
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-dispersible few-layer graphene flakes for selective and rapid ion mercury (Hg2+)-rejecting membranes†
Leyla
Najafi
ab,
Reinier
Oropesa-Nuñez
 ab,
Beatriz
Martín-García
a,
Filippo
Drago
c,
Mirko
Prato
d,
Vittorio
Pellegrini
ab,
Francesco
Bonaccorso
ab and
Sebastiano
Bellani
*ab
ab,
Beatriz
Martín-García
a,
Filippo
Drago
c,
Mirko
Prato
d,
Vittorio
Pellegrini
ab,
Francesco
Bonaccorso
ab and
Sebastiano
Bellani
*ab
aGraphene Labs, Istituto Italiano di Tecnologia, via Morego 30, 16163, Genova, Italy
bBeDimensional Spa, Via Albisola 121, 16163 Genova, Italy. E-mail: s.bellani@bedimensional.it
cNanoChemistry, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy
dMaterials Characterization Facility, Istituto Italiano di Tecnologia, via Morego 30, 16163 Genova, Italy
First published on 29th April 2020
Abstract
Mercury (Hg) is a global highly toxic pollutant released by both anthropogenic and natural sources. Hg decontamination is of the utmost importance for human and ecosystem protection. Here, we propose a novel graphene-based membrane capable of performing rapid and highly selective Hg2+-rejection from water. Functionalized graphene flakes are produced by a non-oxidative, room-temperature and post processing-free “green” method to simultaneously exfoliate graphite into single-/few-layer graphene (SLG/FLG) flakes in water and functionalize them with cationic rhodamine 6G (R6G) via a physisorption process (aromatic ring π–π stacking). The rhodamine 6G-functionalized graphene (R6G-FG) membrane shows a low-density (<0.5 g cm−3) packed laminar structure, where R6G molecules act as spacers between the SLG/FLG flakes. The presence of hydrophilic micro/nanodomains in this low-density structure results in a water permeation rate as high as 789.6 L m−2 h−1 bar−1 (for an 80 μm-thick membrane, R6G-FG mass loading of 3.58 g m−2). Meanwhile, the R6G-FG complexes perform as ion-selective nano-traps for Hg2+, showing almost complete rejection (>99%) for a filtered solution volume normalized to the R6G-FG mass superior to 3 L g−1. The selective rejection capability of the R6G-FG membrane is ruled by competitive adsorption of metal ions and positively charged R6G molecules with different affinity onto the negatively charged graphene surface. Lastly, a washing treatment in alkaline conditions is also proposed for membrane regeneration and reuse. The rationalization of the working mechanism of the R6G-FG membrane is promising for eliminating the “permeability–selectivity trade-offs” often tackled by laminar two-dimensional material membranes.
Introduction
The lack of clean water and sanitation is a pervasive problem afflicting the world today.1,2 This is expected to get worse in the coming years,3 unless efficient water purification methods are developed while minimizing the use of hazardous/toxic chemicals and impact on the environment.4–8 In this scenario, two-dimensional (2D) materials are gaining massive appeal for the realization of novel membranes for filtration,9,10 water purification11,12 and desalination technologies.11–18 As key-cases, graphene based-membranes can provide cost-effective solutions for precise and ultrafast ionic and molecular sieving in aqueous solution.19–24 Recently, other 2D materials, such as transition metal dichalcogenides (TMDs),25–27 boron nitride (BN),28 MXenes29 and layered double hydroxides (LDHs),30,31 have been also reported for molecular and ionic separation membranes.32,33 The effectiveness of 2D material-based membranes originates from their unique laminar structure consisting of nanochannels with tunable sizes32–34 and surface chemistry.35,36 These features can simultaneously provide selective solute rejection34,37,38 and high solvent flux25,39,40 (i.e., water permeation rate under external pressure >500 L m−2 h−1 bar−1 for μm-thick membranes).41,42 The rejection selectivity results from a combination of size-exclusion effects depending on the interlayer spacing of the 2D material flakes,32–34 electrostatic interactions between the charged solute and the charged surface state of the flakes,21,35,36 and adsorption effects (e.g., ion–π interactions43,44 and metal coordination to the flakes’ surface45–47). For the case of water as a solvent, the unusual high flow rate (up to 20 m s−1) has been attributed to both capillary driven force and low-friction flow confined within 2D channels enclosed by hydrophobic, pristine (non-oxidized) regions of graphene.20,48 In contrast, H-bonding interactions between water molecules and O-containing functional groups slow down the water flow between the hydrophilic oxidized graphene flakes.49 Moreover, the interaction between hydrophilic regions and water causes the swelling of the soaked membranes,50,51 which progressively decreases their solute rejection capability.10,52Although the morphological and physicochemical properties of 2D materials hold promise for the development of next-generation ultrathin, high-flux, and energy-efficient membranes for precise ionic and molecular filtration,53,54 a major challenge that practically hinders their implementation is the production of 2D materials at an industrial scale together with controlling their physical/chemical properties at the nanoscale.13,55,56 In this context, liquid phase exfoliation (LPE) methods57–60 have emerged as scalable approaches to exfoliate layered bulk crystals into single-/few-layer flakes in liquid solvents by exploiting cavitation61–65 and shear forces66–71 to break the van der Waals bonds between the adjacent planes of the layered structures. Then, the exfoliated flakes can be deposited or printed on different substrates using well-known solution-processing techniques.72–75 Among LPE techniques, ultrasonication-assisted exfoliation is the most used lab-scale approach because of its easiness.57,59,61 Its optimization also provides guidelines for designing advanced LPE processes with industrial material production rates (up to the order of 1 kg per day, independently of the materials).71,76–78 Subsequently, the produced nanomaterials have to be exploited in the form of interlocked layered structures to effectively act as robust ion and/or molecule filters.9–14 At this stage, the inclusion of different-sized functional groups36,79 can act as both nanometric spacers modulating the nanochannels’ size19,21,80 and active sites for chemical adsorption of solutes,25,81,82 resulting in extraordinary control of the permeation selectivity.83–86 Actually, the possibility of functionalizing 2D materials,87 which intrinsically provide a platform of nanomaterials covering a unique variety of physical/chemical properties,88–91 can create added-value for designing laminar structures with “on-demand” solute-selective removal and solvent permeability.21,53,54 The resulting membrane can be effectively integrated in multifunctional and multi-stage filtration apparatus based on sub-cascade membranes and/or treatment.92–95 This also turns out to minimize fouling invasion,94–97 which reduces the solvent permeability and solute affinity in the end-located nano-filtration blocks.94,95,98–100
Herein, we show a novel membrane based on graphene functionalized with cationic rhodamine 6G – R6G – (hereafter named R6G-FG), a prototypical cost-effective and non-hazardous organic molecule,101,102 for selective Hg2+-rejection from water. It is noteworthy that Hg is one of the heavy metals of most concern, since its non-biodegradability and bio-accumulative pollution are the cause of birth defects, brain damage and diseases in humans and wildlife.103 Mercury is released into the environment by anthropogenic sources in the form of several industry products/byproducts and processes104–107 including gold mining by amalgamation,108 chlor-alkali industry wastewater,109,110 cement and mineral production,111 combustion in coal-fired power plants,112–114 port activities,115 petroleum refineries,116,117 batteries118 and fluorescent lamps.119 In addition, volcanic eruptions,120 deposits of cinnabar121,122 and trace amounts in coal123,124 are relevant natural sources of Hg emission.125,126 In order to face Hg pollution, global agreements have been reached for implementing Hg treaties.127–129 This is spurring the search for novel efficient and rapid methods to remove and recover Hg from water,130–133 where the volatile elemental Hg (Hg0) penetrates via wet precipitation once is oxidized in Hg(II) form.134,135 The rational choice of R6G-based functionalization of graphene for Hg2+ removal is based on the previous use of R6G molecules pre-adsorbed on conductive substrates as fluorescence/surface-enhanced Raman scattering (SERS) chemo-sensors for the selective detection of Hg2+.136–141 R6G-FG was produced by a non-oxidative, room-temperature and post processing-free “green” method to simultaneously exfoliate graphite up to single-/few-layer flakes in water (mild alkaline conditions, pH 8) and functionalize graphene with R6G. This method is effective to obtain graphene flakes with a large contribution of monolayers/bilayers and lateral size comparable to that obtained by conventional LPE methods, e.g., the prototypical ultrasonication-aided exfoliation in N-methyl-2-pyrrolidone (NMP).57,59,61,142,143 Subsequently, selective and rapid Hg2+-rejecting membranes, made of R6G-incorporated graphene interlocked layered structures (R6G-FG membranes), were obtained through facile low-pressure (1 bar) deposition (i.e., vacuum filtration) of the as-produced R6G-FG dispersions. The R6G molecules act as spacers between the graphene flakes and determine the presence of hydrophilic micro/nanodomains, enabling efficient water permeation (up to 789.6 L m−2 h−1 bar−1 for an 80 μm-thick membrane). Meanwhile, R6G/graphene complexes perform as ion-selective nano-traps for Hg2+ (percentage Hg2+ rejection higher than 99%). The ion selective permeation is explained by the Hg-philicity of graphene flakes, which results in adsorption of Hg2+ replacing the R6G molecules. In contrast, the metallophilicity of the graphene flakes for the other investigated metal ions is insufficient to compete with R6G molecules, still saturating the cation-sieving sites of graphene flakes. Lastly, the recyclability of the R6G-FG membranes, i.e., the recovery of their Hg2+ adsorption capability, was attained by washing the membranes in tetrabutylammonium hydroxide 30-hydrate (Bu4NOH·30H2O), representing a novel pH-controlled membrane regeneration method.
Experimental
Production of materials
Rhodamine 6G-functionalized graphene samples were produced in the form of an aqueous dispersion through ultrasonication-assisted LPE of graphite in water, in the presence of R6G molecules and mildly alkaline conditions. Experimentally, 1 g of graphite (+100 mesh, ≥75% min, Sigma Aldrich) and 1.2 mg of R6G powder (99% dye content, Sigma Aldrich) were dispersed in 100 mL of deionized water. The pH of the sample was adjusted to ∼8.0 by adding drops of 1 M KOH solution (Sigma Aldrich). The sample was ultrasonicated in a bath sonicator (Branson 5800 cleaner, Branson Ultrasonics) for 6 h. Afterward, the sample was centrifuged at 500g for 30 min at 15 °C to exploit sedimentation-based separation.142–145 Lastly, ∼80% of the supernatant was collected by pipetting, obtaining the R6G-FG dispersion in water.Characterization of materials
Zeta potential measurements of the as produced R6G-FG in the form of a water dispersion were carried out with a Zetasizer Nano® series (Malvern Instruments) using a disposable capillary cell (DTS 1060) at room temperature for the graphene and R6G-FG water dispersion. Concretely, the electrophoretic mobility was measured and converted into the zeta potential using the Smoluchowski approximation.146 An equilibration time of 3 min was set before each measurement. The zeta potential value was averaged over the values obtained by 3 replicate measurements.Transmission electron microscopy (TEM) images were taken with a JEM 1011 (JEOL) transmission electron microscope operating at 100 kV. Morphological and statistical analysis were carried out using ImageJ software (NIH) and OriginPro 9.1 software (OriginLab), respectively. The lateral dimension of a flake was estimated as its maximum lateral dimension. The samples for the TEM measurements were prepared by drop-casting the R6G-FG dispersions onto carbon-coated Cu grids, subsequently rinsed with deionized water and dried under a vacuum overnight.
Atomic force microscopy (AFM) images were taken using a Nanowizard III (JPK Instruments, Germany) mounted on an Axio Observer D1 (Carl Zeiss, Germany) inverted optical microscope. The AFM measurements were carried out using PPP-NCHR cantilevers (Nanosensors, USA) with a nominal tip diameter of 10 nm. A drive frequency of ∼295 kHz is used. Intermittent contact mode AFM images (512 × 512 data points) of 2.5 × 2.5 μm2 were collected by keeping the working set point above 70% of the free oscillation amplitude. The scan rate for acquisition of images was 0.7 Hz. Height profiles were processed with the JPK Data Processing software (JPK Instruments, Germany) and the data were analyzed with OriginPro 9.1 software. Statistical analysis was carried out by means of Origin 9.1 software on multiple AFM images for each sample. The samples were prepared by drop-casting R6G-FG dispersions onto mica sheets (G250-1, Agar Scientific Ltd, Essex, UK) and dried under a vacuum.
Raman spectroscopy measurements were carried out using a Renishaw microRaman inVia 1000 using a 50× objective, with an excitation wavelength of 532 nm and an incident power on the samples of 1 mW. For each sample, 50 spectra were collected. The samples were prepared by drop casting the R6G-FG and R6G dispersions onto Si/SiO2 substrates and dried under a vacuum.
