect the solubilisation of progesterone.
The saturation solubilities of progesterone and tenofovir disoproxil fumarate were then evaluated in the thermoreversible gels at high concentrations (Fig. 9). Strikingly, progesterone solubility is dramatically enhanced at 37 °C, relative to 25 °C, in the polymer solutions where the effect is small in water alone. This has important implications for drug delivery across a membrane, where liberation of drug from a dosage form is dictated by its thermodynamic activity within that base.44 The greater the degree of saturation, the greater the driving force for liberation. If progesterone is loaded into the solutions at room temperature then applied to the body and warmed then the thermodynamic activity is expected to decrease, thus liberation from the dosage form is expected to be slowed at this higher temperature. Despite the differences in solubilising power and concentration of the two polymers, progesterone saturation solubility in both polymer mixtures at 37 °C was equivalent, at 505.0 ± 7.8 and 504.8 ± 30.2 μg mL−1 for poloxamer 407 and PNIPAM98–PEG122–PNIPAM98, respectively, equivalent to a ca. 45-fold increase relative to the saturation solubility in water (11.3 μg mL−1). The molarities of the two solutions are approximately 16.5 and 18.2 mM for poloxamer 407 and PNIPAM98–PEG122–PNIPAM98 respectively, and the greater solubilising power of PNIPAM98–PEG122–PNIPAM98 is expected to lead to a much larger increase in progesterone solubility in this material. However, nanostructures present at the low concentrations studied for solubilising power calculations (<10 mg mL−1) may not be equivalent to