X-ray photoelectron spectroscopy (XPS) characterization was carried out on a Kratos Axis UltraDLD spectrometer, using a monochromatic Al Kα source (15 kV, 20 mA). The spectra were taken over a 300 × 700 μm2 area. Wide scans were collected with a constant pass energy of 160 eV and energy step of 1 eV. High-resolution spectra were acquired at a constant pass energy of 10 eV and energy step of 0.1 eV. The binding energy scale was referenced to the C 1s peak at 284.8 eV. The spectra were analyzed using the CasaXPS software (version 2.3.17). The samples were prepared by drop-casting R6G dispersions onto Si/SiO2 substrates (LDB Technologies Ltd) and dried under a vacuum.
The steady-state photoluminescence (PL) emission measurements were performed using an Edinburgh Instruments FLS920 spectrofluorometer. The samples were a R6G solution in water at a concentration of 0.2 mM and a 1:100 diluted R6G-FG dispersion, both with and without Hg2+. For the PL experiments with Hg2+, an aqueous solution of Hg2+ (10 ppm) was prepared by dissolving Hg(ClO4)2 salt (Sigma Aldrich, Hg(ClO4)2·xH2O, 98%) in water. From this solution, different aliquots were taken and added to the R6G-FG dispersion in water (∼0.4 mg mL−1). The PL spectra were collected exciting the samples at 400 nm using a Xe lamp coupled to a monochromator and using quartz cuvettes with a path length of 1 cm. The blank (control) measurement was carried out in the same experimental conditions used for the characterization of the aforementioned samples in order to discard any contribution from the solvent. Additionally, before carrying out the PL measurements, the absorbance of the different samples was acquired using a Varian Cary 5000 UV-vis-NIR spectrophotometer. The PL spectra of the samples were then normalized to the absorbance of the samples at the excitation wavelength (400 nm) to allow their comparison.
Fabrication of the membranes
The membranes were produced by depositing the R6G-FG dispersion in water onto a microporous nylon filter (Whatman® membrane filters nylon, 0.2 μm pore size, Sigma Aldrich) through the vacuum filtration process. By controlling the volume of the deposited dispersion, different amounts of material were deposited (mass loading ranging from 17.9 to 71.6 g m−2). The membranes were dried overnight at room temperature before their characterization.Characterization of the membranes
Scanning electron microscopy (SEM) analysis of the membranes was performed using a Helios Nanolab 600 DualBeam microscope (FEI Company) operating at 5 kV and 0.2 nA. The EDX spectra were acquired by combining the microscope (operating at 15 kV and 0.8 nA), an X-Max detector and an INCA system (Oxford Instruments). The samples were imaged without any metal coating or pretreatment. To evaluate the laminar structure of the membranes by the cross section, the membranes were cut with a scalpel and mounted in a tilted sample holder.Atomic force microscopy measurements were taken with the same setup used for the material characterization. However, the AFM mode was QI for morphology and adhesion work. Adhesion work measurements were also carried out with the same AFM setup, but used in the quantitative imaging (QI) mode,147 an AFM mode based on force measurements. V-shaped DNP silicon nitride cantilevers (Bruker, Billerica, MA, USA), with a nominal spring constant of 0.06 N m−1, resonance frequency in air in the 40–70 kHz range and tip typical curvature radius of 20–60 nm were used. The actual spring constant of each cantilever was determined in situ, using the thermal noise method. The acquisition of a large set of force–distance (FD) curves (256 × 256) was performed with a maximum force load of 25 nN and a curve length of 800 nm. 3 × 3 μm2 images were collected. Height profiles were processed with JPK Data Processing software (JPK Instruments, Germany) and the data were analyzed with OriginPro 9.1 software.
Electrical volumetric resistance measurements of the membranes were performed using a four-probe system (Jandel RM3000 Test Unit).
Elemental analysis by inductively coupled plasma optical emission spectroscopy (ICP-EOS) was performed with a ThermoFisher ICAP 6000 Duo inductively coupled plasma optical emission spectrometer. The samples were prepared from stock solutions recovered after passing through the membranes and digesting in HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 (3:1 vol/vol) overnight. The stock solutions, i.e., 10 ppm aqueous solutions of: CaCl2, MnCl2, CoCl2, NiCl2, CuCl2, ZnCl2, CdCl2, and Hg(ClO4)2 (chemicals supplied by Sigma Aldrich), were also measured by ICP-OES. Experimentally, the analyzed solutions were prepared in a 25 mL volumetric flask. Prior to the measurement, 2.5 mL of each sample was diluted with Millipore water to a total volume of 25 mL and stirred by vortexing at 2400 rpm for 10 s. Lastly, the sample was filtered using a PTFE membrane (0.45 μm pore size). Four measurements were performed on each sample to obtain the final averaged values of the metals’ concentration.
:HNO3 (3:1 vol/vol) overnight. The stock solutions, i.e., 10 ppm aqueous solutions of: CaCl2, MnCl2, CoCl2, NiCl2, CuCl2, ZnCl2, CdCl2, and Hg(ClO4)2 (chemicals supplied by Sigma Aldrich), were also measured by ICP-OES. Experimentally, the analyzed solutions were prepared in a 25 mL volumetric flask. Prior to the measurement, 2.5 mL of each sample was diluted with Millipore water to a total volume of 25 mL and stirred by vortexing at 2400 rpm for 10 s. Lastly, the sample was filtered using a PTFE membrane (0.45 μm pore size). Four measurements were performed on each sample to obtain the final averaged values of the metals’ concentration.
Results and discussion
Production and characterization of R6G-FG
Single/few-layer graphene flakes were produced by ultrasonication-assisted LPE of graphite in mild-alkaline water (obtained by adding 1 M KOH to adjust the pH equal to 8) in the presence of R6G molecules (12 mg L−1) (Fig. 1a). By excluding the KOH or the R6G as additives, the exfoliation was not effective, resulting in the precipitation of the aggregates. In agreement with computational148 and experimental studies,148–151 R6G molecules interact with the graphene surface by forming physisorbed complexes (Fig. 1b).148–151 The physisorption behavior originates from the π–π stacking between the aromatic rings of the R6G molecules and graphene,148–152 as well as the electrostatic interaction between positively-charged amino groups of R6G molecules and negatively charged intrinsic functional groups of graphene,148,149i.e., deprotonated carboxyl groups at the edges and/or hydroxyl and epoxy groups on the basal plane for marginal oxidized regions (see the below XPS analysis).153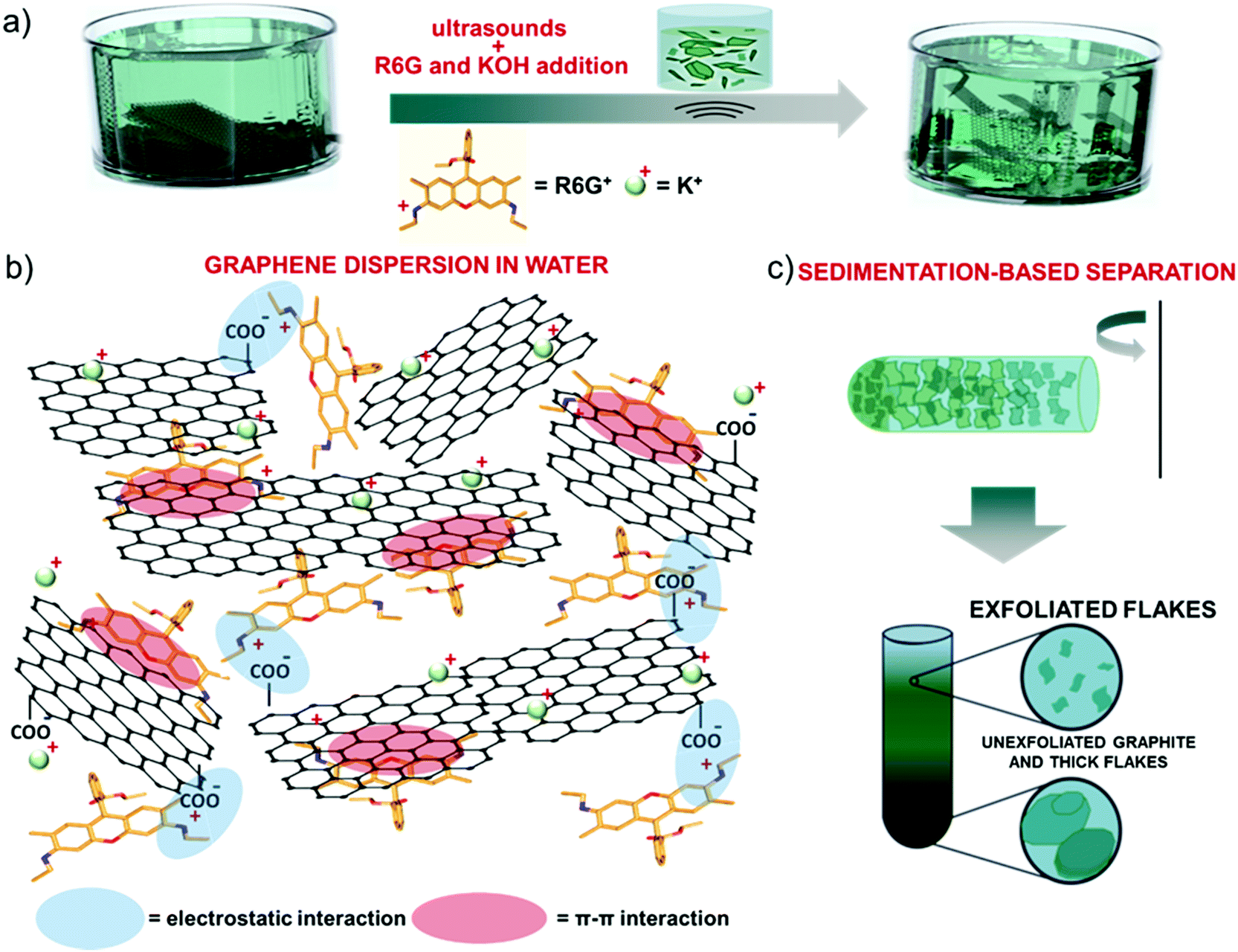 | ||
| Fig. 1 Production of R6G-FG through ultrasonication-assisted LPE in water. (a) Sketch of the exfoliation of graphite through ultrasonication-assisted LPE in water in the presence of R6G molecules (12 mg L−1) and a small amount of KOH (pH = 8). (b) Representation of the physisorption of the R6G molecules on the graphene flakes by electrostatic and π–π interactions. (c) Illustration of the sedimentation-based separation process used to collect the exfoliated sample (R6G-FG dispersion in water). | ||
The electrostatic R6G/graphene interaction is also aided by mild alkaline conditions, as obtained by KOH addition in water. In fact, the alkaline environment completes the dissociation of the R6G from the neutral to the cationic form, thus triggering its electrostatic interaction with the negative surface of graphene, whose deprotonated state is also promoted in alkaline conditions,153 as observed for graphene derivative dispersons.154–156 Furthermore, the carboxyl protons at the edges of graphene first undergo ion exchange with salt cations (K+),149,157 which further eases graphite exfoliation, and thus the graphene/R6G interaction.149,157
Subsequent to the ultrasonication process, sedimentation-based separation based on ultracentrifugation (see the Experimental section) was used to remove unexfoliated materials and thick flakes and collect the supernatant as the exfoliated material in a concentration of ∼0.4 mg mL−1 (Fig. 1c).
The stability of the as-produced aqueous dispersion of graphene flakes was verified by measuring the graphene flake zeta-potential, i.e., the electric potential at the interfacial double layer of dispersed flakes versus a point in the continuous phase away from the interface.158 The measured graphene flake zeta potential is ∼−32 mV, which indicates that the as-produced graphene flake dispersion is electrically stabilized (i.e., electrically repulsive forces between the flakes exceed their mutually attractive van der Waals force).153,159 Transmission electron microscopy and AFM measurements (Fig. 2a and b) show that the exfoliated sample consists of irregularly-shaped wrinkled flakes with a log-normally distributed lateral dimension and thickness of 194.6 nm and 1.0 nm, respectively (Fig. 2c and d). The TEM diffraction pattern of the imaged flakes is shown in Fig. 2e and indicates the crystalline nature of the flakes. The structural properties of the latter have been further investigated by Raman spectroscopy, which is a versatile tool to identify both chemical and physical properties of graphitic materials.160 The typical Raman spectrum of exfoliated graphene flakes shows, as fingerprints, the following peaks:142,161–163 G (E2g phonon at the Brillouin zone centre, ∼1585 cm−1),161,162,164 D (breathing mode of sp2 rings requiring a defect for its activation by double resonance, ∼1380 cm−1),162,164–166 D′ (∼1620 cm−1)167 and 2D (∼2700 cm−1)162 (see the ESI† for a detailed discussion of the origin of the Raman modes). Fig. 2f shows the Raman spectra of the graphite and the as-produced flakes. The Raman spectrum of only R6G is also shown to prove the R6G-functionalization of the graphene flakes, as well as to discriminate the peaks related to graphitic components from those attributed to R6G molecules. For the exfoliated sample, the D and D′ bands increase relatively to the G band compared to the graphite case. The ratio between the intensities of the D and G peaks – I(D)/I(G) – is ∼0.6, which is significantly superior to that of pristine graphite (<0.1). This indicates that few (<5)-layer graphene flakes have been effectively produced by our method, in agreement with AFM analysis.168–171 The analysis of the 2D peak allows the exfoliation of the graphite into few-layer graphene to be further confirmed. In fact, the 2D peak is a single and sharp Lorentzian band centred at ∼2680 cm−1 for single-layer graphene,142,161,162 whereas it is a superposition of multiple components, the main being the 2D1 and 2D2 components, for few-layer graphene.142,161,162 In graphite, the intensity of the 2D2 band is twice the 2D1 band,161,162,172 while the 2D2 progressively decreases with decreasing the number of layers for few-layer graphene.161,162,169,172,173 Therefore, the data shown in Fig. 2f clearly indicate that the exfoliated sample has a few-layer graphene enriched composition,174 since I(2D2) is inferior to I(2D1).161,162,169,172,173 Lastly, the Raman spectrum of the exfoliated sample exhibits additional signatures which are attributed to the R6G bands, in agreement with previous studies.175–179 Notably, some of the bands related to R6G partially overlap those typically observed for graphene including D, G and D′. This results in a broadening of the D, G and D′ bands assigned to graphene, as well as a slight overestimation of the calculated I(D)/I(G). For the sake of comparison, a representative Raman spectrum for graphene flakes produced by conventional LPE in NMP is reported in Fig. S1 (ESI†) to uniquely identify the Raman modes of graphene in the absence of R6G molecules. The chemical composition of the as-produced graphene flakes has been assessed by performing XPS measurements (Fig. 2g). The C 1s spectrum of the flakes can be decomposed into different components. The main one peaks at (284.3 ± 0.2) eV and refers to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (sp2 carbon),180–182 which also results in the corresponding feature at (291.1 ± 0.2) eV due to π–π* interactions.180–182 The component centred at 284.8 eV refers to C–C (sp3)182,183 and it is due to flake edges as well as to environmental contamination (adventitious carbon).184 The other contributions, peaking at binding energies of ∼285.9 eV, 287.2 eV and 289.1 eV, can be ascribed to C–O/C–N, CO and OC–O groups, respectively.185,186 Their origin is ascribed to R6G molecules,185,186 whose presence is confirmed by the peak at 399.4 eV in the N 1s spectrum (inset of Fig. 2g) corresponding to their amine groups. The percentage content of O is inferior to 2.5%, thus proving the high quality of the as-produced graphene flakes. It is noteworthy that the low oxidation of graphene can be also explained by the removal of epoxy and hydroxyl groups attached to the graphene surface by the R6G molecules nearby.148 In more detail, a R6G molecule close to an epoxy/hydroxyl group on graphene can cause the detachment of O/OH species from the graphene flake by forming a N–O bond with the amine group of R6G. Subsequently, the R6G molecule can further interact with graphene through physisorption mechanisms. The formed system decreases the energy of the oxidized graphene by more than 2 eV, while showing an electronic structure of the reduced graphene resembling the one of the pristine graphene.148 The occurrence of an interaction between graphene flakes and R6G molecules was further confirmed by steady-state PL measurements. As shown in Fig. 2h, the PL emission of R6G molecules in aqueous solution is quenched in the presence of graphene. In agreement with previous studies, the PL quenching is linked to the formation of R6G/graphene complexes leading to photo-induced electron transfer from R6G to graphene.150,151,187,188 Overall, the morphological and spectroscopic characterization of the exfoliated sample confirm that the R6G/KOH-assisted LPE of graphite in water is effective for producing high quality R6G-functionalized single-/few-layer graphene flakes.
C (sp2 carbon),180–182 which also results in the corresponding feature at (291.1 ± 0.2) eV due to π–π* interactions.180–182 The component centred at 284.8 eV refers to C–C (sp3)182,183 and it is due to flake edges as well as to environmental contamination (adventitious carbon).184 The other contributions, peaking at binding energies of ∼285.9 eV, 287.2 eV and 289.1 eV, can be ascribed to C–O/C–N, CO and OC–O groups, respectively.185,186 Their origin is ascribed to R6G molecules,185,186 whose presence is confirmed by the peak at 399.4 eV in the N 1s spectrum (inset of Fig. 2g) corresponding to their amine groups. The percentage content of O is inferior to 2.5%, thus proving the high quality of the as-produced graphene flakes. It is noteworthy that the low oxidation of graphene can be also explained by the removal of epoxy and hydroxyl groups attached to the graphene surface by the R6G molecules nearby.148 In more detail, a R6G molecule close to an epoxy/hydroxyl group on graphene can cause the detachment of O/OH species from the graphene flake by forming a N–O bond with the amine group of R6G. Subsequently, the R6G molecule can further interact with graphene through physisorption mechanisms. The formed system decreases the energy of the oxidized graphene by more than 2 eV, while showing an electronic structure of the reduced graphene resembling the one of the pristine graphene.148 The occurrence of an interaction between graphene flakes and R6G molecules was further confirmed by steady-state PL measurements. As shown in Fig. 2h, the PL emission of R6G molecules in aqueous solution is quenched in the presence of graphene. In agreement with previous studies, the PL quenching is linked to the formation of R6G/graphene complexes leading to photo-induced electron transfer from R6G to graphene.150,151,187,188 Overall, the morphological and spectroscopic characterization of the exfoliated sample confirm that the R6G/KOH-assisted LPE of graphite in water is effective for producing high quality R6G-functionalized single-/few-layer graphene flakes.
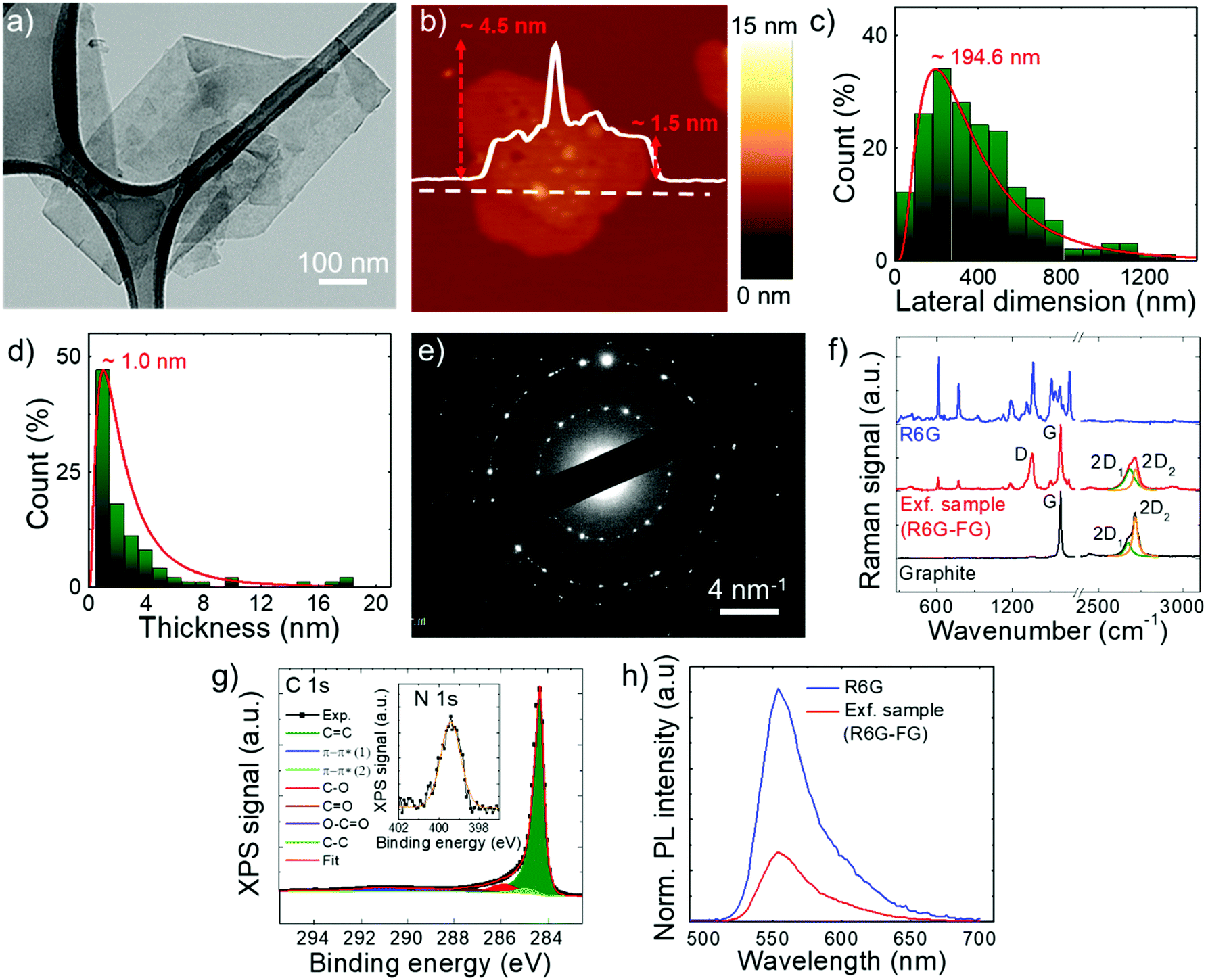 | ||
| Fig. 2 Characterization of as produced R6G-FG flakes. (a and b) Representative TEM and AFM images of the R6G-FG flakes. (c) TEM statistical analysis of the lateral dimension of R6G-FG flakes (calculated on 120 flakes). (d) AFM statistical analysis of the thickness of the R6G-FG flakes (calculated on 120 flakes). (e) Electron diffraction pattern of the TEM image shown in (a), revealing the crystallinity of the R6G-FG flakes. (f) Comparison between the Raman spectra of graphite (black), R6G-FG (red) and R6G molecules (blue). The multi-peak Lorentzian fittings of the 2D mode region of the spectra of graphite and R6G-FG show the contribution of 2D1 (orange) and 2D2 (olive) modes. (g) C 1s XPS spectrum of R6G-FG, together with its deconvolution evidencing the bands ascribed to CC, C–C, C–N, CO and π–π*. The inset shows the N 1s XPS spectrum ascribed to the presence of R6G molecules adsorbed to the surface of graphene flakes. (h) PL spectra of R6G (blue) and R6G-FG (red) in water, normalized to the absorbance of the samples at the excitation wavelength (400 nm). | ||
Fabrication and morphological characterization of graphene-based laminar membranes
The aqueous R6G-FG dispersion was deposited onto a microporous nylon membrane (pore size of 0.2 μm) through a vacuum filtration process to obtain graphene-based laminar membranes (R6G-FG membranes) with mechanical robustness. By controlling the volume of the deposited dispersion, different amounts of material were deposited over the supporting membranes (mass loading ranging from 17.9 to 71.6 g m−2). Noteworthily, functionalization of 2D material-based membranes with dye molecules has been previously reported.25 However, these cases achieved the functionalization by post-deposition treatment of the membranes through a chemical bath in dye solutions over a long time (tens of days).25 Therefore, this approach may be time-consuming for pursuing scalable technologies.Fig. 3a shows a photograph of a representative nylon filter-supported membrane (R6G-FG mass loading of 35.8 g m−2), also illustrating its mechanical flexibility. The surface morphology of the as-prepared membranes has been characterized by SEM and AFM measurements. Fig. 3b shows the top-view SEM image of the R6G-FG membrane reported in Fig. 3a. The image indicates that the membrane has a crumpled, wrinkled, and flake-composed structure.
 | ||
| Fig. 3 Morphological characterization of the R6G-FG membranes. (a) Photograph of the nylon filter-supported R6G-FG membrane, illustrating its mechanical flexibility. (b) Top-view SEM and (c) AFM images of a representative R6G-FG membrane (R6G-FG mass loading of 35.8 g m−2). The Ra value is also reported in (c). (d) Cross-sectional SEM image of the same R6G-FG membrane reported in (b and c), showing its laminar structure. The inset shows the un-tilted cross-section SEM image of the R6G-FG membrane, used for estimating its thickness. | ||
The atomic force microscopy image (Fig. 3c) also confirms such a surface structure, which exhibits an average roughness (Ra) of 92.1 ± 6.3 nm. Fig. 3d reports cross-sectional SEM images of a representative membrane (R6G-FG mass loading of 35.8 g m−2), evidencing a ∼80 μm-thick laminar structure. The corresponding mass density of the membrane is as low as 0.45 g cm3, which is inferior to the one of a reference membrane made of graphene produced by conventional LPE in NMP (0.71 g cm3). The relatively low density of the R6G-FG membrane might be attributed to the presence of R6G molecules, which act as a spacer between the flakes, impeding their restacking during film deposition.76,189 Consequently, the laminar R6G-FG membranes were intended to provide expanded nanochannels for high-flux solute filtration (i.e., high water permeability, as demonstrated below).