those found at 50% (w/v) in the PNIPAM98–PEG122–PNIPAM98. It is known that poloxamer exists as spherical micelles of approximately 10 nm diameter across a wide range of concentrations, however the nanostructure of the PNIPAM98–PEG122–PNIPAM98 materials at 50% (w/v) has never been studied and block copolymers may exist in different phases dependent on concentration.24 It is further suggested that the high concentration of the PNIPAM98–PEG122–PNIPAM98 has driven the micellar transition observed in Fig. 8(iii) at 38 °C to a temperature lower than 37 °C, in line with the depression in Tgel seen at high concentrations by rheometry. The solubilising power of poloxamer 407 (Table 1) would suggest that progesterone solubility would be unaffected by temperature, however behaviour at this high concentration (20% w/v) may differ from that in dilute solution, as has been reported for micelle:water partition coefficients.45
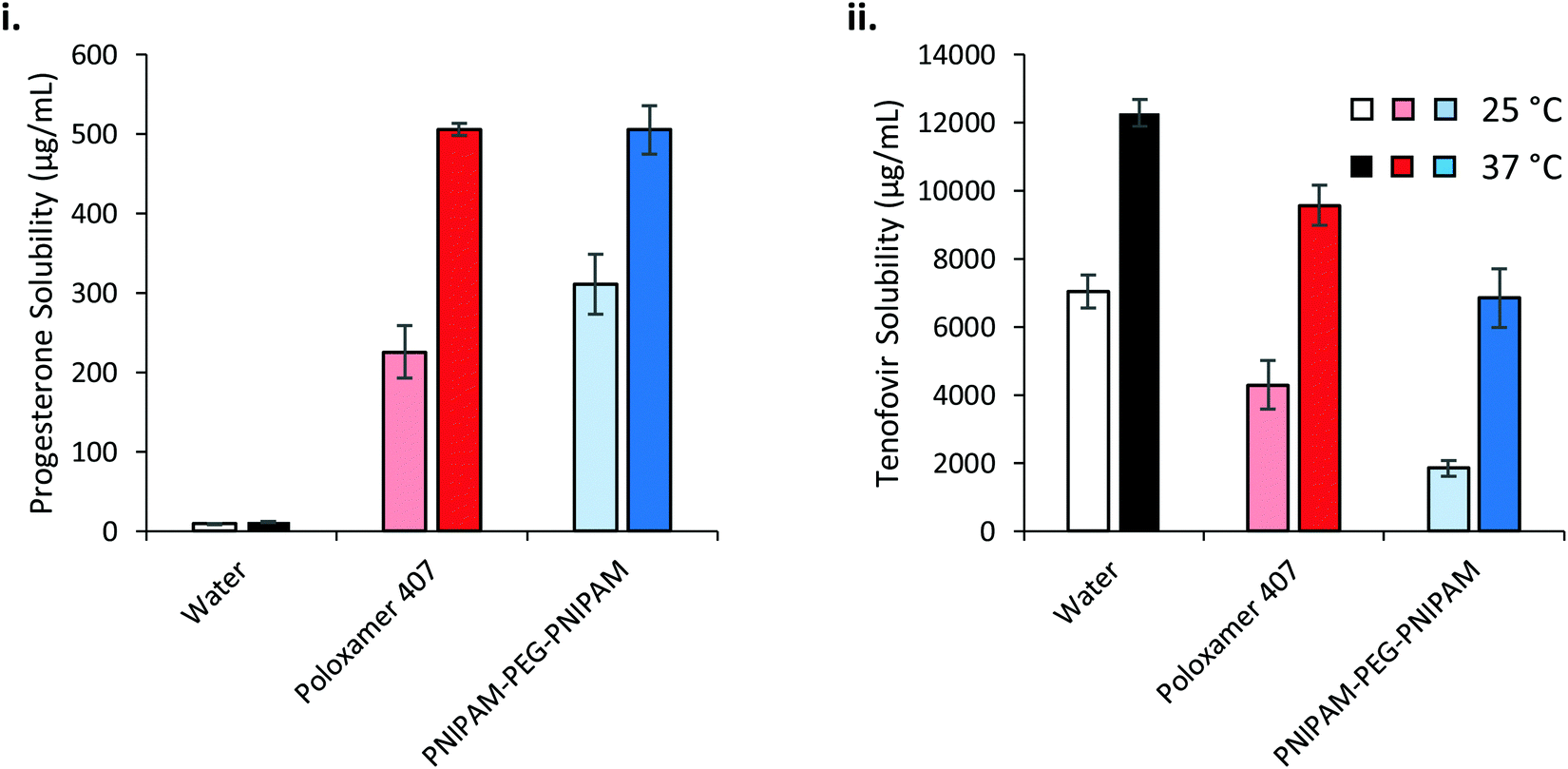 | ||
| Fig. 9 Saturation solubility of progesterone (i) and tenofovir disoproxil fumarate (ii) in 20% (w/v) poloxamer 407 (red) and 50% (w/v) PNIPAM98–PEG122–PNIPAM98 (blue) at 25 and 37 °C (light and dark colours, respectively). | ||
Tenofovir disoproxil fumarate is excluded from the micelles formed by either polymer, and as such the increase in solubility observed in these polymers at elevated temperatures is attributed solely to the improvement in solubility caused by heat alone (Fig. 9(ii)). The reduction in solubility seen in the polymer samples is attributed to the decreased volume fraction of water in these mixtures. Adjusting for the phase volume of water accounts for this depression.