The electrical volumetric resistivity of the R6G-FG membranes is lower than the one of the reference membrane (0.98 × 10−1 Ω cm vs. ∼2.78 × 10−1 Ω cm). These results agree with the marginal presence of oxygen functionalities, which can also act as architectural spacers between the graphene flakes.190 It is noteworthy that both theoretical and experimental studies previously reported that R6G molecules can reduce graphene flakes by removing oxygen functionalities, including epoxy, hydroxyl and carbonyl groups possibly formed in the defective regions of the graphene flakes.148
Adhesion force measurements were carried out with an AFM191,192 in humid (relative humidity – RH – ∼75%) ambient air (assuming the Lennard-Jones force–separation relation193,194) to effectively identify the water accessible sites of the R6G-FG membrane.195 During the measurements, the adhesion forces between the AFM tip and sample surface are dominated by capillary forces,196 which depend on the hydrophilicity/hydrophobicity of the substrate,196–198 as well as its roughness.196,197,199 In addition, chemical specificity (e.g., the presence of functional groups) can strongly affect the pull-off force at the nano/microscale level.196,200–202 Therefore, for the case of the membranes, adhesion force measurements can effectively identify the sites of the R6G-FG membrane accessible to water,195 which are typically expressed by hydrophilic porous nano/microdomains.203–205Fig. 4a and b show the adhesion work maps of a representative R6G-FG membrane and a reference membrane made of graphene produced by conventional LPE in NMP (material mass loading of 35.8 g m−2 for both membranes), respectively. Contrary to the membrane based on graphene produced by LPE in NMP, the R6G-FG membrane manifests elongated nano/microdomains that are more adhesive than the rest of the surface, thus expressing hydrophilic regions. Fig. 4c and d report the adhesion work distribution corresponding to the adhesion work maps. The adhesion work distribution for the R6G-FG membrane can be fitted with two components. The first component, peaking at 6.7 aJ, can be ascribed to the hydrophobic domains of graphene, since it resembles in shape and position the work distribution of the membrane based on graphene produced by LPE in NMP (fitted by a single component peaking at 7.54 aJ). The second component, peaking at 21.5 aJ, lies at an adhesion work higher than the first component, and refers to hydrophilic micro/nanodomains. The latter are expected to facilitate the access of water to the membrane,49 where the water permeation rate is determined by: (1) capillary-driven force; and (2) low-friction flow through the 2D-like channels enclosed by hydrophobic, pristine (non-oxidized) regions of graphene (expanded by R6G molecules acting as spacers).20
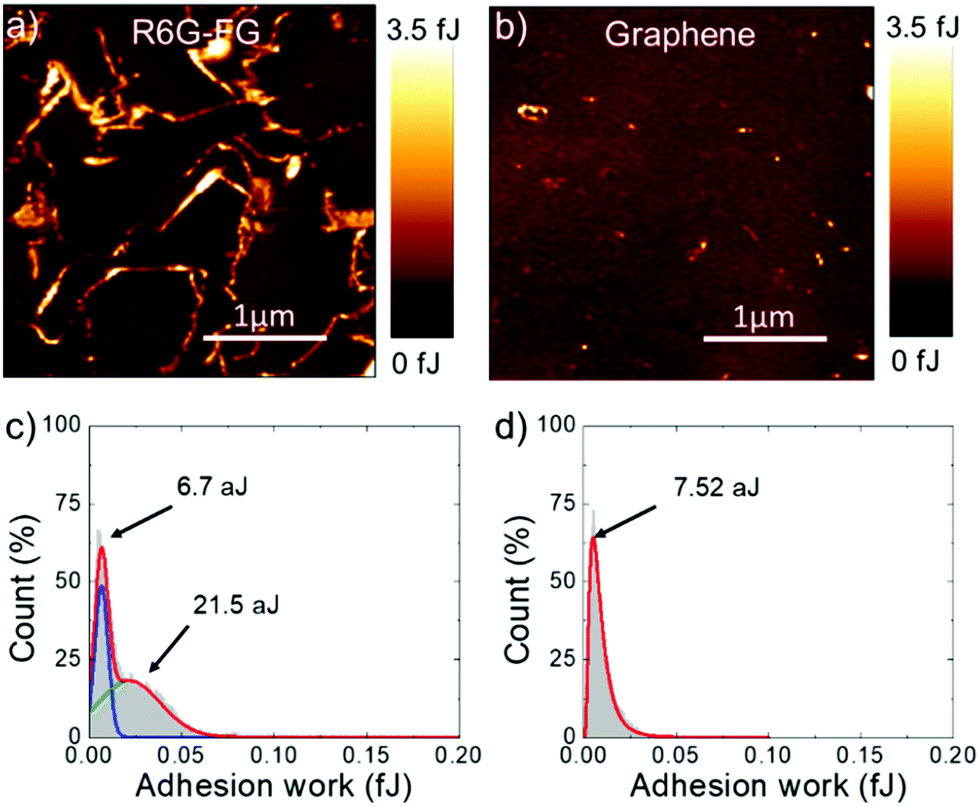 | ||
| Fig. 4 Evaluation of the adhesion forces on the membrane in humid (RH ∼ 75%) ambient air. Adhesion work maps of (a) a R6G-FG membrane (R6G-FG mass loading of 35.8 g m−2) and (b) a membrane made of graphene produced by conventional LPE in NMP (graphene mass loading of 35.8 g m−2) (panel b). (c and d) The adhesion work distributions of the membrane shown in (a) and (b). The corresponding fits are also displayed. | ||
Evaluation of the filtration properties of the graphene-based laminar membranes
The properties and the filtration performance of the as-produced R6G-FG membranes were evaluated in terms of permeability to water (i.e., water permeation rate) and metal ion rejection. The water permeation through the R6G-FG membranes was measured by applying an external pressure in a dead-end filtration mode.206 The water permeation rate was found to be 789.6 L m−2 h−1 bar−1 for an 80 μm-thick membrane (R6G-FG mass loading of 35.8 g m−2). This value significantly surpasses those of other 2D material-based laminar membranes proposed in the relevant literature, as shown in Table 1. In addition, the membrane based on graphene produced by conventional LPE in NMP (mass loading 41.2 g m−2) showed a water permeation rate of 201.58 L m−2 h−1 bar−1, which is significantly inferior to the one measured for the R6G-FG membrane using a comparable active material mass loading (35.8 g m−2). Compared to the R6G-FG membranes, the lower water permeation rate of the membrane based on graphene produced by conventional LPE in NMP is ascribed to the hydrophobicity of the pristine graphene and the absence of spacers (such as R6G molecules in R6GG-FG membranes) between the graphene flakes, which therefore hamper prompt water intercalation. Additional discussion of the high water permeation rate of the R6G-FG membrane is reported in the next section. The ion rejection of the R6G-FG membrane was first evaluated specifically for Hg2+ ICP-OES measurements of filtered Hg(ClO4)2 aqueous solution with a Hg2+ concentration ([Hg2+]) of 10 ppm.| Material | Thickness [μm] | Water permeationa [L m−2 h−1 bar−1] | Ref. |
|---|---|---|---|
| a Values measured in dead-end filtration mode. | |||
| R6G-FG (this work) | 80 | 789.6 | — |
| GO | 0.35 | 1.5 | 207 |
| GO | 2 | 71 | 208 |
| Carbon nanotube-intercalated graphene | n.d. | 11.3 | 40 |
| GO | ∼30 (15 layers) | 27.6 | 23 |
| Nanostrand-channelled GO | 2 | 695 | 42 |
| Corrugated reduced GO membrane | n.d. | 270 | 41 |
| KOH-activated nanoporous graphene | 0.02 | 37 | 209 |
| MoS2 | 1.5 | 245 | 25 |
| MoS2 | 1.7 | 245 | 210 |
| Crystal violet functionalized MoS2 | 6 | 269.5 | 25 |
| WS2 | 500 | 450 | 211 |
| WS2 | 300 | 730 | 211 |
In fact, on the basis of the use of R6G molecules pre-adsorbed on conductive substrates as fluorescence/SERS chemo-sensors for the selective detection of Hg2+,136–141 a distinctive interaction between R6G molecules and Hg2+ was rationally expected to influence the Hg2+ sieving process. Fig. 5a shows the Hg2+ percentage rejection of a R6G-FG membrane with a R6G-FG mass loading of 35.8 g m−2 after passing different volumes of Hg(ClO4)2 solution. A high Hg2+ percentage rejection (>95%) is obtained up to ∼800 L m−2 filtered solution volume normalized to the membrane area. This value can be assumed as the critical filtered solution volume of the membrane, beyond which fading of Hg2+ rejection starts to occur because of saturation of the ion-sieving sites or even a morphology change of the membrane. Fig. 5b shows the Hg2+ percentage rejection of R6G-FG membranes with increasing R6G-FG mass loading (from 17.9 to 71.6 g m−2) after passing a fixed volume normalized to the area of a Hg(ClO4)2 solution of 263.2 L m−2. For a R6G-FG mass loading higher than 25 g m−2, the Hg2+ rejection is superior to 90%, reaching almost complete rejection (>99%) for a R6G-FG mass loading superior to 55 g m−2, which corresponds to a filtered solution volume normalized to the R6G-FG mass superior to 3 L g−1. In contrast, the membrane based on graphene produced by conventional LPE in NMP shows poor Hg2+ rejection capability (∼10%), thus proving the functional role of R6G molecules adsorbed onto the graphene surface to sieve Hg2+. The Hg2+ rejection capability of the R6G-FG membrane was also investigated by energy dispersive X-ray spectroscopy combined with scanning electron microscopy (SEM-EDS). Fig. 5c shows the presence of material deposits on the surface of the membrane with a R6G-FG mass loading of 35.8 g m−2 after the passage of 263.2 L m−2 of the Hg(ClO4)2 solution. The elemental analysis (Fig. 5d–f) reveals that these deposits are formed by Hg. Interestingly, the membrane made of graphene produced by standard LPE in NMP does not show any presence of Hg deposits (Fig. S2, ESI†), in agreement with its poor Hg2+ rejection capability (∼10%, Fig. 5b). The permeation of ionic solutes through R6G-FG membranes was then tested for different alkaline and transition metal ions, i.e., Ca2+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+, and Cd2+. As shown in Fig. 5g, high percentage contents of such ions (∼80% for Ca2+, >50% for Mn2+, Co2+, Ni2+, Cu2+, and Cd2+, and ∼46% for Zn2+) can pass through the membrane, while proving selective rejection for Hg2+. This result evidences that the ion rejection capability of the R6G-FG membrane is not strictly regulated by size-exclusion effects depending on the interlayer spacing of the graphene flakes, but is also ruled by specific chemical processes (see the working mechanism of R6G-FG membranes sub-section below).