The liberation of drugs from the thermoreversible gelators across cellulose membrane was then explored at loading concentrations below the saturation limit (Fig. 10), maintaining sink conditions throughout the experiments. Control solutions of each drug were included to ensure that permeation across cellulose membrane was not rate-limiting, and each of these experiments give a characteristic curve associated with donor depletion in finite dosing experiments.46 Tenofovir disoproxil fumarate release from the two polymer solutions was equivalent at 25 °C. However, drug release was significantly (p < 0.05) enhanced at 37 °C for poloxamer 407 but remained constant in the case of PNIPAM98–PEG122–PNIPAM98. For example, at 8 h the cumulative release at 25 °C was 58.3 ± 1.5% and 61.0 ± 3.8% for PNIPAM98–PEG122–PNIPAM98 and poloxamer 407, respectively, but 53.8 ± 4.3% and 86.6 ± 5.3% for the same samples at 37 °C. As drug release is occurring from a planar dosage form, the Higuchi model was applied to the data. This simple model is based on a linear fit to the fractional drug release with the square-root of time.47 The model gave R2 > 0.98 in all cases, supporting the principle that the gels act as a matrix controlled release system from which Fickian diffusion occurs (Fig. S8, ESI†).47 Heat will enhance diffusion out of the matrix when release is purely diffusion controlled as the Stokes–Einstein equation predicts that the diffusion coefficient of a molecule scales linearly with temperature, when viscosity is constant.48 The control, a solution of tenofovir disoproxil fumarate exhibits this phenomenon. In the poloxamer 407 samples, which are a gel at both temperatures tested, an enhanced release from the gels is attributed to the increased diffusion coefficient, despite the increased viscosity of the system. For PNIPAM98–PEG122–PNIPAM98, a phase transition occurs between the two temperatures tested. The identical release rate at 37 °C relative to 25 °C is attributed to the formation of this gel phase, which provides a more tortuous path for liberation to occur where tenofovir is known to be excluded from the micelles, counteracting the increased diffusion coefficient.47
 | ||
| Fig. 10 Release of 50 μg mL−1 progesterone and 200 μg mL−1 tenofovir disoproxil fumarate from 50% (w/v) PNIPAM98–PEG122–PNIPAM98 (blue) and 20% (w/v) poloxamer 407 (red). A control formulation of the drugs dissolved in a 1:1 water:ethanol mixture (open circles) demonstrates that liberation from the polymer gel is rate-limiting. Please note that for the tenofovir release experiments, the next time point determined for the gels (24 h) contained 100% drug liberation. | ||
Progesterone release from PNIPAM98–PEG122–PNIPAM98 and poloxamer 407 samples was dramatically different than that exhibited by tenofovir disoproxil fumarate. Progesterone release from PNIPAM98–PEG122–PNIPAM98 at 25 °C followed Higuchi kinetics (Fig. S8, ESI†), and 100% of the drug was released after 32 h. Increasing the temperature to 37 °C retarded the release of progesterone significantly, with 100% drug liberated only after 144 h. This release profile did not fit the Higuchi model, but exhibited a good fit (R2: 0.98) to the Korsmeyer–Peppas power law, albeit with an extreme exponent (n = 2.42) designating non-Fickian super case II transport kinetics.49 This retardation of release from PNIPAM98–PEG122–PNIPAM98 at 37 °C is attributed to the formation of the gel phase, which provides microphase separated domains in which drug solubilisation may occur, and from which liberation becomes disfavoured, prolonging the release until 144 h. Additionally, polymer entanglements and an increased tortuosity within the samples may contribute to this effect. Progesterone release from poloxamer 407 was equivalent at 25 and 37 °C, where the material is in the gel phase at both temperatures. Both cases gave Higuchi diffusion-controlled release in the first 60% of the profile (R2 > 