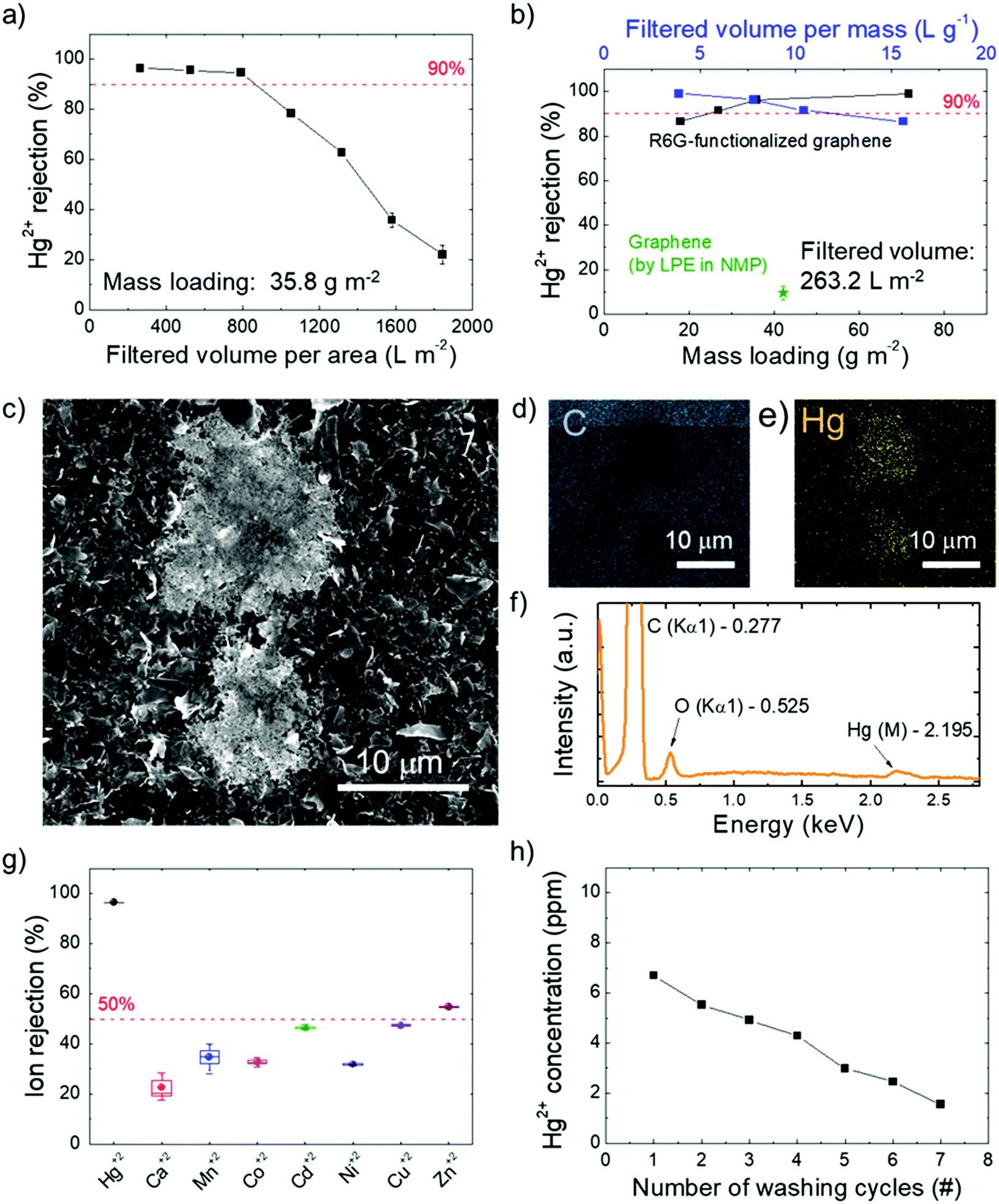 | ||
| Fig. 5 Evaluation of the R6G-FG membrane for metal ion rejection. (a) Hg2+ percentage rejection from Hg(ClO4)2 aqueous solution with a Hg2+ concentration of 10 ppm of the R6G-FG membrane (R6G-FG mass loading of 35.8 g m−2) as a function of the filtered solution volume (normalized to the area of the membrane). The data are the average values obtained by experiments performed on three different equivalently produced membranes. (b) Hg2+ removal percentage rejection of the R6G-FG membrane as a function of the R6G-FG mass loading (bottom x-axis) and filtered solution volume normalized to the mass of R6G-FG (top x-axis). A fixed volume of 263.2 L m−2 was filtered through the membranes. The Hg2+ percentage rejection of a membrane made of graphene produced by conventional LPE in NMP is also reported for comparison. (c) Top-view SEM images of a R6G-FG membrane (R6G-FG mass loading of 35.8 g m−2) after the passage of 263.2 L m−2 of Hg(ClO4)2 solution ([Hg2+] = 10 ppm). (d and e) Elemental mapping of C and Hg for the SEM image and (f) the corresponding EDX spectrum of the R6G-FG membrane imaged in (c). (g) Metal ion percentage rejection of the R6G-FG membrane (R6G-FG mass loading of 35.8 g m−2) for a filtered aqueous solution volume (normalized to the area of the membrane) of 263.2 L m−2 with a 10 ppm metal ion concentration. (h) Hg2+ concentration of the 0.5 M Bu4NOH·30H2O solution passed through a Hg2+ saturated R6G-FG membrane. Hg2+ saturation has been achieved by filtering through the membrane a volume normalized to the membrane area of 1842.4 L m−2 of Hg(ClO4)2 aqueous solution ([Hg2+] = 10 ppm). | ||
The possibility to recycle the membrane is also important for practical application, since it increases the effective critical filtered solution volume (depending on the Hg2+ concentration of the aqueous solution to be filtered). The regeneration of the active sites for Hg2+ rejection was achieved by washing the R6G-FG membrane in 0.5 M Bu4NOH·30H2O. Actually, a similar washing treatment were previously exploited to regenerate the Hg2+-detection capability of R6G-based luminescence sensors,212 as well as the adsorption capacity of R6G-based nanocomposites.213Fig. 5h shows that subsequent washing cycles are effective to desorb Hg2+ from a membrane with a R6G-FG mass loading of 35.8 g m−2 which filtered a solution volume normalized to the membrane area of 1842.4 L m−2 (well above its critical value). After the first washing cycle, ∼80% of the initial Hg2+ rejection capability of the membrane can be successfully restored.
Working mechanism of the R6G-FG membranes
The R6G functionalization of graphene flakes has afforded the realization of a filtration membrane with three functional properties: (1) a high water permeation rate; (2) highly selective metal ion rejection capability for Hg2+ from aqueous solution; and (3) recyclability via chemical washing cycles in alkaline conditions. The high-water permeability (up to 789.6 L m−2 h−1 bar−1 for an 80 μm thick R6G-FG membrane, see Table 1) is first attributed to the low-density (∼0.5 g cm−3) packed laminar structure in the presence of R6G molecules. In fact, the latter act as spacers between the graphene flakes, which can be easily intercalated by water.19 In addition, the peculiar presence of hydrophilic micro/nanodomains, as observed by adhesion force measurements, can further promote the access of water to the membrane.49 Secondly, both capillary-driven force and low-friction flow confined between 2D channels enclosed by hydrophobic, pristine (non-oxidized) regions of graphene can accelerate the passage of water through the membrane.20,214,215The ion selectivity of the R6G-FG membrane for Hg2+ is explained by the competitive adsorption of R6G molecules and metal ions on the surface of the graphene flakes within the membrane (Fig. 6a). At first, R6G molecules are adsorbed onto the graphene surface, forming physiosorbed complexes. As show in the Production and characterization of R6G-FG sub-section, the formation of the complexes originates from the π–π stacking between the aromatic rings of the R6G molecules and graphene,148–152 as well as the electrostatic and non-covalent interactions between positively charged amino groups of R6G molecules and negatively charged intrinsic functional groups of graphene.148,149 Such complexes perform as ion-selective nano-traps for Hg2+. In more detail, during the passage of an aqueous solution with metallic contaminants through the R6G-FG membrane, metal ions, especially heavy metal-based ones, can compete with R6G molecules at being adsorbed onto the surface of the graphene flakes. The capability of graphene to adsorb heavy metal ions has been previously reported to be a consequence of van der Waals and electrostatic interactions between the metal ions and negatively charged oxygen groups of graphene.45–47 The different affinity of metal ions and R6G molecules to graphene rules the competitive adsorption process. For the metals, the affinity to graphene positively correlates with the metal electronegativity and first stability constant (K1) of the associated metal hydroxide.47,216 On one hand, the electronegativity of the metal ions represents the attraction of negative charges, and this plays a crucial role in the metal adsorption to the negatively charged surface of graphene.47,216 On the other hand, K1 of the metal ions determines the speciation of metal ion species, and the formation of hydroxide complexes can alter the order of the metal ions’ affinity to graphene predicted by the metal ions’ electronegativity.216 Consequently, the selective Hg2+ rejection capability of the R6G-FG membranes arises from the high electronegativity (1.9 eV)217 and high stability constant (logK1 = 10.6)218 of Hg2+ compared to those of other metal ions (electronegativity: 1.0 eV for Ca2+, 1.5 eV for Mn2+, 1.8 eV for Co2+, 1.8 eV for Ni2+, 2.0 eV for Cu2+, 1.6 eV for Zn2+ and 1.7 eV for Cd2+;217 logK1: 1.3 for Ca2+, 3.4 for Mn2+, 4.3 for Co2+, 4.1 for Ni2+, 6.5 eV for Cu2+, 5.0 for Zn2+ and 3.9 eV for Cd2+218). Indeed, only the adsorption of Hg2+ is strongly promoted onto graphene, causing the displacement/release of R6G molecules from adsorption sites. In order to further validate the proposed working mechanism of our membranes, PL measurements of the R6G-FG dispersions in water were aquired before and after the addition of Hg(ClO4)2 (Fig. S3, ESI†). The addition of small amounts of a Hg(ClO4)2 aqueous solution with 10 ppm of Hg2+ promotes the quenching of the PL emission of the R6G molecules in R6G-FG. However, a further increase of the Hg2+ leads to a partial recovery of the PL emission, indicating that R6G molecules are released from the graphene, which also effectively quenches the PL of the R6G molecules through physisorption interactions (see Fig. 2h). Thus, these PL measurements further support the competitive adsorption of R6G molecules and metal ions on the surface of the graphene flakes across the membrane.
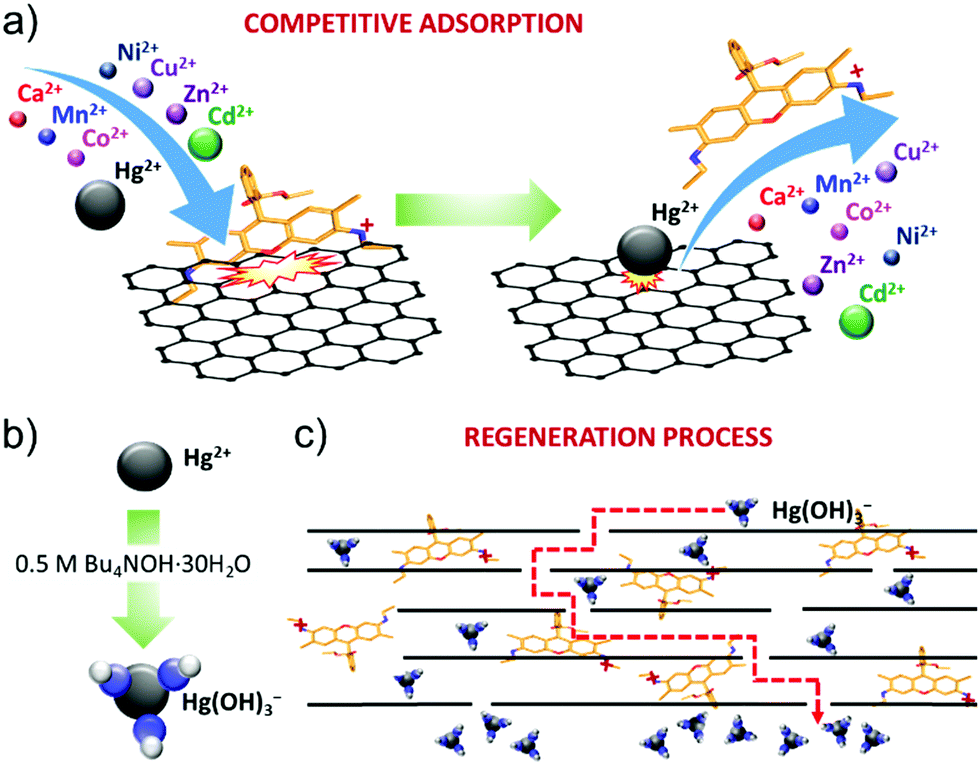 | ||
| Fig. 6 Working mechanism of R6G-FG membranes. (a) Competitive adsorption of metal ions and R6G molecules resulting in selective Hg2+ rejection from aqueous solution. (b) Formation of mercury hydroxide complexes (Hg(OH)3−) in the presence of alkaline conditions (0.5 M Bu4NOH·30H2O). (c) Regeneration of the R6G-FG membrane via washing cycles removing Hg(OH)3−. | ||
Notably, the high water permeability rate of the R6G-FG membranes is compatible with high membrane thicknesses (e.g., 80 μm for a R6G-FG mass loading of 35.8 g m−2), allowing the proposed physico-chemical working mechanism to be effectively exploited. Since the recyclability experiments in Fig. 6b show that the R6G-FG membrane can be regenerated, the released R6G molecules are expected to be trapped between the nano-channels of the membrane. As depicted in Fig. 6b, the regeneration mechanism of the membranes in the presence of highly alkaline conditions (0.5 M Bu4NOH·30H2O) is then attributed to the formation of mercury hydroxide complexes (Hg(OH)3−).219 These negative complexes are electrostatically repulsed by the negative sites of the graphene surfaces.45,46,220 Meanwhile Hg(OH)3− is removed, and trapped R6G molecules can again interact with graphene, sequentially restoring the Hg2+ rejection capability of the membranes. Further optimization of the regeneration process, together with that of the membrane design, is still needed in order to limit the release of R6G molecules during washing cycles.