0.98), which is consistent with prior studied of drug delivery across a membrane from poloxamer gels.8 This behaviour is substantially different than the PNIPAM98–PEG122–PNIPAM98 system which exhibited clear temperature-dependence on drug release, and a switch from Higuchi kinetics to a non-Fickian release mechanisms. Higuchi kinetics following Fickian diffusion may be rationalised below the Tgel where release of drug is controlled by factors such as partitioning and the viscosity of the medium. Super case II kinetics have been reported for chemically cross-linked PNIPAM hydrogels,50 where Fickian diffusion is believed to be combined with polymer relaxation and possible swelling effects. This swelling/relaxation behaviour may combine with nanostructural rearrangement to give rise to the lengthy lag exhibited in the progesterone release studies from PNIPAM98–PEG122–PNIPAM98 at 37 °C, though this was not explicitly studied. It has been demonstrated by DLS that progesterone drives the formation of aggregates even at low temperatures, and the slow depletion of progesterone from the gel may also lead to structural rearrangement. Pure PNIPAM hydrogels also exhibit the long lag time seen for PNIPAM98–PEG122–PNIPAM98 at 37 °C.50 The release of progesterone from PNIPAM98–PEG122–PNIPAM98 was significantly slower than poloxamer 407 at 37 °C. For example, at 72 h, 75.5 ± 1.6% of the entrapped progesterone was delivered from the 20% (w/v) poloxamer 407 sample, where only 25.3 ± 6.7% of the drug had been liberated from PNIPAM98–PEG122–PNIPAM98 (p < 0.0001 by t-test). This gives the PNIPAM98–PEG122–PNIPAM98 an advantage over poloxamer 407 when the sustained release of hydrophobic drugs is required. The total 10 μg progesterone dose was delivered from the 200 μL PNIPAM98–PEG122–PNIPAM98 formulation over 6 days, requiring a 5 mL dose to achieve the approximately 250 μg delivered from the standard Crinone gel over the same period.51
The impact of progesterone and tenofovir disoproxil fumarate (50 μg mL−1) on the rheology of the thermoreversible gelators was assessed by temperature ramp (Fig. S9, ESI†). In the presence of progesterone, the gelation temperature of poloxamer 407 (20%) remained at 25 °C. Whereas the gelation temperature of PNIPAM98–PEG122–PNIPAM98 (50%) was slightly reduced from 35.8 ± 0.4 to 34.2 °C. It is known drug–polymer interactions may decrease the LCST of PNIPAM, which may explain this slight reduction in Tgel.52 The inclusion of progesterone resulted in a decrease in gel strength for poloxamer 407 (20%) from 12.7 ± 0.5 to 10.2 kPa while 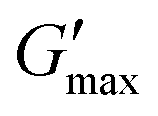 of PNIPAM98–PEG122–PNIPAM98 did not vary, having values of 11.2 ± 1.8 and 10.7 kPa in undoped and progesterone-doped solutions, respectively. Tenofovir disoproxil fumarate did not alter the gelation temperatures of poloxamer 407 (20%) and PNIPAM98–PEG122–PNIPAM98 (50%), having values of 24 and 35.4 °C, respectively. Tenofovir disoproxil fumarate reduced
of PNIPAM98–PEG122–PNIPAM98 did not vary, having values of 11.2 ± 1.8 and 10.7 kPa in undoped and progesterone-doped solutions, respectively. Tenofovir disoproxil fumarate did not alter the gelation temperatures of poloxamer 407 (20%) and PNIPAM98–PEG122–PNIPAM98 (50%), having values of 24 and 35.4 °C, respectively. Tenofovir disoproxil fumarate reduced 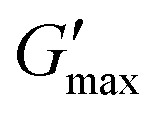 to 9.7 kPa in the poloxamer 407 samples, but the value measured for PNIPAM98–PEG122–PNIPAM98 (10.2 kPa) was within the error of the undoped samples. Overall, the PNIPAM98–PEG122–PNIPAM98 samples retained their ability to act as thermoreversible gelators after the addition of both model drugs.
to 9.7 kPa in the poloxamer 407 samples, but the value measured for PNIPAM98–PEG122–PNIPAM98 (10.2 kPa) was within the error of the undoped samples. Overall, the PNIPAM98–PEG122–PNIPAM98 samples retained their ability to act as thermoreversible gelators after the addition of both model drugs.