Conclusions
In conclusion, a sustainable graphene-based membrane has been proposed for rapid and selective rejection of Hg2+ in aqueous solutions. In order to preclude environmental implications of both the production and the discharge of engineered membranes, a non-oxidative, room-temperature and post processing-free “green” method has been developed to simultaneously exfoliate graphite up to single-/few-layer flakes in water and functionalize graphene with cationic rhodamine 6G (R6G), a prototypical cost-effective and non-hazardous organic molecule. The exfoliated samples consist of R6G-functionalized graphene (R6G-FG) flakes with a log-normally distributed lateral dimension and thickness of 194.6 nm and 1.0 nm, respectively, with a percentage content of O <2.5%. The membranes were then fabricated by depositing the as-produced R6G-FG dispersion onto microporous nylon membranes through vacuum filtration. The R6G-FG membranes show an outstanding water permeation rate of 789.6 L m−2 h−1 bar−1 (for an 80 μm-thick membrane, R6G-FG mass loading of 35.8 g m−2). The high water permeability is attributed to the low-density packed laminar structure in the presence of R6G molecules acting as a spacer between the graphene flakes, as well as to the presence of hydrophilic micro/nanodomains. In addition, the R6G/graphene complexes perform as ion-selective nano-traps for Hg2+, providing high Hg2+ percentage rejection (>95%) up to a critical filtered solution volume (normalized to the membrane area) of ∼800 L m−2 (for a R6G-FG mass loading of 35.8 g m−2), which excludes membrane swelling issues typically occurring in two-dimensional (2D) material-based membranes. For a filtered solution volume of 263.2 L m−2 and a R6G-FG mass loading higher than 30 g m−2, the Hg2+ rejection is superior to 90%, reaching almost complete rejection (>99%) for a R6G-FG mass loading superior to 71.6 g m−2 (filtered solution volume normalized to the R6G-FG mass superior to 3 L g−1). The ion rejection capability of the R6G-FG membrane is strongly selective for Hg2+, as a consequence of the competitive adsorption mechanism between metal ions and R6G molecules with different affinity to interact with the negatively charged surface. Lastly, a washing treatment in highly alkaline conditions has been used to regenerate the Hg2+ rejection capability of the R6G-FG membranes, increasing their effective critical filtered solution volume. Overall, our work rationalizes both the sustainable design and the working mechanism of functionalized graphene-based membranes, providing new insights to overcome the “permeability–selectivity trade-offs” often tackled by laminar 2D material-based membranes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been supported by European Union's Horizon 2020 research and innovation program under grant agreements no. 785219 – GrapheneCore2. We thank Electron Microscopy facility – Istituto Italiano di Tecnologia for the support in TEM data acquisition; Gianmarino Pugliese and Nanochemistry facility – Istituto Italiano di Tecnologia for the support in material characterization; Clean Room facility – Istituto Italiano di Tecnologia for the access to carry out SEM/EDS measurements; and Material Characterization facility – Istituto Italiano di Tecnologia for the access to carry our AFM measurements.Notes and references
- M. A. Montgomery and M. Elimelech, Environ. Sci. Technol., 2007, 41, 17–24 CrossRef PubMed.
- M. Elimelech, J. Water Supply: Res. Technol.–AQUA, 2006, 55, 3–10 CrossRef.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301 CrossRef CAS PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nanoscience and Technology, Macmillan Publishers Ltd, UK, 2009, pp. 337–346 Search PubMed.
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 CrossRef CAS PubMed.
- R. F. Service, Science, 2006, 313, 1088–1090 CrossRef CAS PubMed.
- J. R. Werber, C. O. Osuji and M. Elimelech, Nat. Rev. Mater., 2016, 1, 16018 CrossRef CAS.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 CrossRef CAS PubMed.
- Q. Yang, Y. Su, C. Chi, C. T. Cherian, K. Huang, V. G. Kravets, F. C. Wang, J. C. Zhang, A. Pratt, A. N. Grigorenko, F. Guinea, A. K. Geim and R. R. Nair, Nat. Mater., 2017, 16, 1198 CrossRef CAS PubMed.
- L. Chen, G. Shi, J. Shen, B. Peng, B. Zhang, Y. Wang, F. Bian, J. Wang, D. Li, Z. Qian, G. Xu, G. Liu, J. Zeng, L. Zhang, Y. Yang, G. Zhou, M. Wu, W. Jin, J. Li and H. Fang, Nature, 2017, 550, 380 CrossRef CAS PubMed.
- N. Song, X. Gao, Z. Ma, X. Wang, Y. Wei and C. Gao, Desalination, 2018, 437, 59–72 CrossRef CAS.
- R. K. Joshi, S. Alwarappan, M. Yoshimura, V. Sahajwalla and Y. Nishina, Appl. Mater. Today, 2015, 1, 1–12 CrossRef.
- S. Homaeigohar and M. Elbahri, NPG Asia Mater., 2017, 9, e427 CrossRef CAS.
- A. Boretti, S. Al-Zubaidy, M. Vaclavikova, M. Al-Abri, S. Castelletto and S. Mikhalovsky, npj Clean Water, 2018, 1, 5 CrossRef CAS.
- F. Macedonio, A. Politano, E. Drioli and A. Gugliuzza, Mater. Horiz., 2018, 5, 912–919 RSC.
- Y. C. Woo, L. D. Tijing, W.-G. Shim, J.-S. Choi, S.-H. Kim, T. He, E. Drioli and H. K. Shon, J. Membr. Sci., 2016, 520, 99–110 CrossRef CAS.
- A. Gugliuzza, A. Politano and E. Drioli, Curr. Opin. Chem. Eng., 2017, 16, 78–85 CrossRef.
- E. Drioli, A. Ali and F. Macedonio, Desalination, 2015, 356, 56–84 CrossRef CAS.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- P. Sun, M. Zhu, K. Wang, M. Zhong, J. Wei, D. Wu, Z. Xu and H. Zhu, ACS Nano, 2013, 7, 428–437 CrossRef CAS PubMed.
- Y. Han, Z. Xu and C. Gao, Adv. Funct. Mater., 2013, 23, 3693–3700 CrossRef CAS.
- M. Hu and B. Mi, Environ. Sci. Technol., 2013, 47, 3715–3723 CrossRef CAS PubMed.
- K. Raidongia and J. Huang, J. Am. Chem. Soc., 2012, 134, 16528–16531 CrossRef CAS PubMed.
- W. Hirunpinyopas, E. Prestat, S. D. Worrall, S. J. Haigh, R. A. W. Dryfe and M. A. Bissett, ACS Nano, 2017, 11, 11082–11090 CrossRef CAS PubMed.
- M.-N. Li, X.-F. Sun, L. Wang, S.-Y. Wang, M. Z. Afzal, C. Song and S.-G. Wang, Desalination, 2018, 436, 107–113 CrossRef CAS.
- L. Sun, Y. Ying, H. Huang, Z. Song, Y. Mao, Z. Xu and X. Peng, ACS Nano, 2014, 8, 6304–6311 CrossRef CAS PubMed.
- W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Nat. Commun., 2013, 4, 1777 CrossRef PubMed.
- C. E. Ren, K. B. Hatzell, M. Alhabeb, Z. Ling, K. A. Mahmoud and Y. Gogotsi, J. Phys. Chem. Lett., 2015, 6, 4026–4031 CrossRef CAS PubMed.
- Y. Liu, N. Wang, Z. Cao and J. Caro, J. Mater. Chem. A, 2014, 2, 1235–1238 RSC.
- Y. Liu, N. Wang and J. Caro, J. Mater. Chem. A, 2014, 2, 5716–5723 RSC.
- S. Dervin, D. D. Dionysiou and S. C. Pillai, Nanoscale, 2016, 8, 15115–15131 RSC.
- G. Liu, W. Jin and N. Xu, Angew. Chem., Int. Ed., 2016, 55, 13384–13397 CrossRef CAS PubMed.
- M. I. Walker, K. Ubych, V. Saraswat, E. A. Chalklen, P. Braeuninger-Weimer, S. Caneva, R. S. Weatherup, S. Hofmann and U. F. Keyser, ACS Nano, 2017, 11, 1340–1346 CrossRef CAS PubMed.
- N. Wei, C. Lv and Z. Xu, Langmuir, 2014, 30, 3572–3578 CrossRef CAS PubMed.
- N. Wei, X. Peng and Z. Xu, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 12113 CrossRef PubMed.
- Y. Kang, Z. Zhang, H. Shi, J. Zhang, L. Liang, Q. Wang, H. Ågren and Y. Tu, Nanoscale, 2014, 6, 10666–10672 RSC.
- D. Jang, J. C. Idrobo, T. Laoui and R. Karnik, ACS Nano, 2017, 11, 10042–10052 CrossRef CAS PubMed.
- Y. G. Yan, W. S. Wang, W. Li, K. P. Loh and J. Zhang, Nanoscale, 2017, 9, 18951–18958 RSC.
- Y. Han, Y. Jiang and C. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 8147–8155 CrossRef CAS PubMed.
- L. Qiu, X. Zhang, W. Yang, Y. Wang, G. P. Simon and D. Li, Chem. Commun., 2011, 47, 5810–5812 RSC.
- H. Huang, Z. Song, N. Wei, L. Shi, Y. Mao, Y. Ying, L. Sun, Z. Xu and X. Peng, Nat. Commun., 2013, 4, 2979 CrossRef PubMed.
- G. Shi, Y. Ding and H. Fang, J. Comput. Chem., 2012, 33, 1328–1337 CrossRef CAS PubMed.
- G. Shi, J. Liu, C. Wang, B. Song, Y. Tu, J. Hu and H. Fang, Sci. Rep., 2013, 3, 3436 CrossRef PubMed.
- W. Peng, H. Li, Y. Liu and S. Song, J. Mol. Liq., 2017, 230, 496–504 CrossRef CAS.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Dalton Trans., 2013, 42, 5682–5689 RSC.
- J. Xu, Z. Cao, Y. Zhang, Z. Yuan, Z. Lou, X. Xu and X. Wang, Chemosphere, 2018, 195, 351–364 CrossRef CAS PubMed.
- B. Radha, A. Esfandiar, F. C. Wang, A. P. Rooney, K. Gopinadhan, A. Keerthi, A. Mishchenko, A. Janardanan, P. Blake, L. Fumagalli, M. Lozada-Hidalgo, S. Garaj, S. J. Haigh, I. V. Grigorieva, H. A. Wu and A. K. Geim, Nature, 2016, 538, 222–225 CrossRef CAS PubMed.
- N. Wei, X. Peng and Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 5877–5883 CrossRef CAS PubMed.
- S. Zheng, Q. Tu, J. J. Urban, S. Li and B. Mi, ACS Nano, 2017, 11, 6440–6445 CrossRef CAS PubMed.
- S. Kim, R. Ou, Y. Hu, X. Li, H. Zhang, G. P. Simon and H. Wang, J. Membr. Sci., 2018, 562, 47–55 CrossRef CAS.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T. Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K. Geim and R. R. Nair, Nat. Nanotechnol., 2017, 12, 546–550 CrossRef CAS PubMed.
- B. Mi, Science, 2014, 343, 740–742 CrossRef CAS PubMed.
- W. Kim and S. Nair, Chem. Eng. Sci., 2013, 104, 908–924 CrossRef CAS.
- I. V. Vlassiouk, Nat. Nanotechnol., 2017, 12, 1022 CrossRef CAS PubMed.
- A. Morelos-Gomez, R. Cruz-Silva, H. Muramatsu, J. Ortiz-Medina, T. Araki, T. Fukuyo, S. Tejima, K. Takeuchi, T. Hayashi, M. Terrones and M. Endo, Nat. Nanotechnol., 2017, 12, 1083 CrossRef CAS PubMed.
- F. Bonaccorso, A. Lombardo, T. Hasan, Z. Sun, L. Colombo and A. C. Ferrari, Mater. Today, 2012, 15, 564–589 CrossRef CAS.
- A. Amiri, M. Naraghi, G. Ahmadi, M. Soleymaniha and M. Shanbedi, FlatChem, 2018, 8, 40–71 CrossRef CAS.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun’Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 Search PubMed.
- U. Khan, A. O’Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- C.-J. Shih, A. Vijayaraghavan, R. Krishnan, R. Sharma, J.-H. Han, M.-H. Ham, Z. Jin, S. Lin, G. L. C. Paulus, N. F. Reuel, Q. H. Wang, D. Blankschtein and M. S. Strano, Nat. Nanotechnol., 2011, 6, 439 CrossRef CAS PubMed.
- T. Hasan, F. Torrisi, Z. Sun, D. Popa, V. Nicolosi, G. Privitera, F. Bonaccorso and A. C. Ferrari, Phys. Status Solidi, 2010, 247, 2953–2957 CrossRef CAS.
- L. Najafi, S. Bellani, Re. Oropesa-Nuñez, B. Martín-García, M. Prato, V. Mazánek, D. Debellis, S. Lauciello, R. Brescia, Z. Sofer and F. Bonaccorso, J. Mater. Chem. A, 2019, 7, 25593–25608 RSC.
- W. Zhao, M. Fang, F. Wu, H. Wu, L. Wang and G. Chen, J. Mater. Chem., 2010, 20, 5817–5819 RSC.
- A. E. Del Rio-Castillo, C. Merino, E. Díez-Barra and E. Vázquez, Nano Res., 2014, 7, 963–972 CrossRef.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O’Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O’Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed.
- A. E. Del Rio Castillo, A. Ansaldo, V. Pellegrini and F. Bonaccorso, WO2017089987A1, 2017.
- A. E. Del Rio Castillo, V. Pellegrini, A. Ansaldo, F. Ricciardella, H. Sun, L. Marasco, J. Buha, Z. Dang, L. Gagliani, E. Lago, N. Curreli, S. Gentiluomo, F. Palazon, M. Prato, R. Oropesa-Nuñez, P. S. Toth, E. Mantero, M. Crugliano, A. Gamucci, A. Tomadin, M. Polini and F. Bonaccorso, Mater. Horiz., 2018, 5, 890–904 RSC.
- S. Bellani, F. Wang, G. Longoni, L. Najafi, R. Oropesa-Nuñez, A. E. Del Rio Castillo, M. Prato, X. Zhuang, V. Pellegrini, X. Feng and F. Bonaccorso, Nano Lett., 2018, 18, 7155–7164 CrossRef CAS PubMed.
- F. Bonaccorso, A. Bortolotta, J. N. Coleman and C. Backes, Adv. Mater., 2016, 28, 6136–6166 CrossRef CAS PubMed.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, A. Ansaldo, M. Prato, A. E. Del Rio Castillo and F. Bonaccorso, Adv. Energy Mater., 2018, 8, 1703212 CrossRef.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, A. Ansaldo, M. Prato, A. E. Del Rio Castillo and F. Bonaccorso, Adv. Energy Mater., 2018, 8, 1801764 CrossRef.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, B. Martín-García, M. Prato and F. Bonaccorso, ACS Appl. Energy Mater., 2019, 2, 5373–5379 CrossRef CAS.
- S. Bellani, B. Martín-García, R. Oropesa-Nuñez, V. Romano, L. Najafi, C. Demirci, M. Prato, A. E. Del Rio Castillo, L. Marasco, E. Mantero, G. D’Angelo and F. Bonaccorso, Nanoscale Horiz., 2019, 4, 1077–1091 RSC.
- L. Carbone, A. E. Del Rio Castillo, J. K. Panda, G. Pugliese, A. Scarpellini, F. Bonaccorso and V. Pellegrini, ChemSusChem, 2020, 13, 1593–1602 CrossRef CAS PubMed.
- V. Romano, B. Martín-García, S. Bellani, L. Marasco, J. Kumar Panda, R. Oropesa-Nuñez, L. Najafi, A. E. Del Rio Castillo, M. Prato, E. Mantero, V. Pellegrini, G. D’Angelo and F. Bonaccorso, ChemPlusChem, 2019, 84, 882–892 CrossRef CAS PubMed.
- W. Yu, T. (Yet) Yu and N. Graham, 2D Mater., 2017, 4, 45006 CrossRef.
- D. An, L. Yang, T.-J. Wang and B. Liu, Ind. Eng. Chem. Res., 2016, 55, 4803–4810 CrossRef CAS.
- P. Vandezande, L. E. M. Gevers and I. F. J. Vankelecom, Chem. Soc. Rev., 2008, 37, 365–405 RSC.
- P. Sun, F. Zheng, M. Zhu, Z. Song, K. Wang, M. Zhong, D. Wu, R. B. Little, Z. Xu and H. Zhu, ACS Nano, 2014, 8, 850–859 CrossRef CAS PubMed.
- P. Sun, R. Ma, H. Deng, Z. Song, Z. Zhen, K. Wang, T. Sasaki, Z. Xu and H. Zhu, Chem. Sci., 2016, 7, 6988–6994 RSC.
- A. Striolo, A. Michaelides and L. Joly, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 533–556 CrossRef CAS PubMed.
- I. Vlassiouk, S. Smirnov and Z. Siwy, Nano Lett., 2008, 8, 1978–1985 CrossRef CAS PubMed.
- Y. X. Shen, W. C. Song, D. Ryan Barden, T. Ren, C. Lang, H. Feroz, C. B. Henderson, P. O. Saboe, D. Tsai, H. Yan, P. J. Butler, G. C. Bazan, W. A. Phillip, R. J. Hickey, P. S. Cremer, H. Vashisth and M. Kumar, Nat. Commun., 2018, 9, 2294 CrossRef PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- R. Mas-Ballesté, C. Gómez-Navarro, J. Gómez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 14 CrossRef CAS.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- A. Gupta, T. Sakthivel and S. Seal, Prog. Mater. Sci., 2015, 73, 44–126 CrossRef CAS.
- H. Huang, K. Schwab and J. G. Jacangelo, Environ. Sci. Technol., 2009, 43, 3011–3019 CrossRef CAS PubMed.
- S. S. Gunderson, W. S. Brower, J. L. O’Dell and E. N. Lightfoot, Sep. Sci. Technol., 2007, 42, 2121–2142 CrossRef CAS.
- W. Guo, H.-H. Ngo and J. Li, Bioresour. Technol., 2012, 122, 27–34 CrossRef CAS PubMed.
- P. S. Goh, W. J. Lau, M. H. D. Othman and A. F. Ismail, Desalination, 2018, 425, 130–155 CrossRef CAS.
- A. Grenier, M. Meireles, P. Aimar and P. Carvin, Chem. Eng. Res. Des., 2008, 86, 1281–1293 CrossRef CAS.
- Y. Bessiere, N. Abidine and P. Bacchin, J. Membr. Sci., 2005, 264, 37–47 CrossRef CAS.
- A. Abdelrasoul, H. Doan and A. Lohi, Fouling in Membrane Filtration and Remediation Methods, in Mass Transfer – Advances in Sustainable Energy and Environment Oriented Numerical Modeling, ed. H. Nakajima, InTech, Rijeka, Croatia, 2013 Search PubMed.
- X. Cui and K.-H. Choo, Environ. Eng. Res., 2014, 19, 1–8 CrossRef.
- K. Farahbakhsh, C. Svrcek, R. K. Guest and D. W. Smith, J. Environ. Eng. Sci., 2004, 3, 237–253 CrossRef CAS.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- Handb. Fluoresc. Dye. Probes, 2015.
- M. McNutt, Science, 2013, 341, 1430 CrossRef CAS PubMed.
- N. Lubick and D. Malakoff, Science, 2013, 341, 1443–1445 CrossRef CAS PubMed.
- L. D. Hylander and M. Meili, Crit. Rev. Environ. Sci. Technol., 2005, 35, 1–36 CrossRef CAS.
- E. G. Pacyna, J. M. Pacyna, F. Steenhuisen and S. Wilson, Atmos. Environ., 2006, 40, 4048–4063 CrossRef CAS.
- J. M. Pacyna, K. Sundseth, E. G. Pacyna, W. Jozewicz, J. Munthe, M. Belhaj and S. Aström, J. Air Waste Manage. Assoc., 2010, 60, 302–315 CrossRef CAS PubMed.
- L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2018, 24, 6905–6916 CrossRef CAS PubMed.
- A. G. Bravo, C. Cosio, D. Amouroux, J. Zopfi, P.-A. Chevalley, J. E. Spangenberg, V.-G. Ungureanu and J. Dominik, Water Res., 2014, 49, 391–405 CrossRef CAS PubMed.
- J. M. Esbrí, M. A. López-Berdonces, S. Fernández-Calderón, P. Higueras and S. Díez, Environ. Sci. Pollut. Res., 2015, 22, 4842–4850 CrossRef PubMed.
- Y. Zheng, A. D. Jensen, C. Windelin and F. Jensen, Prog. Energy Combust. Sci., 2012, 38, 599–629 CrossRef CAS.
- J. H. Pavlish, E. A. Sondreal, M. D. Mann, E. S. Olson, K. C. Galbreath, D. L. Laudal and S. A. Benson, Fuel Process. Technol., 2003, 82, 89–165 CrossRef CAS.
- S. X. Wang, L. Zhang, G. H. Li, Y. Wu, J. M. Hao, N. Pirrone, F. Sprovieri and M. P. Ancora, Atmos. Chem. Phys., 2010, 10, 1183–1192 CrossRef CAS.
- S. Tang, L. Wang, X. Feng, Z. Feng, R. Li, H. Fan and K. Li, Fuel, 2016, 180, 194–204 CrossRef CAS.
- S. B. T. Sany, A. Salleh, A. H. Sulaiman, A. Sasekumar, M. Rezayi and G. M. Tehrani, Environ. Earth Sci., 2013, 69, 2013–2025 CrossRef CAS.
- A. Ranjbar Jafarabadi, A. Riyahi Bakhtiyari, A. Shadmehri Toosi and C. Jadot, Chemosphere, 2017, 185, 1090–1111 CrossRef CAS PubMed.
- S. Abdollahi, Z. Raoufi, I. Faghiri, A. Savari, Y. Nikpour and A. Mansouri, Mar. Pollut. Bull., 2013, 71, 336–345 CrossRef CAS PubMed.
- P. Ammann, J. Power Sources, 1995, 57, 41–44 CrossRef CAS.
- M. Asari, K. Fukui and S. Sakai, Sci. Total Environ, 2008, 393, 1–10 CrossRef CAS PubMed.
- J. Nriagu and C. Becker, Sci. Total Environ, 2003, 304, 3–12 CrossRef CAS.
- S. Covelli, J. Faganeli, M. Horvat and A. Brambati, Appl. Geochem., 2001, 16, 541–558 CrossRef CAS.
- H. Edner, P. Ragnarson, S. Svanberg, E. Wallinder, R. Ferrara, B. E. Maserti and R. Bargagli, Sci. Total Environ, 1993, 133, 1–15 CrossRef CAS.
- A. Biswas, J. D. Blum, B. A. Bergquist, G. J. Keeler and Z. Xie, Environ. Sci. Technol., 2008, 42, 8303–8309 CrossRef CAS PubMed.
- B. Toole-O’Neil, S. J. Tewalt, R. B. Finkelman and D. J. Akers, Fuel, 1999, 78, 47–54 CrossRef.
- N. Pirrone, P. Costa, J. M. Pacyna and R. Ferrara, Atmos. Environ., 2001, 35, 2997–3006 CrossRef CAS.
- N. Pirrone, S. Cinnirella, X. Feng, R. B. Finkelman, H. R. Friedli, J. Leaner, R. Mason, A. B. Mukherjee, G. Stracher, D. G. Streets and K. Telmer, Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements and Models, 2009 Search PubMed.
- D. Malakoff, Science, 2013, 341, 1442–1443 CrossRef CAS PubMed.
- N. E. Selin, Environ. Toxicol. Chem., 2013, 33, 1202–1210 CrossRef.
- H. Selin, Glob. Environ. Polit., 2014, 14, 1–19 CrossRef.
- D. A. Atwood and M. K. Zaman, Mercury Removal from Water, in Recent Developments in Mercury Science, ed. D. A. Atwood, Structure and Bonding, Springer, Berlin, Heidelberg, 2006, vol. 120, pp. 163–182 Search PubMed.
- Q. Wang, D. Kim, D. D. Dionysiou, G. A. Sorial and D. Timberlake, Environ. Pollut., 2004, 131, 323–336 CrossRef CAS PubMed.
- A. Shafeeq, A. Muhammad, W. Sarfraz, A. Toqeer, S. Rashid and M. K. Rafiq, Int. Sch. Sci. Res. Innov., 2012, 6, 12–26 Search PubMed.
- A. Azimi, A. Azari, M. Rezakazemi and M. Ansarpour, ChemBioEng Rev., 2017, 4, 37–59 CrossRef CAS.
- F. M. M. Morel, A. M. L. Kraepiel and M. Amyot, Annu. Rev. Ecol. Syst., 1998, 29, 543–566 CrossRef.
- R. P. Mason, W. F. Fitzgerald and F. M. M. Morel, Geochim. Cosmochim. Acta, 1994, 58, 3191–3198 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- E. Sumesh, M. S. Bootharaju, Anshup and T. Pradeep, J. Hazard. Mater., 2011, 189, 450–457 CrossRef CAS PubMed.