The stability of P407 and PNIPAM98–PEG122–PNIPAM98 was assessed in aqueous solution at 4, 25, or 40 °C, reflecting refrigerated storage, storage at room temperature, and an accelerated storage condition, respectively (Fig. 11). Accelerated storage at 40 °C aimed to predict longer-term storage at room temperature. GPC analysis demonstrated that both polymers exhibit small reductions in molecular weight over 12 weeks, with losses accelerated in the PNIPAM98–PEG122–PNIPAM98 sample at elevated temperatures. However, at 4 and 25 °C the reduction in number-average molecular weight of PNIPAM98–PEG122–PNIPAM98 was not statistically significant (p < 0.05). GPC traces (Fig. 11(i)) of poloxamer at weeks 0 and 12 are near-identical, with the accelerated storage condition (red) exhibiting a small increase in a shoulder at low molecular weight. PNIPAM98–PEG122–PNIPAM98 traces exhibit a clear shift to a lower molecular weight under accelerated storage conditions (Fig. 11(ii)). Under all conditions the trace for PNIPAM98–PEG122–PNIPAM98 traces remained monomodal with no shoulder. Thus, hydrolysis was not observed at the ester linkages present between individual polymer blocks, but amide hydrolysis is possible. Poloxamer 407 does not possess hydrolytically unstable ester or amide linkages, and its molecular weight remained constant throughout the study. This is the first report on the long-term stability of a PNIPAM-PEG copolymer in water. Future studies on the stability of the materials should expand this experiment in-line with ICH standards.53 Additionally, it is not known whether the degradation exhibited in the accelerated storage condition will be seen at 25 °C, where typically small extrapolations are made from this data only when the degradation routes have been established.53
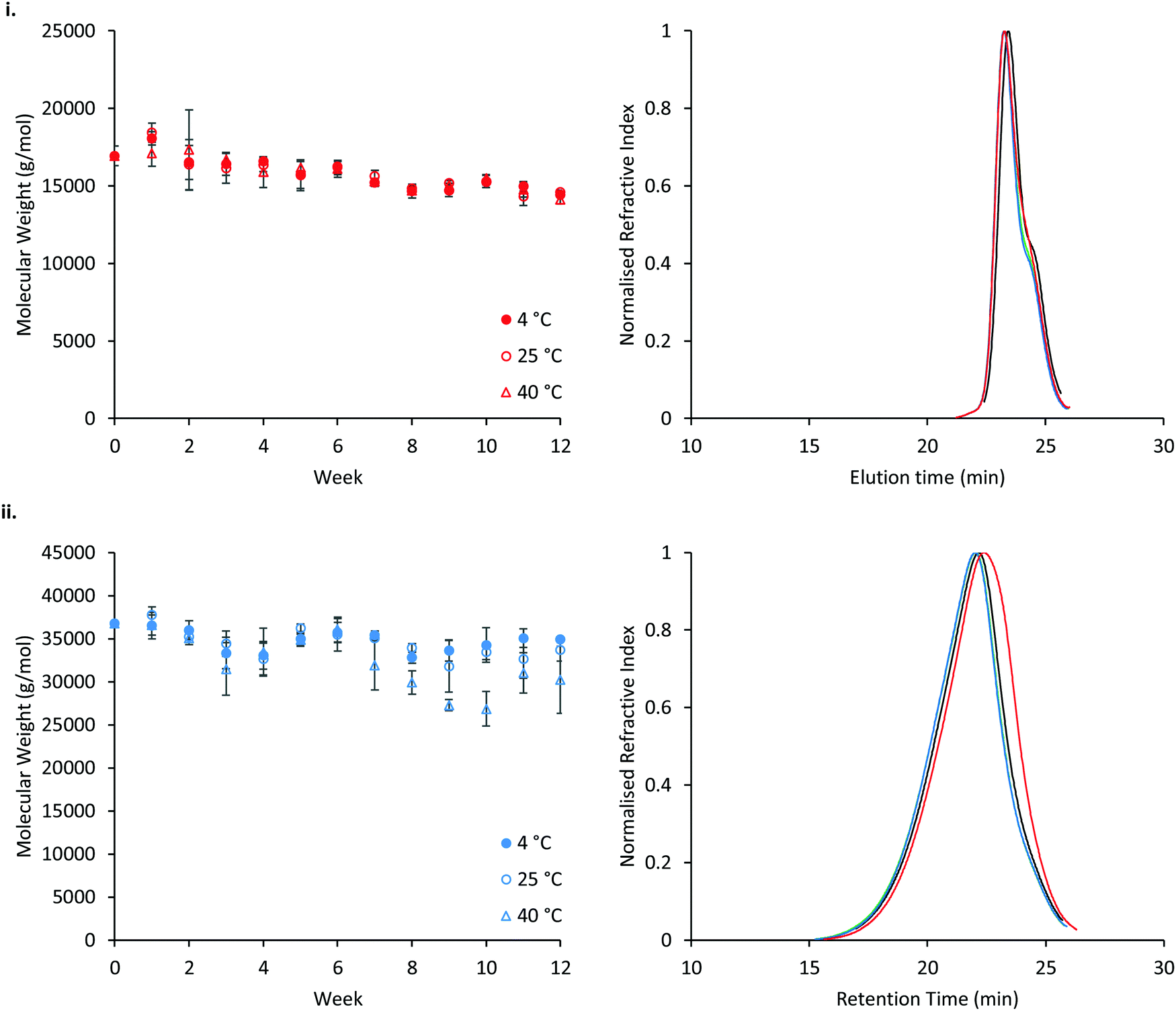 | ||
| Fig. 11 Stability of poloxamer 407 (i) and PNIPAM98–PEG122–PNIPAM98 (ii) at 4, 25 and 40 °C. Variation in the number-average molecular weight is shown on the left. GPC traces are shown on the right, with week 0 shown in black, and week 12 at 4, 25 and 40 °C shown in green, blue and red, respectively. | ||
Conclusions
This study investigates the use of thermoreversible PNIPAM98–PEG122–PNIPAM98 gelators as smart materials for topical administration for the first time, with a critical comparison to poloxamer 407. The low dependence of Tgel on concentration allows PNIPAM98–PEG122–PNIPAM98 to exhibit in situ gelation at concentrations up to 50% (w/v), where Tgel may be finely tuned to temperatures just below 37 °C. Thickening occurred only at temperatures above 28 °C, ensuring that increases in viscosity do not occur at room temperature. This gives the system advantages over poloxamer 407 where values of Tgel are highly concentration dependant. Additionally, of the poloxamer 407 concentrations examined, Tgel typically occurred near or below room temperature (25 °C), making the materials unattractive in warmer climates (e.g. WHO climatic zones II (25 °C), III (30 °C) and IV (30 °C)).25 PNIPAM98–PEG122–PNIPAM98 gels exhibit enhanced mucoadhesion and reduced dissolution compared to poloxamer 407, giving PNIPAM98–PEG122–PNIPAM98 significant advantages for mucosal drug delivery, where these two attributes are linked to retention at the site of administration. PNIPAM98–PEG122–PNIPAM98 was also found to be stable in solution at room temperature over 12 weeks.The solubilisation and release of two drugs relevant to vaginal drug delivery was investigated. Progesterone was selected as a hydrophobic drug, and tenofovir disoproxil fumarate was used as a relatively hydrophilic species. PNIPAM98–PEG122–PNIPAM98 was able to greatly enhance the solubility of progesterone due to inclusion of the drug into polymer micelles, with a greater solubilising power than poloxamer 407, whereas tenofovir disoproxil fumarate was excluded from the micelles in both cases. The release of these two drugs was then evaluated using a Franz diffusion cell system at 25 and 37 °C. Release of progesterone from the poloxamer gels was identical at both temperatures, whereas tenofovir disoproxil was liberated more rapidly at the elevated temperature. PNIPAM98–PEG122–PNIPAM98, however, gave an unusual temperature-responsive retardation of release of both drugs at elevated temperatures, controlling progesterone release over 6 days. This effect may be a means of controlling delivery of other xenobiotics or retaining drug on body surfaces for local effect. Overall, PNIPAM98–PEG122–PNIPAM98 exhibits unique properties as a thermoreversible gelator for drug delivery with advantages over poloxamer 407 and could act as an advanced material for future topical therapies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Centre for Research in Topical Drug Delivery and Toxicology at the University of Hertfordshire funded the studentship of Peter Haddow. The Royal Society are thanked for partial funding of the research (RG160139).References
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS
.