- Z. Wang, M. Yang, C. Chen, L. Zhang and H. Zeng, Sci. Rep., 2016, 6, 29611 CrossRef CAS PubMed.
- H.-Y. Chang, T.-M. Hsiung, Y.-F. Huang and C.-C. Huang, Environ. Sci. Technol., 2011, 45, 1534–1539 CrossRef CAS PubMed.
- X. Ding, L. Kong, J. Wang, F. Fang, D. Li and J. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 7072–7078 CrossRef CAS.
- J. F. Zhang and J. S. Kim, Anal. Sci., 2009, 25, 1271–1281 CrossRef CAS PubMed.
- L. Najafi, S. Bellani, B. Martín-García, R. Oropesa-Nuñez, A. E. Del Rio Castillo, M. Prato, I. Moreels and F. Bonaccorso, Chem. Mater., 2017, 29, 5782–5786 CrossRef CAS.
- A. Ansaldo, P. Bondavalli, S. Bellani, A. E. Del Rio Castillo, M. Prato, V. Pellegrini, G. Pognon and F. Bonaccorso, ChemNanoMat, 2017, 3, 436–446 CrossRef CAS.
- S. Bellani, L. Najafi, A. Capasso, A. E. Del Rio Castillo, M. R. Antognazza and F. Bonaccorso, J. Mater. Chem. A, 2017, 5, 4384–4396 RSC.
- L. Najafi, B. Taheri, B. Martín-García, S. Bellani, D. Di Girolamo, A. Agresti, R. Oropesa-Nuñez, S. Pescetelli, L. Vesce, E. Calabrò, M. Prato, A. E. Del Rio Castillo, A. Di Carlo and F. Bonaccorso, ACS Nano, 2018, 12, 10736–10754 CrossRef CAS PubMed.
- A. Sze, D. Erickson, L. Ren and D. Li, J. Colloid Interface Sci., 2003, 261, 402–410 CrossRef CAS.
- JPK Instruments AG, JPK Instrument Technical Report: QITM Mode-Quantitative Imaging with the NanoWizard® 3 AFM.
- K. Zhang, S. Yu, B. Jv and W. Zheng, Phys. Chem. Chem. Phys., 2016, 18, 28418–28427 RSC.
- X. Wang, S. Zhong, Y. He and G. Song, Anal. Methods, 2012, 4, 360–362 RSC.
- K. Fan, Z. Guo, Z. Geng, J. Ge, S. Jiang, J. Hu and Q. Zhang, Chin. J. Chem. Phys., 2013, 26, 252–258 CrossRef CAS.
- H. Ren, D. D. Kulkarni, R. Kodiyath, W. Xu, I. Choi and V. V. Tsukruk, ACS Appl. Mater. Interfaces, 2014, 6, 2459–2470 CrossRef CAS PubMed.
- X. Zhou, S. He, K. A. Brown, J. Mendez-Arroyo, F. Boey and C. A. Mirkin, Nano Lett., 2013, 13, 1616–1621 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2013, 42, 3222–3233 RSC.
- R. L. D. Whitby, A. Korobeinyk, V. M. Gun’Ko, R. Busquets, A. B. Cundy, K. László, J. Skubiszewska-Ziba, R. Leboda, E. Tombacz, I. Y. Toth, K. Kovacs and S. V. Mikhalovsky, Chem. Commun., 2011, 47, 9645–9647 RSC.
- L. Wu, L. Liu, B. Gao, R. Muñoz-Carpena, M. Zhang, H. Chen, Z. Zhou and H. Wang, Langmuir, 2013, 29, 15174–15181 CrossRef CAS PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- J. Balapanuru, J.-X. Yang, S. Xiao, Q. Bao, M. Jahan, L. Polavarapu, J. Wei, Q.-H. Xu and K. P. Loh, Angew. Chem., Int. Ed., 2010, 49, 6549–6553 CrossRef CAS PubMed.
- T. L. Doane, C. H. Chuang, R. J. Hill and C. Burda, Acc. Chem. Res., 2012, 45, 317–326 CrossRef CAS PubMed.
- D. W. Johnson, B. P. Dobson and K. S. Coleman, Curr. Opin. Colloid Interface Sci., 2015, 20, 367–382 CrossRef CAS.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235 CrossRef CAS PubMed.
- C. Thomsen and S. Reich, Phys. Rev. Lett., 2000, 85, 5214 CrossRef CAS PubMed.
- L. Yang, J. Deslippe, C.-H. Park, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2009, 103, 186802 CrossRef PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 75414 CrossRef.
- C. Y. Su, Y. Xu, W. Zhang, J. Zhao, X. Tang, C. H. Tsai and L. J. Li, Chem. Mater., 2009, 21, 5674–5680 CrossRef CAS.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- M. M. Lucchese, F. Stavale, E. H. M. Ferreira, C. Vilani, M. V. O. Moutinho, R. B. Capaz, C. A. Achete and A. Jorio, Carbon N. Y., 2010, 48, 1592–1597 CrossRef CAS.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- M. S. Dresselhaus, G. Dresselhaus and M. Hofmann, Philos. Trans. R. Soc., A, 2008, 366, 231–236 CrossRef CAS PubMed.
- D. Nuvoli, L. Valentini, V. Alzari, S. Scognamillo, S. B. Bon, M. Piccinini, J. Illescas and A. Mariani, J. Mater. Chem., 2011, 21, 3428–3431 RSC.
- F. Bonaccorso, P.-H. Tan and A. C. Ferrari, ACS Nano, 2013, 7, 1838–1844 CrossRef CAS PubMed.
- L. Jensen and G. C. Schatz, J. Phys. Chem. A, 2006, 110, 5973–5977 CrossRef CAS PubMed.
- Y. Lu, G. L. Liu and L. P. Lee, Nano Lett., 2005, 5, 5–9 CrossRef CAS PubMed.
- R. Lu, A. Konzelmann, F. Xu, Y. Gong, J. Liu, Q. Liu, M. Xin, R. Hui and J. Z. Wu, Carbon N. Y., 2015, 86, 78–85 CrossRef CAS.
- J. A. Dieringer, K. L. Wustholz, D. J. Masiello, J. P. Camden, S. L. Kleinman, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2009, 131, 849–854 CrossRef CAS PubMed.
- N. H. Kim and K. Kim, J. Raman Spectrosc., 2005, 36, 623–628 CrossRef CAS.
- A. Siokou, F. Ravani, S. Karakalos, O. Frank, M. Kalbac and C. Galiotis, Appl. Surf. Sci., 2011, 257, 9785–9790 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon N. Y., 2007, 45, 1558–1565 CrossRef CAS.
- J. Díaz, G. Paolicelli, S. Ferrer and F. Comin, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 8064–8069 CrossRef PubMed.
- J. C. Lascovich, R. Giorgi and S. Scaglione, Appl. Surf. Sci., 1991, 47, 17–21 CrossRef CAS.
- G. Salitra, A. Soffer, L. Eliad, Y. Cohen and D. Aurbach, J. Electrochem. Soc., 2000, 147, 2486–2493 CrossRef CAS.
- R. Buzio, A. Gerbi, S. Uttiya, C. Bernini, A. E. Del Rio Castillo, F. Palazon, A. S. Siri, V. Pellegrini, L. Pellegrino and F. Bonaccorso, Nanoscale, 2017, 9, 7612–7624 RSC.
- H. Sun, A. E. Del Rio Castillo, S. Monaco, A. Capasso, A. Ansaldo, M. Prato, D. A. Dinh, V. Pellegrini, B. Scrosati, L. Manna and F. Bonaccorso, J. Mater. Chem. A, 2016, 4, 6886–6895 RSC.
- E. S. Thrall, A. C. Crowther, Z. Yu and L. E. Brus, Nano Lett., 2012, 12, 1571–1577 CrossRef CAS PubMed.
- S. Sil, N. Kuhar, S. Acharya and S. Umapathy, Sci. Rep., 2013, 3, 3336 CrossRef PubMed.
- S. Bellani, E. Petroni, A. E. Del Rio Castillo, N. Curreli, B. Martín-García, R. Oropesa-Nuñez, M. Prato and F. Bonaccorso, Adv. Funct. Mater., 2019, 29, 1807659 CrossRef.
- C. A. Amadei, A. Montessori, J. P. Kadow, S. Succi and C. D. Vecitis, Environ. Sci. Technol., 2017, 51, 4280–4288 CrossRef CAS PubMed.
- N. F. Martínez and R. García, Nanotechnology, 2006, 17, S167 CrossRef.
- H.-J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- J. E. Lennard-Jones, Proc. Phys. Soc., 1931, 43, 461 CrossRef CAS.
- N. Yu and A. A. Polycarpou, J. Colloid Interface Sci., 2004, 278, 428–435 CrossRef CAS PubMed.
- N. Hilal, H. Al-Zoubi, N. A. Darwish, A. W. Mohamma and M. Abu Arabi, Desalination, 2004, 170, 281–308 CrossRef CAS.
- D. L. Sedin and K. L. Rowlen, Anal. Chem., 2000, 72, 2183–2189 CrossRef CAS.
- M. Farshchi-Tabrizia, M. Kappl and H.-J. Butt, J. Adhes. Sci. Technol., 2008, 22, 181–203 CrossRef.
- J. Melcher, C. Carrasco, X. Xu, J. L. Carrascosa, J. Gómez-Herrero, P. José de Pablo and A. Raman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13655–13660 CrossRef CAS.
- M. Götzinger and W. Peukert, Langmuir, 2004, 20, 5298–5303 CrossRef PubMed.
- E. W. van der Vegte and G. Hadziioannou, Langmuir, 1997, 13, 4357–4368 CrossRef CAS.
- C. D. Frisbie, L. F. Rozsnyai, A. Noy, M. S. Wrighton and C. M. Lieber, Science, 1994, 265, 2071–2074 CrossRef CAS PubMed.
- J. Zlatanova, S. M. Lindsay and S. H. Leuba, Prog. Biophys. Mol. Biol., 2000, 74, 37–61 CrossRef CAS.
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS PubMed.
- S. P. Surwade, S. N. Smirnov, I. V. Vlassiouk, R. R. Unocic, G. M. Veith, S. Dai and S. M. Mahurin, Nat. Nanotechnol., 2015, 10, 459 CrossRef CAS PubMed.
- B. M. Ganesh, A. M. Isloor and A. F. Ismail, Desalination, 2013, 313, 199–207 CrossRef CAS.
- X. Li and J. Li, in Ecyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 1–3 Search PubMed.
- J. Y. Chong, B. Wang and K. Li, Chem. Commun., 2018, 54, 2554–2557 RSC.
- H. Huang, Y. Mao, Y. Ying, Y. Liu, L. Sun and X. Peng, Chem. Commun., 2013, 49, 5963–5965 RSC.
- D. W. Kim, J. Choi, D. Kim and H.-T. Jung, J. Mater. Chem. A, 2016, 4, 17773–17781 RSC.
- L. Sun, H. Huang and X. Peng, Chem. Commun., 2013, 49, 10718–10720 RSC.
- L. Sun, Y. Ying, H. Huang, Z. Song, Y. Mao, Z. Xu and X. Peng, ACS Nano, 2014, 8, 6304–6311 CrossRef CAS PubMed.
- Z. Wang, C. Dong, M. Wang, Y. Chen and G. Wu, Sep. Sci. Technol., 2014, 49, 2721–2727 CrossRef CAS.
- M. Wang, Z. Wang, G. Wu, Y. Zhang and A. Wu, Environ. Chem., 2016, 35, 540–547 CAS.
- J. K. Holt, H. G. Park, Y. Wang, M. Stadermann, A. B. Artyukhin, C. P. Grigoropoulos, A. Noy and O. Bakajin, Science, 2006, 312, 1034–1037 CrossRef CAS PubMed.
- M. Majumder, N. Chopra, R. Andrews and B. J. Hinds, Nature, 2005, 438, 44 CrossRef CAS PubMed.
- S. A. Dastgheib and D. A. Rockstraw, Carbon, 2002, 40, 1843–1851 CrossRef CAS.
- J. A. Dean, Lange's Handbook Of Chemistry, 15th edn, 1999 Search PubMed.
- R. M. Smith, A. E. Martell and R. J. Motekaitis, NIST Stand. Ref. Database 46, 2004.
- L. Chai, Q. Wang, Y. Wang, Q. Li, Z. Yang and Y. Shu, J. Cent. South Univ. Technol., 2010, 17, 289–294 CrossRef CAS.
- X. Wang, Z. Chen and S. Yang, J. Mol. Liq., 2015, 211, 957–964 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00060d |
| This journal is © The Royal Society of Chemistry 2020 |