- K. Al Khateb, E. K. Ozhmukhametova, M. N. Mussin, S. K. Seilkhanov, T. K. Rakhypbekov, W. M. Lau and V. V. Khutoryanskiy, Int. J. Pharm., 2016, 502, 70–79 CrossRef CAS PubMed
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC
- E. Giuliano, D. Paolino, M. Fresta and D. Cosco, Pharmaceutics, 2018, 10, E159 CrossRef PubMed
- M. T. Cook and M. B. Brown, J. Controlled Release, 2018, 270, 145–157 CrossRef CAS PubMed
- M. A. Abou-Shamat, J. Calvo-Castro, J. L. Stair and M. T. Cook, Macromol. Chem. Phys., 2019, 1900173 CrossRef
- C. Wu, T. Liu, B. Chu, D. K. Schneider and V. Graziano, Macromolecules, 1997, 30, 4574–4583 CrossRef CAS
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed
- A. Fakhari, M. Corcoran and A. Schwarz, Heliyon, 2017, 3, e00390 CrossRef PubMed
- M. Teodorescu, I. Negru, P. O. Stanescu, C. Drăghici, A. Lungu and A. Sârbu, React. Funct. Polym., 2010, 70, 790–797 CrossRef CAS
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS
- S. K. Filippov, A. Bogomolova, L. Kaberov, N. Velychkivska, L. Starovoytova, Z. Cernochova, S. E. Rogers, W. M. Lau, V. V. Khutoryanskiy and M. T. Cook, Langmuir, 2016, 32, 5314–5323 CrossRef CAS PubMed
- A. J. De Graaf, K. W. M. Boere, J. Kemmink, R. G. Fokkink, C. F. Van Nostrum, D. T. S. Rijkers, J. Van Der Gucht, H. Wienk, M. Baldus, E. Mastrobattista, T. Vermonden and W. E. Hennink, Langmuir, 2011, 27, 9843–9848 CrossRef CAS PubMed
- H. H. Lin and Y. L. Cheng, Macromolecules, 2001, 34, 3710–3715 CrossRef CAS
- A. J. De Graaf, I. I. Azevedo Próspero Dos Santos, E. H. E. Pieters, D. T. S. Rijkers, C. F. Van Nostrum, T. Vermonden, R. J. Kok, W. E. Hennink and E. Mastrobattista, J. Controlled Release, 2012, 162, 582–590 CrossRef CAS PubMed
- D. Limón, K. Talló Domínguez, M. L. Garduño-Ramírez, B. Andrade, A. C. Calpena and L. Pérez-García, Colloids Surf., B, 2019, 181, 657–670 CrossRef PubMed
- D. H. Owen and D. F. Katz, Contraception, 1999, 59, 91–95 CrossRef CAS PubMed
- M. Garcia, M. P. Beecham, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Eur. Polym. J., 2015, 66, 444–451 CrossRef CAS
- J. Bassi da Silva, S. B. de S. Ferreira, A. V. Reis, M. T. Cook and M. L. Bruschi, Polymers, 2018, 10, 1–19 CrossRef PubMed
- M. Malmsten and B. Lindman, Macromolecules, 1992, 25, 5440–5445 CrossRef CAS
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef
- S. E. Kirkland, R. M. Hensarling, S. D. McConaughy, Y. Guo, W. L. Jarrett and C. L. McCormick, Biomacromolecules, 2008, 9, 481–486 CrossRef CAS PubMed
- A. N. Semenov, J. F. Joanny and A. R. Khokhlov, Macromolecules, 1995, 28, 1066–1075 CrossRef CAS
- R. K. Prud’homme, G. Wu and D. K. Schneider, Langmuir, 1996, 12, 4651–4659 CrossRef
- W. Grimm, Drug Dev. Ind. Pharm., 1993, 19, 2795–2830 CrossRef CAS
- K. Edsman, J. Carlfors and R. Petersson, Eur. J. Pharm. Sci., 1998, 6, 105–112 CrossRef CAS
- M. Castro, D. W. Giles, C. W. Macosko and T. Moaddel, J. Rheol., 2010, 54, 81–94 CrossRef CAS
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed
- D. R. Perinelli, M. Cespi, S. Pucciarelli, L. Casettari, G. F. Palmieri and G. Bonacucina, Colloids Surf., A, 2013, 436, 123–129 CrossRef CAS
- G. Niu, F. Du, L. Song, H. Zhang, J. Yang, H. Cao, Y. Zheng, Z. Yang, G. Wang, H. Yang and S. Zhu, J. Controlled Release, 2009, 138, 49–56 CrossRef CAS PubMed
- N. A. Peppas and Y. Huang, Adv. Drug Delivery Rev., 2004, 56, 1675–1687 CrossRef CAS PubMed
- M. T. Cook and V. V. Khutoryanskiy, Int. J. Pharm., 2015, 495, 991–998 CrossRef CAS PubMed
- J. D. Smart, Adv. Drug Delivery Rev., 2005, 57, 1556–1568 CrossRef CAS PubMed
- V. V. Khutoryanskiy, Macromol. Biosci., 2011, 11, 748–764 CrossRef CAS PubMed
- SPC Crinone 8% Progesterone Vaginal Gel, https://www.medicines.org.uk/emc/product/1283/smpc, accessed 31 October 2019.
- SPC Cyclogest 200 mg, https://www.medicines.org.uk/emc/product/5568/smpc, accessed 31 October 2019.
- SPC Lutigest 100 mg vaginal tablets, https://www.medicines.org.uk/emc/product/3635/smpc, accessed 31 October 2019.
- SPC Utrogestan Vaginal 200 mg Capsules, https://www.medicines.org.uk/emc/product/3244/smpc, accessed 31 October 2019.
- J. M. Smith, R. Rastogi, R. S. Teller, P. Srinivasan, P. M. M. Mesquita, U. Nagaraja, J. M. McNicholl, R. M. Hendry, C. T. Dinh, A. Martin, B. C. Herold and P. F. Kiser, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16145–16150 CrossRef CAS
- P. Alexandridis and T. Alan Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS
- A. M. Bodratti and P. Alexandridis, J. Funct. Biomater., 2018, 9, 11 CrossRef PubMed
- A. R. Tehrani-Bagha and K. Holmberg, Materials, 2013, 6, 580–608 CrossRef CAS PubMed
- A. M. Bodratti and P. Alexandridis, J. Funct. Biomater., 2018, 9, E11 CrossRef PubMed
- F. P. Schwarb, G. Imanidis, E. W. Smith, J. M. Haigh and C. Surber, Pharm. Res., 1999, 16, 909–915 CrossRef CAS PubMed
- P. Singla, S. Chabba and R. K. Mahajan, Colloids Surf., A, 2016, 504, 479–488 CrossRef CAS
- D. Selzer, M. M. A. Abdel-Mottaleb, T. Hahn, U. F. Schaefer and D. Neumann, Adv. Drug Delivery Rev., 2013, 65, 278–294 CrossRef CAS
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm. – Drug Res., 2010, 67, 217–223 CAS
- F. Akomeah, T. Nazir, G. P. Martin and M. B. Brown, Eur. J. Pharm. Sci., 2004, 21, 337–345 CrossRef CAS PubMed
-
M. L. Bruschi, Strategies to Modify the Drug Release from Pharmaceutical Systems, 2015 Search PubMed
- Y. Danyuo, C. J. Ani, A. A. Salifu, J. D. Obayemi, S. Dozie-Nwachukwu, V. O. Obanawu, U. M. Akpan, O. S. Odusanya, M. Abade-Abugre, F. McBagonluri and W. O. Soboyejo, Sci. Rep., 2019, 9, 1–14 CrossRef CAS PubMed
- M. Campana-Seoane, A. Peleteiro, R. Laguna and F. J. Otero-Espinar, Int. J. Pharm., 2014, 477, 495–505 CrossRef CAS PubMed
- D. C. Coughlan and O. I. Corrigan, Int. J. Pharm., 2006, 313, 163–174 CrossRef CAS PubMed
- ICH, Q1A(R2) Stability Testing of New Drug Substances and Products, 2003.
Footnote |
| † Electronic supplementary information (ESI) available: Methods: HPLC methods and validation; calculation of critical micelle concentrations, solubilising powers, and micelle:water partition coefficients. Results: calibration curves, DOSY NMR, additional rheograms, DLS data for poloxamer 407, and mathematical fits to drug release profiles. See DOI: 10.1039/d0ma00080a |
| This journal is © The Royal Society of Chemistry 2020 |